

Abstract
Piezo1 is a mechanosensitive cation channel that is activated by shear stress in endothelial cells (ECs). It has been shown to mediate shear-induced EC responses, including increased calcium influx, and vascular functions, such as vascular tone and blood pressure. Yoda1, a selective Piezo1 activator, has been shown to mimic shear-induced responses in ECs. Since shear-induced calcium influx causes Akt and ERK1/2 activation in ECs, we examined the effects of Yoda1 and the role of Piezo1 on their activation. Here, we show that Yoda1 robustly activates Akt and ERK1/2 in ECs. Additionally, the Piezo1 antagonists, gadolinium and ruthenium red, but not GsMTx4, effectively blocks Yoda1-induced Akt activation. Our results suggest that Yoda1-induced Akt and ERK1/2 activation is not dependent on Piezo1.
Keywords: Yoda1, Piezo1, endothelial cells, Akt, ERK1/2
Introduction
The Piezo proteins, Piezo1/FAM38A and Piezo2/FAM38B, are multi-pass transmembrane proteins that have been identified as mechanically-activated (MA) homomultimeric, pore-forming ion channels [1,2]. Piezo1 is broadly expressed with its highest expression in bladder, colon, kidney, lung, and skin. Piezo2 is highly expressed in bladder, colon, lung, and sensory neurons from dorsal root ganglia. Recent studies have demonstrated that Piezo1, in particular, can mediate MA cationic currents in several different cell types, including endothelial cells (ECs) [3,4]. Additionally, Piezo1 was shown to be critical for calcium influx into ECs as well as EC alignment in response to fluid shear stress [3,4]. Expression of Piezo1 in HEK 293 cells, which lack endogenous Piezo1 and are typically unresponsive to shear stress, evoked shear stress-induced calcium influx, suggesting that Piezo1 channels may directly function as shear stress sensors [3]. Most recently, Piezo1 channels have been proposed as “exercise sensors” that are activated by increased blood flow during whole body physical activity and cause vascular bed-specific vasoconstriction [5].
There are no known endogenous agonists and/or antagonists of Piezo channels to date, but Piezo channels are known to be activated by mechanical stimuli and also by a recently identified synthetic small molecule called Yoda1. Yoda1 was first discovered using a high-throughput screen to test whether chemical compounds exist that can activate Piezo channels. It was determined to be a selective small molecule agonist of Piezo1 based on its ability to induce a calcium response in HEK293 cells that overexpressed Piezo1, but not in vector-transfected cells or in cells that overexpressed Piezo2 [6]. Yoda1 stimulation has since been utilized as an invaluable tool for investigating the activation and regulation of Piezo1. The concept of a Yoda1-like “exercise pill”, which has the ability to specifically target and enhance Piezo1 activity during exercise, has even been introduced [7].
It has been recently demonstrated that Yoda1 can induce responses similar to those activated by fluid shear stress in ECs, such as increased intracellular calcium and nitrate formation [8]. These endothelial responses were shown to be mediated by Piezo1 by using an siRNA targeting approach. Pharmacological inhibition of Piezo1 is an alternative approach that has been utilized in other studies, including the examination of mechanically-activated channel currents in a mouse neuroblastoma cell line [1], the investigation of shear-induced ATP release and calcium influx in RBCs [9], and the study of shear stress-induced endothelial polarization in mesenteric arteries [5]. Using a similar pharmacological approach, we sought to examine the role of Piezo1 on the simultaneous activations of the Akt and ERK1/2 signaling pathways by Yoda1 in ECs.
Materials and Methods
Cell culture
Human coronary artery endothelial cells (HCAECs) from male or female donors were obtained from Lonza (Walkersville, MD) and maintained in complete endothelial growth medium (EGM-2; Lonza) supplemented with 10% heat-inactivated FBS and penicillin-streptomycin. Prior to all experimental procedures, cells were seeded onto glass microscope slides, grown to confluence, and serum-starved overnight in ATP-free endothelial basal medium (EBM-2; Lonza) supplemented with 0.5% BSA. HCAECs within six passages were used for all experiments.
