

Abstract
Cyclin-dependent kinase 4/6 (CDK4/6) specific inhibitors, such as Palbociclib, have shown clinical efficacy, but primary or secondary resistance has emerged as a problem. To develop more effective therapeutic approaches, investigation is needed into the mechanisms of resistance or adaption. Here, it is demonstrated that CDK2 compensates for loss of CDK4 activity to rescue Palbociclib-arrested breast cancer cells, suggesting that inhibition of both kinases is required to achieve durable response. In addition, a novel strategy is described to inhibit tyrosine phosphorylation of p27Kip1 (CDKN1B) and simultaneously inhibit both CDK2 and CDK4. p27Kip1 is a required assembly factor for cyclin-CDK4 complexes, but it must be phosphorylated on residue Y88 to open or activate the complex. The Brk-SH3 peptide, ALT, blocks p27 Y88 phosphorylation, inhibiting CDK4. Non-phosphorylated p27 is no longer a target for ubiquitin-mediated degradation and this stabilized p27 now also inhibits CDK2 activity. Thus, ALT induction inhibits both the kinase that drives proliferation (CDK4) and the kinase that mediates resistance (CDK2), causing a potent and long-lasting cell cycle arrest. ALT arrests growth of all breast cancer subgroups and synergizes with Palbociclib to increase cellular senescence and to cause tumor regression in breast cancer xenograft models. The use of ALT demonstrates that both CDK4 and CDK2 need to be inhibited if long-term efficacy is to be achieved and represents a novel modality to inhibit breast cancer cells.
Keywords: p27 tyrosine phosphorylation, Palbociclib, cdk4, cdk2, ALT, BRK, SH3 domain, cell cycle, breast cancer, drug resistance
Introduction
The G1-S phase cell cycle transition is governed by two cyclin-cdk complexes, cyclin D-cdk4/6 and cyclin E-cdk2. Cyclin D-cdk4 (hereafter D-K4) phosphorylates the G1 gatekeeper Rb, causing the release of S-phase specific transcription factors, such as E2F. E2F causes the transcriptional induction of Cyclin E, which in turn partners with cdk2 to further phosphorylate Rb and irreversibly cause the transition into S-phase (1). Cyclin D1 and cdk4 are overexpressed in a variety of human cancers, and, in mouse models, loss of either prevents the development of certain oncogene-driven tumors (2). Targeting cdk4 activity has been a long-standing goal in the oncology field and, because D-K4 is downstream of most oncogenic signaling pathways, targeting this kinase might prevent the resistance that frequently occurs when cell surface or upstream signal transducers are inhibited. The advent of cdk4 specific inhibitors (cdk4i), such as Palbociclib (PD 0332991, hereafter PD), Abemaciclib, or Ribociclib has demonstrated that cdk4 is a promising target (3). In combination with Letrozole, PD extended median Progression Free Survival (PFS) for metastatic breast cancer patients from 10.2 to 20.2 months (PALOMA trial) (4). However, the overall survival (OS) of patients treated with PD mirrored that seen in patients treated with Letrozole alone, suggesting that resistance to this combination therapy occurs In tissue culture lines, PD- or Ribociclib-mediated arrest did not appear durable either (5-8). While loss of Rb appears to distinguish primary PD-non-responsiveness in cell lines, differences in Ki67, cyclin D, cdk4, and p16 do not appear able to stratify responsive and non-responsive subgroups (9,10).
Cyclin D is a transcriptional target of the MAPK pathway, but after cyclin D partners with cdk4, the dimer is unstable, and rapidly dissociates back into the monomeric forms, unless a third protein, p27Kip1 or p21Cip1, holds the complex together (11). However, p27 binds to D-K4 in two different conformations: a closed and inactivating conformation OR alternatively, an open and activating form. This transition is mediated by the tyrosine (Y) phosphorylation of p27 on residues Y88 (or Y89), which causes a conformational change, opening the D-K4-p27 ternary complex, thus rendering it able to phosphorylate substrates such as Rb (12,13). Non-phosphorylated p27-D-K4 complexes are catalytically inactive because the associated p27 both blocks the ATP binding site on cdk4 and prevents the required Cdk Activating Kinase (CAK) phosphorylation of the cdk4 domain itself (14). p27 Y88 is phosphorylated by the Y kinase Brk (Breast tumor Related Kinase or PTK6, Protein Tyrosine Kinase 6), and interaction between Brk and p27 is mediated though Brk’s SH3 domain and a proline-rich binding site in p27. Addition of Brk SH3-containing peptides in vitro blocks this interaction, preventing p27 Y88 phosphorylation, which in turn causes inhibition of cdk4. Overexpression of a naturally occurring ALTternatively-spliced form of Brk (ALT), which contains Brk’s SH3 domain, but lacks the SH1 kinase domain, also inhibits Brk’s phosphorylation of p27, inhibits cdk4, and causes growth arrest, suggesting that inhibition of p27 Y88 phosphorylation might be an alternative way to target cdk4-dependent tumors (15,16).
In contrast to cdk4, cdk2 does not require p27 to stabilize the interaction with its cyclin; actually cdk2’s phosphorylation of RB is inhibited whenever p27, phosphorylated or not phosphorylated, is associated with the complex. But, even when unable to phosphorylate RB, Y-phosphorylated p27-cyclin E-cdk2 complexes are able to phosphorylate p27 on residue T187, which in vivo, results in decreased p27 stability as it becomes a target for ubiquitin-mediated degradation, reducing p27 association with cdk2, and indirectly activating the cyclin E-cdk2 complex (17). Thus, blocking pY88 might have the added benefit of preventing p27 degradation and stabilizing p27 in the non-phosphorylated form, permitting it to inhibit cdk2 as well as cdk4. The root of resistance to cdk4 inhibiting therapies, such as PD, is unknown, but one candidate that could compensate for loss of cdk4 activity is cdk2, so a therapy that inhibits both kinases at the outset might offer therapeutic advantages.
We sought to examine the mechanism of resistance to PD and to determine whether targeting p27 pY88 would generate a more robust arrest by blocking cdk4 and by inhibiting cdk2 as well. We found that by targeting p27 with ALT, both cdk4 and cdk2 were inhibited, resulting in durable arrest, increased senescence, and tumor regression in animal models. We also found that p27 pY88 directly correlated with cdk4 activity in cell lines, suggesting that this might be a surrogate marker of PD sensitivity that could be used to predict drug response.
Materials and Methods
Antibodies and reagents
Antibodies against Cdk2 (sc-163), Cdk2 Thr 160 (sc-101656), Cdk6 (sc-53638), Cyclin E (sc-481), Cdk4 (sc-260), Rb (sc-102), Rb S780 (sc-12901-R) and Rb S807/811 (sc-16670-R): Santa Cruz Biotechnology. Anti Flag (A2220) and Actin (A2066): Sigma Aldrich. Cyclin D1 (DCS-11), Ki67 (PA1-21520): ThermoFisher Scientific. p27kip1(610241): BD Biosciences. Cleaved Caspase-3 (9661S): Cell Signaling Technology. β-Galactosidase (ab203749): Abcam. The p27 pY88 antibody and its specificity for p27 pY88 was previously described (16). K2i (sc-221409): Santa Cruz Biotechnology. Skp2i C1 (4817): R&D Systems. Palbociclib (PD 0332991) (HY-50767A), Letrozole (HY-14248): MedChem Express. Brki (531000): EMD Millipore. NMS (normal mouse serum) (Invitrogen, 31880) and NRS (normal rabbit serum) (Invitrogen, 31883) were used as isotype controls for monoclonal or polyclonal antibodies respectively.
Cell Culture
MCF7 (ATCC, HTB-22, ER/PR+, Her2-) and MDA MB231 (ATCC, HTB-26, ER/PR-, Her2-) cell lines were cultured at low passage numbers in MEM-EBSS with 10% Fetal Bovine Serum, 1% Non-Essential Amino Acids, 10μg/ml Insulin solution (Sigma Aldrich, I9278) and Penicillin-Streptomycin. T47D (ATCC, HTB-133, ER/PR+, Her2-) and HCC1954 (CRL 2338; ER/PR-, Her2+) were cultured in RPMI 1640 with 10% Fetal Bovine Serum, 1% Non-Essential Amino Acids, 10μg/ml Insulin solution (Sigma Aldrich, I9278) and Penicillin-Streptomycin. Generation of Flag- tagged ALT Doxycycline-inducible lines were generated as per manufacturer’s protocol (Clontech, 631188). TtA (no GOI) cells expressed the Tetracycline transactivator in the absence of Gene of Interest and served a negative control.
Cell proliferation assays
Cells were treated with DMSO or drug combinations in 6-well plates. In all cases, drugs (ie. PD, K2i, Dox) were replenished every 2 d. Cell proliferation and viability were measured by visual counting in a Hemocytometer and by counting with 0.1% Trypan Blue stain, respectively. In order to determine IC50 values, cells were treated with increasing concentrations of PD or Dox. Cell counts were determined to get a dose response curve for each cell line and IC50 values were calculated using GraphPad Prism software. All proliferation experiments were N≥3.
