

Abstract
Chronic inflammation is a common molecular basis underlying a variety of chronic diseases. Accumulating evidence has also suggested that chronic inflammation contributes to the pathogenesis of obesity and diabetes, which have been considered as metabolic diseases. For the past several decades, there has been dramatic progress in understanding the underlying mechanism of adipose tissue dysfunction induced by obesity. Tissue remodeling is one of the histological features of chronic inflammation, in which stromal cells dramatically change in number and cell type. Indeed, adipose tissue remodeling is induced by various stromal cells, and results in the impairment of adipose tissue function, such as adipocytokine production and lipid storage, which leads to systemic metabolic disorder. In addition to adipose tissue, the liver is another example of obesity‐induced tissue remodeling. In the present review, we discuss how obesity induces interstitial fibrosis in adipose tissue and the liver, particularly focusing on the role of macrophages.
Keywords: Chronic inflammation, Obesity, Macrophages
Introduction
Chronic inflammation is a common molecular basis underlying a variety of chronic diseases, such as atherosclerotic disease, autoimmune disease, neurodegenerative disease and cancer. Accumulating evidence has also suggested that chronic inflammation plays a crucial role in obesity and diabetes pathogenesis, which have been considered as metabolic diseases. There are a large number of studies on the effect of excessive energy intake and nutritional imbalance on chronic inflammation and impairment of metabolic homeostasis. In this regard, one of the most important organs is adipose tissue, which senses nutritional conditions in our body and stores the excessive energy as triglyceride. Adipose tissue also regulates inflammatory responses and secretes various bioactive molecules, which are termed ‘adipocytokines’ or ‘adipokines,’ in response to the systemic nutritional status, resulting in a feedback mechanism of metabolic homeostasis1, 2, 3, 4, 5, 6. In obesity, these adipose tissue functions become impaired, which gives rise to systemic metabolic derangements (metabolic syndrome).
In contrast to acute inflammation, typical signs of ‘inflammation,’ such as pain, heat, redness and swelling, are rarely observed in chronic inflammation. In contrast, pro‐inflammatory cytokines and immune cells are a common mechanism of acute and chronic inflammation. In this regard, tissue remodeling is one of the histological features of chronic inflammation, in which stromal cells dramatically change in number and cell type7. In most cases, chronic inflammation finally results in tissue fibrosis and organ dysfunction. In addition to adipose tissue, the liver is another example of obesity‐induced tissue remodeling. Lipid accumulation in the liver or non‐alcoholic fatty liver disease (NAFLD) is a hepatic feature of the metabolic syndrome. In particular, non‐alcoholic steatohepatitis (NASH), characterized by chronic inflammation and pericellular fibrosis, increases the risk of cirrhosis and hepatocellular carcinoma8, 9. In the present review article, we discuss the recent progress in the understanding of the molecular mechanism of obesity‐induced tissue remodeling, especially focusing on adipose tissue and liver.
Obesity‐Induced Adipose Tissue Inflammation
The adipose tissue includes not only lipid‐laden mature adipocytes, but also numerous stromal cells, such as preadipocytes, fibroblasts, endothelial cells and immune cells. During the development of obesity, adipose tissue shows dynamic histological changes characterized by adipocyte hypertrophy, increased angiogenesis, immune cell infiltration and extracellular matrix overproduction2, 6, 10, 11. These changes are reminiscent of ‘vascular remodeling,’ which is the chronic inflammatory responses in atherosclerotic vascular walls. It is well understood that ‘vascular remodeling’ is caused by the complex interactions among various cells, such as vascular endothelial cells, vascular smooth muscle cells, lymphocytes and monocyte‐derived macrophages5, so the dynamic change observed in obese adipose tissue can be referred to as ‘adipose tissue remodeling,’ which should be involved in the pathogenesis of obesity‐induced adipose tissue dysfunction. Furthermore, there is considerable evidence suggesting that adipocytokine production and lipid storage are regulated by the interaction of mature adipocytes and stromal cells in adipose tissue12, 13.