Reagents
Gadolinium chloride and Yoda1 were purchased from Tocris Bioscience (Bristol, UK). Ruthenium Red was purchased from Cayman Chemical (Ann Arbor, MI). GsMTx4 was purchased from both Tocris Bioscience and Alomone Labs (Jerusalem, Israel). Rabbit anti-phospho-Akt (Ser473) (#4060), rabbit anti-phospho-ERK1/2 (T202/Y204) (#9101), and rabbit anti-ERK1/2 (#9102) were obtained from Cell Signaling Technology (Danvers, MA). Goat anti-Akt (sc-1618) was from Santa Cruz Biotechnology (Dallas, Tx).
Western blot analysis
Proteins were separated on NuPAGE 4–12% Bis-Tris gels (Thermo Fisher Scientific) in MOPS SDS running buffer (Thermo Fisher Scientific) and transferred to PVDF membranes (Immobilon-P; Millipore, Temecula, CA). Membranes were blocked for 1 h with 5% BSA in Tris-buffered saline with 0.1% Tween 20 (TBST) and then incubated with a primary antibody for 2h or overnight in 5% BSA-TBST at 4°C. After washing and incubating with horseradish peroxidase-conjugated secondary antibodies for 1h, the membranes were incubated with chemiluminescence substrate (SuperSignal West Pico or West Femto; Thermo Scientific, Rockford, IL). Images were acquired using a C-DiGit Blot Scanner (LI-COR Biosciences, Lincoln, NE).
Statistical analyses
All experimental data are expressed as means ± S.E. from at least three independent experiments. Single comparisons between groups were performed using Student’s t-test, while multiple group comparisons were analyzed using one-way ANOVA with Bonferroni post hoc tests. p values of <0.05 were considered statistically significant.
Results
Yoda1 induces both Akt and ERK1/2 activation in endothelial cells
Yoda1 has been described as a molecule that mimics the effects of flow in ECs, specifically through Piezo1-activated calcium influx. To determine whether Yoda1 can induce the activation of Akt and ERK1/2, two signaling molecules that are both known to be flow-activated and calcium-mediated, we performed a dose response experiment in which human coronary artery endothelial cells (HCAECs) were treated with increasing concentrations of Yoda1 (1.5, 3.0, and 6.0 µM) for 5 min. Indeed, Yoda1induced the activation of both Akt (Fig 1A) and ERK1/2 (Fig 1B) as reflected by their respective increases in phosphorylation levels compared to DMSO-treated control cells. Phosphorylation of both signaling molecules also increased with increasing concentrations of Yoda1. However, for all subsequent experiments, we utilized the lowest concentration of Yoda1 (1.5 µM) at which statistical significance was achieved.
Figure 1. Yoda1-induced signaling in endothelial cells.
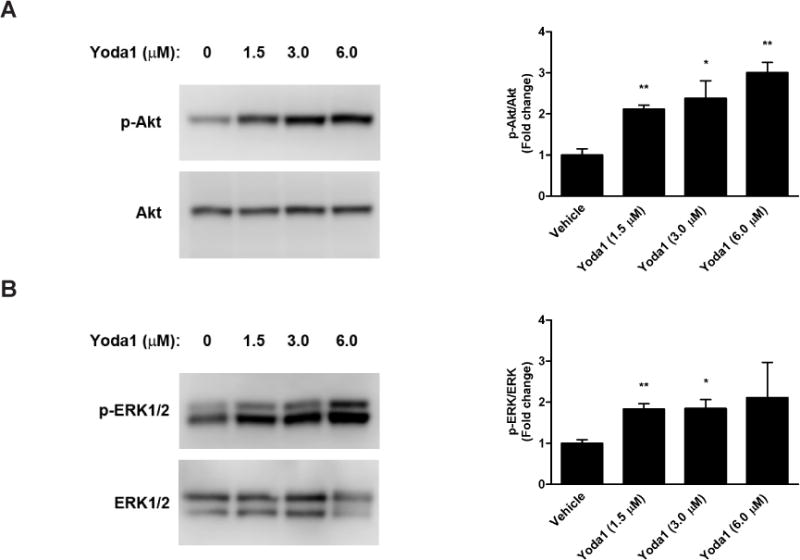
Human coronary artery endothelial cells (HCAECs) were treated with Yoda1 at the indicated concentrations for 5 min. Cell lysates were prepared and analyzed by performing SDS-PAGE followed by immunoblotting. (A) Akt activation was determined by immunoblotting for phosphorylated Akt (S473) and total Akt and performing densitometry of the single band. (B) ERK1/2 activation was determined by immunoblotting for phosphorylated ERK1/2 (T202/Y204) and total ERK1/2 and performing densitometry of the protein doublet. Representative blots from at least 3 independent experiments are shown. Bar graphs represent the quantification as fold change in the ratio of phosphorylated Akt to total Akt (A) or phosphorylated ERK1/2 to total ERK1/2 (B) compared to the DMSO-treated control condition, which is set to 1. The error bars indicate SE. *P<0.05; **P<0.01.