In vitro kinase assays
Performed as described (13,16). Densitometry of immunoblots was performed using Licor Image Studio software to determine Cdk4 activity [(Rb S780/Rb)/cdk4] and Cdk2 activity [(Rb Ser807-811/Rb)/cdk2)].
β-galactosidase staining
Cells were incubated in Fixative Solution (Cell Signaling 9860S) for 12 min. followed by incubation with β-Galactosidase antibodies overnight at 37°C. β-Gal+ cells were counted manually and %-β Gal positive cells were determined (from 100 total).
Knockdown assay
Cells were transfected with p27 siRNA lentiviral (Santa Cruz, sc-29430-V) particles or scrambled control on day 2 in presence of Hexadimethrine bromide to enhance transfection efficiency. Growth media was replenished on day 3 and on day 5, cells were harvested for immunoblot analysis with p27 antibodies to confirm KD or drugs were added. KD cells were plated on day 0, Dox or PD were added on day 2 and proliferation was measured by cell counting.
In vivo xenograft study
Animal studies have been conducted in accordance with the Institutional Animal Care and Use Committee (IACUC). 4-6 weeks old female NOD.CB17-Prkdcscid/NcrCrl mice were purchased from Charles River Breeding Laboratories, implanted with β-Estradiol pellets (SE-121, Innovative Research of America) subcutaneously, and allowed to recover for a week. 0.5×107 MCF7-ALT cells were injected subcutaneously near the 4th mammary fat pad. Tumor development was monitored using digital calipers and volume calculated as [length × (width)2]/2. When tumors reached ~200 mm3 (between 21-31 days post injection), mice were treated daily with Vehicle (50mM Sodium Lactate pH 4), 100mg/kg PD (PD) orally, 13.3mg/kg or 40mg/kg Dox dissolved in drinking water with 1% Sucrose, or the combination of PD and Dox. After day 9, all Dox-treated animals were injected with saline to try to prevent dehydration, until day 19 when the study ended. Tumors were harvested at various time points and were fixed in 4% Paraformaldehyde for 2 days followed by incubation in 70% Ethanol, followed by IHC analysis.
Statistical analysis
Quantification of all immunoblots was performed using the Image Studio Lite software (Licor). In cdk4 and cdk2 in vitro kinase assays, Cdk4 or Cdk2 activity from day 4 or 10 treated cells was normalized to that seen in day 4 or 10 DMSO treated controls, respectively. Outliers were detected using the Thompson Tau test. Mean values were plotted and error bar values were determined using standard deviation. A single factor ANOVA analysis or a two-tailed Student’s t-Test with unequal variance was performed to evaluate the significance of differences between various experimental groups. In order to determine if Dox (ALT) and PD synergize, dose response curves were generated while treating the cells with Dox, PD, or Dox+PD, and the Combination index was calculated as described (18). Cell proliferation was determined by plotting mean values of cell counts for each experimental group and normalizing to values seen in DMSO controls. Error bar values were determined using standard deviation. Significant difference in rate of cell proliferation between PD:ALT and PD:ALT+PD was determined by two tailed Student’s t-Test with unequal variance. Mean mouse tumor volume values were plotted. Error bar values were determined using standard deviation. Significance in the rate of tumor progression between PD:ALT (13.3mg/kg), PD:ALT (40mg/kg) or PD:ALT+PD (13.3mg/kg) was calculated by two tailed Student’s t-Test with unequal variance. Mouse histological analysis was performed by two independent pathologists, quantifying average staining over the entire slide by averaging the values in nonoverlapping high power fields (400×) of viable tumor.
Results
p27 pY88 phosphorylation status correlates with cdk4 activity
Finn and colleagues had stratified a panel of breast cancer cell lines for PD sensitivity and had shown that some lines responded well, with low IC50 values, while others were weak responders, with IC50 values in the 1000 nM range (19). We confirmed PD sensitivity of three representative lines, MCF7, MDA MB231, and HCC1954 corresponding to a high, moderate, or low response, respectively (Fig. 1A), and confirmed that proliferation was inhibited but viability remained constant (Fig. S1A-D). Immunoblot analysis of untreated cells (Fig. 1B) demonstrated that all three lines had similar levels of cdk4 and cyclin D1, consistent with reports that suggested that expression of these proteins was not a predictor of PD response (Quantitation in Fig. S1E) (9,10). Endogenous RB phosphorylation was measured using RB S780 phospho-specific antibodies and standardized to total RB levels (RB S780/RB) (Fig. 1C). MCF-7 cells had the highest levels of phosphorylated RB, consistent with the highest proliferation rate of the three lines, (Fig. 1D).
Figure 1. p27 pY88 correlates with cdk4 activity in breast cancer cells.

(A) Breast cancer cell lines treated with PD to determine IC50 values. (B) Representative immunoblot analysis with cell lysates from cultures without treatment. Quantitation in Fig. S1E. (C) Endogenous Rb S780/RB ratio determined by densitometry of immunoblots in B. (D) Proliferation determined by cell counting. (E, F) Cyclin D1 immunoprecipitated from cells, recombinant Rb and ATP added to perform in vitro cdk4 kinase assays followed by immunoblot analysis. Cdk4 activity [(RbS780/RB ratio)/cdk4] determined by densitometry of immunoblots in E. Activity normalized to that seen in HCC1954 cells. (G,H) p27 immunoprecipitated, followed by immunoblot analysis. pY88/p27 ratio determined by densitometry of immunoblots in H, normalized to that seen in HCC1954 cells. Since p27 levels vary in the cell lines, different amounts of lysates were used to immunoprecipitate similar levels of p27. (I) p27 immunoprecipitated from MCF7 cells +/− 400nM PD, followed by pY88 immunoblot analysis. (J) Relationship between cell proliferation, p27 Y88 phosphorylation and cdk4 activity upon PD treatment. (NRS: Normal rabbit serum. NMS: Normal mouse serum. Data: mean ± SD, All experiments, N≥3).
To directly examine cdk4/6 activity, we performed in vitro kinase assays, immunoprecipitating cyclin D1-associated complexes from lysates, followed by the addition of recombinant RB in the presence of ATP. Immunoprecipitates were subjected to SDS-PAGE electrophoresis and immunoblot analysis with RB S780, Rb and cdk4 antibodies (Fig. 1F) and densitometry allowed us to calculate the RbS780/RB ratio (Fig. 1E). Immunoprecipitates from HCC1954 cells yielded a cdk4 that phosphorylated RB ~2.5 times more than immunoprecipitates from MCF7 cells and cdk4 from MDA MB231 cells was in between. As we had immunoprecipitated a similar level of D-K4 complexes (as detected from associated cdk4 in the cyclin D1 immunoprecipitates) (Fig. 1F), this suggested that the specific activity of cdk4 in HCC1954 cells was higher than that in MCF7 cells. More cdk4 activity in HCC1954 cells would require a higher concentration of PD to achieve an IC50 response, suggesting that cdk4 activity, rather than protein levels, might be a marker of sensitivity.
Since cdk4 activity is dependent on the presence of pY88 p27 in the ternary complex, we immunoprecipitated p27, followed by immunoblot analysis with a p27 pY88 phospho-specific antibody (Fig. 1G-I) (16). Two amounts of MCF7 lysate were used to demonstrate concentration-dependent specificity of both the p27 and the pY88 antibodies (Fig. 1H). We determined the pY88/p27 ratio, and found that HCC1954 lysates had ~3X higher pY88/p27 than MCF7 cells and MDA MB231 cells had levels in between (Fig. 1G). MCF7 cells were treated with 400 nM PD (2X IC50), which completely inhibited proliferation (Fig. S1A), and pY88 was not detectable (Fig. 1I). The level of pY88/p27 decreased proportionally to PD-mediated decreases in cdk4 activity and cell proliferation, suggesting that pY88 was a surrogate marker for cdk4 activity (Fig. 1J).
Cdk2 activity compensates for PD-dependent loss of cdk4 activity in high responders
The PD high responding line, MCF7, was treated with 200 nM PD (IC50 concentration) at day 2, drug was refreshed every 2 days, and proliferation was measured by cell counting, normalizing to proliferation seen in DMSO treated cells (Fig. 2A). Proliferation was arrested by 50% by day 4, but by day 6, cells had adapted to the presence of drug and resumed proliferation (Fig. 2A, blue). When a higher concentration of PD (2X IC50) was used, the arrest was more durable, lasting until day 14, but even then cells started proliferating again in the presence of drug (Fig. 2A, red). PD treatment at either concentration did not affect viability (Fig. S2A). A similar transient arrest in the presence of PD was seen with another ER+, high responding line, T47D (Fig. S2B). Cell lysates were generated from treated MCF7 cells at different timepoints and used in immunoblot analysis (Fig. 2B). PD treatment caused loss of endogenous RB S780 phosphorylation at day 4, but it was detected again by day 6 in spite of replenishing drug. PD treatment did not significantly affect cdk4 or cyclin D1 levels (Fig. 2B). We performed cdk4 in vitro kinase assays, immunoprecipitating either cyclin D1-associated or cdk4-associated complexes from MCF7 (Fig. 2C, S2C). A 50% reduction in cdk4 activity was detected in both day 4 and 10 PD-treated lysates (Fig. 2C lanes 2,4), suggesting that PD was still inhibiting cdk4 at day 10, even when endogenous Rb S780 phosphorylation had been restored (Fig. 2B).
