

Abstract
The primary virulence factor of the skin commensal and opportunistic pathogen, Staphylococcus epidermidis, is the ability to form biofilms on surfaces of implanted materials. Much of this microorganism’s pathogenic success has been attributed to its ability to evade the innate immune system. The primary defense against S. epidermidis biofilm infection consists of complement activation, recruitment and subsequent killing of the pathogen by effector cells. Among pathogen-derived factors, the biofilm exopolysaccharide polysaccharide intercellular adhesion (PIA), as well as the accumulation-associated protein (Aap), and the extracellular matrix binding protein (Embp) have been shown to modulate effector cell-mediated killing of S. epidermidis. Phenol-soluble modulins (PSMs) constitute the only class of secreted toxins by S. epidermidis, at least one type of which (PSMδ) possesses strong cytolytic properties toward leukocytes. However, through selective production of non-cytolytic subtypes of PSMs, S. epidermidis is able to maintain a low inflammatory infection profile and avoid eradication by the host immune system. Taken together, our emerging understanding of the mechanisms behind immune modulation by S. epidermidis elucidates the microorganism’s success in the initial colonization of device surfaces as well as the maintenance of a chronic and indolent course of biofilm infection.
Keywords: Staphylococcus epidermidis, biofilm, polysaccharide intercellular adhesin, accumulation associated protein, extracellular matrix binding protein, phenol-soluble modulins, innate immune system, neutrophil
Introduction
Initially described in 1878, staphylococci are Gram-positive microorganisms that have been implicated in infections involving multiple systems of the human body, including the skin and soft tissue, the skeletal system, the respiratory system, the blood stream, and more recently, infections involving implanted medical devices (Lowy, 1998). Staphylococci are further classified as being coagulase-positive, primarily identifying Staphylococcus aureus (S. aureus), or coagulase-negative (CoNS) (Kloos and Schleifer, 1986). Staphylococcus epidermidis (S. epidermidis) is the most important and best-studied member of the CoNS group (Vuong and Otto, 2002).
While conventionally regarded merely as an innocuous commensal of the human skin, and increasingly recognized for its beneficial role in skin immunity and within the skin microbiota (Naik et al., 2015; Nguyen et al., 2017), S. epidermidis has also emerged as an important human pathogen. This is mostly due to the increased number of implanted prosthetic materials and medical devices (Becker et al., 2014). Lacking most of the many aggressive virulence factors that S. aureus produces, S. epidermidis’ primary virulence mechanism is to form deeply seated microbial communities, known as biofilms, on implanted medical devices surfaces and native host tissues (Otto, 2008, 2009). For example, S. epidermidis is a leading cause of infections on central venous catheters, which occur at a frequency of ∼80,000 annually in the United States alone and may result in severe blood infections (Maki et al., 2006). Furthermore, together with S. aureus, it is the premier pathogen causing prosthetic joint infections (Uckay et al., 2009). Finally, S. epidermidis causes 15–40% of prosthetic valve endocarditis cases (Wang et al., 2007), a less common but very serious infection. All these infections are easily recognized macro- or microscopically to proceed as biofilm-associated. Within biofilms, S. epidermidis are protected from the effects of antimicrobial therapy as well as the host immune system (Vuong and Otto, 2002). Consequently, medical therapy in biofilm-associated infections can be exceptionally challenging (Hoiby et al., 2010), with attempts at infection eradiation often entailing complete removal of the infected foreign body as well as administration of prolonged courses of antimicrobial therapy, approaches that incur risks to patients and excess cost to the health care system (Rogers et al., 2009).
Herein, we attempt to outline our current understanding of the host- and pathogen-derived characteristics that favor effective immune evasion by S. epidermidis, such as those contributing toward the microorganism’s successful seeding, colonization of device surfaces, and securing survival in the form of indolent biofilm communities.
Stages of Biofilm Development
Biofilm development has been modeled to occur in three stages: (1) attachment, (2) proliferation/formation of the matured biofilm, and (3) detachment/dispersal (O’Toole et al., 2000; Otto, 2013). During attachment, staphylococcal surface-attached proteins known as microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) establish non-covalent interactions with device surfaces coated by host proteins and host tissues (Patti et al., 1994; Otto, 2008; Joo and Otto, 2015). After attachment, proliferation and maturation of the biofilm follows, with the production of an extracellular matrix consisting of the staphylococcal biofilm exopolysaccharide, polysaccharide intercellular adhesin (PIA) (Mack et al., 1996), also called poly-N-acetylglucosamine (PNAG), teichoic acids, proteins, and extracellular DNA (eDNA) (O’Toole et al., 2000; Otto, 2008). During this second stage of biofilm expansion, channels and mushroom-shaped structures form to facilitate nutrient delivery to deeper layers of the biofilm. The last stage of biofilm development is characterized by the detachment and subsequent dispersal/dissemination of biofilm clusters to distal sites (Joo and Otto, 2015), a process mostly due to the activity of the surfactant-like phenol-soluble modulin (PSM) peptides (Wang et al., 2011; Otto, 2013; Figure 1).
