

Abstract
Background
The nucleotide reverse transcriptase inhibitor tenofovir is widely administered in a disoproxil prodrug form (tenofovir disoproxil fumarate) for HIV management and prevention. Recently, novel prodrugs tenofovir alafenamide fumarate (TAF) and hexadecyloxypropyl tenofovir (CMX157) have been pursued for HIV treatment while minimizing adverse effects associated with systemic TFV exposure. Dynamic and sensitive bioanalytical tools are required to characterize the pharmacokinetics of these prodrugs in systemic circulation. Two parallel methods have been developed, one to combinatorially quantify TAF and TFV, and a second method for CMX157 quantification, in plasma.
Methods
K2EDTA plasma was spiked with TAF and TFV, or CMX157. Following the addition of isotopically labeled internal standards and sample extraction via solid phase extraction (TAF and TFV) or protein precipitation (CMX157), samples were subjected to liquid chromatographic-tandem mass spectrometric (LC-MS/MS) analysis. For TAF and TFV, separation occurred using a Zorbax Eclipse Plus C18 Narrow Bore RR, 2.1 × 50 mm, 3.5 μm column and analytes were detected on an API5000 mass analyzer; CMX157 was separated using a Kinetex C8, 2.1 × 50 mm, 2.6 μm column and quantified using an API4500 mass spectrometer. Methods were validated according to FDA Bioanalytical Method Validation guidelines.
Results
Analytical methods were optimized for the multiplexed monitoring of TAF and TFV, and CMX157 in plasma. The lower limits of quantification (LLOQs) for TAF, TFV, and CMX157 were 0.03, 1.0, and 0.25 ng/mL, respectively. Calibration curves were generated via weighted linear regression of standards. Intra-and inter-assay precision and accuracy studies demonstrated %CVs ≤ 14.4% and %DEVs ≤ ± 7.95%, respectively. Stability and matrix effects studies were also performed. All results were acceptable and in accordance with the recommended guidelines for bioanalytical methods. Assays were also applied to quantify in vivo concentrations of prodrugs and TFV in a preclinical study post-rectal administration.
Conclusions
Sensitive, specific, and dynamic LC-MS/MS assays have been developed and validated for the multiplexed quantification TAF and TFV, as well as an independent assay for CMX157, in plasma. The described methods meet sufficient throughput criteria to support large research trials.
Keywords: Antiretroviral, LC-MS/MS, Validation, Tenofovir, HIV, Prodrug
Graphical abstract
Introduction
There are approximately 37 million individuals living with HIV worldwide, with an estimated 50% on antiretroviral treatment regimens [1]. While several antiretroviral classes are approved for disease management, the most common regimens include a combinatorial nucleotide reverse transcriptase inhibitor (NRTI) backbone; more recently, there has been a shift to include an integrase strand transfer inhibitor for management in treatment-naïve individuals [1–3]. The adenosine analog tenofovir (TFV), is extensively utilized for both HIV treatment as well as prevention, and is predominantly administered in conjunction with the NRTI emtricitabine (FTC). TFV, like other NRTIs, elicits its antiviral effects via intracellular activation to phosphorylated metabolic moieties [3]. Therefore, it is imperative for TFV to penetrate CD4+ T cells and lymphoid tissues for metabolism to tenofovir-diphosphate (TFV-DP) and subsequent inhibition of viral replication and propagation.
TFV has been predominantly administered as the prodrug tenofovir disoproxil fumarate (TDF) to facilitate increased intestinal absorption and subsequent bioavailability in vivo [2, 4–5]. The disoproxil modification facilitates oral absorption through masking of the negatively charged phosphonic groups located on the nucleotide analog. TDF is degraded to TFV via carboxyesterase and phosphodiesterase enzymes in systemic circulation; prodrug administration increases TFV bioavailability by 25%, resulting in enhanced TFV uptake and metabolic conversion in targeted CD4+ T cells and lymphoid tissues [2, 6–7]. While TDF is administered as Viread®, it is also available in a number of fixed dose formulations for disease management, including Atripla® (300 mg TDF, 200 mg emtricitabine (FTC), and 600 mg efavirenz (EFV)) and Stribild® (150 mg elvitegravir (EVG), 150 mg cobicistat (c), 200 mg FTC and 300 TDF) [3, 8–10]. However, due to potential drug-associated adverse effects, including decreased bone density and renal tubular injury, alternative prodrug forms have been evaluated to enhance intracellular uptake and reduce systemically-associated toxicities [2, 3, 11–12].
Recently, the FDA approved the single tablet formulation, Genvoya®, for HIV treatment as an alternative to Stribild [3, 8–10]. While both Stribild and Genvoya® have the same backbone compounds, Genvoya utilizes 10 mg of the novel TFV prodrug tenofovir alafenamide fumarate (TAF, GS-7340) [3, 8–10, 13]. The addition of the alafenamide group to the antiviral compound prevents non-specific esterase activity, resulting in a more stable, intact, prodrug systemic circulation [11, 14]. TAF passively enters into target cells, where it undergoes ester hydrolysis by the lysosomal enzyme cathepsin A. Activity by cathepsin A results in the production of an unstable metabolite that is ultimately degraded to TFV. TFV may then be effluxed from a target cell, or intracellularly activated via kinase enzymes to generate the phosphorylated moiety [6–7, 11–12]. The improved toxicity profiles and increased intracellular bioavailability observed for TAF has led to more intense interest not only in disease management but also in potential prevention [2–3, 10–13]. Although TAF has demonstrated a significant amount of success in increasing efficacy and reducing toxicity, other prodrugs of TFV are in the pipeline to meet the numerous challenges of HIV treatment and prevention. Additionally, compounds are under evaluation to treat both HIV and hepatitis B virus (HBV), including the TFV lipid conjugate, CMX157 (hexadecyloxypropyl tenofovir) [7, 14–15].
CMX157 is an alkoxylalkyl lysophospholipid conjugate of TFV that was designed for stability in plasma and rapid uptake by cells [14–16]. The stability of CMX157 in plasma prevents TFV accumulation in renal cells via organic anion transporters (OATs) [14]. CMX157 is currently under evaluation as an alternative treatment to TDF for HIV that can achieve antiviral activity while mitigating secondary adverse effects [2, 6–7, 15]. In vitro data support that CMX157 may be effective against drug resistant strains of HIV [2, 14–16]. Further, phase 2 data suggest CMX157 may be a potent alternative for HBV treatment as the lead drug in a combinatorial formulation of other compounds, including other NRTIs [17–18].