Figure 2. Cdk2 activity compensates for loss of cdk4 activity in PD resistant MCF7 cells.
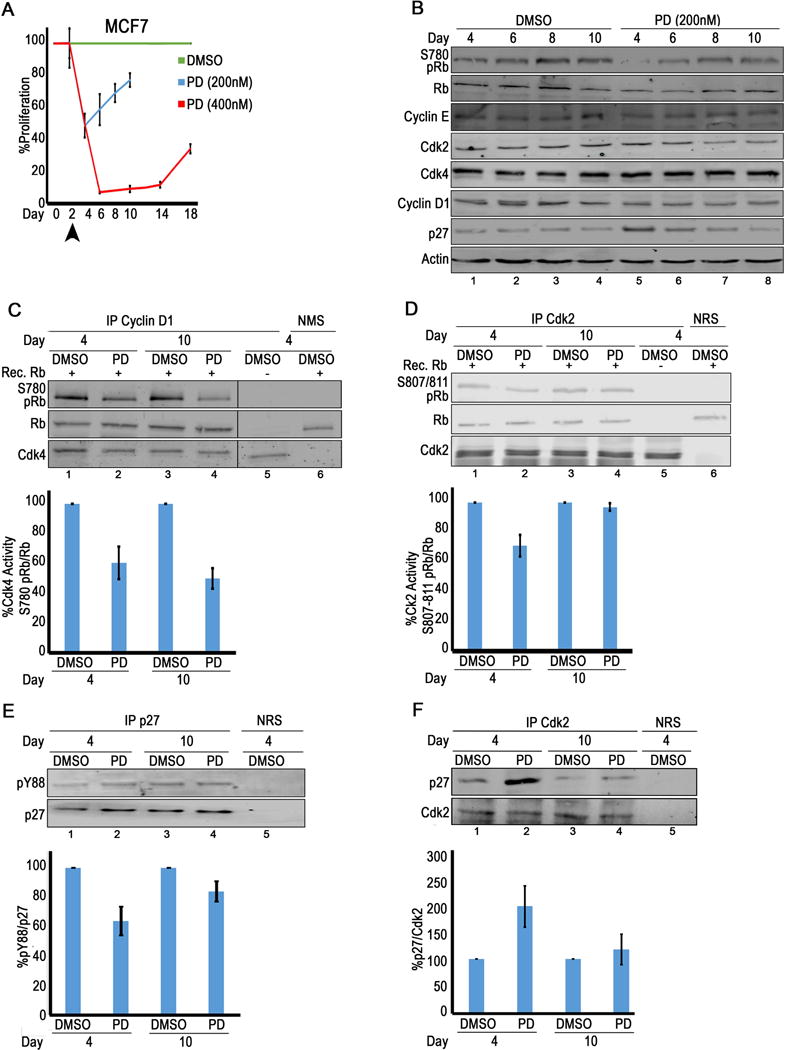
(A) Proliferation of MCF7 cells treated with DMSO or PD. Media with DMSO or drug is refreshed every two days starting at day 2 (arrowhead). (B) Immunoblot analysis with treated MCF7 lysates. (N≥3). (C) Cdk4 activity [(RbS780/RB ratio)/cdk4] determined by densitometry (bottom) of in vitro kinase assay immunoblots (top) (N=5). (D) Cdk2 activity [(RbS7807/811/RB ratio)/cdk2] determined by densitometry (bottom) of in vitro kinase assay immunoblots in (top) (N=5). (E) p27 immunoprecipitated, followed by pY88 immunoblot (top). pY88/p27 ratio determined by densitometry (bottom) (N=3). (F) Cdk2 immunoprecipitated, followed by p27 immunoblot (top). p27 association with cdk2 determined by densitometry (bottom) (N=6).
In the absence of cdk4 and cdk6, cdk2 or cdk1 can phosphorylate RB S780, suggesting that these kinases might compensate for loss of cdk4 activity in the PD-treated cells (20,21). To examine cdk2 activity, cdk2 in vitro kinase assays were performed: exogenous RB and ATP was added to cdk2 immunoprecipitates, and phosphorylation was monitored by immunoblot analysis using the RB S807,811 antibody (Fig. 2D). Densitometry was used to calculate the RBS807,811/RB ratio as a measure of cdk2 activity. While this activity was reduced in lysates from day 4 PD-treated cells relative to DMSO levels, it was increased in lysates from day 10 cells (Fig. 2D lanes 2,4). In vitro, RB phosphorylation is more ubiquitous and cdk2 can also phosphorylate RB at site S780. Similar results were seen in a cdk2 in vitro kinase assay using RbS780 antibodies (Fig. S2D, lanes 1,2 4,5), This suggests that an increase in cdk2 activity could be responsible for the increase in endogenous RB S780 phosphorylation seen in Fig. 2B (lanes 5-8) and the subsequent reentry into S phase (Fig. 2A). Cdk2 immunoprecipitates did not pull down associated cyclin D1 (Fig. S2E) as had been reported by others (5).
Immunoblot analysis demonstrated that while neither cdk2 nor cyclin E increased in the presence of PD (Fig. 2B), p27 levels increased in day 4 PD-treated lysates relative to those in DMSO lysates, but decreased from day 6-10 (Fig. 2B, lanes 5-8). We immunoprecipitated p27, followed by immunoblot analysis with pY88 antibodies (Fig. 2E), to determine the pY88/p27 ratio (Fig. 2E, bottom panel) and found that pY88/p27 decreased by ~50% in the PD-treated lysates relative to levels in DMSO treated cells at day 4, consistent with the ~50% decrease in proliferation. As cells began to proliferate again at day 10, pY88 increased as well (Fig. 2E, lanes 2,4). It has been shown that Y88 phosphorylated p27 associates with cdk2 as well, but in this context p27 becomes a substrate for cdk2-dependent phosphorylation on residue T187.p27 phosphorylation on T187 targets it for ubiquitin-mediated degradation (22) and this might explain the decrease in p27 (Fig. 2B, lanes 5-8). Cdk2 immunoprecipitates were probed by immunoblot analysis for associated p27 (Fig. 2F). While p27 association with cdk2 was increased at day 4 (Fig. 2F, lane 1,2), it’s levels stabilized by day 10 (Fig. 2F lanes 3,4). As p27 is always an inhibitor of cdk2, this data suggest that p27 reduced association at day 10 corresponds to an increase in active, inhibitor-free cdk2, which would be able to compensate for loss of cdk4 activity, phosphorylating RB and restoring proliferation.
PD inhibits both cdk4 and cdk2 in MDA MB231 to cause durable cell cycle arrest
The IC50 concentration for MDA MB231 cells is 400 nM, or ~2X the concentration needed to inhibit MCF7 cells. When either 400 nM or 800 nM PD was added to MDA MB231 cells at day 2, and replenished every 2 days, a 50% or 90% arrest was achieved respectively (Fig. 3A), while viability was unchanged (Fig. S3A). But, at both doses, this arrest was durable and persisted for 10 or 26 days, respectively (Fig. 3A). This suggested that while higher concentrations of PD were required to see response, the arrest achieved was more long-lasting than that seen in MCF7 cells.
Figure 3. Inhibition of cdk4 and cdk2 arrests proliferation durably in the moderate responder MDA MB231.

(A) Proliferation of MDA MB231 cells treated with fresh DMSO or PD every two days starting at day 2 (arrowhead). (B) Immunoblot analysis with treated lysates. (C) Cdk4 activity [(RbS780/RB ratio)/cdk4] determined by densitometry (bottom) of in vitro kinase assay immunoblots (top) (N=4). Lane 5: PD*: 0.4μM PD was exogenously added to the cdk4 immunoprecipitates from DMSO lysates while performing the kinase assay. Lane 6: no Rb added to reaction. (D) Cdk2 activity [(RbS7807/811/RB ratio)/cdk2] determined by densitometry (bottom) of in vitro kinase assay immunoblots (top) (N=4). Lane 5: no Rb added to the reaction. (E) Cdk2 immunoprecipitated, followed by p27 immunoblot (top). p27 association with cdk2 determined by densitometry (bottom). (N=3).
Lysates were generated from MDA MB231 cells treated with 400 nM PD at different time points and subjected to immunoblot analysis (Fig. 3B). Loss of endogenous Rb S780 phosphorylation was seen at days 4-10, consistent with the more durable PD arrest seen in these cells (Fig. 3B, lanes 5-8). Cdk4 and cyclin D1 levels increased in the presence of this high concentration of PD, as had been reported by others (23). Despite this increase, the IC50 concentration of PD inhibited cdk4 activity by 50% as seen in the in vitro kinase assay, and this inhibition was also seen at day 10, consistent with the durability of this response (Fig. 3C lanes 2,4). When Cdk2 activity was examined in cdk2 in vitro kinase assays, we found that PD treatment reduced cdk2 activity by ~40% at day 4 and it remained inhibited at day 10 (Fig. 3D lanes 2,4). This suggested that PD treatment was durable in MDA MB231 cells because cdk2 activity was inhibited, and was unable to compensate for the reduction in cdk4 activity.