FIGURE 1.
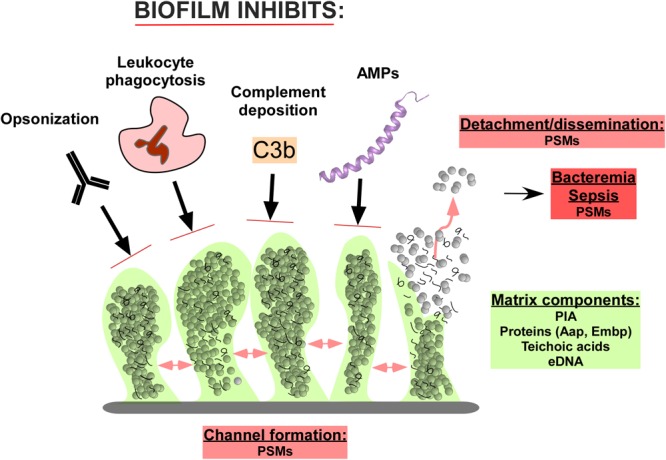
Composition and function of Staphylococcus epidermidis biofilms in immune evasion. The biofilm matrix consists of the polysaccharide intercellular adhesion (PIA) exopolysaccharide, proteins such as accumulation-associated protein (Aap) and extracellular matrix binding protein (Embp), teichoic acids, and extracellular DNA (eDNA). Channels in the biofilm are formed by Phenol-soluble modulins (PSMs), which also lead to cell cluster detachment and ultimately dissemination of the infection. Predominantly in immune-compromised individuals, this can lead to bacteremia and sepsis, in which PSMs also likely play a role. The biofilm structure and matrix provides shelter from host defenses, including the binding of opsonizing immunoglobulins, complement components, and antimicrobial peptides (AMPs). Furthermore, the attack of leukocytes is significantly diminished.
PIA/PNAG
The exopolysaccharide PIA/PNAG is an important and abundant component of the S. epidermidis biofilm matrix. Initially described in S. epidermidis clinical isolates (Mack et al., 1994; Schumacher-Perdreau et al., 1994), PIA is a linear homopolymer of β-1,6-linked N-acetylglucosamine monomers, which is positively charged due to partial deacetylation (Mack et al., 1996; Vuong et al., 2004a). These charged moieties facilitate interactions between components of the biofilm extracellular matrix and the staphylococcal cell wall (Mack et al., 1996; Vuong et al., 2004a). PIA is the product of the icaADBC operon (Heilmann et al., 1996), which encodes three membrane proteins (IcaA, IcaC, IcaD), and an additional protein (IcaB) that is exported and subsequently attached to the staphylococcal cell surface by non-covalent interactions (Vuong et al., 2004a). IcaA, an N-acetylglucosaminyltransferase, along with IcaD, catalyzes the conversion of UDP-N-acetylglucosamine to 10–20-mers of β-1,6-linked poly-N-acetylglucosamine (Gerke et al., 1998). IcaC is needed to further extend the N-acetylglucosamine oligomers, and probably represents a PIA export system (Gerke et al., 1998), while IcaB deacetylates the poly-N-acetylglucosamine molecule (Vuong et al., 2004a), enabling localization of the PIA molecule to the cell surface (Figure 2).
FIGURE 2.
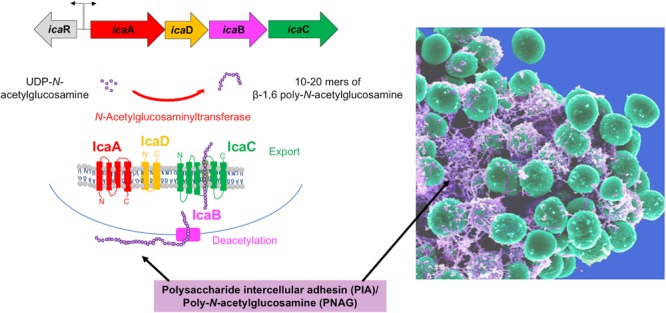
The biofilm exopolysaccharide PIA/PNAG. The PIA biosynthetic locus includes the icaA gene, which codes for an N-acetylglucosamine (GlcNAc) transferase, which adds GlcNAc residues to a growing poly-GlcNAc chain. IcaD assists in this function in an unknown way. The chain is then believed to be exported by IcaC, because in the absence of icaC, polymerization stops at chain lengths of about 10–20 GlcNAc units. IcaB is located at the extracellular cell surface and de-acetylates some of the GlcNAc units, introducing a positive charge in the polymer due to the then unmasked amino groups. This is necessary for PIA surface location (see electron microscopy picture on the right) and functionality in biofilm formation and immune evasion.