Since ground‐breaking reports published in 2003–2004 showed that macrophages infiltrate into obese adipose tissue in mice and humans14, 15, 16, the role of macrophages among the various types of stromal cells in obese adipose tissue has been most intensively studied. The macrophages form a unique histological structure termed crown‐like structure (CLS), where CD11c‐positive macrophages surround dead or dying adipocytes to engulf the dead adipocytes and the residual lipid17. In addition, the heterogeneity of macrophages in obese adipose tissue has also been pointed out: pro‐inflammatory M1 or ‘classically activated’ macrophages, and anti‐inflammatory M2 or ‘alternatively activated’ macrophages18, 19. We and others have shown that chemokines, such as monocyte chemoattractant protein‐1, recruit M1 macrophages from the bone marrow to form CLS20, 21, 22. As pro‐inflammatory cytokines, such as tumor necrosis factor‐α, are produced by M1 macrophages in obese adipose tissue and their number is positively correlated with adiposity and systemic insulin resistance23, 24, CLS is a hallmark of adipose tissue inflammation. Of note, we previously reported that Toll‐like receptor 4 deficiency inhibits the increase of the M1‐to‐M2 ratio of macrophages in diet‐induced obese mice, without affecting the number of CLS25. It is also known that Toll‐like receptor 4‐deficient mice are protected against obesity‐induced insulin resistance26, 27. By contrast, M2 macrophages are observed diffusely in interstitial spaces between adipocytes18, 19. Thus, the M1‐to‐M2 ratio of macrophages, in addition to the number of macrophages, critically regulates obesity‐induced inflammatory changes in adipose tissue.
Paracrine Interaction Between Adipocytes and Macrophages
As chronic inflammation is characterized by sustained interaction of various cell types, it is important to understand how adipocytes and macrophages cross‐talk with each other. We have provided evidence suggesting that a paracrine loop involving mature adipocytes‐derived saturated fatty acids and macrophages‐derived tumor necrosis factor‐α establishes a vicious cycle to exaggerate chronic inflammation28. Indeed, infiltrated macrophages in obese adipose tissue produce tumor necrosis factor‐α, which can induce the production of pro‐inflammatory cytokine and lipolysis of adipocytes. The released high concentrations of free fatty acids, particularly saturated fatty acids, such as palmitate, might induce inflammatory responses in macrophages29, 30. As a molecular mechanism, saturated fatty acids activate the Toll‐like receptor 4 signaling and the integrated stress response in macrophages to produce pro‐inflammatory cytokines31. In contrast, ω‐3 or n‐3 polyunsaturated fatty acids, such as eicosapentaenoic acid (EPA), strongly antagonize the inflammatory effects of saturated fatty acids on macrophages. Consistently, dietary supplementation of EPA effectively reverses the macrophage polarity from M2 to M1 in obese adipose tissue, without affecting the macrophage population32. Thus, the cross‐talk between adipocytes and macrophages maintains chronic inflammatory changes of obese adipose tissue.
Molecular Mechanism of Adipose Tissue Remodeling
Similar to other chronic inflammatory diseases, obesity‐induced chronic inflammation in adipose tissue eventually leads to interstitial fibrosis in rodent models and humans33, 34, 35, 36. As it is known that interstitial fibrosis is a common pathway to end‐stage organ failure, it might cause obesity‐triggered adipose tissue dysfunction. For instance, a recent report suggested that there is a negative correlation between adipose tissue fibrosis and adipocyte diameters in human adipose tissue37, suggesting the role of interstitial fibrosis in adipose tissue expandability during the development of obesity. Interestingly, Khan et al.33 reported that mice lacking collagen VI, which is expressed abundantly in adipose tissue, show uninhibited adipose tissue expansion and substantial improvement in insulin sensitivity when consuming a high‐fat diet. A fundamental function of adipose tissue is to store excessive energy as triglyceride, and the balance of lipogenesis and lipolysis is tightly regulated by hormones, such as insulin, and the sympathetic nervous system in response to nutritional conditions13. Adipose tissue inflammation also regulates this process: macrophage‐derived pro‐inflammatory cytokines can induce insulin resistance as well as lipolysis in adipocytes. In addition, adipose tissue fibrosis might be a novel mechanism of ectopic lipid accumulation, given that it affects the lipid‐storage capacity of adipose tissue. However, the molecular mechanism of adipose tissue fibrosis is still unclear.