Immunoblot analysis of MDA MB231 lysates demonstrated that neither cdk2 nor cyclin E levels changed in the presence of PD, but unlike the situation in MCF7 cells, p27 levels remained high through day 10 of treatment (Fig. 3B, lanes 5-8). p27 association with cdk2 increased in day 4 treated lysates relative to DMSO treated controls, and this association continued to increase in day 10 lysates (Fig. 3E lanes 2,4). The stable association of p27 with cdk2 would result in the continued cdk2 inhibition seen at day 10 (Fig. 3D). This data suggest that the durability of the PD response seen in MDA MB231 cells was due to the fact that at this concentration in this cell line, both cdk4 and cdk2 were inhibited and remained inhibited during treatment. Palbociclib at >10 μM is required to inhibit cdk2 (24), suggesting that these concentrations were too low to inhibit kinase activity directly and instead the association with p27 was responsible.
ALT prevents p27 pY88 and inhibits both cdk4 and cdk2
In MCF-7 cells, a lower concentration of PD was able to inhibit cdk4, but arrest was not durable because cdk2 was able to compensate for loss of cdk4 activity, while in MDA MB231 cells, a higher concentration of PD was needed to inhibit cdk4, but this resulted in the stabilization of p27 and inhibition of cdk2 activity, creating a long-lasting cell cycle arrest. Thus, an inhibitor that blocks both cdk4 and cdk2 in MCF7 cells might produce a more durable response. We had previously shown that blocking p27 Y88 phosphorylation inhibited cdk4 activity, by converting the p27-D-K4 complex into a closed, inactive conformation (12). However, others had shown that blocking pY88 also prevents p27 degradation, permitting it to inhibit cdk2 as well (22). Overexpression of an ALT Brk (ALT), which contains only Brk’s SH3 domain, caused growth arrest (15) and we hypothesized that this might result from direct cdk4 inhibition but also indirect cdk2 inhibition (Fig. 4A).
Figure 4. ALT blocks p27 Y88 phosphorylation and inhibits cdk4 and cdk2 activity.

(A) Cartoon of the ALT:p27 interaction. (Top) Brk with its SH3, SH2 and SH1 (catalytic) domains. ALT only has the SH3 domain. (Bottom left) Brk interacts via its SH3 domain to bind p27 and then phosphorylate it on residue Y88, opening and activating the cdk4 complex. (Bottom right) ALT binds to p27, preventing Brk’s interaction with p27 and blocking its phosphorylation on Y88, closing and inactivating cdk4. (B) MCF7-ALT cells with DMSO, Dox, or PD alone or in combination. (Top) Immunoblot analysis to probe for FLAG-tagged ALT. (Bottom) p27 immunoprecipitates followed by immunoblot with FLAG, p27 or pY88 antibodies. (C) Proliferation of MCF7-ALT treated with DMSO, PD, Dox, or combination refreshed every 2 d. Proliferation of MCF7-TtA (no GOI) in Dox as control. Statistical significance Dox (orange) or Dox+PD (black) relative to PD treatment. (D) Immunoblot analysis with lysates from MCF7 cells treated for 4 or 10 d. Quantitation of %p27 (bottom) and %S780Rb/Rb. Representative immunoblot shown in Fig. S4K. (E) Cdk4 activity [(RbS780/RB ratio)/cdk4] determined by densitometry (bottom) of in vitro kinase assay immunoblots (top) (N=5). (F) Cdk2 activity [(RbS7807/811/RB ratio)/cdk2] determined by densitometry (bottom) of in vitro kinase assay immunoblots (top) (N=5). (G) Cdk2 immunoprecipitated, followed by p27 immunoblot (top). p27 association with cdk2 determined by densitometry (bottom) (N=6). (H) p27 immunoprecipitated, followed by pY88 immunoblot (top). pY88/p27 ratio determined by densitometry (bottom) (N=3). (I) MCF7-ALT cells treated with DMSO, PD (400nM), Dox (86.48μM), Dox+PD or a cdk2 specific inhibitor (K2i) (10μM) in immunoblot analysis with cdk1 and T161cdk1 antibodies. (J) Proliferation of MCF7-ALT p27 KD cells with DMSO, PD, Dox (43.24μM) or the combination of Dox+PD. (Inset) Immunoblot analysis to confirm p27 KD.
A stable cell line of MCF7 that inducibly expressed Flag-tagged ALT in a dose-dependent manner was generated (MCF7-ALT). In presence of Doxycycline (hereafter Dox) in the media, Flag-ALT was induced in MCF7 cells as detected by immunoblot analysis with Flag antibodies (Fig. 4B, top panel). Dose response curves were generated to determine the concentration of Dox needed to achieve a 50% reduction in proliferation after 48 h. of treatment (IC50) and viability was constant (Fig. S4A, E). Immunoblot analysis demonstrated consistent induction of ALT across different experiments Fig. S4J, top) and ALT expression was dependent on Dox dose (Fig. S4J bottom). p27 immunoprecipitations of DMSO, PD, or Dox+PD cells treated for 2 days cells were performed, followed by immunoblot analysis with p27, FLAG and pY88 antibodies (Fig. 4B). Flag-tagged ALT was detected in the Dox treated cells, and pY88 was reduced when ALT was overexpressed (Fig. 4B, lane 3). MCF-7-ALT cells were treated with 200 nM PD (IC50), 43.24μM Dox (IC50), or a combination every two days, and proliferation was monitored by cell counting (Fig. 4C). While MCF7-ALT cells adapted quickly to PD treatment alone, Dox treatment (ALT induction) inhibited proliferation durably up to day 10 (Fig. 4C, orange line). When cells were treated with Dox+PD, a synergistic arrest resulted [with a combination index (CI) below 1 (Fig. 4C, black line, S4M). MCF7-TtA cells, which only contained the tetracycline transactivator and lacked the ALT transgene, were treated with Dox to demonstrate that Dox treatment did not have any effect on proliferation (Fig. 4C, red).
Lysates from MCF7-ALT cells treated with the DMSO, IC50 PD, IC50 Dox, or Dox+PD were harvested at day 4 and 10, and probed by immunoblot analysis (Fig. 4D, S4K). While PD treatment reduced endogenous Rb S780 phosphorylation at day 4 (Fig. 4D top, lane 2), it was restored at day 10 (Fig. 4D top, lane 6). However, when cells were treated with Dox to induce ALT (Fig. 4D top, lanes 3, 7) or treated with Dox+PD (Fig. 4D top, lanes 4, 8), RB phosphorylation was not detected at day 4 or 10. While ALT expression did not increase either cyclin D or cdk4 levels (Fig. S4K), it did affect cdk4 and cdk2 activity as measured directly in in vitro kinase assays (Fig. 4E,F). PD and Dox alone reduced cdk4 kinase activity by ~50% at day 4 (Fig. 4E lanes 2,3), and combination treatment reduced cdk4 activity even further (~ 70%) (Fig. 4E, lane 4). Cdk4 was still inhibited to the same extent in all treatment conditions at day 10 (Fig. 4E, lanes 6, 7, 8 as compared to lanes 2, 3, 4). However, while PD treatment reduced cdk2 kinase activity by ~30% at day 4 (Fig. 4F, lane 2), by day 10 it had increased to the level seen in the DMSO-treated control cells (Fig. 4F lane 6). However, Dox treatment (ALT induction) reduced cdk2 kinase activity by ~50% at day 4 and this reduction was maintained at day 10 (Fig. 4F, lanes 3,7). Dual Dox+PD treatment further reduced cdk2 activity at day 4, and was also durable at day 10 (Fig. 4F, lanes 4,8).
Cyclin E and cdk2 levels did not change under the four treatment conditions (Fig. S4K). However, in the PD-treated lysates at day 10, p27 levels decreased, consistent with the increase in cdk2 activity seen at this timepoint (Fig. 4D bottom, lane 6), but in the Dox or Dox+PD-treated cells, p27 levels were stabilized and did not decrease (Fig. 4D bottom, lanes 7,8). When cdk2 immunoprecipitates from PD-treated lysates were probed for p27 by immunoblot analysis, p27 was associated with cdk2 in day 4 PD-treated cells, but then was lost from the complex at day 10 (Fig. 4G, lanes 2,6). However, p27 remained associated with cdk2 at both day 4 and day 10 in both Dox alone or Dox+PD-treated lysates (Fig. 4G, lanes 3-4,7-8).The ratio of pY88/p27 was decreased relative to DMSO levels at day 4 in PD-treated lysates, as seen before (Fig. 1J), but as cells became resistant to PD treatment at day 10, the ratio of pY88/p27 increased (Fig. 4H, lanes 2, 6). Dox or Dox +PD dual treatment, however, caused a ~50% reduction in pY88 levels at both timepoints (Fig. 4H, lanes 3, 4,7, 8), consistent with ALT-mediated that persisted to day 10. This data suggests that when ALT was induced, it physically associated with p27 (Fig. 4B) and blocked Y88 phosphorylation (Fig. 4H). Since unphosphorylated p27 binds cdk2 in a “closed” conformation, it can no longer be phosphorylated on residue T187, is not targeted for degradation, and can remain associated with and continue to inhibit cdk2 activity, resulting in a durable cell cycle arrest in MCF7 cells.
