

Abstract
DNA provides an ideal substrate for the engineering of versatile nanostructures due to its reliable Watson–Crick base pairing and well-characterized conformation. One of the most promising applications of DNA nanostructures arises from the site-directed spatial arrangement with nanometer precision of guest components such as proteins, metal nanoparticles, and small molecules. Two-dimensional DNA origami architectures, in particular, offer a simple design, high yield of assembly, and large surface area for use as a nanoplatform. However, such single-layer DNA origami were recently found to be structurally polymorphous due to their high flexibility, leading to the development of conformationally restrained multilayered origami that lack some of the advantages of the single-layer designs. Here we monitored single-layer DNA origami by transmission electron microscopy (EM) and discovered that their conformational heterogeneity is dramatically reduced in the presence of a low concentration of dimethyl sulfoxide, allowing for an efficient flattening onto the carbon support of an EM grid. We further demonstrated that streptavidin and a biotinylated target protein (cocaine esterase, CocE) can be captured at predesignated sites on these flattened origami while maintaining their functional integrity. Our demonstration that protein assemblies can be constructed with high spatial precision (within ~2 nm of their predicted position on the platforms) by using strategically flattened single-layer origami paves the way for exploiting well-defined guest molecule assemblies for biochemistry and nanotechnology applications.
Keywords: cocaine esterase, DNA nanotechnology, DNA scaffolding, protein–DNA assembly, single-particle electron microscopy
Graphical abstract
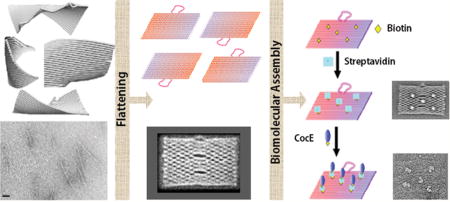
Self-assembled DNA nanostructures have become promising templates for applications ranging from molecular biology to nanomachines and nanorobots.1–7 Early designs of two-dimensional DNA structures with sticky ends were shown to grow into large crystalline structures, illustrating the potential of using DNA to create self-assembling periodic nanostructures.8 Recent advancements in DNA nanotechnology have demonstrated that, in principle, DNA can be engineered into any physical shape using a folding strategy termed “origami”.9–13 The predetermined shapes of these nanostructures allow for the positioning of different reactive groups at desired target locations with nanometer precision, rendering them convenient nanoplatforms for the attachment of guest molecules.14–17 The easily accessible, 7249 nucleotide long, single-stranded bacteriophage M13mp18 strand has generally provided the scaffold of choice to assemble—using hundreds of short “staple” DNA oligonucleotides—DNA origami tiles of defined, albeit size-limited, topology.12,18,19 However, single-molecule fluorescence microscopy and molecular dynamics (MD) simulations have recently revealed substantial conformational flexibility and heterogeneity in M13mp18-based, single-layer, two-dimensional (2D) origami rectangles and suggested that specific corners of a rectangular origami tend to curl.20 Similar rectangular origami tiles have also been characterized to exhibit “molecular threading” of DNA strands and conductance of ions through dynamic openings in the DNA scaffold.21,22 These unintended aberrations in architecture and unexpectedly pronounced dynamic polymorphy of 2D DNA origami led to the development of more complex three-dimensional (3D) DNA nanostructures. While more rigid, 3D DNA structures are also more challenging to design, more expensive to prepare, and offer less surface area per base pair,12,19,23,24 thereby posing limitations to their applications. As an alternative strategy, we sought to broaden the applicability of rectangular 2D origami for assembling guest molecule arrays with nanometer precision by examining their architectural features in more detail and solving their curling problem.
To this end, we have applied negative stain electron microscopy (EM) and single-particle image averaging to examine the architecture of rectangular 2D DNA origami nanoplatforms. We show that origami exhibit pronounced, tight curling along their edges, a problem that can be alleviated by the addition of a low concentration of dimethyl sulfoxide (DMSO). Using streptavidin and cocaine esterase (CocE) for a proof-of-principle, we show that DMSO-flattened DNA origami are platforms suitable for the high-precision, site-specific nanopatterning of proteins.
RESULTS AND DISCUSSION
Previous reports suggest that 2D DNA origami are highly flexible.20–22 However, only few studies have focused on this property, and no systematic interrogation of 2D origami conformational dynamics has been performed. Thus, we sought to explore the topological details of these nanostructures using EM. In accord with previously published observations, solution structures of origami predicted by computer-aided engineering for DNA origami (CanDo)25 reveal a high degree of flexibility (Figure 1a). Although atomic force microscopy (AFM) images of these nanostructures give the impression that the origami are flat and compact in solution (Figure 1b), this appearance is likely a consequence of the experimental procedure, where the origami is flattened by tight binding onto the negatively charged mica surface through an intermediary layer of divalent cations.26,27 The comparably mild deposition of DNA tiles on the hydrophylized carbon support of the EM grid reveals substantial diversity in conformational states (Figure 1c and Supporting Information Figure S1), which presents a challenge when a flat origami nanoplatform is desired. For example, the presence of multiple conformations of DNA nanostructures would result in low-quality image averages in negative stain and cryo-EM experiments. Thus, we sought to determine a method to significantly flatten these structures on the carbon support of commonly used EM grids.
Figure 1.

