

Abstract
The PPARG gene encoding the nuclear receptor PPAR-gamma is activated in bladder cancer, either directly by gene amplification or mutation, or indirectly by mutation of the RXRA gene which encodes the heterodimeric partner of PPAR-gamma. Here we show that activating alterations of PPARG or RXRA lead to a specific gene expression signature in bladder cancers. Reducing PPARG activity, whether by pharmacologic inhibition or genetic ablation, inhibited proliferation of PPARG-activated bladder cancer cells. Our results offer a preclinical proof of concept for PPARG as a candidate therapeutic target in bladder cancer.
Keywords: RXRA, PPARG, Genitourinary cancers: bladder, Nuclear receptors: structure and function
Introduction
Bladder cancer is the fifth most commonly diagnosed cancer in the United States with an annual incidence of 80,000 cases and an annual mortality of 15,000. Most patients are diagnosed with non-invasive/superficial urothelial carcinoma of the bladder and respond well to transurethral resection, Bacillus Calmette-Guérin (BCG) immunotherapy, and regular cystoscopic surveillance, with a 5-year survival rate of 96%. Patients diagnosed with muscle-invasive disease have a poorer prognosis, however, with a 5-year survival rate of 35–70%, depending upon the extent of invasion, regional and systemic metastases, and response to therapy.
Recently several immune checkpoint blockade drugs have been FDA-approved for treatment of urothelial carcinoma, including atezolizumab, an anti-PD-L1 antibody, in 2016 (1); nivolumab and pembrolizumab, anti-PD-1 antibodies, in 2017 (2) ; and avelumab and durvalumab, anti-PD-L1 antibodies, in 2017 (3,4). These approvals mark the first new drugs for metastatic bladder cancer in over 20 years. However, the objective response rates (defined using RECIST v1.1) were relatively low in these clinical trials, with 15–20% overall response rate and 26–28% response rate in PD-L1-positive patients (1,2). Beyond checkpoint inhibitors, a number of therapies targeting specific genetic alterations, including FGFR3 alterations, mTOR pathway alterations, and DNA repair deficiencies associated with ERCC2 alterations (5–8), are under clinical evaluation.
We explored large-scale cancer genome datasets derived from patients with muscle-invasive bladder cancer with the purpose of identifying novel therapeutic targets. Hotspot mutations (p.S427F/Y) in the retinoid X receptor alpha (RXRA) gene are present in ~5% of bladder cancer samples (9,10). RXRA is a well characterized ligand-activated nuclear receptor that serves as a requisite heterodimer partner for ~30 nuclear receptors, including PPARA, PPARG, PPARD, RARA, RARB, VDR, TR, LXR, and PXR (11), suggesting that recurrent mutations in RXRA could impact the formation and/or function of these heterodimers and change the expression of their downstream target genes. Previous reports have shown that cancer samples containing RXRA p.S427F/Y mutations are associated with enhanced expression of genes involved in adipogenesis and lipid metabolism, including ACOX1, ACSL1, ACSL5, FABP4, and HMGS2 (9). These genes are targets of peroxisome proliferator-activated receptor gamma (PPARG), a member of the PPAR subfamily of nuclear receptors.
Interestingly, the PPARG gene is focally amplified in 15% of bladder cancer samples (Supplementary Fig. 1A and (12)). This amplification is strongly correlated with expression of PPARG (Supplementary Fig. 1B) as well as expression of PPARG target genes and luminal differentiation markers such as GATA3, UPK2, ACOX1, and UPK1A (Supplementary Fig. 1C and (13–16)). PPARG is a master regulator of adipocyte differentiation and controls expression of a large set of genes involved in lipid and glucose homeostasis (12). In contrast to PPARG’s well-characterized activity in adipocytes, however, little is known about its function in the urinary bladder and in the pathogenesis of bladder cancer.
The potential risk of bladder cancer upon activation of the PPAR subfamily of nuclear receptors is controversial, and was first suggested following rodent toxicity studies testing anti-diabetic PPAR agonists in which numerous glitazar-class PPARA/PPARG dual agonist compounds were associated with an increased incidence of bladder cancer (17). In terms of more selective PPARG agonists, also anti-diabetic drugs with insulin sensitizing activity, the carcinogenic effect in rodents was also observed with pioglitazone, which has weak PPARA activity (18), but not with rosiglitazone, which is highly selective for PPARG (17,19). In a more sensitive, chemically-induced model using the carcinogen, 4-hydroxybutyl (butyl) nitrosamine (OH-BBN), rosiglitazone was found to potentiate urinary bladder carcinogenesis in rats compared to OH-BBN alone (20). It was originally hypothesized that the effects of PPARA/PPARG dual agonists on promoting bladder cancer was rodent-specific due to indirectly causing calcium crystal formation in the bladder resulting in urolithiasis (21). Numerous studies have since examined the incidence of bladder cancer in humans following the clinical use of these compounds, with some detecting an increased risk and others concluding there is no increased risk (22,23). The most comprehensive retrospective study to date showed an increase in the hazard ratio for bladder cancer with long-term, high-dose treatment with pioglitazone (24).
Based on the combined genetic and pharmacologic evidence, we hypothesized that activation of PPARG is oncogenic in the transitional epithelial cells of the bladder. We evaluated this by investigating the biological impact of ectopic expression of mutant alleles of RXRA and PPARG, pharmacologic ablation of RXRA/PPARG signaling using small molecule perturbagens, and genetic ablation of RXRA and PPARG using CRISPR/Cas9 gene knockouts. Our results demonstrate an oncogenic role for PPARG in the development of luminal bladder cancer, revealing a novel axis that could be exploited for the development of targeted therapies for this disease.
Materials and Methods
Chemicals
Rosiglitazone, pioglitazone, tesaglitazar, GW9662, T0070907, UVI3003 and SR1664 were purchased from Tocris Bioscience (Minneapolis, MN). SR2595 and SR10221 were synthesized according to published methods (25). GW1929, BADGE (Bisphenol A diglycidyl ether), SR202 and GW6471 were purchased from Sigma-Aldrich (St. Louis, MO).
Cell lines
The UM-UC-9 cell line was purchased from Sigma-Aldrich (St. Louis, MO). All other cell lines were obtained from the Cancer Cell Line Encyclopedia (Broad Institute, Cambridge MA), which obtained them from the original source and performed cell line authentication (26). To reduce bias from cell culture medium, all cell lines were maintained in MEMalpha medium supplemented with 10% Tet-system approved fetal bovine serum (FBS) (Clontech).
Ectopic cDNA expression
Wild type ORFs for RXRA and PPARGv1 were obtained from the Genomics Perturbation Platform (Broad Institute, Cambridge MA) in pDONR Gateway cloning vectors. Various mutant alleles were generated using QuikChange Site-Directed Mutagenesis (Agilent, Santa Clara CA). ORFs were then subcloned into lentiviral expression vectors using Gateway LR Clonase and infectious lentiviral particles were generated using standard procedures.
SW780 bladder cancer cell line was transduced with a lentiviral vector encoding the specified RXRA or PPARG ORF under control of constitutive CMV or EF1α promoter, and stable pools were generated following selection for blasticidin-resistance. Cell lines were maintained for at least 7 days following selection prior to expansion for further analysis.
RNA sequencing analysis
RNA was isolated using RNEasy (Qiagen, Germantown, MD) and an RNA sequencing library was prepared using NEBNext Ultra Directional RNA Library Prep Kit for Illumina and NEBNext Multiplex Oligos for Illumina (New England BioLabs). Sample analysis was performed using a MiSeq System and reagent kits (Illumina, San Diego, CA) according to the manufacturers protocol. Sequence data was analyzed using Firehose (Broad Institute, Cambridge MA) to map transcripts and calculate RPKM (SW780 cDNA expression) or TPM (UM-UC-9).