Although there is cell cycle arrest, both 43.24μM (IC50) and 86.48μM (2× the IC50) Dox treated MCF7-ALT cells were viable (Fig. S4A) Frequently, cdk2 inhibitors also inhibit cdk1, due to the high degree of active site homology between these two kinases (25). Lysates from DMSO, 2× IC50 PD, 2× IC50 Dox, and Dox+PD-treated cells at day 4 were probed with anti-cdk1 and T161cdk1, a phospho-specific antibody that recognizes the active form of cdk1 and is routinely used as a surrogate marker for cdk1 activity (26) (Fig. 4I). Cdk1 was still active in PD, Dox or Dox+PD-treated cells. As a control, cells were treated with a cdk2 inhibitor, K2i, and this inhibited T161 phosphorylation by 90% (Fig. 4I). This suggested that ALT-mediated arrest was due to cdk4 and cdk2 specific inhibition, while cdk1 was still active.
To demonstrate that the ALT-mediated arrest was dependent on p27, siRNA was used in the MCF7-ALT cells to knockdown endogenous p27, which was confirmed by immunoblot analysis using p27 antibodies (Fig. 4J, inset). p27 KD cells were treated with DMSO, IC50 PD, IC50 Dox, or Dox+PD and proliferation was monitored by cell counting (Fig. 4J). Neither PD nor Dox treatment of the p27 KD cells affected viability (Fig. S4I) or inhibited proliferation, which was indistinguishable from that seen in the DMSO-treated p27 KD cells (Fig. 4J, blue, grey lines). Dual Dox+PD treatment did retard proliferation slightly from day 2-4, but cells resumed proliferation by day 6 (Fig. 4J, red). These results demonstrated that ALT-mediated and PD-mediated arrest required p27. As a control, Dox+PD dual treatment of parent MCF7-ALT cells that had not received the p27 siRNA were included (Fig. 4J, black line).
Durable arrest requires cdk2 and cdk4 inhibition
Our data suggest that while cdk4 inhibition is required to cause arrest, in order to achieve durability, cdk2 would also have to be inhibited to prevent compensation and resistance. To test this model directly, we treated MCF7-ALT cells with K2i, a cdk2 specific inhibitor, alone or in combination with PD (Fig. 5A). K2i also inhibits cdk1 (Fig. 4I), but does not inhibit cdk4 (27). Dox+PD-treated cells served as a control for durability, demonstrating stable arrest for 10 days (A, black). When MCF7-ALT cells were treated with 0.1 or 1 μM K2i alone, proliferation was only inhibited by ~10 or ~20% respectively from day 4 to 10 (Fig. 5A, orange and purple lines), but when combined with PD, a ~40 or ~50% inhibition, respectively, was observed for almost 10 days (Fig. 5A, red and brown lines), suggesting an additive or synergistic effect. 100% of the K2i treated cells were viable at these concentrations (Fig. S5A). Lysates from day 4 treated cells were used in immunoblot analysis and probed with cdk2 and T160cdk2 phospho-specific antibodies, to detect active cdk2 and confirm that K2i had inhibited its target (Fig. 5B). While cdk2 levels were unchanged, T160cdk2 was reduced in the K2i+PD-treated cells (Fig. 5B, lanes 5-6). PD and K2i treatment each increased p27 levels relative to DMSO controls (Fig. 5B lanes 3-4), and K2i+PD increased p27 expression to an even greater extent (Fig. 5B, lanes 5-6), consistent with the role of cdk2 as a regulator of p27 stability (22,28). Thus, the inhibition of cdk2 at day 4 likely resulted both from inhibition of the cdk2 active site by K2i and increased p27.
Figure 5. Cdk2 compensates for cdk4 activity to cause cell resistance.

Proliferation of MCF7-ALT cells treated every 2 d. with K2i (A) Skp2i (C), LET (D) alone or in combination with 200 nM PD followed by immunoblot analysis on day 4 (B,D,F). (G,I) Proliferation of MDA MB231-ALT (G) and HCC1954-ALT (I) treated with DMSO, PD, Dox or Dox+PD. Proliferation of TtA (no GOI) in Dox as control. Statistical significance relative to PD treatment. (H) Cells treated with Dox to induce ALT and probed by immunoblot with Flag antibodies. (J,K) Treated MDA MB231-ALT cells used in the cdk4 (J) and cdk2 (K) in vitro kinase assays (N= 4). (L) (Left) Proliferation of MDA MB231-ALT p27 KD cells with DMSO, PD (0.4 μM), Dox (21.62μM) or the combination of Dox and PD. (Inset) Immunoblot analysis to confirm p27 KD. (M,N) Proliferation of MDA MB231-ALT cells with DMSO, IC25 PD (0.2μM), Dox (21.62μM) or Dox+PD (M) followed by immunoblot analysis (N).
Since it appeared that a decrease in p27 levels was responsible for the cdk2-dependent rescue of the PD-mediated arrest seen MCF-7 cells (Fig. 2B), we hypothesized that use of a Skp2 inhibitor (Skp2i) (29,30), which should inhibit the ubiquitin ligase required for p27 degradation, would stabilize p27 and inhibit cdk2, and combined Skp2i and PD treatment might phenocopy ALT-mediated inhibition. MCF7-ALT cells were treated with Skp2i C1, alone or in combination with PD (Fig. 5C). 100% of cells were viable in 0.1 and 1 μM Skp2i (Fig. S5B). Both Skp2i treatments reduced proliferation by ~20% (Fig. 5C, purple, orange lines), while the combination of Skp2i+PD reduced proliferation by ~50% (Fig. 5C, red, brown lines). Skp2i (0.1 μM)+PD only inhibited cells up to day 4, and by day 6, proliferation had resumed (Fig. 5C, red), while 1 μM Skp2i+ PD arrested cells up to day 8, but by day 10, these cells too had begun to escape arrest (Fig. 5C, brown). Immunoblot analysis of lysates from day 4 treated cells demonstrated that Skp2i did stabilize p27 levels to a similar extent as that seen with PD mono treatment (Fig. 5D). Dual Skp2i+PD treatment stabilized p27 slightly more (Fig. 5D, lanes 5,6). Both Skp2i mono and dual Skp2i+PD dual treatment reduced cdk2 activity as measured by cdk2T160 immunoblot (Fig. 5D, lanes 3-6).
Letrozole (hereafter LET) in combination with PD is currently a front-line therapy for women with metastatic ER+ breast cancer (31). LET is an aromatase inhibitor, which reduces estrogen levels, and is thought to reduce Cyclin D1 levels, enhancing the efficacy of PD. We treated MCF7-ALT cells every two days with 0.1 or 1 nM LET, PD, or the combination (Fig. 5E). Either concentration of LET alone reduced proliferation by ~ 20% (Fig. 5E, orange, purple), while the combination of either 0.1 or 1 nM LET with PD arrested proliferation by ~50% by day 4 (Fig. 5E, red, brown). The arrest seen with 0.1 nM LET+PD was not durable and cells resumed proliferation by day 6 (Fig. 5E, red), while the arrest with 1 nM LET+PD was durable to day 8 (Fig. 5E, brown). We performed immunoblot analysis to examine its effect on cell cycle proteins (Fig. 5F). We did not see an effect on cyclin D expression (Fig. S5D), but while PD treatment stabilized p27, LET alone did not have any effect (Fig. 5F, lanes 3,4). However, at both concentrations of LET+PD, p27 levels were stabilized (Fig. 5F, lanes 5, 6), and this corresponded to a decrease in cdk2 activity. LET alone did not have an effect on cdk2 activity as measured by T160cdk2 detection, but the combination of LET+ PD significantly reduced cdk2 activity (Fig. 5F, lanes 5, 6).
To determine whether ALT would also arrest cell lines that responded to PD with moderate or poor arrest, additional stable lines that inducibly expressed FLAG-ALT in the presence of Dox were generated (MDA MB231-ALT and HCC1954-ALT) (Fig. 5G-I, S53-G) and dose-response curves were used to determine the appropriate amount of Dox required to achieve an IC50 (Fig. S4C-E). While a higher concentration of PD is needed to inhibit MDA MB231 and HCC1954 than was needed to inhibit MCF7 cells, a similar induction of ALT caused a 50% inhibition and this arrest was durable for 10 days (Fig. 5G,I). This was very striking in HCC1954 cells where 5× more PD (1000 nM) is required to inhibit proliferation by 50%, but a similar level of ALT induction is just as effective and this is induced with half the amount of Dox used for MCF7 (Fig. 5I blue and orange). A synergistic arrest (CI <I) was seen with Dox+PD dual treatment of MDA MB231 cells (Fig. 5G black line, S4M), and cdk4 and cdk2 in vitro kinase assays showed that both cdk4 and cdk2 activity was inhibited more in the dual-treated cells (Fig. 5J, K). While the IC50 concentration of PD blocked pY88 by ~50%, Dox+PD treatment reduced pY88 even more (Fig. S5H, I).