Flexibility of 2D DNA origami rectangles as observed by computational modeling, AFM and EM. (a) Flexibility prediction of DNA tiles using computer-aided engineering for DNA origami (CanDo) software available online (http://cando-dna-origami.org/).25 Snapshots from CanDo-generated movies that show extreme curling of the origami edges. The top panel depicts the top view, whereas the bottom panel depicts the side view. The dotted circles indicate particularly flexible regions. The heat map scale indicates the level of flexibility. (b) Representative AFM images of the DNA origami used in this study. The scale bar is 50 nm. (c) Negative stain EM image showing flexibility and variable folding (highlighted by arrows) of 2D origami. Flat DNA origami tiles (such as the one boxed) were rarely observed. The scale bar is 50 nm.
Although several DNA-binding and intercalating molecules are known to alter the physical properties of DNA, including ethidium bromide (EtBr), doxorubicin, and oxazole yellow dimers (YOYO),28 solvent effects on DNA stability have been poorly explored.29,30 Previous studies have suggested that DMSO helps to denature DNA secondary structures and relaxes DNA helices,29,31 possibly by reducing the concentration of free water around the DNA molecules due to its dehydrating activity.9 It has also long been used in the polymerase chain reaction (PCR) to denature DNA secondary structure.31 We therefore tested whether DMSO may be used to flatten large DNA structures such as 2D origami. To begin, we used a low concentration of DMSO (0.5% v/v) in standard DNA origami buffer (1× TAE-Mg2+) before depositing origami structures on the EM grid. Interestingly, 0.5% (v/v) DMSO can already significantly (~66% of the origami population) flatten the origami structures (Figure 2a and Supporting Information Figure S2), whereas the flattening behavior was extensive (~95% of the origami population) at 2% (v/v) DMSO (Figure 2a and Supporting Information Figures S2). Moreover, while there was no further change observed in origami morphology at up to 10% (v/v) DMSO, the origami structures significantly unraveled at 40% (v/v) DMSO (Figure S2). This approach allowed us to visualize origami by EM with superior resolution compared to AFM images (Figure 2b, compare with Figure 1b). In addition, our raw images resolved the double-stranded DNA loop incorporated into the DNA origami as an asymmetric marker (Figure 2b).10 Thus, we have identified a simple, rapid, and inexpensive strategy to flatten 2D DNA origami directly onto an EM grid without the need for any chemical modification.
Figure 2.

Surface flattening of 2D DNA origami in 2% (v/v) DMSO. (a) Population distribution of origami conformations at 0–2% (v/v) DMSO. A total of ≥350 origami particles from several fields of view were analyzed to generate this plot. (b) Typical EM image of DNA origami in 2% (v/v) DMSO. The arrow highlights a double-stranded DNA marker loop in raw origami images. Scale bar is 50 nm. (c) Representative 2D class averages of negatively stained origami, as shown in (b). Two arrows show the entry and exit points of the double-stranded DNA marker loop. The number of individual origami images contributing in each average is provided in the bottom-right corner of the corresponding image. Scale bar is 50 nm.
To further explore the structure of the 2D DNA nanoplatform, 1619 DMSO-flattened particles were utilized for single-particle classification and averaging to obtain five class averages of origami (Figure 2c). Notably, all five class averages appear similar and were distinguished only by their relative orientation, suggesting that there was no gross conformational heterogeneity after flattening the structures. As a consequence, the crossovers and interwoven nature of the DNA origami were well-resolved in these average images (Figure 2c). Additionally, we were able to visualize topological features produced by different hybridization patterns of the DNA, in particular, two cavities around the middle and one small cavity around the side of the DNA origami (Figure 2c and Supporting Information Figure S3). The loop of the DNA origami was not resolved in the class averages due to its inherent flexibility; however, the entry and exit points of the loop were well-resolved and are indicated with arrows in Figure 2c.
One of the emerging applications of DNA origami in the field of DNA nanotechnology is the site-specific capture and display of guest biomolecules.14–17 This approach permits the study and exploitation of biochemical reactions on a synthetic yet biocompatible designer platform.5,32 For example, there have been several demonstrations that a DNA nanoplatform can be used to study and improve the function of protein enzymes6,14 and provide a landscape to guide molecular nanorobots.5,33 However, a successful implementation of this approach requires a precise spatial organization of protein complexes on the DNA nanostructure. Although AFM is commonly used to characterize DNA nanostructures and the attached biomolecular assemblies, it suffers from mechanical perturbation, excessive surface-binding forces, and tip-limited resolution.3,5 To achieve more detailed snapshots, we investigated whether DMSO-mediated flattening of DNA origami could be used to obtain images of regularly patterned protein–origami complexes by EM. To demonstrate a proof-of-principle, we designed a DNA origami with five biotins (incorporated during assembly using biotin-modified staple strands) displayed on one of its faces (Figure S3, Supporting Information).20 Four biotins are located in a rectangular arrangement of ~41 × 25 nm dimensions, and the fifth lies in the center of this rectangle (Figures S3 and S4, Supporting Information). These biotins were previously used to immobilize the origami to a streptavidin-modified surface.20 Upon addition of streptavidin to the preflattened DNA origami on the EM grid, we observed up to five streptavidin molecules bound to each origami (Figure 3a, left panel). Surprisingly, almost all (~91%) origami bound four or five streptavidin molecules (Figure S5, Supporting Information), suggesting that the origami nanoplatforms preferentially land on the grid with their biotin-modified face pointing toward the solution. This observation is consistent with a previous report wherein 90–95% of similar origami were oriented with their biotin-modified surface projecting toward the solution.34 Interestingly, the remaining ~9% of nanoplatforms showed no streptavidin binding, which is likely attributed to an inaccessible biotin-modified surface that faces down toward the carbon surface. Two-dimensional class averages from 1004 images of origami with streptavidin on top yielded a nicely resolved structure with five streptavidin molecules captured in the expected geometry (Figure 3a, right panel, and Figure S4, Supporting Information). Interesting, while we were able to resolve individual DNA double helices in the averages (Figure 3b), the streptavidin molecules appear blurry (Figure 3a, right panel). We attribute this effect to the random orientation of the tetrameric streptavidin complexes around their single attachment sites, leading to blurring in the average images where the DNA origami pattern dominates the particle alignment. When we compared the interstreptavidin distances experimentally measured from these images with distances estimated from the origami map, we found that the streptavidin molecules reside within 2 nm of their predicted positions (Figure 3c and Supporting Information Figure S4). These results demonstrate that DNA origami structures can be used to position biomolecular assemblies with high spatial accuracy (Figure 3). It is possible that the remaining localization error of 2 nm is due to averaging of the random orientations of streptavidin molecules. Alternatively, we cannot rule out that the origami tiles still maintain some flexibility after surface flattening with DMSO, which may contribute to the localization error. Another possible source of error may be the flexibility of the six-carbon long (C6) biotin linker.
