

Abstract
Gut microbiota dysbiosis reduces expression of GLP‐1 receptor (GLP‐1R) and neuronal nitric oxide synthase (nNOS) in the enteric nervous system and hampers GLP‐1‐induced nitric oxide (NO) production through a pattern recognition receptor (PRR)‐dependent mechanism, hence preventing activation of the gut–brain–periphery axis for control of insulin secretion and gastric emptying.
Gut hormonal substances released in response to nutrients include incretins, gastric inhibitory polypeptide and glucagon‐like peptide‐1 (GLP‐1). Together, incretins are responsible for 50–60% of postprandial insulin secretion. Oral glucose administration leads to greater insulin release from pancreatic islets than intravenous glucose loading that yields equivalent blood glucose levels; this phenomenon is called the incretin effect. It has been reported that the incretin effect is markedly impaired in individuals with type 2 diabetes mellitus. The impaired incretin effect in type 2 diabetes patients has been found to be characterized by an almost complete loss of insulin secretion in response to gastric inhibitory polypeptide and a reduced insulinotropic potency of GLP‐1. Several studies reported a marked reduction in postprandial plasma GLP‐1 levels in type 2 diabetes patients compared with healthy individuals, suggesting that a lower level of GLP‐1 secretion might contribute to the impaired incretin effect in type 2 diabetes. However, a meta‐analysis of 22 trials showed that patients with type 2 diabetes do not generally show reduced GLP‐1 secretion in response to an oral glucose tolerance test or meal test, although deteriorating glycemic control might be associated with reduced GLP‐1 secretion1. In any case, supraphysiological doses of GLP‐1 are effective in type 2 diabetes patients; thus, GLP‐1 receptor (GLP‐1R) agonism might represent an attractive pharmacological strategy for diabetes treatment. However, it is observed in clinical practice that GLP‐1‐based therapies do not always achieve the intended therapeutic outcomes in all patients with type 2 diabetes. Furthermore, some patients have to discontinue treatment because of a lack of efficacy, which defines a state of GLP‐1 resistance.
Accumulating evidence has shown that differences in the structure, function and diversity of the human gut microbiota have implications for human health and disease. Recent studies have shown that the immensely diverse gut microbiota contributes to the pathogenesis of obesity and type 2 diabetes2. Microbial metabolites regulate metabolism in different tissues. Gram‐negative bacteria‐derived lipopolysaccharide promotes macrophage recruitment and polarization in white adipose tissue, inducing inflammation through Toll‐like receptor 4. Short‐chain fatty acids, which are fermentation products of the gut microbiota, serve as an energy source in enterocytes and also stimulate intestinal gluconeogenesis. Secondary bile acids (lithocholic acid and deoxycholic acid) derived from the microbiota promote thermogenesis in brown adipose tissue, thus increasing energy expenditure. The gut microbiota induces signaling through farnesoid X receptor in the liver and intestine by metabolizing a natural farnesoid X receptor antagonist, tauro‐beta‐muricholic acid, reducing bile acid synthesis and altering fatty acid metabolism. Short‐chain fatty acids and secondary bile acids (lithocholic acid and deoxycholic acid) are involved in stimulating GLP‐1 secretion from enteroendocrine L‐cells.
In a recent study, Grasset et al.3 showed that gut microbiota dysbiosis caused GLP‐1 resistance in two types of high‐fat diet‐fed mice with impaired glucose tolerance. They clearly showed that the dysbiosis reduced expression of GLP‐1R and neuronal nitric oxide synthase (nNOS) in the enteric nervous system and hampered GLP‐1‐induced nitric oxide (NO) production through a pattern recognition receptor‐dependent mechanism, hence preventing activation of the gut–brain–periphery axis for control of insulin secretion and gastric emptying (Figure 1). These results provide new insight into the role of the gut microbiota in maintaining normal metabolic conditions by regulating GLP‐1 biology.
Figure 1.
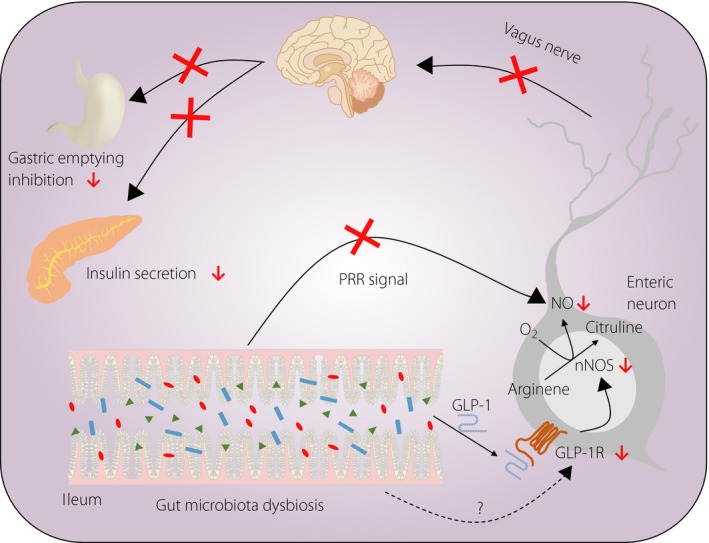
Glucagon‐like peptide‐1 (GLP‐1) resistance caused by gut microbiota dysbiosis. Gut microbiota dysbiosis reduces expression of GLP‐1 receptor (GLP‐1R) and neuronal nitric oxide synthase (nNOS) in the enteric nervous system, and hampers GLP‐1‐induced nitric oxide (NO) production through a pattern recognition receptor (PRR)‐dependent mechanism, hence preventing activation of the gut–brain–periphery axis for control of insulin secretion and gastric emptying. [Colour figure can be viewed at wileyonlinelibrary.com]
One of the remarkable findings of the study was that GLP‐1 unresponsiveness was far more severe in lean diabetic mice fed a high‐fat carbohydrate‐free diet (HFD) than in obese diabetic mice fed a high‐carbohydrate HFD. Because meal composition significantly affects the gut microbiota, the difference in the extent of GLP‐1 sensitivity between the two mouse models might have resulted from variations in the ileum bacteria. Venn analyses of pathway modules showed that there were 25 common pathways enriched in both the high‐carbohydrate HFD‐fed and HFD‐fed groups, and 19 pathway modules uniquely enriched in the HFD‐fed mice compared with the high‐carbohydrate HFD‐fed mice. Using prediction algorithms, Grasset et al.3 suggested that bacterial pathways related to amino acid metabolism and transport system modules could be involved in GLP‐1 resistance. However, the specific mechanism by which gut microbiota dysbiosis in HFD‐fed mice reduces GLP‐1R expression in the enteric nervous system remains to be elucidated. Furthermore, it should be noted that the dysbiosis observed in HFD‐fed mice was possibly specific to the extreme meal composition applied in that study; thus, the severe GLP‐1 resistance observed in HFD‐fed diabetic mice is not necessarily recapitulated in human diabetes.