Recently, we showed that macrophage‐inducible C‐type lectin (Mincle, Clec4e or Clecsf9) is a novel regulatory factor of adipose tissue fibrosis38. Mincle is a type II membrane protein in macrophages, and functions as a pathogen sensor that recognizes Mycobacterium tuberculosis and certain types of fungi39, 40, 41. Recent evidence also suggests that Mincle senses cell death to induce sterile inflammation42. In obese adipose tissue, Mincle expression is localized to the macrophages constituting CLS, where dead or dying adipocytes are surrounded by macrophages. When Mincle is activated by currently unknown endogenous ligands, which are probably released from dead or dying adipocytes, Mincle signaling can activate fibroblast or induce myofibroblast formation resulting in interstitial fibrosis in adipose tissue. Indeed, obesity‐induced adipose tissue fibrosis is attenuated in Mincle‐deficient mice, which leads to less ectopic lipid accumulation in the liver. For the next step, it is important to identify the cellular origin of myofibroblasts in obese adipose tissue. In this regard, Iwayama et al.43 reported that activation of platelet‐derived growth factor receptor‐α signaling perturbs differentiation of preadipocytes to induce myofibroblast formation. As activated Mincle signaling in macrophages markedly increases expression of platelet‐derived growth factor and transforming growth factor‐β, it is interesting to know how myofibroblast formation is regulated by Mincle (Figures 1 and 2).
Figure 1.
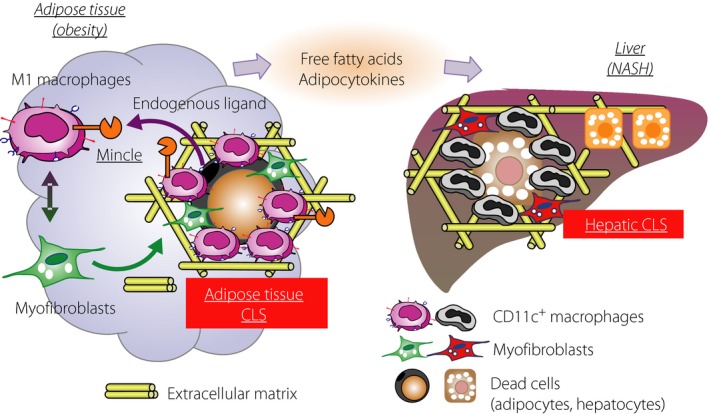
Role of crown‐like structure (CLS) in obesity‐induced ‘metabolic’ tissue remodeling in adipose tissue and liver. Mincle, a novel sensor for cell death, is exclusively expressed in the CD11c‐positive macrophages constituting CLS in obese adipose tissue. When activated by currently unknown endogenous ligands, Mincle potently induces myofibroblast formation and interstitial fibrosis. There is a similar histological structure termed hepatic CLS in the liver during the disease progression from simple steatosis to non‐alcoholic steatohepatitis (NASH). Thus, CLS plays a critical role in driving ‘metabolic’ tissue remodeling.
Figure 2.
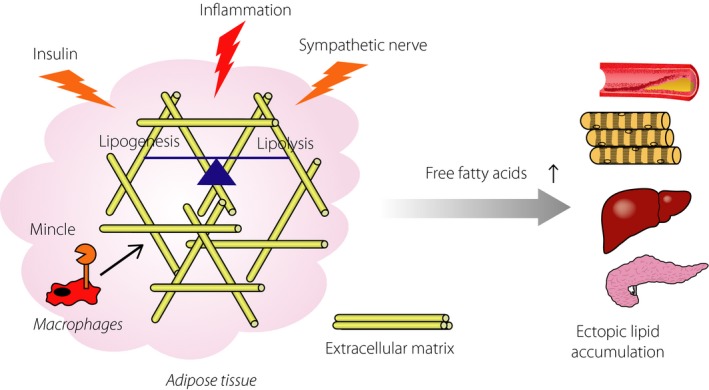
Role of adipose tissue fibrosis in ectopic lipid accumulation. To store excessive energy as triglyceride is a fundamental function of adipose tissue, which is tightly regulated by hormones, such as insulin, and the sympathetic nerve. In addition, recent evidence has suggested that chronic inflammation is involved in this process. Particularly, interstitial fibrosis limits adipose tissue expandability, which finally leads to increased ectopic lipid accumulation.