Figure 3.

Visualization of streptavidin binding to DNA origami. (a) Raw EM image (left panel) and representative class averages (right panel) of negative stained DNA origami with bound streptavidin. Inset shows the experimental strategy. In the right panel, the number of individual origami images contributing to each average is provided in the bottom-right corner of the corresponding image. Scale bar is 50 nm. (b) Averaged density of streptavidin (circled) bound to DNA origami. The origami structure is resolved down to each DNA double-helix (numbered 1–24). Scale bar is 50 nm. (c) Localization accuracy of streptavidin molecules on DNA origami. A comparison of the interstreptavidin distances (center-to-center distances of the circles enclosing the streptavidin molecules) between the estimation (from DNA origami map, numbers outside parentheses) and the actual measurement (numbers in parentheses) shows that biomolecules are localized with ~2 nm accuracy.
The highly efficient capture of streptavidin on the 2D DNA scaffold, together with the detailed average images, suggests a possible utility for this platform in structural studies of proteins. Therefore, we aimed to explore the potential of this technique by asking whether, in principle, any biotinylated protein can be immobilized and studied. As a proof-of-principle, we sought to capture cocaine esterase (CocE) molecules on these flattened origami structures in a site-specific fashion. CocE, a homodimeric bacterial enzyme with molecular weight of ~130 kDa, is the most efficient protein catalyst for the hydrolysis of cocaine.35–37 In this experiment, streptavidin and biotinylated CocE molecules were added sequentially onto preflattened DNA origami (containing five biotins, Figure 4a) on an EM grid. Taking advantage of the biotin–streptavidin interaction and the four biotin-binding sites of streptavidin, we successfully assembled CocE molecules onto these DNA nanostructures and visualized the complex by negative stain EM (Figure 4b). Most origami tiles captured 3–5 CocE molecules (Figure 4c). Each streptavidin molecule captured only one CocE molecule (binding of up to three CocE molecules may be expected as only one of the four binding sites of the tetrameric streptavidin is used for binding to the origami). The observation of one-to-one binding between CocE and streptavidin is most likely due to the steric hindrance imposed by the large CocE dimer, which is over twice as large as the streptavidin tetramer.
Figure 4.

Cocaine esterase captured on DNA origami by biotin–streptavidin sandwiching. (a) Schematic of the experimental strategy. (b) Typical negative stain images of the CocE–origami complex. Several representative DNA origami tiles with five CocE molecules (bright spots) are shown. (c) Distribution of the number of CocE molecules per 2D DNA origami. A total of 355 origami particles were analyzed. (d) Representative class averages of the CocE–origami complex. The number of individual origami images contributing to each average is provided in the bottom-right corner of the corresponding image. Scale bars are 50 nm.
When individual CocE–origami particle images (n = 866) were averaged, five CocE molecules were observed at the predicted positions (Figure 4d). However, averages of streptavidin–CocE origami (Figure 4d) are less well resolved due to the lack of a fixed orientation of the biotinylated CocE when bound to streptavidin, which is clearly evident in the raw images (Figure 4b). The biotinylation of CocE was not precisely site-directed so that the biotin moiety could be placed on one of six possible locations on the CocE dimer (see Materials and Methods section). This random placement likely accounts for the variability and poor averaging. Although outside the scope of this study, we predict that the protein arrangement can be further directed by employing a monovalent binding site such as provided by monovalent streptavidin38,39 to capture biotin–CocE complexes. To test CocE for functional activity upon origami immobilization, we compared hydrolysis of cocaine by origami-bound and free CocE molecules (Materials and Methods) and found that free and origami-bound CocE exhibits similar catalytic efficiency (kcat/KM; Figure S6, Supporting Information), indicating that functionality is well retained. Furthermore, given that DMSO-flattened DNA origami were successfully used to capture both streptavidin- and biotin-modified CocE, it appears that this reagent does not perturb the functional behavior of protein guest molecules.