For UM-UC-9 RNA sequencing experiment, the resulting reads were used to calculate transcript abundance in units of TPM (transcripts per million) (27), which were then adjusted using TMM normalization (28) for comparison. Log fold-change and Mann-Whitney test significance was used to identify differentially expressed genes between the agonist and inverse-agonist-treated samples.
Western blot analysis
Cells were grown in 6-well plates and harvested using Complete Lysis-M with protease and phosphatase inhibitors (Roche Applied Science, Germany). Western blots were performed using standard protocols with semi-dry transfer Trans-Blot® SD (Bio-Rad Hercules, CA), Li-Cor Odyssey Blocking buffer, and imaging with LiCor Odyssey Imaging System (LI-COR Lincoln, NE). The anti-PPARG C26H12, anti-PPARG 81B8, anti-FABP4 D25B3, and anti-CEACAM5 CB30 antibodies were obtained from Cell Signaling Technology (Beverly, MA). The anti-ACSL5 ab57210 and anti-HMCGS2 EPR8642 antibodies were obtained from Abcam (Cambridge, MA). The anti-VCL (vinculin) V9264 antibody was obtained from Sigma-Aldrich (St. Louis MO). All primary antibodies were tested at 1:1000 dilutions, with the exception of anti-VCL, which was tested at a 1:5000 dilution. Secondary goat anti-mouse 926-68020, and goat-anti-rabbit 926-32211 antibodies were obtained from Li-Cor Biosciences (Lincoln, NE) and used at 1:15,000 dilutions.
RT112-FABP4-NLucP reporter gene assay
Reporter cell line was generated by engineering the NanoLuc gene into the 3’ UTR of FABP4 in RT112/84 cells using CRISPR/Cas9 guided genome engineering. Single cell clones were isolated and the clone with the widest dynamic range selected for use. Assays were performed by seeding 384-well plates with 10,000 cells per well in MEMalpha containing 10% FBS and dosing compounds at indicated concentration using the HP D300 digital dispenser (HP/Tecan). 18–24 hours after dosing with compound, cells were assayed using NanoGlo Luciferase Assay Reagent (Promega Madison, WI) and plates were read using an EnVision Multilabel Reader (PerkinElmer Waltham, MA).
Biochemical assays
The LanthaScreen TR-FRET PPAR gamma Competitive Binding Assay and LanthaScreen TR-FRET PPAR gamma Coactivator Assay were obtained from ThermoFisher Scientific (Waltham, MA). Assays were performed according to the manufacturer’s protocol. In order to assay inverse-agonism, the TR-FRET PPAR gamma Coactivator assay was modified by the use of fluorescently labeled corepressor peptides (NCoR1 ID2 peptide, SMRT ID2 peptide) in place of TRAP220 coactivator peptide to convert from agonist mode (coactivator recruitment) into inverse-agonist mode (corepressor recruitment).
Proliferation assays
To enable cell-counting experiments, cell lines were transduced with a lentiviral vector encoding nuclear-targeted GFP, TagGFP2-H2B (Evrogen, Moscow, Russia), and stable pools generated following selection for puromycin-resistance. Cell lines were maintained for at least 7 days following selection prior to expansion and seeding into 96-well or 384-well plates for further analysis. For kinetic proliferation assays, 96-well plates (n = 4 per condition) were imaged and counted every two hours using IncuCyte Zoom (Essen BioScience, Ann Arbor MI). Media and compounds were replaced approximately every 3–4 days. For end-point assays to measure dose-response, cells were plated in 384-well plates, dosed with compound, and upon reaching approximately 70–90% confluence in control wells, cells were either counted using fluorescence imaging (IncuCyte Zoom) or incubated with CyQuant (ThermoFisher, Waltham, MA) at the indicated time and plates were read with a fluorescent plate reader.
CRISPR/Cas9 genetic dependency studies
Cell lines were first transduced with a lentiviral vector encoding hSpCas9 under control of a tetracycline-inducible CMV promoter (CMV-TO, ThermoFisher Scientific) and stable pools generated following selection for blasticidin-resistance. Following confirmation of regulated Cas9 expression by Western blot analysis, the cells were transduced with a lentiviral vector encoding an sgRNA under control of a tetracycline-inducible H1 promoter (H1-TO, ThermoFisher Scientific) and double-stable pools were generated following selection for puromycin-resistance. In addition to providing for regulated sgRNA expression, these lentiviral vectors also constitutively express one of three nuclear-targeted fluorescent proteins to enable unambiguous identification of transduced cells in subsequent cell counting experiments: YFP-expressing vectors were used for sgRNAs targeting PPARG; CFP-expressing vectors were used for sgRNAs targeting non-essential control genes, and RFP-expressing vectors were used for sgRNAs targeting essential control genes. This approach is described in more detail elsewhere (Strathdee et al., manuscript in preparation).
We evaluated 6–8 different sgRNAs per gene of interest using Western blot analysis in order to identify 2–3 highly active, doxycyline-inducible guides targeting PPARG (sgPPARG-3: GTCTTCTCAGAATAATAAGG, sgPPARG-6: GTTTCAGAAATGCCTTGCAG) for use in our experiments. We used KIF11 (sgKIF11-3: GGTGGTGGTGAGATGCAGGT) as an essential control gene, and an sgRNA targeting PPARG intronic sequence (sgPPARG-21: GATACTGCTGCATTAGACCAG) was used as a non-essential controls.
Following generation of stable pools of for each sgRNA were combined in equivalent numbers within replicate wells of 6-well plate and doxycycline was added to one of the replicates to induce Cas9 and sgRNA. Cells were passaged every 3–5 days and one replicate maintained under doxycycline induction, with second replicate maintained in the absence of doxycycline. During each passage, four replicate wells were passaged into 96-well plates for fluorescent imaging and fixed with methanol for imaging 1–3 days after passage into 96-well plates. Changes in relative abundance of cells containing the on-test sgRNA, non-essential sgRNA, and essential sgRNA were thus followed by comparing relative abundance of cells based on fluorescent label (Yellow, Cyan, Red) through serial passages for a period of 28 days. Cells with stably transduced TREx inducible vectors were all maintained continuously in MEMalpha medium containing 10% Tet-system approved FBS (Clontech).
Data availability
Bladder cancer incidence and survival data was obtained from Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, et al. November 2015 SEER data submission, posted to the SEER web site, April 2016. SEER Cancer Statistics Review, 1975–2013. National Cancer Institute <http://seer.cancer.gov/csr/1975_2013/>.
The provisional TCGA muscle-invasive urothelial carcinoma data is available from the Broad Institute TCGA Genome Data Analysis Center. Analysis-ready standardized TCGA data from Broad GDAC Firehose 2016_01_28 run. https://doi.org/10.7908/C11G0KM9. CCLE Affymetrix U133+2 arrays mRNA expression data is available through https://portals.broadinstitute.org/ccle/ (26). RNA sequencing data is available through the National Center for Biotechnology Information BioProject accession # PRJNA396067.
Results
PPARG signaling is activated by ectopic expression of RXRA p.S427F/Y and PPARG p.T447M mutant alleles
To assess the biological effects of mutations in RXRA and PPARG, we ectopically expressed cDNAs encoding wild-type and mutant alleles of each gene in the SW780 bladder cancer cell line, which is wild-type for RXRA and PPARG (26). RXRA hotspot mutant alleles p.S427F and p.S427Y, as well as a PPARG mutant allele found in RT4 cells, p.T447M in PPARG isoform 1, NP_005028, also commonly referred to as PPARG p.T475M in isoform 2, NP_056953.2, were selected for initial testing. We performed RNA sequencing on parental SW780 cells in comparison with cells expressing wild-type or mutant PPARG or RXRA alleles. SW780 cells with ectopic expression of RXRA p.S427F/Y and PPARG p.T447M mutant alleles demonstrated up-regulation of canonical PPARA and PPARG target genes such as ACSL5, HMGCS2, and FABP4 (Fig. 1A). Immunoblot assays confirmed significant up-regulation of several of the corresponding proteins in SW780 cells expressing mutant RXRA and PPARG alleles (Fig. 1B) even though parental SW780 cells appear to have elevated expression of PPARG and PPARG target genes when compared to other bladder cancer cell lines (Fig. 1C). These data are consistent with the observations of increased expression of these PPAR targets in patient bladder tumors bearing RXRA hotspot mutations (9).