In light of the recent approval of TAF as part of a combinatorial therapy for HIV management, and a growing interest in prodrugs with more targeted antiviral activity, robust bioanalytical assays are required to characterize prodrug, drug, and metabolite pharmacokinetics. There are few bioanalytical assays describing TAF quantification in plasma; published assays have significant limitations, including insufficient analytical sensitivities for prodrug quantification from approved oral formulations (10 mg daily), non-multiplexing with TFV, large sample volumes, and long-analytical run times [3, 19–21]. There is substantially less bioanalytical data described in the literature with regard to CMX157 quantification. Thus, we describe the development, validation, and implementation of sensitive liquid chromatographic-tandem mass spectrometric (LC-MS/MS) methods for the multiplexed quantification of TAF and TFV in plasma, as well as plasma CMX157 quantification. The described assays have lower limits of quantification that are sufficiently sensitive to support preclinical studies characterizing TFV and prodrug pharmacokinetics using an enema-based drug delivery system.
Experimental
Chemicals
The reference standard TAF (C21H29N6O5P•C4H4O4) was obtained as a gift from Gilead Sciences (Gilead, Foster City, CA). The reference standard TFV monohydrate (C9H14N5O4P•H2O) was obtained from the NIH AIDS Research and Reference Reagent Program (NIH, Germantown, MD). The TFV internal standard, 13C5-TFV (13C5C4H14N5O4P) was obtained from Moravek Biochemicals (Moravek, Brea, CA). The reference standards CMX157 (C26H52N5O5P) and its isotopically labeled internal standard, CMX157-d6 (C28H46D6N5O5P), were generously provided as a gift from Chimerix (Chimerix, Durham, NC). The structures for the aforementioned compounds are depicted in Figure 1. Human plasma K2EDTA, as well as hemolyzed and lipemic plasma lots were obtained from Bioreclamation (Bioreclamation IVT, West Berry, NY). Water, methanol, acetonitrile, 2-propanol, and acetone (all Optima LC/MS UHPLC grade), as well as HPLC grade dimethyl sulfoxide, ammonium hydroxide and glacial acetic acid, were all obtained from Fisher Scientific (Fair Lawn, NJ). Ammonium bicarbonate, ammonium acetate, formic acid and trifluoroacetic acid were obtained from Sigma-Aldrich (Saint Louis, MO).
Figure 1.
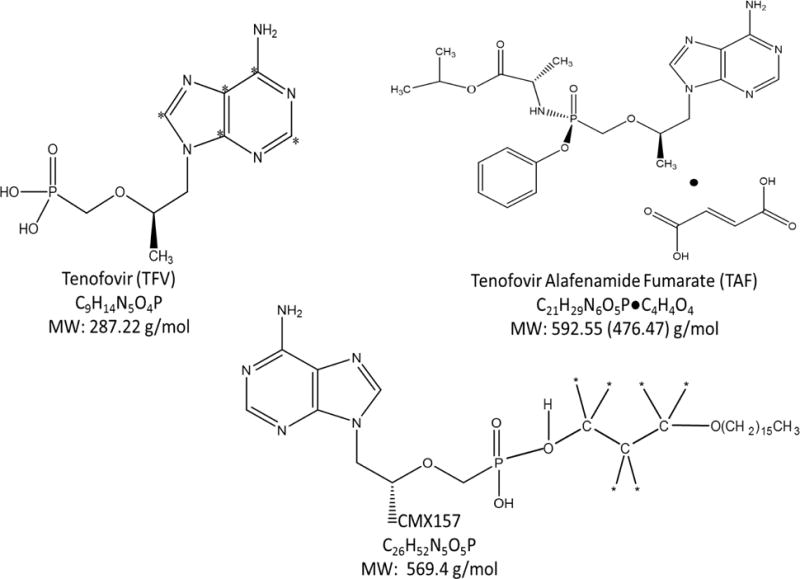
Chemical structures for TAF (C21H29N6O5P•C4H4O4), TFV monohydrate (C9H14N5O4P•H2O), and CMX157 (C26H52N5O5P) are depicted. Asterisks denote 13C- and deuterium locations in isotopically labeled internal standards 13C5-TFV and CMX157-d6.
Preparation of reagents and standards
For the multiplexed quantification of TAF and TFV, stock concentrations were corrected for loss of fumarate (TAF) and the presence of the monohydrate (TFV). Stock solutions were prepared in DMSO (TAF) and water (TFV) at concentrations of 1000 μg/mL and 941 μg/mL, respectively, and working stock solutions were prepared from these stocks. Calibration standards containing final TAF/TFV concentrations of 0.030/1.00, 0.06/2.00, 0.250/5.00, 1.00/10.0, 5.00/25.0, 25.0/50.0, 75.0/100, and 150/200 ng/mL were prepared by spiking human K2EDTA plasma with appropriate volumes of working stock solutions. Quality control (QC) materials were prepared from independently weighed drug master stocks and spiked into K2EDTA plasma. QC materials were prepared at the lower limits of quantification (LLOQs), as well as low, mid and high levels. Final QC concentrations for TAF/TFV at the LLOQ, low, mid and high levels were 0.030/1.00, 0.090/3.00, 3.00/20.0 and 130/170 ng/mL, respectively. The internal standard, 13C5-TFV, was used for quantification of both TAF and TFV. An internal standard stock was prepared in water to a final concentration of 1.00 mg/mL, and was ultimately diluted to a working stock concentration of 50 ng/mL to be added during sample preparation.
For CMX157 quantification, a stock solution of CMX157 was prepared in 80:20 methanol:water with 0.2% ammonium hydroxide at a concentration of 500 μg/mL. Working stock solutions were prepared in 60:40 100 mM ammonium acetate:acetonitrile and spiked into K2EDTA plasma for final calibrator concentrations of 0.250, 1.00, 10.0, 25.0, 75.0, 125, 150, and 200 ng/mL. QC materials were prepared from an independently weighed master stock to generate QC concentrations at 0.250 (LLOQ), 0.750 (low), 50.0 (mid), and 160 (high) ng/mL. A stock solution of the isotopically labeled internal standard, CMX157-d6, was prepared in 80:20 methanol:water with 0.2% ammonium hydroxide to a concentration of 500 μg/mL, and was diluted in 60:40 100 mM ammonium acetate:acetonitrile to a working stock concentration of 75 ng/mL to be added during sample preparation.
Sample Preparation
TAF and TFV were extracted from plasma via mixed mode cation exchange solid phase extraction. One hundred microliters of plasma was combined with an equal volume of internal standard and 300 μL of 1.0% trifluoroacetic acid. This mixture was added to a Waters Oasis MCX SPE plate (Waters, Milford, MA). Extraction occurred under vacuum conditions; samples were washed once with 500 μL of 1.0% trifluoroacetic and ultimately eluted with 500 μL 5.0% ammonium hydroxide in methanol. Eluents were evaporated to dryness, reconstituted in a 150 μL 80:20 water:acetonitrile solution containing 0.1% formic acid, and introduced into the LC-MS system for analysis.