Our DNA origami contain 12 additional, each 20 nucleotides long, protruding single-stranded DNA appendages (5′-CCTCTCACCCACCATTCATC-3′) on the face opposite the biotin-modified side (Figure 5a and Supporting Information Figure S3).20 These appendages are extensions of a subset of staple strands used during DNA origami assembly and may assist in orienting the biotin face-up during deposition on the EM grid. To test this hypothesis, we used these protruding DNA extensions to capture complementary, biotin-modified, DNA oligonucleotides (5′-GATGAATGGTGGGTGAGAGG/Bio/-3′). To ensure that the protruding linkers are indeed accessible to capture complementary biotin-modified DNA oligonucleotides, the resulting hybridization was first visualized by fluorescence microscopy (Materials and Methods). The rate constant for capturing equivalent, but fluorophore-labeled, DNA oligonucleotides (5′-/Cy3/GATGAATGGTGGGTGAGAGG-3′) onto the origami surface was determined to be 0.52 min−1 (Figure S7, Supporting Information) under our experimental conditions (Materials and Methods), in agreement with a published report suggesting that the protruding DNA linkers are readily available for efficient hybridization.20 We then prehybridized our origami nanoplatforms with the biotin-modified complementary oligonucleotides (Figure 5a) and purified them by Sephacryl-300 gel filtration to remove excess oligonucleotides (Figure S8, Supporting Information). As expected, while the addition of the complementary DNA strands alone did not have any observable effect on the appearance of the negative stained origami, after addition of streptavidin, we observed a variable number of streptavidin molecules on each origami nanoplatform (Figure 5b). Depending on which face of the origami is pointing toward the solution, either 5 or 12 streptavidin molecules may be expected to be observed at the predetermined positions (Figure 5a, left and right panels). When experimentally quantified, however, we found between 6 and 14 streptavidin molecules per origami (Figure 5c). Most strikingly, ~35% of the origami population showed more than 12 streptavidin per origami (Figure 5c), which is possible only when the biotin face points up (toward the solution) and a subset of DNA extensions from the other face thread through the scaffold (Figure 5a, middle panel; threading of three DNA linkers is shown as an example). These appendages likely get trapped upon hybridization with the complementary biotinylated oligonucleotides. Such threading is in accord with a recent study suggesting that single-stranded DNA can thread from one face of a similar single-layer origami through the interhelix pores of the scaffold to the other face.18 We note that our individual EM images readily identify which specific DNA extensions thread.
Figure 5.

EM visualization of threading on DNA origami nanoplatforms. (a) Schematic of the experimental strategy and expected results. Yellow and red arrows highlight the biotin- (top) and DNA linker-modified (bottom) faces on the left and right panel, respectively. Similarly, the biotins on the top and bottom faces of the origami are highlighted as yellow and red diamonds, respectively. The middle panel depicts threading of DNA oligonucleotides (blue) from the bottom to the top face. (b) Negative stain image of streptavidin molecules (bright spots) attached to the origami structures via surface biotins and/or biotinylated DNA oligonucleotides. (c) Distribution of streptavidin molecules per DNA origami. A total of 156 individual origami images were analyzed for the distribution histogram. (d) Representative origami images that show evidence of threading. Five streptavidin molecules bound to surface biotins are indicated by yellow circles. The binding of streptavidin to the threaded DNA linkers via prehybridized biotin-modified oligonucleotides is highlighted by red arrows (threading of 3, 5, and 7 DNA linkers are highlighted as “5 + 3”, “5 + 5”, and “5 + 7”, respectively). (e) Streptavidin captured on an origami face with 12 DNA linkers. Bottom row of (d) and (e) shows the most likely arrangement of streptavidin on DNA origami. Scale bars are 50 nm.
In support of this interpretation of our data, in addition to five streptavidin molecules observed in a rectangular arrangement (as in Figure 3b), we observed streptavidin molecules outside of this core pattern (indicated as 5 + x in Figure 5d; where x represents the number of threaded DNA linkers). However, due to the inherent flexibility of DNA linkers and the consequently variable origami position that the linkers can thread through (as threading through any nearby holes of the origami framework is possible), the pattern was often ambiguous when several DNA linkers were threaded. Therefore, it was not possible to accurately quantify the origami population with a specific 5 + x pattern. Furthermore, the binding of 12 streptavidin molecules in a 4 × 3 pattern (Figure 5e) was rare (<7%), an observation consistent with the idea that the face of the 2D origami directly modified with biotins, and not the face hybridized with biotinylated complementary oligonucleotides, preferably orients toward solution.34 Notably, in AFM experiments of 2D origami lacking our DNA extensions, Rothemund observed a nearly even distribution between those facing up and those facing down.10 As a control experiment, we redesigned the DNA origami by removing the 12 protruding DNA linkers while keeping the five biotins on the other face of the origami. We then quantified the origami facing up and facing down using our streptavidin binding assay. We found that in this experiment the population of streptavidin-bound DNA origami decreased to ~61% (Figure S9, Supporting Information) compared to ~92% of the population labeled with streptavidin when the protruding linkers were present. These observations indicate that protruding DNA extensions on origami have affinity toward the carbon surface to anchor the DNA-modified face toward the carbon grid.
CONCLUSIONS
Using negative stain EM, we revealed a substantial diversity in conformational states of 2D DNA origami nanoplatforms. We have shown that addition of an organic solvent (2% (v/v) DMSO) to the buffer significantly reduces the conformational polymorphism of 2D origami structures. Using negative stain EM and image averaging, we obtained 2D DNA origami images of much improved resolution, enabling us to visualize the DNA double helices and other structural features such as DNA networks and holes within the structures. Our approach represents a simple, rapid, and inexpensive strategy to flatten 2D DNA origami on an EM grid without the need for chemical modification. Compared to their 3D counterparts, 2D DNA origami structures offer several advantages, such as simplicity of design, high yield of assembly, and larger available surface area on which to immobilize biomolecular assemblies. As representative examples, we demonstrated that streptavidin and cocaine esterase can be site-specifically captured on these flattened DNA origami at the desired positions. The use of EM to study protein assemblies precisely immobilized on 2D DNA origami structures may inspire novel strategies to exploit such assemblies for biochemistry and nanotechnology applications. For example, in the future, DNA origami scaffolds may be employed to site-specifically capture and visualize the architecture and dynamics of macromolecular complexes by single-particle cryo-EM.