Figure 1. PPARG signaling is activated by ectopic expression of RXRA p.S427F/Y and PPARG p.T447M mutant alleles.
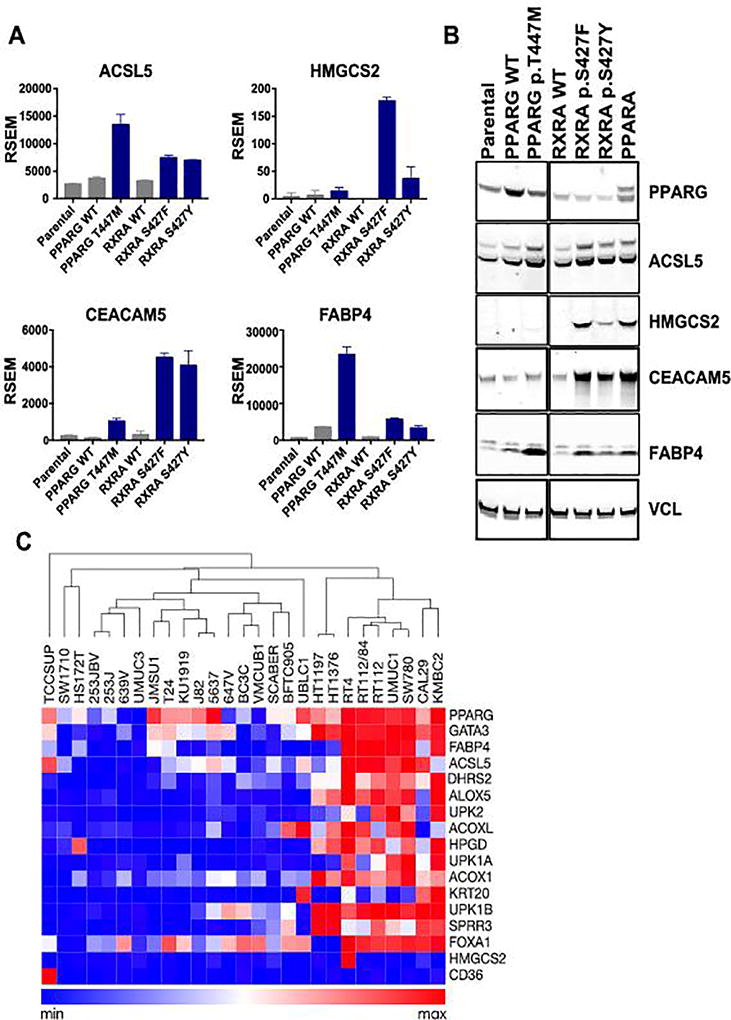
A, RNA expression (RSEM) of selected genes that were differentially expressed when comparing parental SW780 cells, cells with ectopic expression of wild-type RXRA and PPARG, and cells expressing mutant alleles of RXRA and PPARG. B, Immunoblot analysis of lysates from SW780 cells ectopically expressing RXRA and PPARG wild-type and activating mutant alleles. C, Heatmap of Cancer Cell Line Encyclopedia (CCLE) gene-centric RMA-normalized mRNA expression data (26) across bladder cancer cell lines indicating luminal differentiation markers and PPARG target genes. The samples were grouped by hierarchical clustering of columns using the Morpheus software package (https://software.broadinstitute.org/morpheus/).
We also noted a discrete set of genes up-regulated by RXRA p.S427F/Y, such as HMGSC2 and CEACAM5, which were minimally regulated by any of the PPARG alleles. In follow-up immunoblot studies, we included wild-type PPARA and observed that both HMGCS2 and CEACAM5 were selectively upregulated upon ectopic expression of PPARA, but not PPARG (Fig. 1B), indicating that these are likely PPARA targets. We also observed more pronounced effects of RXRA p.S427F compared to the p.S427Y allele on gene expression of HMGCS2, FABP4, and others by RNA sequencing (Fig. 1A), confirmed on the protein level by immunoblot assays (Fig. 1B). This may indicate a stronger phenotype for RXRA p.S427F, which is consistent with the higher frequency of this allele in bladder cancer patients (9). Ectopic expression of other RXRA mutant alleles, selected based on apparent clusters (eg. p.P231L, p.E233K), and sequence proximity to S427 (p.R421C, p.R421H, p.R426H, p.G429Y) from pan-cancer genomics analysis (29), had no activity in these assays (Supplementary Fig. 2).
PPARG inverse-agonists decrease target gene expression in PPARG-activated cell lines
The study of the phenotype of PPARG-activating genome alterations is facilitated by a wide variety of compounds that modulate PPARG activity, including agonists, antagonists, and inverse agonists. PPARG agonists increase recruitment of coactivators such as CREBBP, PGC1α (PPARGC1A), and MED1 to the RXRA-PPARG complex, leading to increased expression of target genes (Supplementary Fig. 3 - top) (30). PPARG inverse-agonists recruit co-repressors such as NCOR1, NCOR2, and HDAC3, leading to a decrease in basal expression of target genes (Supplementary Fig. 3 – bottom) (30). In contrast, PPARG antagonists have minimal effects on basal receptor function but are able to prevent both agonists and inverse-agonists from binding the receptor, thereby blocking their effects (Supplementary Fig. 3) (30).
To evaluate the pharmacological inhibition of PPARG in bladder cancer cell lines, we tested the impact of known PPARG modulating compounds in gene expression assays. In preliminary experiments across a panel of PPARG-activated bladder cancer cell lines (Fig. 1C) we found that dosing bladder cancer cell lines with T0070907, a PPARG inverse-agonist (31), but not GW9662, a PPARG antagonist (32), was able to reduce expression of the canonical PPARG target gene, FABP4 (Supplementary Fig. 4). Although these compounds have similar potency and selectivity profiles and share a remarkably similar chemical structure (Supplementary Table 1), they behaved quite differently in cells.
We engineered an FABP4 reporter cell line to enable higher throughput assays with which to evaluate PPARG transactivation. Briefly, the NanoLuc luciferase reporter gene was inserted into the 3’-UTR of the FABP4 gene in the PPARG-activated RT112/84 bladder cancer cell line using CRISPR/Cas9 mediated homology-directed repair (Supplementary Fig. 5). Since many compounds that target nuclear receptors are selective modulators which have context-dependent activity profiles (30), we used this reporter cell line to characterize the activity of 13 previously characterized PPARG agonists, antagonists, and inverse-agonists (18,25,31–40). The full PPARG agonists, including rosiglitazone, pioglitazone, tesaglitazar, and GW1929, were able to increase the basal activity of the PPARG reporter from 5.5 to 7.4 fold in this assay (Figure 2A, indicated in red). The partial agonists, including UVI3003 and SR1664, increased the basal activity of the reporter from 1.4 to 3.1 fold (Fig. 2A, indicated in black). The antagonists, including BADGE, SR202, GW9662, and SR2595, had minimal detectable effect on this unstimulated reporter (Fig. 2A, indicated in light blue). The PPARA-selective inverse agonist GW6471 exerted a modest inhibitory effect. Finally, the two inverse-agonists tested, T0070907 and SR10221, reduced basal PPARG reporter activity by 85 to 88 percent (Fig. 2A, indicated in dark blue and green). Interestingly, the closely related structural analogs (Supplementary Table 1) of these compounds, GW9662 and SR2595, respectively, were essentially neutral antagonists in this assay.