Accumulation-Associated Protein (Aap) and Extracellular Matrix Binding Protein (Embp)
PIA does not appear to be absolutely required for S. epidermidis biofilm formation, as S. epidermidis isolates from biofilm-associated catheter and prosthetic joint infections were found to be negative for the ica genes (Francois et al., 2003; Chokr et al., 2007; Rohde et al., 2007). In S. epidermidis ica(-) strains, intercellular adhesion has been shown to be mediated through proteinaceous components, of which two of the best-defined are the accumulation-associated protein (Aap) (Hussain et al., 1997) and the extracellular matrix binding protein (Embp) (Christner et al., 2010). However, PIA-dependent biofilms appear to be more structured and robust than those dependent on proteins (Schommer et al., 2011).
Accumulation-associated protein is a 240-kDa surface-bound protein consisting of two domains, designated as A and B. Proteolytic cleavage of domain A induces a conformation change in the protein, enabling domain B to mediate polymerization of Aap into fibrils, leading to aggregation and biofilm formation in the PIA-negative S. epidermidis clinical strain 5179 (Rohde et al., 2005; Conrady et al., 2008; Conlon et al., 2014).
Extracellular matrix binding protein is a 1-MDa protein surface protein that has been implicated as an intercellular adhesin that mediates S. epidermidis biofilm formation. Embp is composed of 59 found-in-various-architecture (FIVAR) domains and 38 G-related albumin-binding (GA) domains (Christner et al., 2010). In vitro, the FIVAR domains were observed to bind to fibronectin, thus suggesting a role in the initial attachment phase of biofilm development. Furthermore, like the FIVAR domains described in the S. aureus protein Fmtb, which interacts with N-acetylglucosamine, the FIVAR domains of Embp in S. epidermidis are hypothesized to also bind to PIA (Christner et al., 2010). However, it appears that FIVAR domains alone are insufficient for biofilm aggregation (Christner et al., 2010).
Phenol Soluble Modulins (PSMs)
Phenol soluble modulins (PSMs) were first described in 1999 in S. epidermidis (Mehlin et al., 1999). Later it was found that S. epidermidis produces six PSM peptides, PSMα, PSMβ1, PSMβ2, PSMδ, PSM𝜀, and δ-toxin (Otto et al., 2004; Yao et al., 2005). Next to δ-toxin, β-PSMs are the primary PSMs produced in S. epidermidis (Yao et al., 2005; Cheung et al., 2010). Besides these PSMs, which are all core genome-encoded, there is one PSM, PSM-mec, which is encoded within some types of the methicillin resistance-conferring mobile genetic element, SCCmec (Queck et al., 2009). S. epidermidis β-PSMs (Wang et al., 2011) have been shown to be key effector molecules in biofilm structuring and dissemination (Otto, 2013), but investigation in S. aureus indicates all PSMs have similar capacities (Periasamy et al., 2012). The general mechanism by which PSMs contribute to biofilm structuring and dispersal is believed to be the disruption of non-covalent (hydrophobic, electrostatic) interactions between biofilm matrix macromolecules (Otto, 2013). The PSM structuring effect occurs independently of the mode of biofilm formation (PIA-dependent or -independent) (Wang et al., 2011).
Components of the Innate Immune Response in Staphylococcal Infections
In immunocompetent hosts, the primary innate immune response against planktonic staphylococcal infections involve the complement system as well as effector cells (Foster, 2005). The complement system’s primary role is to recruit effector molecules that label staphylococci and mark them for destruction by effector cells. In addition, the activated complement system produces a cell-killing membrane attack complex. Complement fixation occurs through the classical, alternative, and lectin pathways. While the alternative and lectin pathways are part of the innate immune system, activation of the classical pathway requires binding of an antibody to an antigen on the staphylococcal surface. Activation of these pathways leads to the generation of C3a, a pro-inflammatory chemoattractant, which recruits phagocytes to the infection site. While the alternative pathway seems to play a smaller role in host defense against S. epidermidis infections (Fredheim et al., 2011), the classical and lectin pathways are believed to be necessary for rapid killing of planktonic S. epidermidis by effector cells (Kristian et al., 2008).
Neutrophils (or, polymorphonuclear leukocytes, PMNs) have been identified as the primary effector cells in the innate immune defense against staphylococcal infection. Activation of neutrophils might involve: (1) recognition of pathogen-associated molecular patterns (PAMPs) on bacterial surfaces by the cellular recognition receptors (Toll-like receptors), (2) deposition of opsonins on the bacterial surface, or (3) the direct action of complement system (Rigby and DeLeo, 2012). Leukocyte responses involve direct contact of the staphylococci with the host immune cells, and are channeled via pattern recognition receptor-dependent pathways (Flannagan et al., 2015). While the role of these pathways in staphylococcal infection have primarily been established in S. aureus, there is evidence that the cellular recognition receptor, Toll-like receptor 2 (TLR2), plays a significant role in S. epidermidis bloodstream infection (Strunk et al., 2010). However, how exactly pathogen recognition receptors impact S. epidermidis blood infection remains unknown.