CMX157 was isolated from plasma via protein precipitation. Briefly, 50 μL of sample was combined with 50 μL of internal standard and 100 μl of water onto an Agilent Captiva 0.45 μm filter plate (Agilent, Wilmington, DE). Following a 5 min incubation, precipitation occurred using 500 μL acetonitrile with 0.25% ammonium hydroxide. Post-elution, eluents were evaporated to dryness and reconstituted in 100 microliters of 60:40 0.25% ammonium hydroxide in acetonitrile:5 mM ammonium bicarbonate in water, and introduced into the LC-MS system for analysis.
Separation and Instrument Acquisition Parameters
TAF and TFV samples were analyzed using a Waters Acquity UPLC system (Waters, Milford, MA) interfaced with an API5000 mass analyzer (SCIEX, Foster City, CA). Analytes were chromatographically separated using a Zorbax Eclipse Plus C18 Narrow Bore RR, 2.1 × 50 mm, 3.5 μm column (Agilent, Wilmington, DE) using a mobile phase system consisting of 0.1% formic acid in water (mobile phase A, MPA) and 0.1% formic acid in acetonitrile (mobile phase B, MPB). The total analytical run time was 4.0 minutes. TAF and TFV were quantified on an API5000 operated in positive ionization and selective reaction monitoring modes, monitoring transitions m/z 477.3→176.0 (TAF), m/z 288.1→176.1 (TFV) and m/z 293.1→181.1 (13C5-TFV IS).
CMX157 samples were analyzed using a Shimadzu Nexera XR LC system (Shimadzu, Kyoto, Japan) interfaced with an API4500 mass analyzer (SCIEX). CMX157 was separated using a Kinetex C8, 2.1 × 50 mm, 2.6 μm column (Phenomenex, Torrance, CA) maintained at 50 °C and a mobile phase system consisting of 5 mM ammonium bicarbonate in water (MPA), 0.25% ammonium hydroxide in methanol with 0.05% DMSO, pH 8.5 (MPB), and 80% methanol, 20% acetone, and 0.3% DMSO for the strong wash (mobile phase C, MPC). The total analytical run time was 5.50 minutes. The mass spectrometer operated in negative ionization and selective reaction monitoring modes, and drug and internal standard were monitored at transitions m/z 568.4→134.0 (CMX157) and m/z 574.1→134.0 (CMX157-d6), respectively. Full chromatographic and mass spectrometer conditions are described in Supplemental Table 1.
Data Evaluation
Data were acquired using Analyst® 1.6.2 Software (Build 8489; SCIEX). Calculations for validation assessment, which includes precision, accuracy, stability, and matrix effects, were performed using Microsoft Office Excel 2010 and 2013. Outliers were defined by Grubbs’ Outlier Test.
Method Validation
Bioanalytical methods were validated in accordance with the recommendations endorsed by FDA Guidance for Industry, Bioanalytical Method Validation guidelines [22]. Assays were tested and validated for intra- and inter-assay precision and accuracy, calibration curve analysis, selectivity and matrix effects; further stability, carryover, and dilutional integrity challenges were also performed.
Precision & Accuracy Studies
Intra-assay precision was determined through the analysis of six independent replicates of QC materials extracted from plasma. The observed means, SDs and %CVs were calculated at the aforementioned LLOQ, as well as low, mid, and high QC levels. Inter-assay precision was evaluated for all QC levels over three independent experiments; observed values were quantified from run-specific calibration curves. Accuracy was determined as the %DEV from theoretical concentrations at each QC level. Both intra-and inter-assay accuracy were determined from six independent replicates within an experiment and between three independent experiments, respectively.
Calibration Curve Analysis
Calibration curve standards were prepared and evaluated at the beginning and end of each analytical run. Calibration curves for TAF, TFV, and CMX157 were generated based on the peak area ratios of the analytes to the respective internal standards using a weighted 1/x2 linear regression.
Dilutional Integrity
Extended linearity beyond the primary analytical measuring range was also established. Plasma was spiked with TFV and its prodrugs at concentrations three times above assay upper limits of quantification (ULOQ): TAF [450 ng/mL], TFV [600 ng/mL] and CMX157 [600 ng/mL]. Samples were diluted up to 20-fold with drug-free plasma in quadruplicate, prepared, and analyzed. Further, mid and high QC samples (n=4) were diluted up to five-fold with drug-free plasma and analyzed to determine if a volume-limited sample could also be analyzed using the described assays. The %DEV at each dilutional level was determined to evaluate acceptability, which was defined as < ± 15% of target concentrations.
Stability Assessment
Several stability challenges were performed to characterize the quantification of TAF, TFV and CMX157 under several conditions. Challenges were conducted using low, mid, and high QC levels for each analyte. Injection matrix stability was performed by re-analyzing extracted and reconstituted QC material (replicates of 6) that was maintained in an autosampler at 4°C for 48 (CMX157) or 72 (TAF, TFV) h. Sample matrix studies were performed by incubating plasma QC samples in quadruplicate at ambient temperature (22-24 °C) for 24 (TAF, TFV) to 72 h (CMX157); room-temperature incubated samples were tested in parallel with freshly thawed calibrators and QCs. Freeze-thaw challenges were conducted by subjecting QC samples in quadruplicate to two or three freeze-thaw cycles at -80°C; challenged specimens were prepared and tested in parallel with freshly thawed calibrators and QCs. Long term stability was evaluated by spiking freshly thawed plasma at the low, mid, and high QC levels, which were considered Day 0; these samples were compared against QC specimens stored at -80°C from 161 (CMX157) to 244 (TAF, TFV) days. Matrix stability was further tested by comparing QCs prepared in lipemic plasma and hemolyzed plasma to QCs prepared in K2EDTA plasma in quadruplicate. For the aforementioned assessments, challenged QCs were compared against freshly thawed samples and a percent difference (%DIF) was determined. Percent differences were calculated as the difference between challenges and reference QC results divided by reference QC concentrations, and multiplied by 100. Percent differences < ± 15% were deemed acceptable.
Signal to Noise Ratio, Carryover, and Cross-Talk Characterization
Signal to noise ratios (selectivity) were determined by processing blank plasma samples alongside the LLOQ of the assay. The peak height threshold was set to zero, and peak areas of blank plasma were compared to analyte intensities and peak areas at analyte-specific lower limits of quantification. Signal to noise ratios at assay LLOQs of ≥ 20% as compared to blank were considered acceptable. Carryover was determined by running a sequence of samples at the LLOQ in replicates of four, samples at the ULOQ in replicates of four, and blank plasma extracts in replicates of four. Comparison of peak areas between LLOQ samples and post-ULOQ samples were conducted; carryover was defined as minimal if peak areas of post-ULOQ blanks ≤ 20% of the peak areas at the LLOQ. Cross talk was similarly determined by injecting five blank plasma samples, five blank plasma samples spiked only with the internal standard, and finally five ULOQ plasma samples. Contribution of assay internal standards (13C5-TFV and CMX157-d6) to TAF, TFV or CMX157 total ion chromatograms was deemed negligible if peak area intensities of internal standard-spiked samples yielded analyte peak areas ≤ 20% of the peak areas at the LLOQ.