Figure 2. PPARG inverse-agonists decrease basal activity by inducing active repression.
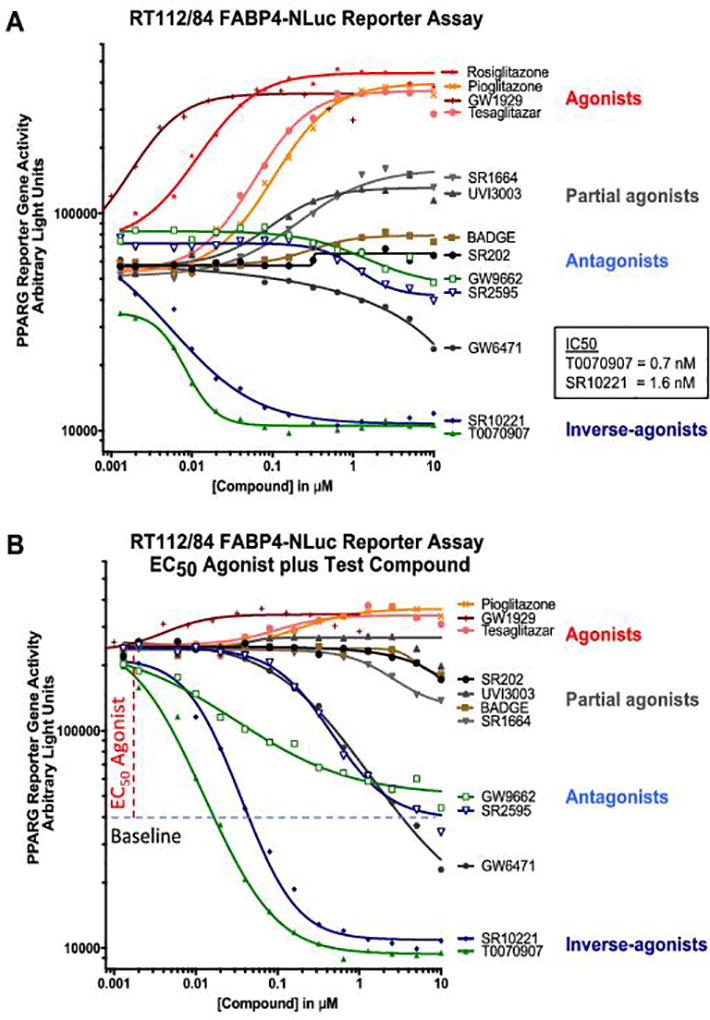
PPARG-activated RT112/84 bladder cancer cell line engineered to contain the NanoLuc gene in the 3’ UTR of a canonical PPARG target gene, FABP4, was used to profile the effect of compounds on PPARG-dependent gene transactivation. A, Dose-response testing of panel of PPARG modulators in the RT112/84 FABP4-NLucP reporter assay under unstimulated conditions to evaluate activity of agonists and inverse-agonists. B, Antagonist mode dose-response testing of panel of PPARG modulators in the RT112/84 FABP4-NLucP reporter assay performed by combined dosing of the PPARG agonist, rosiglitazone at the EC50, 20nM, in combination with a dose-response for test compounds, as indicated.
To evaluate antagonist and inverse-agonist activity in more detail, compounds were tested in the presence of a PPARG agonist, rosiglitazone, for their impact on ligand-activated PPARG reporter activity. Here, the agonists gave little additional stimulation (Fig. 2B, indicated in black). The antagonists GW9662 and SR2595 decreased the agonist-induced signal back to the original baseline (Fig. 2B, indicated in light blue), whereas the inverse-agonists T0070907 and SR10221 further reduced the agonist-induced signal 80–90% below baseline (Fig. 2B, indicated in dark blue and green). These data align with quantitative PCR analysis of FABP4 gene expression in 5637 and UM-UC-9 cells treated with PPARG antagonists and inverse-agonists (Supplementary Fig. 6) and establish that the reporter assay accurately represents the effects of PPARG modulators on target gene expression in bladder cancer cell lines.
We next evaluated the effects of long-term dosing on the transcriptional profile of the PPARG-amplified cell line, UM-UC-9. Briefly, UM-UC-9 cells were treated for 7 days with various PPARG modulators dosed at 500 nM and gene expression was analyzed using RNA sequencing. Many canonical PPARG target genes were amongst the top differentially expressed genes that were upregulated with PPARG agonists and downregulated with inverse-agonists, for example FABP4 and UCP1 (Supplementary Fig. 7). Genes that were more abundantly expressed upon treatment with inverse-agonists included ALPP, SPINK4, and ALDH1A3 (Supplementary Fig. 7). Hierarchical clustering of the gene expression signatures indicated that GW9662 and SR2595 cluster with vehicle-treated controls. Therefore, the PPARG inverse-agonists, T0070907 and SR10221, are clearly biologically distinct from the antagonists, GW9662 and SR2595, in bladder cancer cells. While both SR2595 and SR10221 were described as PPARG inverse-agonists when tested in mouse 3T3-L1 cells (25), our studies using human bladder cancer cells suggest that SR10221, but not SR2595, behaves as an inverse-agonist in these cells (Fig. 2 and Supplementary Figs. 6–7). In summary, we have characterized the in vitro pharmacological properties of known PPARG modulators and have identified two distinct chemotypes with bona-fide inverse-agonist activity in PPARG-activated bladder cancer cells.
PPARG inverse-agonists induce a repressive complex through inducing interactions with co-repressor NCOR2
To determine effects on interactions between PPARG and co-regulators, we tested PPARG modulators in two TR-FRET biochemical interaction assays for agonism and inverse-agonism. Agonism was quantitated using a co-activator assay, which measured the interaction between the PPARG ligand-binding domain and a fluorescently-labeled peptide from TRAP220/MED1. Full agonists, such as rosiglitazone, tesaglitazar, GW1929, and pioglitazone, enhanced interaction between PPARG and MED1 coactivator peptide, resulting in an increase in fluorescent signal from 2.2 to 3.0 fold (Fig. 3A, indicated in red). The antagonists and partial agonists, BADGE, SR202, GW9662, UVI3003, and SR1664 had minimal effect in this assay (Fig. 3A, indicated in gray and light blue). The PPARA-selective inverse-agonist GW6471, PPARG antagonist SR2595, and PPARG inverse-agonists, T0070907, and SR10221, exerted a modest inhibitory effect of 40–60%. (Fig. 3A, indicated in dark blue and gray).
Figure 3. PPARG inverse-agonists induce a repressive complex by blocking interactions with coactivators and inducing interactions with co-repressor NCOR2.

A TR-FRET assay was used to evaluate the ligand-dependent interactions between Terbium-labeled PPARG ligand binding domain (PPARG-LBD) and fluorescein-labeled peptides derived from nuclear receptor coregulators, as indicated. A, Dose-response testing of a panel of PPARG modulators in an agonist biochemical model evaluating in a PPARG-LBD - TRAP220 (MED1) co-activator peptide interaction assay to measure agonist-induced interactions, or inverse-agonist induced decrease in interactions. B, Dose-response testing of a panel of PPARG modulators in an inverse-agonist biochemical model evaluating PPARG LBD – SMRT (NCOR2) co-repressor interactions.
To evaluate inverse-agonism biochemically, we measured ligand-dependent interactions between PPARG ligand-binding domain and corepressor peptides from NCoR/NCOR1 and SMRT/NCOR2. The majority of PPARG partial-agonists and antagonists had minimal effect on the NCOR2 assay signal (Fig. 3B). PPARG inverse-agonists, T0070907 and SR10221 (Fig. 3B, indicated in dark blue), increased signal 3–6 fold, while antagonists GW9662 and SR2595 (Fig. 3B, indicated in light blue), induced only a small increase in signal of 1.7-fold. A decrease in signal of 50–60% was observed with the rosiglitazone, tesaglitazar, and GW9129 agonists (Fig. 3B, indicated in red).