To demonstrate that ALT-mediated arrest was dependent on p27, siRNA was used in the MDA MB231-ALT cells to knockdown endogenous p27 and was confirmed by immunoblot analysis using p27 antibodies (Fig. 5L, right panel). p27 KD did not alter viability (Fig. S5K). p27 KD cells were treated with DMSO, the IC50 concentration of PD or Dox, or the combination and proliferation was monitored (Fig. 5L). PD or Dox treatment of the p27 KD cells only inhibited proliferation by 20% as compared to the 50% inhibition seen with PD or Dox treatment in the parent MDA MB231-ALT cells (Fig. 5L vs. 5G). Dual Dox+PD treatment did retard proliferation of p27 KD cells by 50% at day 6 (Fig. 5L), but was not as effective as Dox+PD treatment in the p27+ cells (Fig. 5G, black line) where 80% inhibition was detected, suggesting that p27 was required for ALT-mediated inhibition in MDA MB231 cells. Knockdown using scrambled siRNA did not alter either PD or Dox-mediated arrest (Fig. 5K,L).
We hypothesized that ALT induction might permit a lower, more pharmacological concentration of PD to be used to elicit arrest. MDA MB231-ALT cells were treated with Dox, 200 nM PD (IC25), or Dox+PD, and proliferation was monitored by cell counting (Fig. 5M). When 200 nM PD (IC25) used, only a 30% inhibition was seen at day 4, and by day 6, proliferation had resumed (Fig. 5M blue). Immunoblot analysis of treated lysates demonstrated that at this lower concentration, PD treatment was unable to block endogenous RB S780 phosphorylation at day 4, and cdk2 was still active as measured by T160cdk2 phosphorylation (Fig. 5N, lane 2). Dox treatment inhibited proliferation by 50% and endogenous Rb S780 phosphorylation was reduced (Fig. 5M, N, lane 3, 7). However, in Dox+PD (200 nM) treated cells, proliferation continued to decrease up to day 10 (~70%), suggesting a small additive effect with the addition of this low, non-inhibiting concentration of PD (Fig. 5M, black). Immunoblot analysis of Dox+PD-treated lysates harvested at day 4 and 10 demonstrated that endogenous RB S780 phosphorylation and cdk2 activity was inhibited as well (Fig. 5N, lanes 4, 8).
ALT + PD combine to cellular senescence
To determine how sustainable Alt-mediated arrest was, cells were treated with a higher concentration of Dox (2× IC50, which induced more ALT (Fig. S4K), a higher concentration of PD (2× IC50) or the combination (Fig. 6A,D,G). In the presence of this increased concentration of PD, proliferation was arrested to ~10-20% in all cell lines. In the MCF7 line, cells became resistance to PD at day 14 (Fig. 6A blue), while MDA MB231 and HCC1954 cells were stably arrested for >26 days (Fig. 6D, G blue). In the presence of increased Dox alone, MCF7 cells were arrested to day 14, although at these extended time points escape eventually occurs (Fig. 6A orange). MDA MB231 cells escaped Dox-mediated arrest around day 14, while HCC1954 cells were arrested for >26 days (Fig. 6D, G orange). However, for all cell lines, Dox+PD treatment arrested cells for 30 days (Fig. 6A,D,G black). Cells remained viable during this time (Fig. S6A-C).
Figure 6. ALT+PD treatment induces durable arrest and increases senescence.

Proliferation of MCF7-ALT (A), MDA MB231-ALT (D) or HCC1954-ALT (G) cells treated with DMSO, 2× ICD50 PD, 2× ICD50 Dox or Dox+PD. MCF7-ALT (B), MDA MB231-ALT (E) or HCC1954 (H) cells were treated with DMSO or drugs for 6 days, trypsinized and re-plated in fresh drug-free media (DMSO*, PD*, etc. denotes the prior treatment conditions). Replated cells were counted. (C, F, I) % viability determined by Trypan Blue stain. MCF7-ALT (J) and MDA MB231-ALT (K) cells treated for 6 days with DMSO or drugs were stained with β-Galactosidase.
Cells that had been treated with these different combinations (DMSO, Dox, PD or Dox+PD) for six days and were arrested (Fig. 6A,D,G), were then trypsinized and replated in fresh, drug-free media (designated DMSO*, Dox*, PD* or Dox+PD*) (Figs. 6B,C,E,F,H,I). Proliferation was monitored by cell counting (Figs 6B,E,H) and viability of these pre-treated cells was measured by trypan blue staining (Fig. 6C,F,I). Upon replating in drug-free media, PD* treated MCF7 cells resumed proliferation, demonstrating that PD arrest was reversible (Fig. 6B, green, blue). Re-entry back into cycle was delayed in the Dox* treated (ALT induced) cells (Fig. 6B, orange), but the replated Dox+PD* dual treated cells were unable to resume proliferation even after 10 days (Fig. 6B, black). Viability was reduced in the Dox+PD* treated cells, but ~50% of the cells were alive and remained non-proliferative (Fig. 6C). MCF7 cells arrested for 6 days were stained for beta-galactosidase and an increase in beta-gal+ cells was detected in the Dox+PD-treated cells (Fig. 6J), suggesting that these non-proliferative cells were actually senescent and could not reenter cell cycle. Similar results were seen with another ER+ breast cancer cell line, T47D, suggesting that ALT+PD dual therapy induced senescence (Fig. S6D-G; β-gal data not shown).
HCC1954-ALT and MDA MB231-ALT cells were assayed for their ability to proliferate in the replating assay. Dox* and Dox+PD*-treated HCC1954 cells did not regain proliferation post replating, even though the cells remained viable (Fig. 6H,I). All of the drug treatment groups in the MDA MB231-ALT cells were retarded in their ability to reestablish proliferation, but did eventually reenter cycle (Fig. 6E). The replated MDA MB231 cells were viable (Fig. 6F), and when beta-gal expression was analyzed, we found that PD treatment alone produced a significant number of beta-gal+ cells, which was not increased significantly in the presence of ALT (Fig. 6K). Together, data suggest that PD treatment was reversible in MCF7 and HCC1954 cells, but the combination of ALT+PD rendered these cells unable to recover from arrest. As shown in the MCF7 line, the inability to renter cycle could be attributed to the induction of senescence (Fig. 6J). In the MDA MB231 cells. all treatments impaired but did not abrogate the ability to escape from arrest.
ALT + PD reduces tumors in xenograft models
Because ALT+PD dual treatment induced senescence in MCF7 cells, we hypothesized that instead of just stopping tumor growth, dual treatment might cause tumor regression in a breast cancer animal model (Fig. 7A). We injected MCF7-ALT cells into the 4th mammary gland of NOD/SCID female mice that had an estrogen release pellet subcutaneously implanted, tumors were observed within 5 weeks, and treatment was started when each animal’s tumor reached 200 mm3 (Day 1). Some animals were dosed with either 13.33 mg/kg Doxlow or 40 mg/kg Doxhigh in their drinking water to induce the ALT transgene in vivo, and we had shown that an increase in the Dox concentration corresponded to an increase in ALT expression (Fig. S4K). Animals were treated with 100 mg/kg PD as a mono therapy or in combination with the Dox-containing water. Tumor growth was monitored every other day using digital calipers and plotted as the change in tumor volume (Fig. S7A). Tumor growth continued in the vehicle- treated animals and most animals had tumors >3000 mm3 by day 17 of treatment, and were removed from the experiment (Fig. 7A, green). Tumor growth in the PD-treated animals was slowed compared to vehicle-treated animals (not statistically significant), but no tumor regression was seen in any treated animals, and tumors >2500 mm3 were detected by day 17, when animals were removed from the experiment (Fig. 7A, blue). Tumor growth was significantly reduced at day 11 in the animals fed the two concentrations of Dox (Fig. 7A, orange, red, *). In the animals receiving Doxhigh, tumor regression was observed for the first 5 days of treatment, before starting to increase in size again at day 7 (Fig. 7A, red). Tumor regression was not seen in the Doxlow treated animals, and in both cases by day 17 of treatment, Dox treated animals had tumors > ~1500 mm3 (Fig. 7A, orange, red, *). Tumors in the dual treated animals (Doxhigh+PD and Doxlow +PD), however, showed actual tumor regression within 2 days of treatment, which continued up to 19 days for Doxlow+PD animals (Fig. 7A, purple, black). Doxhigh+PD animals became dehydrated around day 9 due to the Dox in the drinking water and had to be euthanized (Fig. 7A, black). This data demonstrated that dual treatment produced a unique result: instead of just slowing tumor growth, tumors underwent regression.