In a previous report, it was shown that long‐term HFD intake caused a remarkable loss of intestinal myenteric neurons in mice as a result of a lack of energy substrates, reduced acetylcholine synthesis, membrane deterioration and oxidative stress4. Grasset et al.3 showed that gut microbiota dysbiosis in HFD‐fed mice is responsible for the loss of enteric neurons, impaired NO production and subsequent GLP‐1 resistance. It has been generally assumed that NO produced by nNOS in enteric neurons has protective effects that outweigh any damaging effect of this free radical. Therefore, it is possible that the impaired NO production as a result of reduced nNOS expression in enteric neurons is attributable to an extensive loss of intestinal myenteric neurons in HFD‐fed mice.
Grasset et al.3 showed that insulin secretion and gastric emptying inhibition in response to GLP‐1 were impaired by vagotomy or by cisplatin‐induced enteric neuropathy. In contrast, energy intake in vagotomized mice and cisplatin‐treated mice was comparable with that in control mice, showing that insulin secretion and gastric emptying inhibition in response to GLP‐1 are tightly dependent on a functional gut–brain–periphery axis, whereas food intake regulation is not. However, HFD‐fed mice were resistant not only to GLP‐1‐induced insulin secretion and gastric emptying inhibition, but also to GLP‐1‐induced suppression of food intake. Therefore, it is supposed that the arcuate and paraventricular nuclei, the hypothalamic feeding centers in which GLP‐1R is expressed, could be involved in GLP‐1 unresponsiveness in HFD‐fed mice. However, GLP‐1R expression and GLP‐1 responsiveness in the arcuate and paraventricular nuclei under high‐fat dietary perturbations have not yet been examined.
It has been reported that l‐arginine is a GLP‐1 secretagogue, and that improvement of glucose tolerance by oral l‐arginine administration is dependent on GLP‐1R signaling5. Interestingly, in HFD‐fed mice with severe GLP‐1 resistance, glucose‐induced insulin secretion was increased by acute and chronic treatment with l‐arginine, a substrate of nNOS, suggesting improved GLP‐1 sensitivity. These findings raise the intriguing possibility that l‐arginine supplementation might benefit glucose tolerance by improving both the postprandial GLP‐1 response and GLP‐1 sensitivity in type 2 diabetes patients.
Gut microbiota is attracting attention as a potential target for therapeutic intervention. A deeper understanding of how gut microbiota dysbiosis contributes to the pathophysiology of obesity and diabetes might provide a novel therapeutic approach for diabetes and some clues to overcome the difficulties in diabetes treatment.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This work was supported by Scientific Research Grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Nobuya Inagaki received research grants from Astellas Pharma Inc., Taisho Toyama Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Takeda Pharmaceutical Co. Ltd., Daiichi Sankyo Co., Ltd., MSD, Sanofi, Dainippon Sumitomo Pharma Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Eli Lilly Japan K.K., Shiratori Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., JT, Pfizer, Nippon Boehringer Ingelheim Co., Ltd., Sanwa Kagaku Kenkyusho Co., Ltd., Kissei Pharmaceutical Co., Ltd., AstraZeneca and the Japan Diabetes Foundation.
References
- 1. Calanna S, Christensen M, Holst JJ, et al Secretion of glucagon‐like peptide‐1 in patients with type 2 diabetes mellitus: systematic review and meta‐analyses of clinical studies. Diabetologia 2013; 56: 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arora T, Bäckhed F. The gut microbiota and metabolic disease: current understanding and future perspectives. J Intern Med 2016; 280: 339–349. [DOI] [PubMed] [Google Scholar]
- 3. Grasset E, Puel A, Charpentier J, et al A specific gut microbiota dysbiosis of type 2 diabetic mice induces GLP‐1 resistance through an enteric NO‐dependent and gut‐brain axis mechanism. Cell Metab 2017; 25: 1075–1090. [DOI] [PubMed] [Google Scholar]
- 4. Voss U, Sand E, Olde B, et al Enteric neuropathy can be induced by high fat diet in vivo and palmitic acid exposure in vitro . PLoS ONE 2013; 8: e81413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clemmensen C, Smajilovic S, Smith EP, et al Oral L‐arginine stimulates GLP‐1 secretion to improve glucose tolerance in male mice. Endocrinology 2013; 154: 3978–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]