Activation of Akt, but not ERK1/2, by Yoda1 is abrogated by gadolinium (Gd3+)
To determine whether Piezo1 activation is required for Yoda1-induced activation of Akt and ERK1/2 by ECs, we selectively blocked Piezo1 with gadolinium (Gd3+), one of the most commonly-used pharmacological inhibitors of mechanically-activated channels [1,9,10,11]. Addition of Yoda1 (using DMSO as the vehicle) to HCAECs at 1.5 µM for 5 min caused an increase in phosphorylated Akt (83% increase) compared to vehicle-treated cells (Fig. 2A). For cells pre-treated with Gd3+ at 30 µM for 30 min, the level of Akt phosphorylation induced by Yoda1 was virtually unchanged (2% decrease) compared to vehicle-treated cells that were not pre-treated. However, there was still an increase in Yoda1-induced Akt phosphorylation (62% increase) when compared to vehicle-treated cells that were pre-treated, which is due to lower baseline phospho-Akt levels (39% decrease) caused by Gd3+ pretreatment alone. Similarly, Yoda1 stimulation resulted in increased phospho-ERK1/2 (94% increase) levels in control cells (Fig. 2B). Although phospho-ERK1/2 levels in cells pre-treated with Gd3+ prior to Yoda1 stimulation were down (68% increase) when compared to control cells with no pre-treatment, they were increased (90% increase) when more appropriately compared to pre-treated control cells. This is due to the fact that Gd3+ alone caused a slight decrease in phosphorylated ERK1/2 (12% decrease).
Figure 2. Inhibition of Piezo1 using gadolinium.
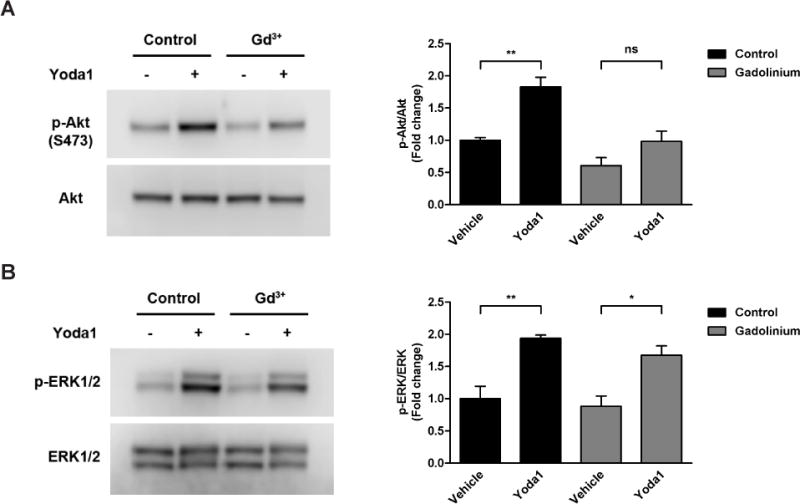
HCAECs were pre-incubated with vehicle control or gadolinium (Gd3+) at 30 µM for 30 min prior to stimulation with Yoda1 (1.5 µM) for 5 min. Immunoblotting was then performed on cell lysates using antibodies against phosphorylated Akt (S473) and total Akt to assess the level of Akt activation (A) or against phosphorylated ERK1/2 (T202/Y204) and total ERK1/2 to assess the level of ERK1/2 activation (B). Representative blots from 3 independent experiments are shown. Bar graphs represents the quantification as fold change in the ratio of phosphorylated Akt to total Akt (A) or phosphorylated ERK1/2 to total ERK1/2 (B) compared to the control static condition, which is set to 1. The error bars indicate SE. *P<0.05; **P<0.01.