MATERIALS AND METHODS
Origami Design, Synthesis, and Purification
We used 2D DNA origami formed by self-assembly of the bacteriophage M13mp18 with 207 single-stranded staple strands published elsewhere.20 In our one-pot assembly, all modified DNA oligonucleotides (that is, biotinylated staple strands in five specific positions of the origami and staple strands with and without an additional 5′-sequence, referred to as a DNA extension 5′-CCTCTCACCCACCATTCATC, to occupy 12 specific positions in the origami, Figure S3, Supporting Information) and unmodified DNA oligonucleotides were all mixed with M13mp18 scaffold DNA in 3-fold molar excess in 1× TAE-Mg2+ (Tris-acetate-EDTA, pH 8.0; 40 mM Tris base, 20 mM acetic acid, 2 mM EDTA, 12.5 mM Mg(CH3COO)2) buffer. The DNA mixture was annealed by decreasing the temperature from 94 to 25 °C over 12 h using a thermocycler (Eppendorf). To remove excess DNA oligonucleotides, the origami sample was purified using a custom-made Sephacryl-300 (Sigma) gel filtration column (Figure S8, Supporting Information) as previously described,40 with the slight modification that a 0.6 mL Eppendorf tube (Fisher) was used after poking a tiny hole at its bottom, instead of a commercial column. These custom-made columns rendered a high yield of recovery (67–90%, Figure S8b, Supporting Information). The removal of excess staple strands and complementary biotin-modified DNA oligonucleotides was confirmed by agarose gel electrophoresis (Figure S8, Supporting Information). In addition to excess DNA oligonucleotides, other debris observed in the negative stain images of unpurified DNA origami was efficiently removed in this purification step. The purified origami were stored in the same buffer at 4 °C until needed. The purified origami sample was directly tested by negative stain EM at a concentration of 5 nM.
Real-Time Visualization of Binding of Cy3-Labeled DNA Oligonucleotides to the Complementary Capture DNA Oligonucleotides on DNA Origami
s A 100 pM solution of DNA origami in 1× TAE-Mg2+ buffer, pH 7.4, was flowed into the channel of a streptavidin-coated slide, prepared as described,20,41 and allowed to bind via the biotin–streptavidin interaction for 5 min. Excess origami were flushed out with 300 µL of 1× TAE-Mg2+ buffer. While the fluorescence of Cy3 was monitored at the slide surface using a TIRF microscope (Figure S7a, Supporting Information), a solution of 30 nM Cy3-labeled DNA oligonucleotide in 1× TAE-Mg2+ supplemented with an oxygen scavenger system42 (containing 2.5 mM 3,4-dihydroxybenzoic acid, Sigma; 1 mM Trolox, Acros; and 25 nM protocatechuate dioxygenase, Sigma-Aldrich) was injected into the flow channel with a dead time of less than 10 s. To limit photobleaching, a shuttered illumination scheme was used with 0.5 s exposures separated by 29.5 s dark periods, as described.41 The Cy3-labeled DNA oligonucleotide was injected during the dark period. The mean fluorescence signal from all origami (n = 68) in a field of view was plotted as a function of time (Figure S7b, Supporting Information) and fit with a single-exponential function to extract a rate constant as described.41
Prediction of DNA Origami Shape
The three-dimensional solution shapes of DNA origami were predicted by computer-aided engineering for DNA origami (CanDo) (http://cando-dnaorigami.org/).25 The default geometric parameters for B-form DNA (0.34 nm axial rise per base pair, 2.25 nm diameter of a helix, 10.5 base pairs per turn) and mechanical properties (stretching, bending, and torsional stiffness of 1100 pN, 230 pN nm2, and 460 pN nm2, respectively) were used.13,25
Sample Preparation for EM
Protein samples were prepared for EM using a conventional negative staining protocol.43 For DNA origami, 3 µL of 5 nM of DNA origami in 1× TAE-Mg2+ buffer and 0–40% (v/v) DMSO was applied to a freshly glow-discharged carbon-coated grid, incubated for 2 min at room temperature, and excess sample was removed by blotting with filter paper from one side of the grid. The grid was washed with buffer and stained with 0.75% (w/v) of uranylformate (UF). For positioning streptavidin molecules, DNA origami were applied on the grid first and excess sample was removed by blotting after 1–2 min. Streptavidin was added immediately afterward and incubated for an additional minute to allow for the streptavidin–biotin interaction. After excess streptavidin was blotted away, the grid was washed with 1× TAE-Mg2+ buffer supplemented with 2% (v/v) DMSO and stained using 0.75% (w/v) UF. To visualize CocE binding, the DNA origami were incubated on a carbon-coated grid for 2 min followed by sequential addition of streptavidin and biotinylated CocE. Finally, the complex was washed with 1× TAE-Mg2+ supplemented with 2% (v/v) DMSO to remove unbound protein and stained with 0.75% (w/v) UF.
Electron Microscopy
All images were collected at room temperature with a Tecnai T12 transmission electron microscope operated at a voltage of 120 kV. Images were recorded at a nominal magnification of 50 000× and a defocus value of −1.0 µm on a Gatan US4000 CCD camera. All images were binned over 2 × 2 pixels to obtain a pixel size of 4.16 Å on the specimen level.