Figure 7. ALT treatment reduces tumor volumes in vivo.

(A) NOD/SCID mice injected with MCF7-ALT cells. When tumor volume reached ~200 mm3 (Day 1), mice were treated daily with Vehicle, PD (100 mg/kg), 2 concentrations of Dox (Dox(H)= 40 mg/kg or Dox(L)= 13.3. mg/kg), Dox(H)+PD or Dox(L)+PD. % change in tumor volume plotted. (Data: mean ±SE, *p<0.005 relative to PD vehicle-treated animals, individual animals plotted in Fig. S7A). (B,C) IHC on mouse tissue samples from animals harvested at day 9-11. Flag, pink at 100X (B), 600X (C). In C, note the increase in non-viable cells (picnotic, dark, small nuclei). (D) Viable tumor cells normalized to vehicle, as determined from viability in H&E X total tumor volume. (E) % of pY88+ viable tumor cells determined from (F, pY88 single stain, green) and (G, pY88/p27 dual stain, red/brown).
Tumors were excised from animals treated for 9-11 days, fixed, embedded in paraffin and subjected to IHC (Fig. 7B-G, S7C-D). Histological examination demonstrated that tumors from the Doxlow, Doxhigh and Dox+PD animals all had induced FLAG-ALT as seen both by the presence of bright pink cells and a distinct pink blush in all viable tumor cells as compared to surrounding mouse tissue, while the vehicle and PD-treated samples were FLAG negative (Fig. 7B, C). The most significant histological finding was that most tumors in all groups had large areas of necrosis, with only patches of viable tumor cells (H&E, data not shown). The total viable cells (as determined from H&E stains X total tumor volume, normalized to vehicle control) demonstrated that viability decreased with PD, Doxhigh and Dox+PD treatment (Fig. 7D). We looked for beta-gal and cleaved caspase (CC) expression, as markers of senescence and apoptosis, respectively. Increases in both CC and beta-gal were detected in the Dox and Dox+PD-treated animals, relative to the vehicle or PD-treated animals, but the only a small percentage of total viable cells were beta-gal or CC positive, and positive cells were most prominent in the viable area boarding the necrotic areas (Fig. S7B, C). Given that we only examined a single and late time point, it is possible that we missed a window when more cells in the treated groups were undergoing senescence and/or apoptosis and that much of that nonviable tumor was cleared away in the Dox or Dox+PD treated animals.
To determine whether ALT induction in the xenograft model blocked pY88, we stained sections with pY88 antibodies either as a single stain (Fig. 7F, S7, green) or as part of a dual pY88/p27 double staining assay (Fig. 7G, pink/brown). p27 (brown) was not detectably different in the different treatment conditions. We calculated the % of viable cells that were pY88+ and found that while 20% of cells in the vehicle were pY88+, almost no detectable staining was seen in the Doxhigh or Dox+PD treated animals, suggesting that ALT expression had reduced pY88 (Fig. 7E). Interestingly, a larger percentage of viable cells in the PD and Dox(low)-treated animals stained positive for pY88 than was seen in vehicle treated animals.
Discussion
We have shown that while ER+ breast cancer cell lines are inhibited by PD in culture, they quickly adapt because they permit the degradation of p27 and a subsequent increase in cdk2 activity, allowing compensatory phosphorylation of Rb and passage into S phase. Using in vitro Rb kinase assays, we demonstrate the effect of PD on cdk4 and cdk2 activity, as opposed to its effect on just expression. While arrest might be dependent on a drug’s ability to inhibit cdk4, durability is dependent on the ability to also inhibit cdk2. Inhibition of cdk2 alone was insufficient but any combination that inhibited both cdk4 and cdk2, including use of K2i, Skp2i, or LET in combination with PD, caused arrest and increased durability. While we could have verified the role of cdk2 in PD-mediated durability by manipulation of cdk2 directly via knockout or knockdown, we feared that this would alter the balance of p27 association and hinder interpretation. In all cases, the limiting factor in achieving a long-lasting arrest was cdk2 inhibition, supporting the idea that cdk2 specific inhibitors in combination with PD would have the most clinical efficacy. In moderately responding cells like MDA MB231, which require higher concentrations to achieve an IC50 arrest, PD stabilized p27 and inhibited cdk2 on its own, causing a durable arrest. To date, most cdk2 inhibitors also inhibit cdk1, which in turn causes loss of viability and associated toxicity. We have demonstrated the feasibility of targeting p27 pY88 as a non-toxic modality to inhibit both cdk4 and cdk2. We have shown that ALT binds to p27 in vivo and blocks Y88 phosphorylation, locking the p27-cyclin D-cdk4 ternary complex into the closed, off conformation. But ALT expression also stabilized p27 itself, which in turn caused cdk2 inhibition, resulting in a durable growth arrest, demonstrating that this may represent a new approach to deal with the drug resistance that occurs when only cdk4 is inhibited.
The stabilization of p27 seen in the presence of ALT inhibits cdk2, but cdk1 remains active. This discrimination distinguishes pY blocking therapy from most cdk2is, which do not show this preference and for which toxicity has prevented their clinical use. Cells become resistant to ALT monotherapy at extended time frames (>14 days), and it is unclear whether at these time points, cdk1 eventually compensates for loss of cdk4 and cdk2 and permits progression. Increased concentrations of ALT might have resulted in even longer-term stable arrest. Several other studies have looked at resistance in ER+ lines developing in the presence of long term, very high, less clinically relevant PD concentrations, and in these conditions, amplification of cdk6 or cyclin E or loss of Rb was detected (5,7). Cyclin E amplification has also been shown to be responsible for PD-mediated resistance in pancreatic ductal adenocarcinoma lines, which a priori one might have felt would be responsive to PD given their near universal loss of p16 and overexpression of Ras (33). However, given the short time course of our experiments and the fact that we used lower, more clinically relevant concentrations of PD in MCF7 and T47D cells, it is unlikely that we are tracking genetic changes, but rather epigenetic modification. This is significant because any ability to escape PD-mediated arrest at the onset of treatment would set the stage for additional proliferation and the subsequent acquisition of future genetic changes to lock in the adaption, converting the tumor ultimately to full resistance. Patients treated with PD are on treatment for three weeks, and then removed for one week, which could allow expansion of small populations of adapted clones. This suggests that inhibiting both the kinase that drives tumor progression (cdk4) as well as the kinase that will permit escape (cdk2) at the onset would provide advantages.
While ALT expression reduces both cdk4 and cdk2 activity, the ALT+PD combination reduces both activities even more and causes a synergistic arrest, increases senescence, and prevents cells from recovering when drugs were removed. While this may be a result of cdk4 and cdk2 combined inhibition, we cannot exclude the possibility that this synergy may be mediated via other molecular mechanisms. However, arrest was not seen in the p27 KD cells, and inhibition of cdk2 induces senescence by directly blocking the function of the transcription factor c-Myc (34). Interestingly, PD-mediated arrest was not detected in the p27KD cells either. While p27KD reduces the concentration of the assembled p27-cyclin D1-cdk4 ternary complex, removing PD’s target, it would also release cdk2, which has been shown competent to drive proliferation in the absence of cdk4 (21).
Senescent cells irreversibly exit the cell cycle and become targets for removal by the host immune system. When we induced ALT in the xenograft model, we saw tumor reduction, which lasted >17 days, even in the setting of a compromised immune system. This suggests that while PD treatment is cytostatic, slowing down growth kinetics, ALT or ALT+PD causes some degree of cytotoxicity. We did see increased beta-gal and CC expression, but primarily we detected much smaller tumor volumes with large regions of necrosis. ALT induction in large, previously untreated tumors or even PD-resistant tumors (day 17) caused rapid tumor reduction within 1 day, suggesting that we may have missed documenting ALT’s direct effect with our histology experiments by using tumors from animals treated >9 days. Our experiments did not measure overall survival as animals were continually removed from the study for histological analysis or due to dehydration, and additional studies will be required to address this. Recently, several studies have suggested that in order for PD to cause an irreversible senescent exit from the cell cycle, additional targets must be inhibited. Loss of MDM2 or inhibition of ROS and autophagy have all been shown to increase the synergistic and senescence-inducing potential of PD. Our data is consistent with this and suggests that inhibition of cdk2 may be upstream of some of these alternative targets. It also suggests that use of ALT, with its putative one target toxicity, may circumvent differences in genetic background responsible for inducing senescence in the presence of PD (35,36).