Matrix Effects
Matrix effects, as well as extraction efficiency and processing efficiency were determined as per the approach described by Matuszewski and colleagues [23], for all analytes. Three sets of QC samples were prepared in six independent lots of drug-free human plasma. An un-extracted set was prepared at low, mid and high QC concentrations in the absence of matrix. Post-extracted samples were prepared by spiking analytes at the aforementioned concentrations into post-extracted plasma. Finally, a pre-extracted set was prepared in plasma and processed as previously described. Raw peak areas for the analytes and their respective internal standards, were analyzed to determine matrix effects (comparison of post-extracted to un-extracted samples), extraction efficiency (comparison of pre-extracted to post-extracted samples) and processing efficiency (comparison of pre-extracted to un-extracted samples).
Assay Implementation in Biological Samples
In support of a preclinical animal study to characterize TFV prodrug pharmacokinetics, mice were rectally dosed with molar equivalents of TAF (3.28 mg/mL TAF; n=10) or CMX157 (3.72 mg/mL CMX157; n=5) in an enema formulation comprised of normal saline (Quality Biologicals, Gaithersburg, MD). Mice were subsequently sacrificed and blood was collected at 15 min and 4 h for TAF and CMX157 analysis.
Results
Method Development
Several drug-specific aspects were evaluated during assay development. Notably, studies were conducted to assess the potential to include quantification of all TFV prodrugs (including TDF) in conjunction with TFV in a single method to enhance assay flexibility. However, consistent with biological observations, whole blood stability spiking challenges demonstrated lack of ex vivo TDF stability, with degradation occurring as quickly as 5 minutes post-spiking. Contrary to these observations, TAF and CMX157 were significantly more stable in whole blood at all challenged time points; up to 1 h for TAF and 8 h for CMX157 (data not shown). Due to the conjugated hydrophobic moieties associated with CMX157, the prodrug could not be chromatographically resolved using the same conditions that were optimal for TAF and TFV. Most notably, CMX157 was not reproducibly eluted from an octodecyl (C18) stationary phase, and was associated with substantial carryover using the chromatographic system optimal for TAF and TFV separation. The highly organic and basic mobile phase system required for optimal separation and elution of CMX157 was further unsuitable for TAF and TFV.
Using aforementioned chromatographic conditions and LC system, both TAF and TFV were resolved in a single assay without compromising analytical sensitivity. Due to the unavailability of an isotopically labeled standard for TAF, two surrogate molecules were assessed as an alternative internal standard. Both des-methyl-TDF and isotopically labeled TFV were evaluated in the assay. The des-methyl-TDF had an average %CV that was twice the %CV of the isotopically labeled TFV, leading des-methyl-TDF to give greater variability in final concentrations compared to the isotopically labeled TFV internal standard. In the combinatorial quantification of TAF and TFV, TAF and TFV eluted at 2.21 min and 1.31 min, respectively. In the assay for CMX157 quantification, the compound and its internal standard eluted at 0.89 min (Figure 2).
Figure 2.
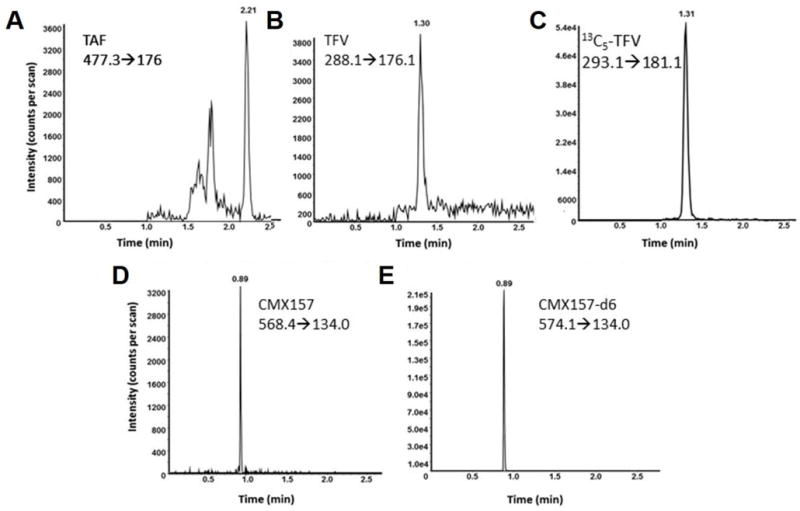
Representative chromatograms of TAF, TFV, CMX157 and respective internal standards. (A) Plasma-spiked with TAF at 0.090 ng/mL eluted at 2.21 min; the transition monitored was m/z 477.3→176.0. (B) Plasma-spiked with TFV at a concentration of 3.0 ng/mL was monitored at m/z 288.1→176.1 and eluted at 1.31 min. (C) 13C5-TFV added to a sample at a final concentration of 50 ng/mL, eluted at 1.31 min and the m/z 293.1→181.1 transition was monitored. (D) Plasma-spiked CMX157 at a concentration of 0.75 ng/mL eluted at 0.89 min, and was monitored at a transition of m/z 568.4→134.0. (E) The CMX157 internal standard, CMX157-d6, at a concentration of 75 ng/mL, eluted at the same retention time as the molecule; the transition was of m/z 574.1→134.0.
Precision & Accuracy Studies
Intra- and inter-assay precision and accuracy studies were performed at analyte LLOQs, as well as low, mid and high QC levels. For all analytes, intra- and inter-assay precision ranged from 2.91% to 14.4% and intra- and inter-assay accuracy ranged from -7.95% to 7.76%, respectively. All observed results were acceptable as per FDA recommendations (%CVs and %DEVs ≤ ± 20% at the LLOQ and ≤ ± 15% at low, mid, and high QC levels). A summary of intra- and inter-assay precision and accuracy results is included in Table 1.
Table 1.