EM Image Averaging
Individual origami were manually excised from EM images using e2helixboxer.py of the EMAN 2.1 software suite.44 Reference-free alignment and classification were carried out using the EMAN 1.9 image processing suite.45 For DNA origami, 1619 particle projections were grouped into five classes. For the DNA origami–streptavidin complex, 1004 particle projections were grouped into 10 classes. For complexes of DNA origami, streptavidin, and CocE, 866 particles were grouped into 50 classes.
Atomic Force Microscopy
The DNA origami sample (2 µL, 5 nM) in 1× TAE-Mg2+ buffer was deposited on a freshly cleaved mica surface (Ted Pella, Inc.) to adsorb for 2 min. After adsorption, 60 µL of the same 1× TAE-Mg2+ buffer was added to the Z-scanner and mica. Imaging was carried out at room temperature using the ScanAsyst in fluid mode (Bruker, Dimension FastScan AFM, probe type: ScanAsyst-fluid+). AFM images were processed with NanoScope Analysis software (Bruker, version 1.40).
Purification of Cocaine Esterase
BL21 Escherichia coli cells expressing recombinant CocE were pelleted, resuspended in 1× TBS, pH 7.4, supplemented with protease inhibitors (3 mg/mL each of leupeptin and lima bean or soybean trypsin inhibitor), and lysed using a French press (Thermo Scientific). CocE was enriched using Talon metal chelate affinity chromatography (Clontech Laboratories, Inc.), followed by anion-exchange chromatography on a Q-Sepharose fast-performance liquid chromatography column (GE Healthcare). CocE was eluted from the Q-Sepharose column with a 150–450 mM NaCl linear gradient in buffers containing 50 mM Tris-HCl, pH 8.0. The peak fractions were pooled and concentrated to 1 mg/mL using Centricon-30 concentrators (Millipore), snap frozen in liquid nitrogen, and stored at −80 °C.
Binding Cocaine Esterase to DNA Origami and Activity Assay
CocE was biotinylated at cysteine residues using thiol coupling by mixing protein and HPDP-biotin (Sigma-Aldrich Corp., St. Louis, MO) (HPDP: N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide), at a 1:10 ratio in 1× PBS buffer, pH 6.8, and incubated at room temperature for 1 h. Because there are three free cysteines per monomer, conjugation of up to six biotins is expected for each CocE dimer. Unbound biotin was removed using a Zeba spin desalting column (Thermo Scientific, Rockford, IL). Streptavidin and DNA origami were mixed at a 10:1 molar ratio and allowed to bind at room temperature for 30 min. CocE–biotin was then added at a 50:1 molar ratio (CocE/DNA origami) and incubated at room temperature for 30 min. Using the amount of CocE in the reaction as the initial concentration, the sample was diluted to 50 ng/mL of CocE for an activity assay.
Cocaine (Mallinckrodt, Hazelwood, MO) stocks were generated at 200, 100, 50, 25, 10, 5, 1, and 0.5 µM using PBS, pH 7.4. Each concentration of cocaine was added to a 96-well UV-permeable plate (Costar, Lowell, MA). An equal volume of CocE (at 50 ng/mL) was added to each well of the plate to initiate the reaction. The absorbance of cocaine was monitored at 240 nm for 20 min with readings every 10 s using a SpectraMax Plus 384 UV plate reader (Molecular Devices, Sunnyvale, CA) run by SOFTmax Pro software. The decrease in absorbance over time was converted to decrease in concentration using cocaine's extinction coefficient. Kinetic parameters of cocaine hydrolysis (Vmax and KM) were determined using Prism software (GraphPad Software Inc., San Diego, CA).
Supplementary Material
Acknowledgments
This work was funded by an MCubed award from the University of Michigan and by Department of Defense MURI Award W911NF-12-1-0420. The authors gratefully acknowledge H. Yan for providing DNA origami, and the assistance of M. Su in some EM experiments.
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Supporting Information Available: Conformational polymorphism of rectangular 2D DNA origami; DMSO-assisted flattening of DNA origami; mapping the DNA origami design to a class average EM image; distance measurements between the capture DNA oligonucleotides and biotins; distribution of streptavidin on 2D DNA origami; enzyme activity of origami bound CocE; binding of Cy3-labeled complementary DNA oligonucleotides to DNA origami; agarose gels and purification of DNA origami by gel filtration; and determining the face-up and face-down population by streptavidin binding to the DNA origami lacking 12 protruding DNA linkers. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b01841.