Ruthenium red (RR) effectively blocks Yoda1-induced activation of Akt
We next investigated the effects of ruthenium red (RR), another well known Piezo1 antagonist [1,2,9]. As expected, Yoda1 treatment resulted in a robust increase in Akt phosphorylation in control HCAECs (2-fold increase)(Fig. 3A). However, in cells pre-treated with RR (30 µM; 30 min), Akt phosphorylation induced by Yoda1 addition was drastically reduced compared to either vehicle-treated control cells with no pre-treatment (28% increase) or RR pre-treated cells to which DMSO was added (31% increase). Yoda1 treatment caused a 2.1-fold increase in ERK1/2 phosphorylation in control cells in the absence of RR (Fig. 3B). In the presence of RR, ERK1/2 was not as robustly activated by Yoda1, but was still augmented when compared to either vehicle-treated cells with no pre-treatment (1.8-fold increase) or vehicle-treated cells pre-treated with RR (1.7-fold).
Figure 3. Inhibition of Piezo1 using ruthenium red.
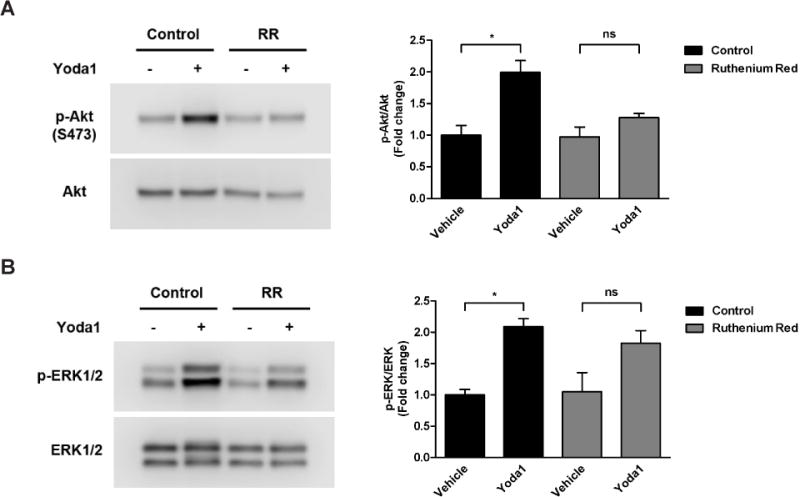
HCAECs were pre-incubated with vehicle control or ruthenium red (RR) at 30 µM for 30 min prior to stimulation with Yoda1 (1.5 µM) for 5 min. Immunoblotting was then performed on cell lysates using antibodies against phosphorylated Akt (S473) and total Akt to assess the level of Akt activation (A) or against phosphorylated ERK1/2 (T202/Y204) and total ERK1/2 to assess the level of ERK1/2 activation (B). Representative blots from 3 independent experiments are shown. Bar graphs represents the quantification as fold change in the ratio of phosphorylated Akt to total Akt (A) or phosphorylated ERK1/2 to total ERK1/2 (B) compared to the control static condition, which is set to 1. The error bars indicate SE. *P<0.05.
GsMTx4, a potent blocker of Piezo1, does not inhibit Yoda1-induced Akt or ERK1/2 phosphorylation
To test the inhibitory effects of the most selective and potent Piezo1 channel blocker to date, we pre-treated HCAECs with the spider venom-derived peptide, Grammostola spatulata mechanotoxin 4 (GsMTx4)[12], at 10 µM for 30 min prior to stimulation with Yoda1. Surprisingly, our results showed that pre-treatment with GsMTx4 had no significant difference on Yoda1-induced phosphorylation of either Akt (Fig. 4A) or ERK1/2 (Fig. 4B). Specifically, Yoda1 stimulation resulted in increased Akt phosphorylation in the absence (3.4-fold increase) and presence (2.9-fold increase) of GsMTx4 pretreatment when compared to their respective controls (Fig. 4A). Likewise, Yoda1-induced phospho-ERK1/2 levels were dramatically elevated both with (3.2-fold increase) and without (3.9-fold increase) GsMTx4 pre-treatment (Fig. 4B).