Once activated, neutrophil-mediated killing involves reactive oxygen species as well as non-oxygen-dependent processes involving antimicrobial peptides (AMPs), such as defensins and cathelicidins, and antimicrobial proteins, such as lysozyme (Hancock and Diamond, 2000; Nauseef, 2007). Again, much of this is general knowledge obtained in other bacteria, but a significant role of AMP resistance mechanisms, such as proteolysis by the SepA protease (Cheung et al., 2010) and PIA-mediated resistance on the cell surface (Vuong et al., 2004b) (see below), as well as sensing of the presence of AMPs by the S. epidermidis ApsRSX system (Li et al., 2007), have been demonstrated to contribute to S. epidermidis survival in neutrophils and biofilm infection.
The Innate Immune Response to S. epidermidis Biofilm Infection
As compared to S. aureus, infections by S. epidermidis are characterized by proceeding with decidedly less inflammation and overall morbidity. This is also true for biofilm-associated infections. Specific mechanisms that S. epidermidis employs to limit the inflammatory response will be discussed in the following. On the other hand, there are specific interactions between S. epidermidis and the host that can be described as pro-inflammatory, which will also be discussed. These may be less important given the overall outcome.
Early research has suggested that biofilm formation protects S. epidermidis from phagocytosis by effector cells (Johnson et al., 1986; Heinzelmann et al., 1997). When compared to the response against planktonic infection, the innate immune response against S. epidermidis biofilm infection has been characterized as being less pronounced (Cerca et al., 2006; Kristian et al., 2008; Schommer et al., 2011; Thurlow et al., 2011; Hanke et al., 2012, 2013; Spiliopoulou et al., 2012). However, vigorous induction of the complement system has been described (Kristian et al., 2008). Yet, deposition of C3b and IgG on S. epidermidis biofilm surfaces was paradoxically diminished, when compared to the planktonic mode of growth, suggesting that subsequent activation and killing of S. epidermidis biofilm by effector cells might also be impaired (Kristian et al., 2008).
Diminished activation of leukocytes has been described in S. epidermidis biofilm infection and seems to involve multiple mechanisms. First, observation of a blunted response to pro-inflammatory compounds by macrophages after exposure to S. epidermidis biofilm suggests that interference of signaling by cellular recognition receptors might partially contribute to the observed quiescent immune response toward biofilms, when compared to the planktonic mode of growth (Schommer et al., 2011). Additionally, when rabbit polyclonal PIA/PNAG antiserum was utilized as opsonin, fewer deaths of S. epidermidis derived from disrupted biofilms were detected, when compared to their isogenic planktonic form (Cerca et al., 2006), implicating opsonin deposition as one aspect of innate immune response that may be modulated in S. epidermidis biofilm infection.
Moreover, it has been hypothesized that sufficient contact of the bacteria with components of the host immune system is necessary for efficient activation of effector cells (Schommer et al., 2011). In fact, deficient uptake of bacteria by macrophages and reduced generation of an NF-κB-mediated macrophage inflammatory response have been described in S. epidermidis biofilm (Riber et al., 1995; Schommer et al., 2011), phenotypes probably linked to the immune evasion effect of PIA (see below) and biofilm formation per se. Additionally, selective modulation of the immune response seems to occur, as discriminatory activation of the weakly pro-inflammatory J774A.1 macrophage by S. epidermidis biofilms was reported (Schommer et al., 2011). Once consumed by macrophages, biofilm-derived S. epidermidis appears to be able to survive more effectively within these effector cells, than their isogenic planktonic counterpart (Spiliopoulou et al., 2012).
The Role of PIA/PNAG
The exopolysaccharide PIA represents a particularly important constituent of the microorganism’s immune evasion strategies (Foster, 2005; Otto, 2009). When compared to the isogenic 1457 ica(-) M10 strain, S. epidermidis 1457 is killed by PMNs less efficiently (Vuong et al., 2004b; Kristian et al., 2008), likely by reducing phagocytosis. Reduced uptake has been shown directly for macrophages (Schommer et al., 2011). This is likely mostly due to PIA being a preeminent biofilm constituent and a molecule that leads to the formation of bacterial aggregates. However, there are also more specific interactions of PIA with the immune system that have been reported. Some of them are probably due to PIA forming a sort of positively charged “capsule” around S. epidermidis, which is a general mechanism to shield the bacteria from immune recognition. In accordance with that notion, differences in PMN-mediated killing have been attributed to decreased availability of opsonizable surfaces in ica(+) biofilm (Cerca et al., 2006) and differences in PIA-mediated opsonization mechanisms (Kristian et al., 2008). Furthermore, PIA appears to prevent neutrophil attacks when cell clusters are disintegrated (Vuong et al., 2004b).