REFERENCES AND NOTES
- 1.Bhatia D, Surana S, Chakraborty S, Koushika SP, Krishnan Y. A Synthetic Icosahedral DNA-Based Host–Cargo Complex for Functional in vivo Imaging. Nat. Commun. 2011;2:339. doi: 10.1038/ncomms1337. [DOI] [PubMed] [Google Scholar]
- 2.Bath J, Turberfield AJ. DNA Nanomachines. Nat. Nanotechnol. 2007;2:275–284. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]
- 3.Jungmann R, Scheible M, Simmel FC. Nanoscale Imaging in DNA Nanotechnology. WIREs Nanomed. Nanobiotechnol. 2012;4:66–81. doi: 10.1002/wnan.173. [DOI] [PubMed] [Google Scholar]
- 4.Seeman N. An Overview of Structural DNA Nanotechnology. Mol. Biotechnol. 2007;37:246–257. doi: 10.1007/s12033-007-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lund K, Manzo AJ, Dabby N, Michelotti N, Johnson-Buck A, Nangreave J, Taylor S, Pei R, Stojanovic MN, Walter NG, Winfree E, Yan H. Molecular Robots Guided by Prescriptive Landscapes. Nature. 2010;465:206–210. doi: 10.1038/nature09012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu J, Yang YR, Johnson-Buck A, Liu M, Liu Y, Walter NG, Woodbury NW, Yan H. Multi-Enzyme Complexes on DNA Scaffolds Capable of Substrate Channelling with an Artificial Swinging Arm. Nat. Nanotechnol. 2014;9:531–536. doi: 10.1038/nnano.2014.100. [DOI] [PubMed] [Google Scholar]
- 7.Michelotti N, Johnson-Buck A, Manzo AJ, Walter NG. Beyond DNA Origami: The Unfolding Prospects of Nucleic Acid Nanotechnology. WIREs Nanomed. Nanobiotechnol. 2012;4:139–152. doi: 10.1002/wnan.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winfree E, Liu F, Wenzler LA, Seeman NC. Design and Self-Assembly of Two-Dimensional DNA Crystals. Nature. 1998;394:539–544. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 9.Studer D, Humbel B, Chiquet M. Electron Microscopy of High Pressure Frozen Samples: Bridging the Gap between Cellular Ultrastructure and Atomic Resolution. Histochem. Cell Biol. 2008;130:877–889. doi: 10.1007/s00418-008-0500-1. [DOI] [PubMed] [Google Scholar]
- 10.Rothemund PWK, Folding DN. A. to Create Nanoscale Shapes and Patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 11.Han D, Pal S, Nangreave J, Deng Z, Liu Y, Yan H. DNA Origami with Complex Curvatures in Three-Dimensional Space. Science. 2011;332:342–346. doi: 10.1126/science.1202998. [DOI] [PubMed] [Google Scholar]
- 12.Douglas SM, Dietz H, Liedl T, Hogberg B, Graf F, Shih WM. Self-Assembly of DNA into Nanoscale Three-Dimensional Shapes. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castro CE, Kilchherr F, Kim D-N, Shiao EL, Wauer T, Wortmann P, Bathe M, Dietz H. A Primer to Scaffolded DNA Origami. Nat. Methods. 2011;8:221–229. doi: 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- 14.Fu J, Liu M, Liu Y, Woodbury NW, Yan H. Interenzyme Substrate Diffusion for an Enzyme Cascade Organized on Spatially Addressable DNA Nanostructures. J. Am. Chem. Soc. 2012;134:5516–5519. doi: 10.1021/ja300897h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakata E, Liew FF, Uwatoko C, Kiyonaka S, Mori Y, Katsuda Y, Endo M, Sugiyama H, Morii T. Zinc-Finger Proteins for Site-Specific Protein Positioning on DNA-Origami Structures. Angew. Chem. 2012;124:2471–2474. doi: 10.1002/anie.201108199. [DOI] [PubMed] [Google Scholar]
- 16.Ke Y, Lindsay S, Chang Y, Liu Y, Yan H. Self-Assembled Water-Soluble Nucleic Acid Probe Tiles for Label-Free RNA Hybridization Assays. Science. 2008;319:180–183. doi: 10.1126/science.1150082. [DOI] [PubMed] [Google Scholar]
- 17.Kuzyk A, Laitinen KT, Törmä P. DNA Origami as a Nanoscale Template for Protein Assembly. Nanotechnology. 2009;20:235305. doi: 10.1088/0957-4484/20/23/235305. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Z, Liu Y, Yan H. Organizing DNA Origami Tiles into Larger Structures Using Preformed Scaffold Frames. Nano Lett. 2011;11:2997–3002. doi: 10.1021/nl201603a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nangreave J, Han D, Liu Y, Yan H. DNA Origami: A History and Current Perspective. Curr. Opin. Chem. Biol. 2010;14:608–615. doi: 10.1016/j.cbpa.2010.06.182. [DOI] [PubMed] [Google Scholar]
- 20.Johnson-Buck A, Nangreave J, Kim D-N, Bathe M, Yan H, Walter NG. Super-Resolution Fingerprinting Detects Chemical Reactions and Idiosyncrasies of Single DNA Pegboards. Nano Lett. 2013;13:728–733. doi: 10.1021/nl304415b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu N, Czajkowsky DM, Zhang J, Qu J, Ye M, Zeng D, Zhou X, Hu J, Shao Z, Li B, Fan C. Molecular Threading and Tunable Molecular Recognition on DNA Origami Nanostructures. J. Am. Chem. Soc. 2013;135:12172–12175. doi: 10.1021/ja403863a. [DOI] [PubMed] [Google Scholar]