Intra- and Inter- Precision and Accuracy
| QC Level | Intra - Assay Precision and Accuracya
|
Inter - Assay Precision and Accuracyb
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean [ng/ml] |
SD [ng/ml] |
%CV | %DEV | Mean [ng/ml] |
SD [ng/ml] |
%CV | %DEV | ||||
| TAF
|
|||||||||||
| LLOQ [0.030 ng/mL] |
0.0307 | 0.00410 | 13.3 | 2.39 | 0.0321 | 0.00450 | 13.9 | 7.04 | |||
| Low [0.090 ng/mL] |
0.0846 | 0.00850 | 10.0 | -5.96 | 0.0881 | 0.0127 | 14.4 | -2.07 | |||
| Mid [3.00 ng/mL] |
3.11 | 0.230 | 7.38 | 3.56 | 3.22 | 0.200 | 6.15 | 7.43 | |||
| High [130 ng/mL] |
120 | 8.60 | 7.20 | -7.95 | 123 | 10.7 | 8.65 | -5.13 | |||
|
|
|||||||||||
| TFV
|
|||||||||||
| LLOQ [1.00 ng/mL] |
1.03 | 0.100 | 9.64 | 3.27 | 1.01 | 0.107 | 10.6 | 0.680 | |||
| Low [3.00 ng/mL] |
2.91 | 0.320 | 11.0 | -3.00 | 2.80 | 0.269 | 9.58 | -6.52 | |||
| Mid [20.0 ng/mL] |
18.6 | 1.10 | 5.93 | -7.25 | 18.5 | 1.05 | 5.68 | -7.50 | |||
| High [170 ng/mL] |
174 | 5.06 | 2.91 | 2.35 | 172 | 7.99 | 4.66 | 0.980 | |||
|
|
|||||||||||
| CMX157
|
|||||||||||
| LLOQ [0.25 ng/mL] |
0.265 | 0.0290 | 11.1 | 6.07 | 0.269 | 0.0310 | 11.4 | 7.76 | |||
| Low [0.75 ng/mL] |
0.735 | 0.0480 | 6.54 | -1.98 | 0.734 | 0.0540 | 7.42 | -2.13 | |||
| Mid [50.0 ng/mL] |
52.1 | 2.34 | 4.50 | 4.17 | 50.1 | 2.39 | 4.76 | 0.211 | |||
| High [160 ng/mL] |
171 | 5.32 | 3.10 | 7.08 | 167 | 5.87 | 3.52 | 4.31 | |||
n=6 for each level of QC; representative data from a single analytical run
n=18 for all QC levels; complex precision and accuracy from three analytical runs
Calibration Curve Analysis
Standard curves for all analytes were generated using a weighted linear 1/x2 regression analysis. The average regression for the analytes was ≥ 0.9963 (TAF), ≥ 0.9958 (TFV), and ≥ 0.9987 (CMX157).
Dilutional Integrity
Plasma samples were spiked 3xULOQ for TAF [450 ng/mL], TFV [600 ng/mL], and CMX157 [600 ng/mL] and diluted up to 20-fold with drug free plasma, yielding a %DEV range of -0.71% to 10.8%. Volume-limited sample set %DEVs, in which mid and high QC levels were diluted up to five-fold, were -8.38% to 5.97% (data not shown).
Stability Challenges
Several stability challenges were evaluated during validation, including injection matrix (48 h CMX157 and 72 h TAF and TFV), sample matrix (24 h TAF and TFV and 72 h CMX157), two or three-freeze-thaw cycles, long term storage at -80°C, and finally evaluation of hemolyzed and lipemic plasma compared to K2EDTA plasma. TAF appears unstable for 72 h in injection matrix at 4°C (data not shown) if compared to the previously run curve, but did meet the acceptability criteria when the entire sample set was run again continuously. The %DIFs for TAF and TFV with the re-analyzed curve ranged from −8.44% to 5.57%. CMX157 was found to be stable for 48 h at 4°C in injection matrix, with %DIFs ranging from -9.02% to 4.66%. The %DIFs for sample matrix analysis ranged from -14.5% to 8.45% (24 h TAF & TFV and 72 h CMX157) and three freeze-thaw stability challenged samples ranged from -0.399% to 6.68% for CMX157. While TFV was stable for three-freeze thaw cycles (data not shown) TAF was only stable for two freeze-thaw cycles with %DIF for both drugs that ranged from -15.2% to 3.04%. Long term storage at -80°C was assessed after 161 (CMX157) and 244 (TAF and TFV) days and the %DIF was ≤ ±10.5%. Further, the presence of hemolysis did not impact drug quantification. Hemolyzed plasma compared to QCs in K2EDTA plasma ranged from -9.38% to 8.16% difference for the three analytes (data not shown). Lipemia has no effect on the quantification of the TAF and CMX157 as the %DIF ranged from -14.2% to 6.72, but there was an impact on TFV quantification (%DIF ranged from 9.57% to 18.4%).
Signal to Noise Ratio, Carryover, and Cross-Talk Characterization
For TAF, TFV and CMX157, peak areas of blank samples were ≤ 20% of peak areas observed at analyte LLOQs; no interfering peaks were observed in drug-free plasma at the elution times of TAF (2.21 min), TFV (1.31 min) and CMX157 (0.89 min) (Supplemental Figure 1). Additionally, peak areas in blank sample post-analysis of a specimen at the ULOQ were ≤ 20% of the LLOQ peak areas for TAF and TFV; the peak area of the detected peak for CMX157 was slightly greater than 20%. In this scenario, there was increased baseline intensity across the chromatographic run in the post-injection blank sample, resulting in integration of a peak. However, based on overall peak shape and intensity above baseline, the signal observed in the post-injection blank was not considered carryover as it did not meet standards for peak shape (data not shown). There was negligible (≤20% peak area of LLOQ) contribution of internal standards 13C5-TFV and CMX157-d6 to their respective analytes.
Matrix Effects
To characterize potential ion enhancement or suppression of the analytes, the peak areas were measured in un-extracted, pre-extracted, and post-extracted samples for TAF, TFV and CMX157 and their respective internal standards. The matrix effects observed for TAF and TFV were relative, and ion suppression was observed for the analyte CMX157, indicating that endogenous compounds co-elute with the analyte in the protein precipitation extraction. However, comparable ion suppression was also observed for CMX157-d6, thereby reducing the matrix effects that could impact quantification of the analyte. The overall recovery efficiency for TAF, TFV, CMX157, and their respective internal standards was ≥80%, and the %DIF in recovery efficiency between the analytes and the internal standards was <15% at any QC level. The observations made for the overall processing efficiency of the analytes were similar to the observations made for matrix effects. A summary of matrix effects for TAF and TFV and CMX157 are detailed in Table 2.
Table 2.
Matrix Effects of TAF, TFV, and CMX157 in human K2EDTA-plasma
| QC Level | Matrix Effects (%)a
|
Recovery Efficiency (%)b
|
Processing Efficiency (%)c
|
|||
|---|---|---|---|---|---|---|
| Analyte | Internal Standard | Analyte | Internal Standard | Analyte | Internal Standard | |
| TAF
|
||||||
| Low QC [0.090 ng/mL] | 82.7 | 85.4 | 94.1 | 83.1 | 77.8 | 71.0 |
| Mid QC [3.00 ng/mL] | 79.6 | 94.2 | 90.1 | 80.8 | 71.7 | 76.2 |
| High QC [130 ng/mL] | 80.4 | 93.5 | 98.0 | 84.9 | 78.8 | 79.3 |
|
|
||||||
| TFV | ||||||
| Low QC [3.00 ng/mL] | 98.2 | 85.4 | 83.4 | 83.1 | 81.9 | 71.0 |
| Mid QC [20.0 ng/mL] | 99.3 | 94.2 | 80.7 | 80.8 | 80.2 | 76.2 |
| High QC [170 ng/mL] | 99.2 | 93.5 | 81.2 | 84.9 | 80.5 | 75.5 |
|
|
||||||
| CMX157
|
||||||
| Low QC [0.750 ng/mL] | 10.3 | 5.70 | 87.1 | 96.4 | 9.00 | 5.50 |
| Mid QC [50.0 ng/mL] | 6.30 | 4.40 | 98.7 | 102 | 6.20 | 4.50 |
| High QC [160 ng/mL] | 6.00 | 4.10 | 105 | 116 | 6.40 | 4.80 |
ME% = Peak area of (post-extracted samples/un-extracted samples) * 100
RE% = Peak area of (pre-extracted samples/post-extracted samples) * 100
PE% = Peak area of (pre-extracted samples/un-extracted samples) * 100
Assay Implementation in Biological Samples
To evaluate the fidelity of the described LC-MS/MS assays, methods were applied to a preclinical study aimed at understanding the pharmacokinetics of TFV and its prodrugs using a topical drug delivery system. Molar equivalents of TAF (3.28 mg/mL) or CMX157 (3.72 mg/mL) were formulated in a normal saline enema solution. Mice were dosed rectally with 50 μL of drug containing solution; samples were collected 15 min and 4 h after dosing.