In addition to the PPARG-NCOR2 interaction assay described above, we measured interactions between PPARG and a peptide from the corepressor NCOR1. In the PPARG-NCOR1 interaction assay, T0070907 induced signal 6–8-fold, while the antagonist GW9662 induced signal 2.5-fold and inverse agonist, SR10221, induced signal less than 2-fold, and antagonist, SR2595, had no effect (data not shown). The potent inverse-agonist activity of SR10221 in cellular assays (Fig. 2 and Supplementary Figs. 6–7) and biochemical PPARG-LBD - NCOR2 assay (Fig. 3B), but not PPARG-LBD - NCOR1 interaction assay (Fig. 3B), suggests that NCOR2 may be the functional co-repressor mediating PPARG inverse-agonist activity in bladder cancer cells.
PPARG inverse-agonists inhibit the proliferation of PPARG-activated cell lines
To determine whether PPARG modulators affect the proliferation and/or viability of PPARG-activated bladder cancer cell lines, we performed a direct cell counting-based assay. This assay likely avoids potential artifacts associated with the use of an ATP content-based assay, given that PPAR modulators are known to regulate cellular metabolic activity.
Both inverse-agonists, T0070907 and SR10221, significantly reduced proliferation of UM-UC-9 cells, compared to DMSO control (P<0.001) (Fig. 4A, indicated in green), with calculated IC50 values of 39 nM and 16 nM. In contrast, both antagonists tested, GW9662 and SR2595, had no significant effects on cell proliferation (Fig. 4A, indicated in gray). These IC50 values align well with the calculated IC50s from both the cell-based reporter gene assay (Fig. 2A) and the biochemical co-repressor assay (Fig. 3B). They are also close to the respective reported biochemical IC50s against PPARG, and two orders of magnitude below that reported for activity against PPARA and PPARD (31), suggesting that the observed anti-proliferative effects in UM-UC-9 cells are due to downregulation of PPARG target genes.
Figure 4. PPARG inverse-agonists, but not antagonists, inhibit proliferation of PPARG-activated bladder cancer cell lines.
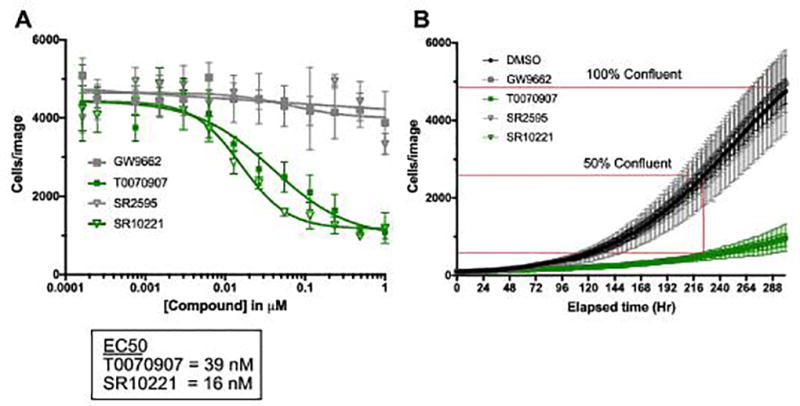
A, proliferation assay measuring dose-dependent effect of PPARG modulators on cell number in UM-UC-9 cells using high content imaging to count fluorescently-labeled nuclei. B, kinetic proliferation assay measuring the effect of 100nM modulator on growth rate over time in UM-UC-9 with graphical representation of percent of control calculation. Fluorescently labeled nuclei were counted using IncuCyte Zoom every 2 hours (n= 4 replicates per condition). Data are represented as mean ± SD.
We expanded this assay to include an additional 8 representative bladder cancer cell lines, including cell lines with PPARG amplification, RXRA p.S427F mutation, activated gene signature, and control cell lines with low level expression of PPARG and target genes. As we could not always accurately calculate an IC50 value, we used an alternative quantitative endpoint assay, which measures the relative number of cells in the DMSO vehicle control compared to treatment with 100 nM of each compound with analysis performed at the time required for the cells to reach 50% confluency in the DMSO control. Representative data for the UM-UC-9 cell line are shown in Figure 4B, and is tabulated for the tested bladder cancer cell lines in Table 1. Similar to the full dose-response assay (Fig. 4A), the T0070907 and SR10221 inverse-agonists reduce proliferation by 81% and 80% relative to control, whereas the GW9662 and SR2595 antagonists have no effect. This significant (P<0.01) preferential sensitivity to PPARG inverse-agonists, but not antagonists, is maintained across all of the PPARG-activated cell lines in the panel, including the RXRA p.S427F cell line, HT-1197 (41), the PPARG-amplified cell lines, 5637 and UM-UC-9, and the PPARG-activation gene signature cell lines Cal29, UM-UC-1, and RT112/84 (Table 1). The SW1710, UM-UC-3, and KU19.19 cells lines that did not exhibit high expression of PPARG or target genes (Fig. 1C) were insensitive to PPARG inverse-agonists and antagonists (Table 1). We obtained similar results with these cell lines in parallel studies using clonogenic assays to quantify colony forming ability (Supplementary Figure 8). These data reveal that proliferation of the PPARG-activated subset of bladder cancer cell lines, but not control bladder cancer cell lines, is dependent on PPARG activity, and inhibition of this activity with the inverse-agonists, T0070907 or SR10221, results in decreased proliferative potential.
Table 1.
Relative proliferation rate of bladder cancer cell lines treated with PPARG modulators.
| Cell Line | Pathway Alteration | GW9662 | SR2595 | T0070907 | SR10221 |
|---|---|---|---|---|---|
| HT-1197 | RXRA p.S427F | 127 | 78 | 48** | 64* |
| 5637 | PPARG amplified | 93 | 117 | 51** | 73** |
| UM-UC-9 | PPARG amplified | 108 | 97 | 19** | 20** |
| RT112/84 | PPARG signature | 98 | 101 | 69** | 80** |
| UM-UC-1 | PPARG signature | 96 | 84 | 47** | 62** |
| Cal29 | PPARG signature | 97 | 86 | 55** | 74* |
| SW1710 | Not altered | 99 | 91 | 96 | 81 |
| KU19.19 | Not altered | 85 | 91 | 95 | 91 |
| UM-UC-3 | Not altered | 92 | 102 | 91 | 96 |
Relative proliferation rate of bladder cancer cell lines treated with PPARG inhibitors at 100nM compared to DMSO vehicle, reported as percent of vehicle control (POC), in long-term kinetic proliferation assays. Percent of DMSO control was calculated by determining the relative number of cells for treatment versus DMSO control at the timepoint where DMSO control reached 50% confluence (see Fig. 4B for graphical representation) by counting fluorescently labeled nuclei (IncuCyte Zoom). Significant differences in cell number for test sample relative to DMSO control were calculated using two-way ANOVA with Dunnett’s multiple comparison test(*P<0.01, **P <0.001).