Animal experiments as well as observations from human S. epidermidis biofilm infections have largely recapitulated an overall attenuated immune response to biofilm infection due to wildtype S. epidermidis strains when compared to those derived from the isogenic ica(-) isolates (Fey et al., 1999; Rupp et al., 1999a,b; Vuong et al., 2004a). Levels of inflammatory cytokines (including TNF-α, IL-6, IFN-γ) and CD11b expression in human whole blood were higher in infections with S. epidermidis 1457 ica(-) M10 than those with the isogenic wildtype strain (Fredheim et al., 2011). Moreover, skin and soft tissue inflammatory changes surrounding subcutaneous catheter insertion sites were found to be more severe in mice infected with wildtype S. epidermidis compared to the ica(-) strain (Kristian et al., 2008), which correlates with in vitro findings of differential activation of the complement pathways (Kristian et al., 2008). Furthermore, levels of IL-6 in blood cultures of ica(+) S. epidermidis isolated from neonates were lower as compared to that of an ica(-) control strain (Hartel et al., 2008). Finally, there was a significant correlation between low levels of C-reactive protein (CRP) and in vitro biofilm-forming capacity in S. epidermidis isolates from neonate blood infection (Klingenberg et al., 2005).
Moreover, PIA confers protection against the action of host AMPs (Vuong et al., 2004b). The mechanism behind the protective effect of PIA against AMPs is thought to involve repulsion between the cationic PIA and the commonly cationic AMPs. However, a similar effect was reported in the same study for the anionic AMP dermcidin, suggesting that different mechanisms of PIA-mediated resistance to AMPs also exist (Vuong et al., 2004a; Otto, 2006).
Polysaccharide intercellular adhesion has also been reported to act in a pro-inflammatory fashion. For example, it has been implicated in the activation of the complement system during infection, based on the comparison of wild-type 1457 and isogenic ica(-) M10 strains (Fredheim et al., 2011). This phenotype was also achieved using a PIA preparation in the same study. These results await verification by a demonstration of a specific receptor-mediated mechanism.
The Role of Aap and Embp
In contrast to PIA, which forms a meshwork of extracellular matrix that embeds adjacent S. epidermidis cells, Aap is covalently attached to the S. epidermidis surface through its LPXTG motif and extends radially to form tuffs of fibrils (Rohde et al., 2005; Banner et al., 2007). Embp is also found on the S. epidermidis surface, forming a proteinaceous matrix (Schommer et al., 2011). Like in a predominantly PIA-dependent biofilm, biofilms dependent on Aap or Embp protect S. epidermidis from J774A.1 macrophage phagocytosis (Christner et al., 2010; Schommer et al., 2011).
The Role of PSMs
Complete genome analyses of S. epidermidis have shown that besides from their crucial roles in S. epidermidis biofilm structuring and dissemination, PSMs are the only gene products with cytolytic properties in S. epidermidis (Zhang et al., 2003; Gill et al., 2005), with the PSMδ of the α-type PSM subclass discovered as the first highly potent cytolysin produced by S. epidermidis (Cheung et al., 2010, 2014). PSMδ has been shown to be highly cytolytic against human neutrophils (Cheung et al., 2010). Nevertheless, when culture filtrates of S. epidermidis were examined, low levels of neutrophil lysis were measured (Cheung et al., 2010), and this is thought to be a consequence of selective production of PSMs by S. epidermidis, favoring production of the non-cytolytic β-type PSMs over the α-type PSMs, with the exception of the δ-toxin (PSMγ) (Cheung et al., 2010). This strategy results in a low inflammatory profile and likely contributes toward S. epidermidis’ successful immune evasion and subsequent colonization of device surfaces (Cheung et al., 2010).
The most frequent and serious complication of biofilm-associated infection on indwelling medical devices is bloodstream infection that arises from hematogenous seeding of dispersed biofilm clusters, which can develop into full-blown sepsis. As the most frequent cause of biofilm infections on such devices, S. epidermidis is also a leading cause of hospital-associated bloodstream infections, in particular in neonates (Cheung and Otto, 2010; Becker et al., 2014). Believed for the longest time to be due to an over-reacting immune response to invariant bacterial surface structures such as lipoteichoic acids and lipopeptides (Bochud and Calandra, 2003), a recent study has implicated PSM-mec, which is highly expressed in many methicillin-resistant isolates of S. epidermidis (Qin et al., 2017). Like other PSMs, PSM-mec is pro-inflammatory due to activation of the formyl peptide receptor 2 (Kretschmer et al., 2010). Most likely, other PSMs have similar effects, which remain to be determined.
Finally, it needs to be mentioned that there are reports on the presence of other toxins in S. epidermidis, such as a pathogenicity-island encoded enterotoxin, but this has to be considered a very rare exception (Madhusoodanan et al., 2011). Reports claiming much more widespread presence of toxins in S. epidermidis have to be regarded as due to inappropriate species identification.