Utilizing the aforementioned LC MS/MS methods, the measured TAF concentrations ranged from below the quantitation limit (BQL) to 85.4 pg/mL for the 5 mice treated with the TAF enema formulation at the 15-min time point, which were within the established analytical measuring range (AMR). All TAF concentrations were BQL at the 4 h time point. TFV, on the other hand, was above the quantitation limit (AQL) at 15 min time point but fell within the established AMR at the 4 h time point. The five CMX157 treated mice had detectable TFV concentrations previously measured utilizing a previously validated TFV method with a lower limit of quantification of 0.31 ng/mL. TFV concentrations ranged from 0.357 to 1.02 ng/mL at the 4 h time point; the corresponding CMX157 concentrations measured ranged from 0.381 to 3.63 ng/mL (Table 3). The results of these preclinical samples demonstrate an inverse relationship between the prodrug and the parent drug. Additional studies conducted in our group looking at oral formulations of TAF demonstrate, unsurprisingly, systemic higher TAF concentrations as compared to TFV (data not shown).
Table 3.
TAF, TFV, and CMX157 concentrations in preclinical mouse samples post-colonic administration of drugs.
| Mouse | Time (Hour) | TFV [ng/mL] | TAF [ng/mL] |
|---|---|---|---|
| 1 | 0.25 | AQL | BQL |
| 2 | 0.25 | AQL | BQL |
| 3 | 0.25 | AQL | 0.0321 |
| 4 | 0.25 | AQL | 0.0313 |
| 5 | 0.25 | AQL | 0.0854 |
|
| |||
| 1 | 4 | 11.6 | BQL |
| 2 | 4 | 6.49 | BQL |
| 3 | 4 | 19.1 | BQL |
| 4 | 4 | 8.05 | BQL |
| 5 | 4 | 15.1 | BQL |
|
| |||
| Mouse | Time (Hour) | TFV [ng/mL] | CMX157 [ng/mL] |
|
| |||
| 1 | 4 | 1.02 | 3.63 |
| 2 | 4 | 0.533 | 1.33 |
| 3 | 4 | 0.745 | 0.381 |
| 4 | 4 | 0.373 | 0.707 |
| 5 | 4 | 0.640 | 1.71 |
TFV AQL: >250 ng/mL
TAF BLQ: <0.03 ng/mL
Discussion
The methods presented for the quantification of TFV and its prodrugs are sensitive and can support in vivo studies to better evaluate drug pharmacokinetics. Although the presented TAF and TFV assay uses a larger sample volume than previously published methods, and involves a more complex sample extraction approach, this highly sensitive method is the first to describe the multiplexed analysis of both TAF and its metabolic product, TFV [19–21]. Conversely, the assay presented for the quantification of CMX157 utilizes a small sample volume with a simple and robust sample preparation as well as a sensitive, dynamic analytical measuring range. With growing interest in prodrugs such as TAF, the ability to multiplex quantification of both the alafenamide conjugate in conjunction with TFV can provide additional information on antiviral agent pharmacokinetics. Many of the reports describing TAF utility in disease management primarily measure TFV as an endpoint. However, recent reports, including those from Custodio and colleagues report Cmax concentrations for TAF and TFV at 199 ng/mL and 9.5 ng/mL, respectively, following a single 25 mg dose of TAF. While the described TAF upper limit of quantification (ULOQ) is below reported Cmax concentrations observed with 25 mg TAF dosing, extended linearity studies were conducted to ensure quantification at higher dosing regimens of TAF [24]. We developed our assays to maximize analytical sensitivity; since TAF is orally administered at <10% of an oral TDF dose (300 mg), the assay was validated to ensure prodrug quantification later PK time points. Additionally, the described assays were developed and validated to facilitate testing flexibility for both oral and topical delivery systems of TFV prodrugs.
Topical delivery of compounds results in enhanced localized drug concentrations, while substantially reducing systemic concentrations. This has been demonstrated in a number of studies using topical delivery systems, including TFV-containing intravaginal rings, pericoital gels, and films [25–29]. This is further reflected in ongoing preclinical studies evaluating alternative delivery systems for TFV and its prodrugs (Table 3). Unlike oral formulations, which would be associated with plasma TAF/TFV ratios >1, colonic administration of the prodrug results in lower TAF concentrations being released into systemic circulation, resulting in a TAF/TFV ratio <1, as observed in the described preclinical mouse studies. This is due to probable TAF uptake and bioconversion by localized tissue, with lower concentrations of unchanged prodrug reaching systemic circulation. Additionally, unlike oral regimens, topical TAF was administered at equivalent molar concentrations, thereby preventing direct comparisons with oral TAF/TFV gradients. Although systemic TAF concentrations were only detectable in 60% of plasma samples (concentrations ≤ 0.085 ng/mL), localized tissue TFV-DP concentrations were quantifiable, suggesting prodrug uptake, bioconversion and metabolism at later time points (data not shown). The described efforts are part of ongoing studies evaluating prodrug performance in this delivery system. The described assay demonstrates the capacity to quantify TFV and its prodrug following rectal delivery; in the preclinical mouse study, TAF bioconversion occurs rapidly, with high TFV drug concentrations observed as soon as 15 min post-rectal administration, and quantifiable concentrations > 6.49 ng/mL 4 h post-dose. However, the inability to quantify TAF in all samples collected proximal to rectal delivery is a limitation of the system, suggesting that alternative dosing strategies or model systems may be required to fully characterize TFV prodrug pharmacokinetics.
Although there are ongoing clinical trials evaluating the pharmacokinetics and safety profiles of CMX157, our group has evaluated the potential utility of topical CMX157 in preclinical studies. In mice that were dosed rectally with CMX157, we were able to measure concentrations of both prodrug and TFV and in systemic circulation. Further, localized tissue tenofovir-diphosphate concentrations (TFV-DP) were measured and found to be quantifiable (data not shown), showing a potential for drug delivery and in vivo bioactivation in a mouse model. Additional studies in both mouse and macaque models for both TAF and CMX157 prodrugs are ongoing.