PPARG-activated cell lines are genetically dependent on PPARG
To test for PPARG dependency in PPARG-activated cell lines with an orthogonal approach, we performed CRISPR/Cas9 knockout studies using a high-precision multicolor competition dependency assay (Strathdee et al., manuscript in preparation). Briefly, we first validated that guide RNAs against PPARG were able to significantly diminish PPARG expression by Western immunoblot after normalization to loading control vinculin, VCL (Fig. 5A, B). We next compared the relative effect of knockout of PPARG on cell proliferation to knockout of an essential gene (KIF11) and to knockout of a non-essential gene (HPRT or PPIB) or PPARG intron control. The sgRNAs targeting these genes were cloned into lentiviral vectors that co-express one of three different fluorescent proteins, YFP, RFP, or CFP, and which allowed for unambiguous identification of transduced cells in complex pools: PPARG knockout cells were labeled with YFP, essential control knockout cells were labeled with RFP, and non-essential (or PPARG intron) control knockout cells were labeled with CFP. In the competition format, replicate pools of cells are generated at the beginning of the experiment in which each gene knockout/color are at equivalent abundance. Changes in relative abundance of each population are monitored during progressive serial passage by counting fluorescent nuclei using high-content imaging. Relative changes are plotted as Percent of Control (POC) of the normalized prevalence of on-test sgRNAs as a percent of non-essential sgRNA control targeting an intron of PPARG.
Figure 5. Cell lines with PPARG activation are dependent on PPARG.
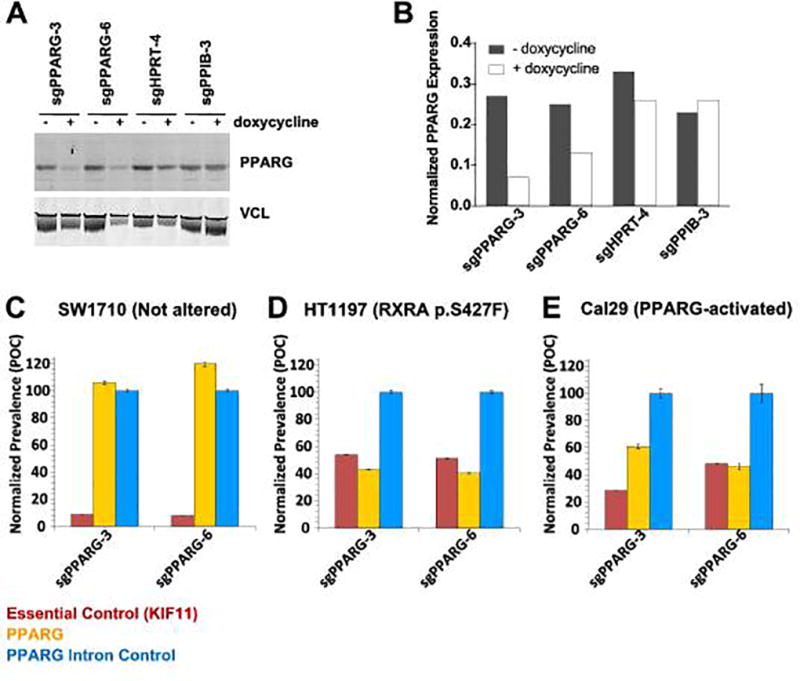
A, PPARG sgRNA’s 3, and 6 knock out PPARG protein in 5637 cells. A, Western immunoblot of PPARG and loading control VCL. B, Quantification of PPARG Western blot signal after normalization to VCL, reported as ratio of integrated fluorescence intensity K counts and was performed using Li-Cor Odyssey Application Software version 3.0.30 (Li-Cor, Lincoln NE). C–E, CRISPR/Cas9 competition screen to measure relative proliferation of cells harboring sgRNA targeting PPARG (yellow), non-essential control of PPARG intron (cyan), and essential control gene KIF11 (red). Cells lines were infected with lentivirus encoding both fluorescent marker and sgRNA prior to pooling cells for assay. Cell lines include B, HT-1997 (RXRA p.S427F), C, Cal 29 (PPARG-activated) and D, SW1710 (not altered, neutral control). Data are represented as POC, normalized prevalence of test sgRNAs as a percent of the non-essential PPARG intron control sgRNA (mean ± SD).
Analysis of three bladder cancer cell lines showed a clear PPARG dependency in PPARG-activated cells. The growth of the SW1710 cell line, which shows low PPARG/FABP4 expression (Fig. 1C), is insensitive to PPARG knockout (Fig. 5C). In contrast, HT-1197 the only known cell line with an RXRA p.S427F mutation (41), exhibited a strong PPARG-dependency in this CRISPR competition assay (Fig. 5D). The Cal29 cell line, with a highly activated PPARG signaling pathway (Fig. 1), also exhibits clear PPARG dependency (Fig. 5E). In preliminary experiments we determined that the RT4 cell line supported inadequate hCas9 expression for use in gene knockout experiments, which is unfortunate since it is the only cell line available with a PPARG p.T447M mutation. Note that we did not include any cell lines containing PPARG focal amplifications in order to avoid the well-known copy number sensitivity artifact inherent to the CRISPR/Cas9 platform (42,43). We conclude that from the above analyses that PPARG-activated bladder cancer cell lines are dependent upon a functional PPARG.
Discussion
Recently, genomic analysis of bladder cancer revealed that the PPARG signaling pathway is significantly activated in tumors, and that this can be driven by either RXRA p.S427F/Y mutations or PPARG focal amplifications (9,15). Here we confirm that these alterations, in addition to PPARG p.T447M mutation which may be emerging as a new hotspot (Supplementary Figure 10), activate the PPARG signaling pathway, and that cell lines with the corresponding mutations are genetically-dependent on PPARG and are also sensitive to pharmacological inactivation using PPARG inverse-agonists.
One model to explain this data is that these alterations confer ligand-independent activation of PPARG. In the case of RXRA p.S427F/Y and PPARG p.T447M mutations this could be achieved by the gain of hydrophobic interactions that lock PPARG helix 12 into the active conformation, phenocopying the agonist-induced state in the absence of ligand. Co-crystal structures of RXRA/PPARG (e.g. PDB ID: 1FM6 and PDB ID: 5JI0) (15,44) show that the S427 position of RXRA is located in the dimerization interface with PPARG in the ligand-activated state, directly adjacent to both the T447 residue and c-terminus Y477 residue of PPARG. This positioning is also conserved in homology models of RXRA/PPARA (45). RXRA p.S427F/Y mutations may also disrupt interactions between RXRA and its other heterodimer partners (eg. RARA, VDR, TR), further shifting equilibrium of RXRA further towards PPARs (15).
It has been established that other mechanisms can lead to ligand-independent activation of PPARG. For example, signaling by insulin through the actions of MAP kinases leads to phosphorylation of PPARG in the AF-1 domain which can lead to ligand-independent activation (46). Insulin-dependent PPARG activation is not sensitive to inhibition by the PPARG antagonist GW9662, whereas ligand-driven activation by PPARG agonist, cigitazone, is sensitive to GW9662 (47). Here we show that PPARG-activated bladder cancer cells, through PPARG amplification, RXRA p.S427F mutation, or other unknown mechanisms, are similarly not responsive to antagonists, including GW9662 and SR2595, but are sensitive to inverse-agonists. Since PPARG inverse-agonists induce a conformational change in the ligand-binding domain to actively recruit co-repressors to the complex, these could overcome ligand-independent signaling, as our studies show.
The impact of PPARG on bladder cancer signaling is supported by the evidence that PPARA/PPARG dual agonists cause bladder cancer in rodents, although there are conflicting epidemiological data that PPARA/PPARG agonists are associated with increased rates of disease in humans (20,22–24,48).
PPARG as a therapeutic target in bladder cancer can be seen as analogous to targeting androgen receptor in prostate cancer or estrogen receptor in breast cancer. One of the hallmarks of luminal cancers is the expression of a ligand-activated nuclear receptor. Therapeutic targeting of the nuclear receptors in patients with these cancers can be a very effective therapeutic approach as in the example of targeting ESR1-positive luminal breast cancer with anti-estrogens and AR-positive prostate cancer with androgen deprivation therapy. PPARG agonists upregulate expression of luminal differentiation markers UPK1A, UPK1B, and KRT20 in primary rat urothelial cells (49). These same genes, plus GATA3, and FOXA1 are the key luminal markers of bladder cancer from human patients (13,14) and bladder cancer cell lines (16). The lineage-defining role of GATA3, FOXA1, and PPARG in luminal bladder cancer is reminiscent of luminal breast cancer, in which coordinated expression of GATA3, FOXA1, and ESR1 enable chromatin remodeling and regulate luminal gene expression programs (50,51). Whereas in prostate cancer, FOXA1 and GATA2 (52) coordinately regulate the activity AR (53) and distribution and selectivity for AR response elements.