ALT is a naturally-occurring molecule, suggesting that nature has already performed its own high throughput screen and determined that this is a bona fide way to bind to p27 and inhibit pY88. Use of small peptides spanning the region of p27 that binds to the Brk SH3 domain also acted as competitive inhibitors and blocked pY88 (16). However, SH3 domains show significant homology and use of a p27 peptide might block many other Y kinases. We have shown that ALT had no effect on proliferation when p27 expression was reduced by KD, suggesting that while ALT itself may have other targets, its ability to regulate proliferation occurs via p27. Use of small molecule Brk inhibitor (Brki) did not produce durable arrest nor cause an irreversible exit from the cell cycle in the replating assay (Fig. S6H,I). As Brk is upstream in the signaling pathway, it is possible that inhibition was not complete or that other factors are able to quickly compensate for its loss, supporting the idea that targeting downstream at the p27-cdk4-cdk2 core may be more beneficial. Unlike PD, which is most efficacious in ER/PR+, Her2- lines, we found that a similar level of ALT induction inhibited a variety of lines with different receptor status: basal, ER/PR-,HER2+ (HCC1954), luminal ER/PR+, Her2- (MCF7, T47D), and triple negative (MDA MB 231). Differences were noted in terms of ALT’s ability to induce senescence or cause permanent cell cycle exit in these cell types, suggesting that genetic and epigenetic signatures of ALT-responsive cells must be determined.
We have demonstrated that pY88 correlates with cdk4 activity in multiple cell lines, and have shown that lines with high pY88 and high cdk4 activity required high concentrations of PD, while lines with lower pY88 and cdk4 activity required lower concentrations of PD. In cell lines, PD treatment inhibits pY88 in a dose-dependent fashion. In the xenograft model, however, we found that animals whose tumors continued to grow in the presence of treatment (PD or ALTlow) regained pY88 and actually had an increase in pY88+ cells, suggesting that this might also correlate with secondary drug resistance. Ki67 was not able to distinguish between these cells, suggesting that pY88 might serve as a surrogate marker for PD or any cdk4-inhibitory therapy’s responsiveness (including ALT).
In our xenograft system, the ALT-transgene was induced when animals were fed Dox, and this strategy was a proof of concept efficacy study to examine the effect when ALT was expressed in every tumor cell. Clearly, for the peptide ALT to be used as a therapy it needs to be effectively delivered and shown to be non-toxic in vivo. Combination therapy with high levels of PD and several other classes of targeted therapies, including PI3K or autophagy inhibitors (36,37), have also shown increased efficacy in tissue culture and animal models, but our study with ALT has demonstrated synergy at low, clinically relevant concentrations of PD. Additional work will be required to translate it into a clinical compound, but this proof of principle study has shown that ALT as a mono therapy or in combination with PD has several advantageous properties that make it worth investigating. Use of ALT has also clearly demonstrated the requirement to inhibit both cdk4 and cdk2 if long-term efficacy is to be achieved.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications.
Modulating tyrosine phosphorylation of p27 impacts both proliferative (CDK4) and resistance (CDK2) mechanisms in breast cancer and suggests that phospho-p27 status may serve as a biomarker for patients that are responsive to CDK4/6 inhibition.
Acknowledgments
We thank Safraz Hamid, Khurran Sharique and Mengru Li for expert technical assistance. This work was supported by CCLM Research Fund and NIH R01CA201536. All of the data was independently verified by the SUNY Compliance Management Committee.
References
- 1.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–46. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016;6(4):353–67. doi: 10.1158/2159-8290.CD-15-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin Cancer Res. 2014;20(13):3379–83. doi: 10.1158/1078-0432.CCR-13-1551. [DOI] [PubMed] [Google Scholar]
- 4.Cadoo KA, Gucalp A, Traina TA. Palbociclib: an evidence-based review of its potential in the treatment of breast cancer. Breast Cancer (Dove Med Press) 2014;6:123–33. doi: 10.2147/BCTT.S46725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016;76(8):2301–13. doi: 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu F, Korc M. Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther. 2012;11(10):2138–48. doi: 10.1158/1535-7163.MCT-12-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang C, Li Z, Bhatt T, Dickler M, Giri D, Scaltriti M, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2016 doi: 10.1038/onc.2016.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang YX, Sicinska E, Czaplinski JT, Remillard SP, Moss S, Wang Y, et al. Antiproliferative effects of CDK4/6 inhibition in CDK4-amplified human liposarcoma in vitro and in vivo. Mol Cancer Ther. 2014;13(9):2184–93. doi: 10.1158/1535-7163.MCT-14-0387. [DOI] [PubMed] [Google Scholar]
- 9.DeMichele A, Clark AS, Tan KS, Heitjan DF, Gramlich K, Gallagher M, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res. 2015;21(5):995–1001. doi: 10.1158/1078-0432.CCR-14-2258. [DOI] [PubMed] [Google Scholar]
- 10.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 11.Bockstaele L, Coulonval K, Kooken H, Paternot S, Roger PP. Regulation of CDK4. Cell Div. 2006;1:25. doi: 10.1186/1747-1028-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blain SW. Switching cyclin D-Cdk4 kinase activity on and off. Cell Cycle. 2008;7(7):892–8. doi: 10.4161/cc.7.7.5637. [DOI] [PubMed] [Google Scholar]
- 13.James MK, Ray A, Leznova D, Blain SW. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol. 2008;28(1):498–510. doi: 10.1128/MCB.02171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray A, James MK, Larochelle S, Fisher RP, Blain SW. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol Cell Biol. 2009;29(4):986–99. doi: 10.1128/MCB.00898-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brauer PM, Zheng Y, Evans MD, Dominguez-Brauer C, Peehl DM, Tyner AL. The alternative splice variant of protein tyrosine kinase 6 negatively regulates growth and enhances PTK6-mediated inhibition of beta-catenin. PLoS One. 2011;6(3):e14789. doi: 10.1371/journal.pone.0014789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel P, Asbach B, Shteyn E, Gomez C, Coltoff A, Bhuyan S, et al. Brk/Protein tyrosine kinase 6 phosphorylates p27KIP1, regulating the activity of cyclin D-cyclin-dependent kinase 4. Mol Cell Biol. 2015;35(9):1506–22. doi: 10.1128/MCB.01206-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, et al. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128(2):281–94. doi: 10.1016/j.cell.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res. 2004;10(23):7994–8004. doi: 10.1158/1078-0432.CCR-04-1087. [DOI] [PubMed] [Google Scholar]
- 19.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aleem E, Kiyokawa H, Kaldis P. Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol. 2005;7(8):831–6. doi: 10.1038/ncb1284. [DOI] [PubMed] [Google Scholar]
- 21.Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118(4):493–504. doi: 10.1016/j.cell.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128(2):269–80. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 23.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29(28):4018–32. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 24.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–38. [PubMed] [Google Scholar]
- 25.Coxon CR, Anscombe E, Harnor SJ, Martin MP, Carbain BJ, Golding BT, et al. Cyclin-Dependent Kinase (CDK) Inhibitors; Structure-Activity Relationships and Insights into the CDK-2 Selectivity of 6-Substituted 2-Arylaminopurines. J Med Chem. 2016 doi: 10.1021/acs.jmedchem.6b01254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Zhang Y, Xu S, Li W, Chen Z, Wang Z, et al. Prognostic significance of G2/M arrest signaling pathway proteins in advanced non-small cell lung cancer patients. Oncol Lett. 2015;9(3):1266–72. doi: 10.3892/ol.2015.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye W, Blain SW. S phase entry causes homocysteine-induced death while ataxia telangiectasia and Rad3 related protein functions anti-apoptotically to protect neurons. Brain. 2010;133(Pt 8):2295–312. doi: 10.1093/brain/awq139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitrea DM, Yoon MK, Ou L, Kriwacki RW. Disorder-function relationships for the cell cycle regulatory proteins p21 and p27. Biol Chem. 2012;393(4):259–74. doi: 10.1515/hsz-2011-0254. [DOI] [PubMed] [Google Scholar]
- 29.Pavlides SC, Huang KT, Reid DA, Wu L, Blank SV, Mittal K, et al. Inhibitors of SCF-Skp2/Cks1 E3 ligase block estrogen-induced growth stimulation and degradation of nuclear p27kip1: therapeutic potential for endometrial cancer. Endocrinology. 2013;154(11):4030–45. doi: 10.1210/en.2013-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol. 2012;19(12):1515–24. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med. 2016;375(20):1925–36. doi: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 32.Mahmoud KA, Krug M, Wersig T, Slynko I, Schachtele C, Totzke F, et al. Discovery of 4-anilino alpha-carbolines as novel Brk inhibitors. Bioorg Med Chem Lett. 2014;24(8):1948–51. doi: 10.1016/j.bmcl.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Witkiewicz AK, Borja NA, Franco J, Brody JR, Yeo CJ, Mansour J, et al. Selective impact of CDK4/6 suppression on patient-derived models of pancreatic cancer. Oncotarget. 2015;6(18):15788–801. doi: 10.18632/oncotarget.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hydbring P, Bahram F, Su Y, Tronnersjo S, Hogstrand K, von der Lehr N, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci U S A. 2010;107(1):58–63. doi: 10.1073/pnas.0900121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kovatcheva M, Liao W, Klein ME, Robine N, Geiger H, Crago AM, et al. ATRX is a regulator of therapy induced senescence in human cells. Nat Commun. 2017;8(1):386. doi: 10.1038/s41467-017-00540-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916. doi: 10.1038/ncomms15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonelli MA, Digiacomo G, Fumarola C, Alfieri R, Quaini F, Falco A, et al. Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia. 2017;19(8):637–48. doi: 10.1016/j.neo.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.