While the described methods were developed and validated to optimize flexibility in support of clinical studies, and have been successfully applied to a topical pharmacokinetic study, this work does have limitations. Notably, due to the enhanced lipophilicity of the CMX157 prodrug, aggressive re-equilibration and a strong organic mobile phase system was required for chromatographic elution and mitigation of carryover. Due to the stark differences in polarity between the prodrug and the more hydrophilic TFV, a dual method for prodrug and product quantification could not be achieved. Therefore, for studies in which both CMX157 and TFV are required, two independent specimen preparations and analytical methods are required. For our combinatorial TAF and TFV assay, the analytical measuring range of TAF may appear to be too narrow, requiring additional testing of samples near Cmax. While this may occur in a subset of samples, the frequency of samples that could exceed the assay ULOQ is dependent on dosage, route of administration, and time of collection relative to dosing. Dilutional analysis testing further supports specimen dilution to extend assay linearity, largely circumventing the limitation.
Of note, while the assay was validated using human plasma, preclinical studies were performed in a mouse model. Although peak area intensities of our internal standards were comparable between species, it is noted that enzymatic activity and prodrug conversion via esterases may not be conserved between mouse and human models. Studies performed during validation indicated that while TFV and CMX157 peak area intensities were comparable between species, TAF was less stable in mouse plasma as compared to human matrices, suggesting differential enzyme activity (data not shown). Lastly, and not innate to the bioanalytical method, is the lower circulating concentrations of TAF in plasma with colonic administration. TAF was rapidly converted into TFV in less than 15 minutes as demonstrated by the in vivo mouse samples that were dosed rectally, generating undetectable TAF concentrations and AQL TFV concentrations utilizing the presented TAF and TFV assay. An oral dose, and/or alternate route of administration, of TAF may generate slightly different results with higher circulating prodrug concentrations in vivo.
Conclusions
Two LC-MS/MS methods have been developed and validated; one for the quantification of TAF and TFV, and the other for CMX157 in plasma in accordance with FDA recommendations. The novel method CMX157 involves a streamlined, low volume requirement, and dynamic analytical measuring range, which meets the sensitivity needs in supporting clinical studies focused on drug pharmacokinetics. Further, the method for TAF and TFV is more sensitive than previously published methods and multiplexes the two analytes into one simplified assay.
Supplementary Material
Highlights.
Development of an LC-MS/MS method for TFV prodrug quantification in tissue
Validation of LC-MS/MS assays following FDA guidelines
In vivo measurement of tenofovir alafenamide fumarate (TAF) and hexadecyloxypropyl tenofovir (CMX157) in preclinical study samples
Acknowledgments
The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Tenofovir. The reagent TAF was acquired through a material transfer agreement with Gilead Sciences. CMX157 and the isotopically labeled internal standard CMX157-d6 were generous gifts from Chimerix. This study was supported by the US National Institutes of Health (NIH), including the National Institute of Allergy and Infectious Diseases (NIAID) under the program project grant 1U19AI113127-01. We also acknowledge Randall Lanier and Tim Tippin (Chimerix), and Jim Rooney (Gilead Sciences) for their thoughtful review of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest.
Contributor Information
Pamela Hummert, Department of Medicine, Johns Hopkins University School of Medicine, 1800 Orleans Street, Sheikh Zayed Tower, B1020-G, Baltimore, MD, 21287, USA.
Teresa L. Parsons, Department of Medicine, Johns Hopkins University School of Medicine, 1800 Orleans Street, Sheikh Zayed Tower, B1020-G, Baltimore, MD, 21287, USA.
Laura M. Ensign, Department of Opthamology, Johns Hopkins University School of Medicine, 4940 Eastern Avenue, Mason F. Lord Center Tower, Baltimore, MD, 21224, USA.
Thuy Hoang, Department of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine, 4940 Eastern Avenue, Mason F. Lord Center Tower, Baltimore, MD, 21224, USA.
Mark A. Marzinke, Departments of Medicine and Pathology, Johns Hopkins University School of Medicine, 1800 Orleans Street, Sheikh Zayed Tower, B1020-G, Baltimore, MD, 21287, USA.
References
- 1.amfAR. Statistics: worldwide from UNAIDS Fact sheet July 2017. http://www.amfar.org/worldwide-aids-stats/ (Accessed 22 August 2017)
- 2.Cory TJ, Midde NM, Rao PSS, Kumar S. Investigational reverse transcriptase inhibitors for treatment of HIV. Expert Opinion on Investigational Drugs. 2015;24:1219–1228. doi: 10.1517/13543784.2015.1058357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Department of Health and Human Services Panel on Antiretroviral Guidelines for Adults and Adolescents. AIDSinfo: Guidelines for the use of antiretroviral agents in HIV-1 infected adults and adolescents. National Institutes of Health; Bethesda, MD, USA: 2017. [Google Scholar]
- 4.Friend DR, Clark JT, Kiser PF, Clark MR. Multipurpose prevention technologies: products in development. Antiviral Research. 2013;100:S39–S47. doi: 10.1016/j.antiviral.2013.09.030. [DOI] [PubMed] [Google Scholar]
- 5.Kearney BP, Flaherty JF, Shah J. Tenofovir disoproxil fumerate; clinical pharmacology and pharmacokinetics. Clin Pharmacokinet. 2004;43:595–612. doi: 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- 6.Giesler KE, Liotta DC. Next-generation reduction sensitive lipid conjugates of tenofovir: antiviral activity and mechanism of release. J Med Chem. 2016;59:10244–10252. doi: 10.1021/acs.jmedchem.6b01292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giesler KE, Marengo J, Liotta DC. Reduction sensitive lipid conjugates of tenofovir: synthesis, stability, and antiviral activity. J Med Chem. 2016;59:7097–7110. doi: 10.1021/acs.jmedchem.6b00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markowitz M, Zolopa A, Squires K, Ruane P, Coakley D, Kearney B, Zhong L, Wulfsohn M, Miller MD, Lee WA. Phase I/II study of the pharmacokinetics, safety and antiretroviral activity of tenofovir alafenamide, a new prodrug of the HIV reverse transcriptase inhibitor tenofovir, in HIV-infected adults. J Antimicrob Chemother. 2014;69:1362–1369. doi: 10.1093/jac/dkt532. [DOI] [PubMed] [Google Scholar]