The steroid hormone receptors ESR1 and AR are also in the nuclear receptor superfamily, however, distinct from PPARG, they utilize high affinity ligands for signaling. Endogenous production of estrogen in breast tissue, and testosterone / dihyrotestosterone in the testes, are required for receptor activation. Therefore, in the context of ligand-activation, antagonists of ESR1 and AR are effective therapies for primary cancers. Another effective strategy in breast and prostate cancer are therapies leading to inhibition of ligand production such as the use of aromatase inhibitors for treatment of breast cancer and androgen deprivation therapy for prostate cancer. In contrast, PPARG does not have a high-affinity endogenous ligand and is considered a lipid sensor, with low affinity for its ligands (54). Therefore, mechanisms leading to ligand-independent activation of PPARG, and not ligand-dependent signaling, appear to be the primary driver of PPARG activity and will require a different pharmacological properties than in the case of targeting ligand-activated ESR1 or AR with a pure antagonist.
Patients treated with anti-estrogen and anti-androgen therapy commonly develop resistance, with common mechanisms being somatic alterations that lead to either ligand hypersensitivity or ligand-independent signaling. In the case of ESR1, recurrent mutations at Y537 mutations lead to ligand-independent signaling (Robinson et al 2013) and due to being in the ligand-binding domain, also confer resistance to ESR1 antagonists. In the case of AR, gene amplification leads to ligand hypersensitivity, whereas point mutations and alternative splicing can lead to ligand-independent activation (55). In order to attenuate ligand-independent activation of ESR1, a new class of compounds was developed that lead to receptor degradation. Selective estrogen receptor degraders have had promising clinical outcomes, with fulvestrant reaching clinical approval and providing a new hope for ER+ breast cancer patients refractory to SERMs and aromatase inhibitors. Based on the fact that there appears to be high level of ligand-independent PPARG signaling in bladder cancer, we focused efforts on validating inverse-agonists as a candidate therapeutic strategy. However, a selective PPARG-destabilizer could be another promising therapeutic approach worth exploring.
We have demonstrated a genetic and pharmacologic dependence on PPARG in PPARG-activated, luminal bladder cancer cell lines. Our studies provide a well-defined patient population and clear therapeutic hypothesis. Since PPARG agonists rosiglitazone and pioglitazone are used for the treatment of diabetes by a mechanism of sensitizing cells to insulin, lowering blood glucose, and lowering lipid levels (56), one may predict that a PPARG inverse-agonist may have an opposing effect, eliciting symptoms of diabetes. Furthermore, patients with deleterious PPARG mutations have an increased risk for diabetes and can exhibit lipodystrophy and insulin resistance (57). While there are concerns for on-mechanism complications for PPARG inverse-agonists as a potential therapy for bladder cancer, there is renewed hope that rigorous testing and a deep understanding of emerging PPARG biology and pharmacology (25) can overcome these hurdles through selective receptor modulation. Recent advances have highlighted a potential role for oncogenic activation of PPARG in bladder cancer to regulate inflammatory cytokines, thereby regulating immune cell infiltration and immunosurveillance (15). Further studies of PPARG in bladder cancer will help to evaluate whether PPARG inverse agonists can complement conventional and emerging therapies including other genomically defined therapeutic targets such as FGFR inhibitors, in addition to immune checkpoint blockade.
Supplementary Material
Acknowledgments
The authors would like to thank Xiaoyang Zhang, Josh Francis, Chandra Pedamallu, Patrick McCarren, Chris Lemke, Aruna Ramachandran Robert Hilgraf, Stuart Schreiber, and Todd Golub for their helpful discussions and guidance.
Financial support: Financial support for this work was provided by Bayer AG (Berlin, Germany) (J.Goldstein, A.Berger, J.Shih, F.Duke, L.Furst, A.Cherniack, M.Meyerson, and C.Strathdee) and the National Cancer Institute under grant 5R35CA197568-02 (M. Meyerson).
Conflict of interest disclosure statement;
The Broad Institute and Dana-Farber Cancer Institute have filed a patent application for aspects of the work described in this paper. Financial support for this work was provided by Bayer AG (Berlin, Germany). M. Meyerson is a member of the Scientific Advisory Board and equity holder of Origimed.
References
- 1.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. The Lancet. 2016;387:1909–20. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18:312–22. doi: 10.1016/S1470-2045(17)30065-7. [DOI] [PubMed] [Google Scholar]
- 3.Apolo AB, Infante JR, Balmanoukian A, Patel MR, Wang D, Kelly K, et al. Avelumab, an Anti-Programmed Death-Ligand 1 Antibody, In Patients With Refractory Metastatic Urothelial Carcinoma: Results From a Multicenter, Phase Ib Study. J Clin Oncol. 2017;35:2117–24. doi: 10.1200/JCO.2016.71.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol. 2016;34:3119–25. doi: 10.1200/JCO.2016.67.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012;338:221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu D, Plimack ER, Hoffman-Censits J, Garraway LA, Bellmunt J, Van Allen E, et al. Clinical Validation of Chemotherapy Response Biomarker ERCC2 in Muscle-Invasive Urothelial Bladder Carcinoma. JAMA Oncology. 2016;2:1094. doi: 10.1001/jamaoncol.2016.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4:1140–53. doi: 10.1158/2159-8290.CD-14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagle N, Grabiner BC, Van Allen EM, Hodis E, Jacobus S, Supko JG, et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014;4:546–53. doi: 10.1158/2159-8290.CD-13-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013;45:1459–63. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans Ronald M, Mangelsdorf David J. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–66. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–66. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biton A, Bernard-Pierrot I, Lou Y, Krucker C, Chapeaublanc E, Rubio-Perez C, et al. Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 2014;9:1235–45. doi: 10.1016/j.celrep.2014.10.035. [DOI] [PubMed] [Google Scholar]
- 14.Choi W, Porten S, Kim S, Willis D, Plimack Elizabeth R, Hoffman-Censits J, et al. Identification of Distinct Basal and Luminal Subtypes of Muscle-Invasive Bladder Cancer with Different Sensitivities to Frontline Chemotherapy. Cancer Cell. 2014;25:152–65. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korpal M, Puyang X, Jeremy Wu Z, Seiler R, Furman C, Oo HZ, et al. Evasion of immunosurveillance by genomic alterations of PPARγ/RXRα in bladder cancer. Nature Communications. 2017;8 doi: 10.1038/s41467-017-00147-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warrick JI, Walter V, Yamashita H, Chung E, Shuman L, Amponsa VO, et al. FOXA1, GATA3 and PPAR Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Sci Rep. 2016;6:38531. doi: 10.1038/srep38531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Hage J. Toxicity Profiles of Peroxisome Proliferator-Activated Receptor (PPAR) Agonists and Preclinical Safety Profile for Muraglitazar. FDA Report CDER. 2005 [Google Scholar]
- 18.Sakamoto J, Kimura H, Moriyama S, Odaka H, Momose Y, Sugiyama Y, et al. Activation of human peroxisome proliferator-activated receptor (PPAR) subtypes by pioglitazone. Biochem Biophys Res Commun. 2000;278:704–11. doi: 10.1006/bbrc.2000.3868. [DOI] [PubMed] [Google Scholar]