Figure 4. Inhibition of Piezo1 using GsMTx4.
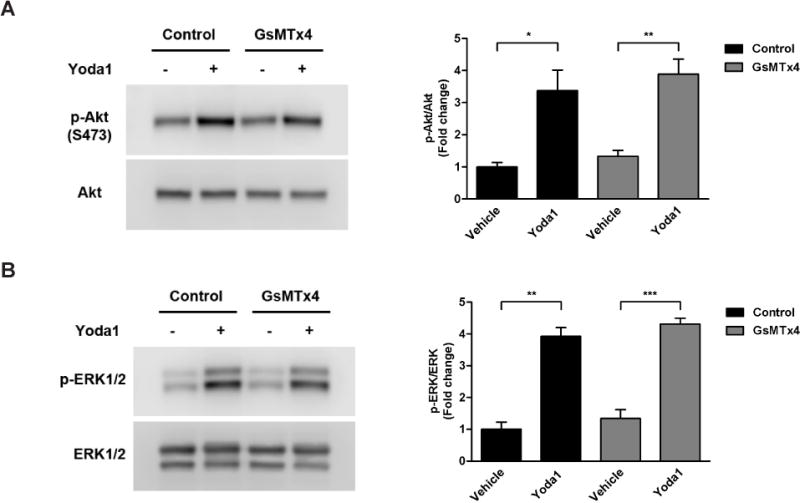
HCAECs were pre-incubated with vehicle control or the Piezo1 channel inhibitor, GsMTx4 (10 µM), for 30 min prior to stimulation with Yoda1 (1.5 µM) for 5 min. Immunoblotting was then performed on cell lysates using antibodies against phosphorylated Akt (S473) and total Akt to assess the level of Akt activation (A) or against phosphorylated ERK1/2 (T202/Y204) and total ERK1/2 to assess the level of ERK1/2 activation (B). Representative blots from 3 independent experiments are shown. Bar graphs represents the quantification as fold change in the ratio of phosphorylated Akt to total Akt (A) or phosphorylated ERK1/2 to total ERK1/2 (B) compared to the control static condition, which is set to 1. The error bars indicate SE. *P<0.05; **P<0.01; ***P<0.001.
Discussion
It is widely accepted that calcium (Ca2+) is an important signaling molecule in cells. In endothelial cells, changes in intracellular Ca2+ can lead to the production and release of vasoactive factors, such as nitric oxide (NO), that affect a wide range of vascular functions, including EC permeability and vascular tone. The Akt and ERK1/2 signaling pathways, which mediate cell proliferation and survival, are also activated by changes in intracellular Ca2+. Membrane ion channels play a critical role in regulating this influx of Ca2+ and the specific activation and opening of Piezo channels, whether through mechanical (i.e. shear stress) or chemical means (i.e. Yoda1), can trigger an influx of Ca2+ causing its various downstream signaling pathways to also be initiated. Our results showed that both Akt and ERK1/2 phosphorylation are increased in endothelial cells in response to Yoda1 treatment, suggesting that Piezo channels have been activated.
It has recently been shown that Akt phosphorylation is induced in human umbilical artery endothelial cells (HUAECs) exposed to 1 µM Yoda for 5 minutes [8]. Moreover, it was shown that Yoda1-induced Akt phosphorylation was strongly reduced by knockdown of Piezo1 with siRNA, which implies that Piezo1 mediates this response in HUAECs. Although our results confirm that Yoda1 can induce Akt activation in ECs regardless of its origin, we could not observe a significant decrease in Akt phosphorylation in cells pre-treated with the Piezo1 antagonist, GsMTx4, prior to exposure to Yoda1. It should be noted that here that siRNA knockdown of Piezo1 has been reported to decrease the expression levels of eNOS (~40% decrease) in ECs [3]. Consequently, decreased eNOS expression may cause a decrease in NO production and release from cells in response to Yoda1 stimulation. Since NO is known to activate PI3K-Akt [13,14], a decrease in phosphorylated Akt in Yoda-induced ECs in which Piezo1 is knocked down via siRNA may be partly attributed to a decrease in eNOS expression.