- 9.Angione SA, Cherian SM, Ӧzdener AE. A review of the efficacy and safety of genvoya® (elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide) in the management of HIV-1 infection. Journal of Pharmacy Practice May. 2017;30:1–6. doi: 10.1177/0897190017710519. [DOI] [PubMed] [Google Scholar]
- 10.Pozniak A, Arribas JR, Gathe J, Gupta SK, Post FA, Bloch M, Avihingsanon A, Crofoot G, Benson P, Lichtenstein K, Ramgopal M, Chetchotisakd P, Custodio JM, Abram ME, Wei X, Cheng A, McCallister M, SenGupta D, Fordyce MW. Switching to tenofovir alafenamide, coformulated with elvitegravir, cobicistat, and emtricitabine, in HIV-infected patients with renal impairment: 48-week results from a single-arm, multicenter, open-label phase 3 study. J Acquir Immune Defic Syndr. 2016;71:530–537. doi: 10.1097/QAI.0000000000000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Clercq E. Tenofovir alafenamide (TAF) as the successor tenofovir disoproxil fumarate (TDF) Biochemical Pharmacology. 2016;119:1–7. doi: 10.1016/j.bcp.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Bam RA, Yant SR, Cihlar T. Tenofovir alafenamide is not a substrate for renal organic anion transporters (OATs) and does not exihibit OAT-dependent toxicity. Antiviral Therapy. 2014;19:687–692. doi: 10.3851/IMP2770. [DOI] [PubMed] [Google Scholar]
- 13.Ray AS, Fordyce MW, Hitchcock MJM. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Research. 2016;125:63–70. doi: 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Hostetler KY. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: current state of the art. Antiviral Research. 2009;82:A84–A98. doi: 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanier RE, Ptak RG, Lampert BM, Keilholz L, Hartman T, Buckheit RW, Mankowski MK, Osterling MC, Almond MR, Painter GR. Development of hexadecyloxypropyl tenofovir (CMX157) for treatment of infection caused by wild-type and nucleoside/nucleotide-resistant HIV. Antimicrol Agents and Chemother. 2010;54:2901–2909. doi: 10.1128/AAC.00068-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Painter GR, Almond MR, Trost LC, Lampert BM, Neyts J, De Clercq E, Korba BE, Aldern KA, Beadle JR, Hostetler KY. Evaluation of hexadecyloxypropyl-9-R-[2-(Phosphonomethoxy)Propyl]-Adenine, CMX157, as a potential treatment for human immunodeficiency virus type 1 and hepatitis B virus infections. Antimicrob Agents and Chemother. 2007;51:3505–3509. doi: 10.1128/AAC.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shire NJ. Cure strategies for hepatitis B virus: the promise of immunotherapy. Clinical Pharmacology and Drug Development. 2017;6:186–194. doi: 10.1002/cpdd.317. [DOI] [PubMed] [Google Scholar]
- 18.Tajiri K, Shimizu Y. New horizon for radical cure of chronic hepatitis B virus infection. World J Hepatol. 2016;8:863–873. doi: 10.4254/wjh.v8.i21.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sluis-Cremer N, Wainberg MA, Schinazi RF. Resistance to reverse transcriptase inhibitors used in the treatment and prevention of HIV-1 infection. Future Microbiol. 2015;10:1773–1782. doi: 10.2217/fmb.15.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunawardana M, Remedios-Chen M, Miller CS, Fanter R, Yang F, Marzinke MA, Hendrix CW, Bellveau M, Moss JA, Smith TJ, Baum MM. Pharmacokinetics of long-acting tenofovir alafenamide (GS-7340) sub-dermal implant for HIV prophylaxis. Antimicrob Agents and Chemother. 2015;59:3913–3919. doi: 10.1128/AAC.00656-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zack J, Chuck S, Chu H, Graham H, Cao H, Tijerina M, West S, Fang L, Quirk E, Kearney B. Bioequivalence of rilpivirine/emtricitabine/tenofovir alafenamide single-tablet regimen. J Bioquiv Availab. 2016;8:049–054. [Google Scholar]
- 22.Department of Health and Human Services UF, Center for Drug Evaluation and Research, Center for Veterinary Medicine. Guidance for Industry: Bioanalytical Method Validation, FDA; Rockville, MD, USA: 2012. [Google Scholar]
- 23.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effects in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 24.Custodio JM, Fordyce M, Garner W, Vimal M, Ling KHJ, Kearney BP, Ramanathan S. Pharmacokinetics and safety of tenofovir alafenamide in HIV-uninfected subjects with severe renal impairment. Antimicrob Agents and Chemother. 2016;60:5135–5140. doi: 10.1128/AAC.00005-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marrazzo JM, Ramjee G, Richardson BA, Gomez K, Mogdi N, Nair G, Palanee T, Nakabiito C, van der Straten A, Noguchi L, Hendrix CW, Dai JY, Ganesh S, Mkhize B, Taljaard M, Parikh UM, Piper J, Mâsse B, Grossman C, Rooney J, Schwartz JL, Watts H, Marzinke MA, Hillier SL, McGowan IM, Chirenje ZM, VOICE study team Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2015;372:509–518. doi: 10.1056/NEJMoa1402269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mcgowan I, Cranston RD, Duffill K, Siegel A, Engstrom JC, Nikiforov A, Jacobson C, Rehman KK, Elliott J, Khanukhova E, Abebe K, Mauck C, Spiegel HM, Dezzutti CS, Rohan LC, Marzinke MA, Hiruv H, Hendrix CW, Richardson-Harman N, Anton PA. A phase 1 randomized, open label, rectal safety, acceptability, pharmacokinetic, and pharmacodynamics study of three formulations of tenofovir 1% gel (the CHARM-01 study) PLoS One. 2015;10:e0125363. doi: 10.1371/journal.pone.0125363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdool Karim Q, Abdool Karim SS, Frolich JA, Grobler AC, Baxter C, Mansoor LE, Kharsany ABM, Sibeko S, Mlisana KP, Omar Z, Gengiah TN, Maarschalk S, Arulappan N, Mlotshwa M, Morris L, Taylor D, CAPRISA 004 Trial group Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010;329:1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cranston RD, Lama JR, Richardson BA, Carballo-Diéquez A, Kunjara Na Ayudha RP, Liu K, Patterson KB, Leu CS, Galaska B, Jacobson CE, Parikh UM, Marzinke MA, Hendrix CW, Johnson S, Piper JM, Grossman C, Ho KS, Lucas J, Pickett J, Bekker LG, Chariyalertsak S, Chitwarakorn A, Gonzales P, Holtz TH, Lui AY, Mayer KH, Zorilla C, Schwartz JL, Rooney J, McGowan I, MTN-017 Protocol Team MTN-017: a rectal phase 2 extended safety and acceptability study of tenofovir reduced-glycerin 1% gel. Clin Infect Dis. 2017;64:614–620. doi: 10.1093/cid/ciw832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruane PJ, DeJesus E, Berger D, Markowitz M, Bredeek UF, Callebaut C, Zhong L, Ramanathan SS, Rhee M, Fordyce MW, Yale K. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide as 10-day monotherapy in HIV-1-positive adults. JAIDS. 2013;63:449–455. doi: 10.1097/QAI.0b013e3182965d45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.