- 19.Tannehill-Gregg SH, Sanderson TP, Minnema D, Voelker R, Ulland B, Cohen SM, et al. Rodent carcinogenicity profile of the antidiabetic dual PPAR alpha and gamma agonist muraglitazar. Toxicol Sci. 2007;98:258–70. doi: 10.1093/toxsci/kfm083. [DOI] [PubMed] [Google Scholar]
- 20.Lubet RA, Fischer SM, Steele VE, Juliana MM, Desmond R, Grubbs CJ. Rosiglitazone, a PPAR gamma agonist: potent promoter of hydroxybutyl(butyl)nitrosamine-induced urinary bladder cancers. Int J Cancer. 2008;123:2254–9. doi: 10.1002/ijc.23765. [DOI] [PubMed] [Google Scholar]
- 21.Dominick MA, White MR, Sanderson TP, Van Vleet T, Cohen SM, Arnold LE, et al. Urothelial carcinogenesis in the urinary bladder of male rats treated with muraglitazar, a PPAR alpha/gamma agonist: Evidence for urolithiasis as the inciting event in the mode of action. Toxicol Pathol. 2006;34:903–20. doi: 10.1080/01926230601072327. [DOI] [PubMed] [Google Scholar]
- 22.Lewis JD, Habel LA, Quesenberry CP, Strom BL, Peng T, Hedderson MM, et al. Pioglitazone Use and Risk of Bladder Cancer and Other Common Cancers in Persons With Diabetes. Jama. 2015;314:265. doi: 10.1001/jama.2015.7996. [DOI] [PubMed] [Google Scholar]
- 23.Tseng CH. Pioglitazone and bladder cancer in human studies: is it diabetes itself, diabetes drugs, flawed analyses or different ethnicities? J Formos Med Assoc. 2012;111:123–31. doi: 10.1016/j.jfma.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Tuccori M, Filion KB, Yin H, Yu OH, Platt RW, Azoulay L. Pioglitazone use and risk of bladder cancer: population based cohort study. BMJ. 2016:i1541. doi: 10.1136/bmj.i1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marciano DP, Kuruvilla DS, Boregowda SV, Asteian A, Hughes TS, Garcia-Ordonez R, et al. Pharmacological repression of PPARgamma promotes osteogenesis. Nat Commun. 2015;6:7443. doi: 10.1038/ncomms8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology. 2016;34:525–7. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 28.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–64. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 31.Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, et al. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277:19649–57. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- 32.Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41:6640–50. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- 33.Brown KK, Henke BR, Blanchard SG, Cobb JE, Mook R, Kaldor I, et al. A novel N-aryl tyrosine activator of peroxisome proliferator-activated receptor-gamma reverses the diabetic phenotype of the Zucker diabetic fatty rat. Diabetes. 1999;48:1415–24. doi: 10.2337/diabetes.48.7.1415. [DOI] [PubMed] [Google Scholar]
- 34.Cesario RM, Klausing K, Razzaghi H, Crombie D, Rungta D, Heyman RA, et al. The rexinoid LG100754 is a novel RXR:PPARgamma agonist and decreases glucose levels in vivo. Mol Endocrinol. 2001;15:1360–9. doi: 10.1210/mend.15.8.0677. [DOI] [PubMed] [Google Scholar]
- 35.Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature. 2010;466:451–6. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cronet P, Petersen JF, Folmer R, Blomberg N, Sjoblom K, Karlsson U, et al. Structure of the PPARalpha and -gamma ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure. 2001;9:699–706. doi: 10.1016/s0969-2126(01)00634-7. [DOI] [PubMed] [Google Scholar]
- 37.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–6. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 38.Rieusset J, Touri F, Michalik L, Escher P, Desvergne B, Niesor E, et al. A new selective peroxisome proliferator-activated receptor gamma antagonist with antiobesity and antidiabetic activity. Mol Endocrinol. 2002;16:2628–44. doi: 10.1210/me.2002-0036. [DOI] [PubMed] [Google Scholar]
- 39.Wright HM, Clish CB, Mikami T, Hauser S, Yanagi K, Hiramatsu R, et al. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J Biol Chem. 2000;275:1873–7. doi: 10.1074/jbc.275.3.1873. [DOI] [PubMed] [Google Scholar]
- 40.Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–7. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 41.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Research. 2017;45:D777–D83. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016;6:914–29. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 2016;6:900–13. doi: 10.1158/2159-8290.CD-16-0178. [DOI] [PubMed] [Google Scholar]
- 44.Gampe RT, Jr, Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, et al. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cell. 2000;5:545–55. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- 45.Venalainen T, Molnar F, Oostenbrink C, Carlberg C, Perakyla M. Molecular mechanism of allosteric communication in the human PPARalpha-RXRalpha heterodimer. Proteins. 2010;78:873–87. doi: 10.1002/prot.22613. [DOI] [PubMed] [Google Scholar]
- 46.Zhang B, Berger J, Zhou G, Elbrecht A, Biswas S, White-Carrington S, et al. Insulin- and Mitogen-activated Protein Kinase-mediated Phosphorylation and Activation of Peroxisome Proliferator-activated Receptor. Journal of Biological Chemistry. 1996;271:31771–4. doi: 10.1074/jbc.271.50.31771. [DOI] [PubMed] [Google Scholar]
- 47.Al-Rasheed NM, Chana RS, Baines RJ, Willars GB, Brunskill NJ. Ligand-independent activation of peroxisome proliferator-activated receptor-gamma by insulin and C-peptide in kidney proximal tubular cells: dependent on phosphatidylinositol 3-kinase activity. J Biol Chem. 2004;279:49747–54. doi: 10.1074/jbc.M408268200. [DOI] [PubMed] [Google Scholar]
- 48.Turner RM, Kwok CS, Chen-Turner C, Maduakor CA, Singh S, Loke YK. Thiazolidinediones and associated risk of bladder cancer: a systematic review and meta-analysis. Br J Clin Pharmacol. 2014;78:258–73. doi: 10.1111/bcp.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varley CL, Southgate J. Effects of PPAR agonists on proliferation and differentiation in human urothelium. Exp Toxicol Pathol. 2008;60:435–41. doi: 10.1016/j.etp.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 50.Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23:12–22. doi: 10.1101/gr.139469.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, Brown M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007;67:6477–83. doi: 10.1158/0008-5472.CAN-07-0746. [DOI] [PubMed] [Google Scholar]
- 52.Zhao JC, Fong KW, Jin HJ, Yang YA, Kim J, Yu J. FOXA1 acts upstream of GATA2 and AR in hormonal regulation of gene expression. Oncogene. 2016;35:4335–44. doi: 10.1038/onc.2015.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin HJ, Zhao JC, Wu L, Kim J, Yu J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat Commun. 2014;5:3972. doi: 10.1038/ncomms4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poulsen L, Siersbaek M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. 2012;23:631–9. doi: 10.1016/j.semcdb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC) Transl Androl Urol. 2015;4:365–80. doi: 10.3978/j.issn.2223-4683.2015.05.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes. 1996;45:1661–9. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- 57.Semple RK, Chatterjee VK, O'Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116:581–9. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bladder cancer incidence and survival data was obtained from Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, et al. November 2015 SEER data submission, posted to the SEER web site, April 2016. SEER Cancer Statistics Review, 1975–2013. National Cancer Institute <http://seer.cancer.gov/csr/1975_2013/>.
The provisional TCGA muscle-invasive urothelial carcinoma data is available from the Broad Institute TCGA Genome Data Analysis Center. Analysis-ready standardized TCGA data from Broad GDAC Firehose 2016_01_28 run. https://doi.org/10.7908/C11G0KM9. CCLE Affymetrix U133+2 arrays mRNA expression data is available through https://portals.broadinstitute.org/ccle/ (26). RNA sequencing data is available through the National Center for Biotechnology Information BioProject accession # PRJNA396067.