The fact that Yoda1 can activate Piezo1 in the absence of other cellular components other than a cell membrane [2], suggests that it may directly interact with and activate Piezo1. However, this does not preclude Yoda1 from interacting and activating non-Piezo channels, particularly in endothelial cells. It is possible that Yoda1 activates Akt through Piezo1-independent mechanisms and that both gadolinium and ruthenium red can block Yoda1-induced phosphorylation of Akt because they are broader spectrum inhibitors than GsMTx4. For example, Gd3+ has been demonstrated to block other stretch-activated ion channels [15] as well as transient receptor potential (TRP) channels, such as the TRPML3 channel [16]. Ruthenium red is also known to block TRP channels and is a well described antagonist of ryanodine receptors, a class of intracellular Ca2+ release channel expressed by skeletal and cardiac muscle [17]. All except two of the 28 identified TRP channels allow Ca2+ influx and at least 19 isoforms are expressed in vascular endothelial cells [18]. Therefore, it is possible that in addition to activating Piezo1 in ECs, Yoda1 also activates one or more TRP channels, which leads to increased intracellular Ca2+, which in turn contributes to the activation of both Akt and ERK1/2.
To our knowledge, this is the first study to show that Yoda1 stimulation increases ERK1/2 phosphorylation in ECs or in any cell type for that matter. However, the lack of a substantial blocking effect of three widely-used antagonists against Piezo1 (i.e. ruthenium red, gadolinium, and GsMTx4) suggests that Yoda1-induced ERK1/2 phosphorylation is not mediated specifically through this channel. Our findings differ from that of two recent studies. First, it has been demonstrated that ruthenium red significantly inhibits ERK1/2 activation in dental stem cells by low-intensity pulsed ultrasound (LIPUS) and therefore concluded that it is regulated by Piezo1 [19]. Secondly, it has been reported that stretch induces calcium-dependent activation of ERK1/2 in epithelial cells that is Piezo1-mediated, since it can be blocked by gadolinium addition [20]. One likely explanation is that Yoda1 differs in its ability to activate Piezo1 and potentially other channels than either LIPUS or cell stretching. Our data suggests that Yoda1 has the ability to stimulate ERK1/2 activation in ECs through other calcium channels and/or mechanisms.
A previous study showed that at a concentration of 1.5 µM, which was the concentration used in the present study, Yoda1 does not trigger a Ca2+ response in human embryonic kidney (HEK) 293T cells transfected with human Piezo1 [6]. In fact, there was no appreciable increase in Ca2+ influx in human Piezo1-transfected HEK293T cells until they were stimulated with Yoda1 concentrations at and above 6 µM, which implies that activation of Piezo1 by Yoda1 is concentration-dependent. In another study, Yoda1 at a lower concentration (1 µM) was shown to induce a robust Ca2+ response in human umbilical endothelial cells (HUVECs)[8]. This discrepancy may be explained by the possibility that Piezo1 was not activated by Yoda1 in the latter study, and that there are inherent differences in the endogenous expression of other ion channels that control Ca2+ influx in ECs compared to HEK293T cells, thereby allowing Yoda1 to still cause a Ca2+ response. TRPV4 is one such Ca2+-permeable channel that is highly expressed by ECs, but not expressed by HEK293T [21,22]. Interestingly, TRPV4 has been shown to be activated by shear stress and to play a regulatory role in vascular tone [23,24]. TRPM7 is another example of a Ca2+-permeable channel that is expressed in ECs [25], but not reported to be expressed by HEK293T cells. Additionally, it has been shown to rapidly accumulate at the plasma membrane in response to shear stress [26]. Despite ECs having low levels of endogenous TRPM7-like current that is not increased by shear stress [26], TRPM7 channels have been implicated in regulating cell survival [27,28] and cell proliferation via the ERK signaling pathway [25,29].