Novel Animal Model of NASH
Recently, we found that there is an adipose tissue CLS‐like histological structure in the liver of NASH in mice and humans17. The ‘two‐hit’ hypothesis has been proposed to explain NASH progression from steatosis: (i) the first hit – excessive lipids accumulation in the liver; and (ii) the second hit – additional pathogenic stimuli, such as oxidative stress, endotoxins, pro‐inflammatory cytokines and lipotoxicity. However, the pathogenesis of NASH is still unclear; it is partly because of the limited availability of suitable animal models that reflect a liver condition of human NASH44. For instance, methionine and choline‐deficient diet‐fed mice develop steatosis and mild fibrosis, without obesity and insulin resistance. In addition, chemically‐induced liver fibrosis is not accompanied by metabolic abnormalities, such as obesity, insulin resistance and hepatic steatosis. In this regard, our NASH model using melanocortin‐4 receptor (MC4R) deficient (MC4R‐KO) mice is unique, as MC4R‐KO mice fed a high‐fat diet show not only obesity, insulin resistance and dyslipidemia, but also a liver condition similar to human NASH36. They also develop multiple liver tumors after a longer period of time. It is known that MC4R expression is restricted to the hypothalamus and other brain regions, and MC4R messenger ribonucleic acid is undetectable in the liver and the adipose tissue. Based on this point of view, it is likely that the hepatic phenotype in high‐fat diet‐fed MC4R‐KO mice results from loss of function of MC4R in the brain rather than in the liver itself. Accordingly, MC4R‐KO mice would be a unique mouse model of NASH, and this model is useful for investigating the molecular mechanism of human NASH.
Hepatic CLS‐Mediated Liver Fibrosis in NASH
Using our NASH model, we found hepatic CLS in the liver of MC4R‐KO mice, in which CD11c‐positive macrophages surround dead or dying hepatocytes with large lipid droplets17. Hepatic CLS shares most of the histological features of adipose tissue CLS: hepatic CLS is composed of CD11c‐positive macrophages, and surrounded by myofibroblasts and extracellular matrices. The number of hepatic CLS is positively correlated with the extent of liver fibrosis17. Furthermore, hepatic CLS is a cellular source that produces pro‐inflammatory cytokines and fibrogenic mediators. As hepatic CLS formation is observed before the development of NASH, it is likely that hepatic CLS is involved in the disease progression from simple steatosis to NASH. It is noteworthy that hepatic CLS is observed in other animal models of NASH including dietary deficiency of methionine and choline, and long‐term feeding of a high‐fat diet. Hepatic CLS is also useful to evaluate the effect of interventions to prevent or treat NASH. For instance, we previously reported using MC4R‐KO mice that pirfenidone, a clinically available antifibrotic agent for idiopathic pulmonary fibrosis, effectively prevents the development of NASH45. Interestingly, treatment with pirfenidone strongly inhibited hepatic CLS formation and the following inflammation and fibrosis, without affecting hepatic steatosis. These observations led us to speculate that hepatic CLS might be a site of action of pirfenidone. In contrast, EPA treatment suppressed hepatic steatosis along with hepatic CLS formation and liver fibrosis in MC4R‐KO mice46. In addition to the prophylactic protocol, EPA showed the effect in the therapeutic protocol, in which EPA was administered after MC4R‐KO mice developed NASH. In this protocol, there was a marked decrease in the number of hepatic CLS after EPA treatment. Although hepatocyte death is considered a pathogenic feature of NASH47, it remains to be elucidated how hepatocyte death leads to liver fibrosis. These findings suggest that hepatic CLS drives hepatocyte death‐induced chronic inflammation and pericellular fibrosis in the development of NASH.
Clinical Relevance of Hepatic CLS in NASH
It is important to assess the clinical implication of hepatic CLS in human NASH. Notably, hepatic CLS was observed in liver biopsy specimens from patients with NAFLD/NASH17. On the basis of the NAFLD Activity Score, the number of hepatic CLS was positively associated with the score for ballooning degeneration, a hallmark for hepatocyte injury. Furthermore, the number of hepatic CLS was the highest in the patients with a NAFLD Activity Score of 5. These clinical findings are consistent with our experimental data showing that hepatocyte death triggers hepatic CLS formation, thereby inducing inflammation and fibrosis. Interestingly, hepatic CLS was rarely observed in patients with chronic viral hepatitis, whose serum AST and ALT concentrations were roughly equal to those in patients with NAFLD/NASH. Therefore, hepatic CLS would be a NASH‐specific pathological mechanism. As a future direction, it is interesting to translate the findings based on basic research using our NASH model into clinical practice. For instance, secreted factors derived from hepatic CLS might be a novel biomarker to predict the development of NASH or reflect the disease activity of NASH. Histological examinations focusing on hepatic CLS might also be helpful to evaluate the clinical usefulness of non‐invasive diagnostic tools, such as ultrasound elastography and magnetic resonance elastography.