Conclusion and Outlook
In summary, by modulating multiple aspects of the host innate immune response using secreted exopolysaccharides, peptides, and other proteinaceous biofilm components, S. epidermidis is able to orchestrate an overall low inflammatory profile, escape killing by the host innate immune system, and persist on the surfaces of implanted devices (Figure 1). While often considered innocuous partly due to its low inflammatory infection profile, it is precisely the low immunogenicity that makes S. epidermidis difficult to diagnose at initial stages of infection. Often, S. epidermidis biofilm infections are diagnosed at the third stage of biofilm infection, when infection sequelae are more severe and difficult to manage and treat, suggesting a need for a more complete basic understanding of the mechanisms behind this relatively quiescent immune response, so that safer avenues of therapy as well as novel approaches to prevention might be pursued.
Presently, our knowledge on the pro-inflammatory capacity of S. epidermidis in comparison to S. aureus during infection is poor. Future research should include an in-depth analysis of how only recently discovered pro-inflammatory molecules of S. epidermidis, such as the PSMs, contribute to inflammation and immune priming. This research will also benefit from a comparison with recent advances in the understanding of how S. epidermidis primes the skin immune system during the commensal state (Naik et al., 2015; Linehan et al., 2018).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, U.S. National Institutes of Health.
References
- Banner M. A., Cunniffe J. G., Macintosh R. L., Foster T. J., Rohde H., Mack D., et al. (2007). Localized tufts of fibrils on Staphylococcus epidermidis NCTC 11047 are comprised of the accumulation-associated protein. J. Bacteriol. 189 2793–2804. 10.1128/JB.00952-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K., Heilmann C., Peters G. (2014). Coagulase-negative staphylococci. Clin. Microbiol. Rev. 27 870–926. 10.1128/CMR.00109-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochud P. Y., Calandra T. (2003). Pathogenesis of sepsis: new concepts and implications for future treatment. BMJ 326 262–266. 10.1136/bmj.326.7383.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerca N., Jefferson K. K., Oliveira R., Pier G. B., Azeredo J. (2006). Comparative antibody-mediated phagocytosis of Staphylococcus epidermidis cells grown in a biofilm or in the planktonic state. Infect. Immun. 74 4849–4855. 10.1128/IAI.00230-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung G. Y., Joo H. S., Chatterjee S. S., Otto M. (2014). Phenol-soluble modulins–critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 38 698–719. 10.1111/1574-6976.12057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung G. Y., Otto M. (2010). Understanding the significance of Staphylococcus epidermidis bacteremia in babies and children. Curr. Opin. Infect. Dis. 23 208–216. 10.1097/QCO.0b013e328337fecb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung G. Y., Rigby K., Wang R., Queck S. Y., Braughton K. R., Whitney A. R., et al. (2010). Staphylococcus epidermidis strategies to avoid killing by human neutrophils. PLoS Pathog. 6:e1001133. 10.1371/journal.ppat.1001133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chokr A., Leterme D., Watier D., Jabbouri S. (2007). Neither the presence of ica locus, nor in vitro-biofilm formation ability is a crucial parameter for some Staphylococcus epidermidis strains to maintain an infection in a guinea pig tissue cage model. Microb. Pathog. 42 94–97. 10.1016/j.micpath.2006.09.001 [DOI] [PubMed] [Google Scholar]
- Christner M., Franke G. C., Schommer N. N., Wendt U., Wegert K., Pehle P., et al. (2010). The giant extracellular matrix-binding protein of Staphylococcus epidermidis mediates biofilm accumulation and attachment to fibronectin. Mol. Microbiol. 75 187–207. 10.1111/j.1365-2958.2009.06981.x [DOI] [PubMed] [Google Scholar]
- Conlon B. P., Geoghegan J. A., Waters E. M., Mccarthy H., Rowe S. E., Davies J. R., et al. (2014). Role for the A domain of unprocessed accumulation-associated protein (Aap) in the attachment phase of the Staphylococcus epidermidis biofilm phenotype. J. Bacteriol. 196 4268–4275. 10.1128/JB.01946-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady D. G., Brescia C. C., Horii K., Weiss A. A., Hassett D. J., Herr A. B. (2008). A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc. Natl. Acad. Sci. U.S.A. 105 19456–19461. 10.1073/pnas.0807717105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey P. D., Ulphani J. S., Gotz F., Heilmann C., Mack D., Rupp M. E. (1999). Characterization of the relationship between polysaccharide intercellular adhesin and hemagglutination in Staphylococcus epidermidis. J. Infect. Dis. 179 1561–1564. 10.1086/314762 [DOI] [PubMed] [Google Scholar]