Taken together, our results indicate that Yoda1 activates Akt and ERK1/2 signaling pathways in ECs through mechanisms that are not entirely dependent on Piezo1 activation. Yoda1 is non-specific in its activation of Piezo1 in ECs with possibly many other unidentified targets. This finding has important implications as Yoda1 and/or Yoda1-like chemical compounds have been proposed as a way to increase blood flow to different tissues or organs in the body through the specific activation of Piezo1 during and perhaps in the absence of physical activity. Future studies on the effects of Yoda1 on the potential activation of other targets, such as the aforementioned TRP channels, are still necessary and may provide further insights on the suitability of Yoda1 in particular as an “exercise pill” that can be administered and taken to mimic exercise and whether it would actually be beneficial.
Supplementary Material
Highlights.
Yoda1 induces activation of both Akt and ERK1/2 in endothelial cells (ECs).
Gadolinium and ruthenium red both block Yoda1-induced Akt phosphorylation.
GsMTx4 does not block Yoda1-induced Akt or ERK1/2 phosphorylation.
Yoda1-induced activation of Akt and ERK1/2 in ECs are Piezo1-independent.
Acknowledgments
None
Grants
This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute MERIT Award R37 HL040696 to J.A.F.
Abbreviations
- EC
endothelial cells
- Gd3+
gadolinium
- RR
ruthenium red
- GsMTx4
Grammostola spatulata mechanotoxin 4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: N.G.P. and J.A.F. conceived study and designed experiments; N.G.P. performed experiments, analyzed data, and wrote manuscript; N.G.P. and J.A.F. edited, revised, and approved final version of manuscript.
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- 1.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, P A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, Kim SE, Schmidt M, Mathur J, Dubin AE, Montal M, Patapoutian A. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature. 2012;483:176–181. doi: 10.1038/nature10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, Sedo A, Hyman AJ, McKeown L, Young RS, Yuldasheva NY, Majeed Y, Wilson LA, Rode B, Bailey MA, Kim HR, Fu Z, Carter DA, Bilton J, Imrie H, Ajuh P, Dear TN, Cubbon RM, Kearney MT, Prasad RK, Evans PC, Ainscough JF, Beech DJ. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515:279–282. doi: 10.1038/nature13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li YS, Chien S, Patapoutian A. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci USA. 2014;111:10347–10352. doi: 10.1073/pnas.1409233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rode B, Shi J, Endesh N, Drinkhill MJ, Webster PJ, Lotteau SJ, Bailey MA, Yuldasheva NY, Ludlow MJ, Cubbon RM, Li J, Futers TS, Morley L, Gaunt HJ, Marszalek K, Viswambharan H, Cuthbertson K, Baxter PD, Foster R, Sukumar P, Weightman A, Calaghan SC, Wheatcroft SB, Kearney MT, Beech DJ. Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat Commun. 2017;8 doi: 10.1038/s41467-017-00429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Syeda R, Xu J, Dubin AE, Coste B, Mathur J, Huynh T, Matzen J, Lao J, Tully DC, Engels IH, Petrassi HM, Schumacher AM, Montal M, Bandell M, Patapoutian A. Chemical activation of the mechanotransduction channel Piezo1. eLife. 2015;4 doi: 10.7554/eLife.07369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beech DJ. Endothelial Piezo1 channels as sensors of exercise. J Physiol. 2017 doi: 10.1113/JP274396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J Clin Invest. 2016;126:4527–4536. doi: 10.1172/JCI87343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cinar E, Zhou S, DeCourcey J, Wang Y, Waugh RE, Wan J. Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc Natl Acad Sci USA. 2015;112:11783–11788. doi: 10.1073/pnas.1507309112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caldwell RA, Clemo HF, Baumgarten CM. Using gadolinium to identify stretch-activated channels: technical considerations. Am J Physiol. 1998;275:C619–621. doi: 10.1152/ajpcell.1998.275.2.C619. [DOI] [PubMed] [Google Scholar]
- 11.Yang XC, Sachs F. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science. 1989;243:1068–1071. doi: 10.1126/science.2466333. [DOI] [PubMed] [Google Scholar]