Conclusion
For the past several decades, there has been dramatic progress in understanding the underlying mechanism of chronic inflammation in lifestyle‐related diseases, such as obesity, diabetes and NAFLD/NASH. In the present review, we discussed the recent progress of this research field, particularly focusing on how obesity‐induced chronic inflammation leads to interstitial fibrosis in adipose tissue and the liver. CLS is the site of the cross‐talk between parenchymal cells and stromal cells, including macrophages, thereby inducing persistent inflammation and interstitial fibrosis. In other words, CLS might function to drive ‘metabolic’ tissue remodeling. Better understanding of the molecular mechanism underlying obesity‐induced chronic inflammation would pave the way to developing a novel therapeutic strategy for lifestyle‐related diseases, such as obesity, diabetes and NAFLD/NASH.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (16H05171, 16KT0110, 16K08732, 17K19686 and 17H05500), and Japan Agency for Medical Research and Development (CREST). This work was also supported by research grants from Takeda Science Foundation, Takeda Medical Research Foundation and the Joint Usage/Research Program of Medical Research Institute, and Tokyo Medical and Dental University.
J Diabetes Investig 2018;9: 256–261
References
- 1. Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res 2005; 96: 939–949. [DOI] [PubMed] [Google Scholar]
- 2. Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006; 444: 860–867. [DOI] [PubMed] [Google Scholar]
- 3. Kadowaki T, Yamauchi T, Kubota N, et al Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 2006; 116: 1784–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matsuzawa Y, Funahashi T, Nakamura T. Molecular mechanism of metabolic syndrome X: contribution of adipocytokines adipocyte‐derived bioactive substances. Ann N Y Acad Sci 1999; 892: 146–154. [DOI] [PubMed] [Google Scholar]
- 5. Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol 2009; 6: 399–409. [DOI] [PubMed] [Google Scholar]
- 6. Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 2008; 118: 2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Suganami T, Ogawa Y. Adipose tissue macrophages: their role in adipose tissue remodeling. J Leukoc Biol 2010; 88: 33–39. [DOI] [PubMed] [Google Scholar]
- 8. de Alwis NM, Day CP. Non‐alcoholic fatty liver disease: the mist gradually clears. J Hepatol 2008; 48(Suppl 1): S104–S112. [DOI] [PubMed] [Google Scholar]
- 9. Lade A, Noon LA, Friedman SL. Contributions of metabolic dysregulation and inflammation to nonalcoholic steatohepatitis, hepatic fibrosis, and cancer. Curr Opin Oncol 2014; 26: 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nishimura S, Manabe I, Nagasaki M, et al Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes 2007; 56: 1517–1526. [DOI] [PubMed] [Google Scholar]
- 11. Nishimura S, Manabe I, Nagasaki M, et al In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest 2008; 118: 710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest 2011; 121: 2094–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suganami T, Tanaka M, Ogawa Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr J 2012; 59: 849–857. [DOI] [PubMed] [Google Scholar]
- 14. Weisberg SP, McCann D, Desai M, et al Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112: 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu H, Barnes GT, Yang Q, et al Chronic inflammation in fat plays a crucial role in the development of obesity‐related insulin resistance. J Clin Invest 2003; 112: 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clement K, Viguerie N, Poitou C, et al Weight loss regulates inflammation‐related genes in white adipose tissue of obese subjects. FASEB J 2004; 18: 1657–1669. [DOI] [PubMed] [Google Scholar]
- 17. Itoh M, Kato H, Suganami T, et al Hepatic crown‐like structure: a unique histological feature in non‐alcoholic steatohepatitis in mice and humans. PLoS ONE 2013; 8: e82163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007; 117: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lumeng CN, DelProposto JB, Westcott DJ, et al Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008; 57: 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamei N, Tobe K, Suzuki R, et al Overexpression of monocyte chemoattractant protein‐1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem 2006; 281: 26602–26614. [DOI] [PubMed] [Google Scholar]
- 21. Kanda H, Tateya S, Tamori Y, et al MCP‐1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006; 116: 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ito A, Suganami T, Yamauchi A, et al Role of CC chemokine receptor 2 in bone marrow cells in the recruitment of macrophages into obese adipose tissue. J Biol Chem 2008; 283: 35715–35723. [DOI] [PubMed] [Google Scholar]