- Flannagan R. S., Heit B., Heinrichs D. E. (2015). Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathogens 4 826–868. 10.3390/pathogens4040826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster T. J. (2005). Immune evasion by staphylococci. Nat. Rev. Microbiol. 3 948–958. 10.1038/nrmicro1289 [DOI] [PubMed] [Google Scholar]
- Francois P., Tu Quoc P. H., Bisognano C., Kelley W. L., Lew D. P., Schrenzel J., et al. (2003). Lack of biofilm contribution to bacterial colonisation in an experimental model of foreign body infection by Staphylococcus aureus and Staphylococcus epidermidis. FEMS Immunol. Med. Microbiol. 35 135–140. 10.1016/S0928-8244(02)00463-7 [DOI] [PubMed] [Google Scholar]
- Fredheim E. G., Granslo H. N., Flaegstad T., Figenschau Y., Rohde H., Sadovskaya I., et al. (2011). Staphylococcus epidermidis polysaccharide intercellular adhesin activates complement. FEMS Immunol. Med. Microbiol. 63 269–280. 10.1111/j.1574-695X.2011.00854.x [DOI] [PubMed] [Google Scholar]
- Gerke C., Kraft A., Sussmuth R., Schweitzer O., Gotz F. (1998). Characterization of the N-acetylglucosaminyltransferase activity involved in the biosynthesis of the Staphylococcus epidermidis polysaccharide intercellular adhesin. J. Biol. Chem. 273 18586–18593. 10.1074/jbc.273.29.18586 [DOI] [PubMed] [Google Scholar]
- Gill S. R., Fouts D. E., Archer G. L., Mongodin E. F., Deboy R. T., Ravel J., et al. (2005). Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187 2426–2438. 10.1128/JB.187.7.2426-2438.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. E., Diamond G. (2000). The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 8 402–410. 10.1016/S0966-842X(00)01823-0 [DOI] [PubMed] [Google Scholar]
- Hanke M. L., Angle A., Kielian T. (2012). MyD88-dependent signaling influences fibrosis and alternative macrophage activation during Staphylococcus aureus biofilm infection. PLoS One 7:e42476. 10.1371/journal.pone.0042476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke M. L., Heim C. E., Angle A., Sanderson S. D., Kielian T. (2013). Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J. Immunol. 190 2159–2168. 10.4049/jimmunol.1202348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartel C., Osthues I., Rupp J., Haase B., Roder K., Gopel W., et al. (2008). Characterisation of the host inflammatory response to Staphylococcus epidermidis in neonatal whole blood. Arch. Dis. Child. Fetal Neonatal Ed. 93 F140–F145. [DOI] [PubMed] [Google Scholar]
- Heilmann C., Schweitzer O., Gerke C., Vanittanakom N., Mack D., Gotz F. (1996). Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol. Microbiol. 20 1083–1091. 10.1111/j.1365-2958.1996.tb02548.x [DOI] [PubMed] [Google Scholar]
- Heinzelmann M., Herzig D. O., Swain B., Mercer-Jones M. A., Bergamini T. M., Polk H. C., Jr. (1997). Phagocytosis and oxidative-burst response of planktonic Staphylococcus epidermidis RP62A and its non-slime-producing variant in human neutrophils. Clin. Diagn. Lab. Immunol. 4 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoiby N., Bjarnsholt T., Givskov M., Molin S., Ciofu O. (2010). Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 35 322–332. 10.1016/j.ijantimicag.2009.12.011 [DOI] [PubMed] [Google Scholar]
- Hussain M., Herrmann M., Von Eiff C., Perdreau-Remington F., Peters G. (1997). A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect. Immun. 65 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson G. M., Lee D. A., Regelmann W. E., Gray E. D., Peters G., Quie P. G. (1986). Interference with granulocyte function by Staphylococcus epidermidis slime. Infect. Immun. 54 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo H. S., Otto M. (2015). Mechanisms of resistance to antimicrobial peptides in staphylococci. Biochim. Biophys. Acta 1848 3055–3061. 10.1016/j.bbamem.2015.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg C., Aarag E., Ronnestad A., Sollid J. E., Abrahamsen T. G., Kjeldsen G., et al. (2005). Coagulase-negative staphylococcal sepsis in neonates. Association between antibiotic resistance, biofilm formation and the host inflammatory response. Pediatr. Infect. Dis. J. 24 817–822. 10.1097/01.inf.0000176735.20008.cd [DOI] [PubMed] [Google Scholar]
- Kloos W., Schleifer K. H. (1986). “Staphylococcus,” in Bergey’s Manual of Systematic Bacteriology eds Sneath P. H. A., Mair N. S., Sharpe M. E., Holt J. G. (Baltimore, MD: Williams & Wilkins; ). [Google Scholar]
- Kretschmer D., Gleske A. K., Rautenberg M., Wang R., Koberle M., Bohn E., et al. (2010). Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 7 463–473. 10.1016/j.chom.2010.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristian S. A., Birkenstock T. A., Sauder U., Mack D., Gotz F., Landmann R. (2008). Biofilm formation induces C3a release and protects Staphylococcus epidermidis from IgG and complement deposition and from neutrophil-dependent killing. J. Infect. Dis. 197 1028–1035. 10.1086/528992 [DOI] [PubMed] [Google Scholar]