- 12.Bae C, Sachs F, Gottlieb PA. The mechanosensitive ion channel Piezo1 is inhibited by the peptide GsMTx. Biochemistry. 2011;50:6295–6300. doi: 10.1021/bi200770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujita M, Imadome K, Endo S, Shoji Y, Yamada S, Imai T. Nitric oxide increases the invasion of pancreatic cancer cells via activation of the PI3K-AKT and RhoA pathways after carbon ion irradiation. FEBS Lett. 2014;588:3240–3250. doi: 10.1016/j.febslet.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 14.Tejedo JR, Cahuana GM, Ramírez R, Esbert M, Jiménez J, Sobrino F, Bedoya FJ. nitric oxide triggers the phosphatidylinositol 3-kinase/Akt survival pathway in insulin-producing RINm5F cells by arousing Src to activate insulin receptor substrate-1. Endocrinology. 2004;145:2319–2327. doi: 10.1210/en.2003-1489. [DOI] [PubMed] [Google Scholar]
- 15.Ruknudin A, Sachs F, Bustamante JO. Stretch-activated ion channels in tissue-cultured chick heart. Am J Physiol. 1993;264:H960–972. doi: 10.1152/ajpheart.1993.264.3.H960. [DOI] [PubMed] [Google Scholar]
- 16.Nagata K, Zheng L, Madathany T, Castiglioni AJ, Bartles JR, García-Añoveros J. The varitint-waddler (Va) deafness mutation in TRPML3 generates constitutive, inward rectifying currents and causes cell degeneration. Proc Natl Acad Sci USA. 2008;105:353–358. doi: 10.1073/pnas.0707963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu L, Tripathy A, Pasek DA, Meissner G. Ruthenium red modifies the cardiac and skeletal muscle Ca(2+) release channels (ryanodine receptors) by multiple mechanisms. J Biol Chem. 1999;274:32680–32691. doi: 10.1074/jbc.274.46.32680. [DOI] [PubMed] [Google Scholar]
- 18.Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97:853–863. doi: 10.1161/01.RES.0000187473.85419.3e. [DOI] [PubMed] [Google Scholar]
- 19.Gao Q, Cooper PR, Walmsley AD, Scheven BA. Role of Piezo Channels in Ultrasound-stimulated Dental Stem Cells. J Endod. 2017;43:1130–1136. doi: 10.1016/j.joen.2017.02.022. [DOI] [PubMed] [Google Scholar]
- 20.Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF, Krishnegowda V, Rosenblatt J. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature. 2017;543:118–121. doi: 10.1038/nature21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilius B, Droogmans G, Wondergem R. Transient receptor potential channels in endothelium: solving the calcium entry puzzle? Endothelium. 2003;10:5–15. doi: 10.1080/10623320303356. [DOI] [PubMed] [Google Scholar]
- 22.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2:695–702. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 23.Gao X, Wu L, O’Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and-independent pathways. J Biol Chem. 2003;278:27129–27137. doi: 10.1074/jbc.M302517200. [DOI] [PubMed] [Google Scholar]
- 24.Köhler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T, Hoyer J. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. 2006;26:1495–1502. doi: 10.1161/01.ATV.0000225698.36212.6a. [DOI] [PubMed] [Google Scholar]
- 25.Inoue K, Xiong ZG. Silencing TRPM7 promotes growth/proliferation and nitric oxide production of vascular endothelial cells via the ERK pathway. Cardiovasc Res. 2009;83:547–557. doi: 10.1093/cvr/cvp153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oancea E, Wolfe JT, Clapham DE. Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res. 2006;98:245–253. doi: 10.1161/01.RES.0000200179.29375.cc. [DOI] [PubMed] [Google Scholar]
- 27.Cheng H, Feng JM, Figueiredo ML, Zhang H, Nelson PL, Marigo V, Beck A. Transient receptor potential melastatin type 7 channel is critical for the survival of bone marrow derived mesenchymal stem cells. Stem Cells Dev. 2010;19:1393–1403. doi: 10.1089/scd.2009.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wykes RC, Lee M, Duffy SM, Yang W, Seward EP, Bradding P. Functional transient receptor potential melastatin 7 channels are critical for human mast cell survival. J Immunol. 2007;179:4045–4052. doi: 10.4049/jimmunol.179.6.4045. [DOI] [PubMed] [Google Scholar]
- 29.Baldoli E, Maier JA. Silencing TRPM7 mimics the effects of magnesium deficiency in human microvascular endothelial cells. Angiogenesis. 2012;15:47–57. doi: 10.1007/s10456-011-9242-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.