- 23. Alkhouri N, Gornicka A, Berk MP, et al Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem 2010; 285: 3428–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wueest S, Rapold RA, Schumann DM, et al Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest 2010; 120: 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ichioka M, Suganami T, Tsuda N, et al Increased expression of macrophage‐inducible C‐type lectin in adipose tissue of obese mice and humans. Diabetes 2011; 60: 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi H, Kokoeva MV, Inouye K, et al TLR4 links innate immunity and fatty acid‐induced insulin resistance. J Clin Invest 2006; 116: 3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suganami T, Mieda T, Itoh M, et al Attenuation of obesity‐induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll‐like receptor 4 mutation. Biochem Biophys Res Commun 2007; 354: 45–49. [DOI] [PubMed] [Google Scholar]
- 28. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol 2005; 25: 2062–2068. [DOI] [PubMed] [Google Scholar]
- 29. Lee JY, Sohn KH, Rhee SH, et al Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase‐2 mediated through Toll‐like receptor 4. J Biol Chem 2001; 276: 16683–16689. [DOI] [PubMed] [Google Scholar]
- 30. Suganami T, Tanimoto‐Koyama K, Nishida J, et al Role of the Toll‐like receptor 4/NF‐kappaB pathway in saturated fatty acid‐induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol 2007; 27: 84–91. [DOI] [PubMed] [Google Scholar]
- 31. Iwasaki Y, Suganami T, Hachiya R, et al Activating transcription factor 4 links metabolic stress to interleukin‐6 expression in macrophages. Diabetes 2014; 63: 152–161. [DOI] [PubMed] [Google Scholar]
- 32. Itoh M, Suganami T, Satoh N, et al Increased adiponectin secretion by highly purified eicosapentaenoic acid in rodent models of obesity and human obese subjects. Arterioscler Thromb Vasc Biol 2007; 27: 1918–1925. [DOI] [PubMed] [Google Scholar]
- 33. Khan T, Muise ES, Iyengar P, et al Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 2009; 29: 1575–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu J, Divoux A, Sun J, et al Genetic deficiency and pharmacological stabilization of mast cells reduce diet‐induced obesity and diabetes in mice. Nat Med 2009; 15: 940–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mutch DM, Tordjman J, Pelloux V, et al Needle and surgical biopsy techniques differentially affect adipose tissue gene expression profiles. Am J Clin Nutr 2009; 89: 51–57. [DOI] [PubMed] [Google Scholar]
- 36. Itoh M, Suganami T, Nakagawa N, et al Melanocortin 4 receptor‐deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am J Pathol 2011; 179: 2454–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Divoux A, Tordjman J, Lacasa D, et al Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 2010; 59: 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanaka M, Ikeda K, Suganami T, et al Macrophage‐inducible C‐type lectin underlies obesity‐induced adipose tissue fibrosis. Nat Commun 2014; 5: 4982. [DOI] [PubMed] [Google Scholar]
- 39. Wells CA, Salvage‐Jones JA, Li X, et al The macrophage‐inducible C‐type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol 2008; 180: 7404–7413. [DOI] [PubMed] [Google Scholar]
- 40. Ishikawa E, Ishikawa T, Morita YS, et al Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C‐type lectin Mincle. J Exp Med 2009; 206: 2879–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schoenen H, Bodendorfer B, Hitchens K, et al Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose‐dibehenate. J Immunol 2010; 184: 2756–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamasaki S, Ishikawa E, Sakuma M, et al Mincle is an ITAM‐coupled activating receptor that senses damaged cells. Nat Immunol 2008; 9: 1179–1188. [DOI] [PubMed] [Google Scholar]
- 43. Iwayama T, Steele C, Yao L, et al PDGFRalpha signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev 2015; 29: 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Varela‐Rey M, Embade N, Ariz U, et al Non‐alcoholic steatohepatitis and animal models: understanding the human disease. Int J Biochem Cell Biol 2009; 41: 969–976. [DOI] [PubMed] [Google Scholar]
- 45. Komiya C, Tanaka M, Tsuchiya K, et al Antifibrotic effect of pirfenidone in a mouse model of human nonalcoholic steatohepatitis. Sci Rep 2017; 7: 44754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Konuma K, Itoh M, Suganami T, et al Eicosapentaenoic acid ameliorates non‐alcoholic steatohepatitis in a novel mouse model using melanocortin 4 receptor‐deficient mice. PLoS ONE 2015; 10: e0121528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feldstein AE, Canbay A, Angulo P, et al Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003; 125: 437–443. [DOI] [PubMed] [Google Scholar]