- 22.Plesa C, Ananth AN, Linko V, Gülcher C, Katan AJ, Dietz H, Dekker C. Ionic Permeability and Mechanical Properties of DNA Origami Nanoplates on Solid-State Nanopores. ACS Nano. 2014;8:35–43. doi: 10.1021/nn405045x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ke Y, Bellot G, Voigt NV, Fradkov E, Shih WM. Two Design Strategies for Enhancement of Multilayer-DNA-Origami Folding: Underwinding for Specific Intercalator Rescue and Staple-Break Positioning. Chem. Sci. 2012;3:2587–2597. doi: 10.1039/C2SC20446K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ke Y, Ong LL, Shih WM, Yin P. Three-Dimensional Structures Self-Assembled from DNA Bricks. Science. 2012;338:1177–1183. doi: 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D-N, Kilchherr F, Dietz H, Bathe M. Quantitative Prediction of 3D Solution Shape and Flexibility of Nucleic Acid Nanostructures. Nucleic Acids Res. 2012;40:2862–2868. doi: 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fay MJ, Walter NG, Burke JM. Imaging of Single Hairpin Ribozymes in Solution by Atomic Force Microscopy. RNA. 2001;7:887–895. doi: 10.1017/s1355838201002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pastré D, Piétrement O, Fusil S, Landousy F, Jeusset J, David M-O, Hamon L, Le Cam E, Zozime A. Adsorption of DNA to Mica Mediated by Divalent Counterions: A Theoretical and Experimental Study. Biophys. J. 2003;85:2507–2518. doi: 10.1016/s0006-3495(03)74673-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaurasiya KR, Paramanathan T, McCauley MJ, Williams MC. Biophysical Characterization of DNA Binding from Single Molecule Force Measurements. Phys. Life Rev. 2010;7:299–341. doi: 10.1016/j.plrev.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonner G, Klibanov AM. Structural Stability of DNA in Nonaqueous Solvents. Biotechnol. Bioeng. 2000;68:339–344. doi: 10.1002/(sici)1097-0290(20000505)68:3<339::aid-bit12>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 30.Ke F, Luu YK, Hadjiargyrou M, Liang D. Characterizing DNA Condensation and Conformational Changes in Organic Solvents. PLoS One. 2010;5:e13308. doi: 10.1371/journal.pone.0013308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardjasa A, Ling M, Ma K, Yu H. Investigating the Effects of DMSO on PCR Fidelity Using a Restriction Digest-Based Method. J. Exp. Microbiol. Immunol. 2010;14:161–164. [Google Scholar]
- 32.Gietl A, Holzmeister P, Grohmann D, Tinnefeld P. DNA Origami as Biocompatible Surface to Match Single-Molecule and Ensemble Experiments. Nucleic Acids Res. 2012;40:e110. doi: 10.1093/nar/gks326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michelotti N, de Silva C, Johnson-Buck AE, Manzo AJ, Walter NG. A Bird's Eye View: Tracking Slow Nanometer-Scale Movements of Single Molecular Nano-Assemblies. Methods Enzymol. 2010;475:121–148. doi: 10.1016/S0076-6879(10)75006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voigt NV, Torring T, Rotaru A, Jacobsen MF, Ravnsbaek JB, Subramani R, Mamdouh W, Kjems J, Mokhir A, Besenbacher F, Gothelf KV. Single-Molecule Chemical Reactions on DNA Origami. Nat. Nanotechnol. 2010;5:200–203. doi: 10.1038/nnano.2010.5. [DOI] [PubMed] [Google Scholar]
- 35.Larsen NA, Turner JM, Stevens J, Rosser SJ, Basran A, Lerner RA, Bruce NC, Wilson IA. Crystal Structure of a Bacterial Cocaine Esterase. Nat. Struct. Biol. 2002;9:17–21. doi: 10.1038/nsb742. [DOI] [PubMed] [Google Scholar]
- 36.Turner JM, Larsen NA, Basran A, Barbas CF, Bruce NC, Wilson IA, Lerner RA. Biochemical Characterization and Structural Analysis of a Highly Proficient Cocaine Esterase. Biochemistry. 2002;41:12297–12307. doi: 10.1021/bi026131p. [DOI] [PubMed] [Google Scholar]
- 37.Collins GT, Brim RL, Narasimhan D, Ko M-C, Sunahara RK, Zhan C-G, Woods JH. Cocaine Esterase Prevents Cocaine-Induced Toxicity and the Ongoing Intravenous Self-Administration of Cocaine in Rats. J. Pharmacol. Exp. Ther. 2009;331:445–455. doi: 10.1124/jpet.108.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howarth M, Chinnapen DJF, Gerrow K, Dorrestein PC, Grandy MR, Kelleher NL, El-Husseini A, Ting AY. A Monovalent Streptavidin with a Single Femtomolar Biotin Binding Site. Nat. Methods. 2006;3:267–273. doi: 10.1038/NMETHXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whorton MR, Bokoch MP, Rasmussen SGF, Huang B, Zare RN, Kobilka B, Sunahara RK. A Monomeric G Protein-Coupled Receptor Isolated in a High-Density Lipoprotein Particle Efficiently Activates Its G Protein. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajendran A, Endo M, Sugiyama H. Current Protocols in Nucleic Acid Chemistry. John Wiley & Sons Inc.; New York: 2001. DNA Origami: Synthesis and Self-Assembly; pp. 12.9.1–12.9.18. [DOI] [PubMed] [Google Scholar]
- 41.Johnson-Buck A, Nangreave J, Jiang S, Yan H, Walter NG. Multifactorial Modulation of Binding and Dissociation Kinetics on Two-Dimensional DNA Nanostructures. Nano Lett. 2013;13:2754–2759. doi: 10.1021/nl400976s. [DOI] [PubMed] [Google Scholar]
- 42.Aitken CE, Marshall RA, Puglisi JD. An Oxygen Scavenging System for Improvement of Dye Stability in Single-Molecule Fluorescence Experiments. Biophys. J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohi M, Li Y, Cheng Y, Walz T. Negative Staining and Image Classification – Powerful Tools in Modern Electron Microscopy. Biol. Proced. Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: An Extensible Image Processing Suite for Electron Microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated Software for High-Resolution Single-Particle Reconstructions. J. Struct. Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.