- Li M., Lai Y., Villaruz A. E., Cha D. J., Sturdevant D. E., Otto M. (2007). Gram-positive three-component antimicrobial peptide-sensing system. Proc. Natl. Acad. Sci. U.S.A. 104 9469–9474. 10.1073/pnas.0702159104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan J. L., Harrison O. J., Han S. J., Byrd A. L., Vujkovic-Cvijin I., Villarino A. V., et al. (2018). Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell 172 784–796. 10.1016/j.cell.2017.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy F. D. (1998). Staphylococcus aureus infections. N. Engl. J. Med. 339 520–532. 10.1056/NEJM199808203390806 [DOI] [PubMed] [Google Scholar]
- Mack D., Fischer W., Krokotsch A., Leopold K., Hartmann R., Egge H., et al. (1996). The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J. Bacteriol. 178 175–183. 10.1128/jb.178.1.175-183.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack D., Nedelmann M., Krokotsch A., Schwarzkopf A., Heesemann J., Laufs R. (1994). Characterization of transposon mutants of biofilm-producing Staphylococcus epidermidis impaired in the accumulative phase of biofilm production: genetic identification of a hexosamine-containing polysaccharide intercellular adhesin. Infect. Immun. 62 3244–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhusoodanan J., Seo K. S., Remortel B., Park J. Y., Hwang S. Y., Fox L. K., et al. (2011). An enterotoxin-bearing pathogenicity Island in Staphylococcus epidermidis. J. Bacteriol. 193 1854–1862. 10.1128/JB.00162-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki D. G., Kluger D. M., Crnich C. J. (2006). The risk of bloodstream infection in adults with different intravascular devices: a systematic review of 200 published prospective studies. Mayo Clin. Proc. 81 1159–1171. 10.4065/81.9.1159 [DOI] [PubMed] [Google Scholar]
- Mehlin C., Headley C. M., Klebanoff S. J. (1999). An inflammatory polypeptide complex from Staphylococcus epidermidis: isolation and characterization. J. Exp. Med. 189 907–918. 10.1084/jem.189.6.907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S., Bouladoux N., Linehan J. L., Han S. J., Harrison O. J., Wilhelm C., et al. (2015). Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 520 104–108. 10.1038/nature14052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef W. M. (2007). How human neutrophils kill and degrade microbes: an integrated view. Immunol. Rev. 219 88–102. 10.1111/j.1600-065X.2007.00550.x [DOI] [PubMed] [Google Scholar]
- Nguyen T. H., Park M. D., Otto M. (2017). Host response to Staphylococcus epidermidis colonization and infections. Front. Cell Infect. Microbiol. 7:90. 10.3389/fcimb.2017.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole G., Kaplan H. B., Kolter R. (2000). Biofilm formation as microbial development. Annu. Rev. Microbiol. 54 49–79. 10.1146/annurev.micro.54.1.49 [DOI] [PubMed] [Google Scholar]
- Otto M. (2006). Bacterial evasion of antimicrobial peptides by biofilm formation. Curr. Top. Microbiol. Immunol 306 251–258. 10.1007/3-540-29916-5_10 [DOI] [PubMed] [Google Scholar]
- Otto M. (2008). Staphylococcal biofilms. Curr. Top. Microbiol. Immunol. 322 207–228. 10.1007/978-3-540-75418-3_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. (2009). Staphylococcus epidermidis–the ‘accidental’ pathogen. Nat. Rev. Microbiol. 7 555–567. 10.1038/nrmicro2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. (2013). Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu. Rev. Med. 64 175–188. 10.1146/annurev-med-042711-140023 [DOI] [PubMed] [Google Scholar]
- Otto M., O’Mahoney D. S., Guina T., Klebanoff S. J. (2004). Activity of Staphylococcus epidermidis phenol-soluble modulin peptides expressed in Staphylococcus carnosus. J. Infect. Dis. 190 748–755. 10.1086/422157 [DOI] [PubMed] [Google Scholar]
- Patti J. M., Allen B. L., Mcgavin M. J., Hook M. (1994). MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48 585–617. 10.1146/annurev.mi.48.100194.003101 [DOI] [PubMed] [Google Scholar]
- Periasamy S., Joo H. S., Duong A. C., Bach T. H., Tan V. Y., Chatterjee S. S., et al. (2012). How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. U.S.A. 109 1281–1286. 10.1073/pnas.1115006109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Da F., Fisher E. L., Tan D. C., Nguyen T. H., Fu C. L., et al. (2017). Toxin mediates sepsis caused by methicillin-resistant Staphylococcus epidermidis. PLoS Pathog. 13:e1006153. 10.1371/journal.ppat.1006153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queck S. Y., Khan B. A., Wang R., Bach T. H., Kretschmer D., Chen L., et al. (2009). Mobile genetic element-encoded cytolysin connects virulence to methicillin resistance in MRSA. PLoS Pathog. 5:e1000533. 10.1371/journal.ppat.1000533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riber U., Espersen F., Kharazmi A. (1995). Comparison of adherent and non-adherent staphylococci in the induction of polymorphonuclear leukocyte activation in vitro. APMIS 103 439–446. 10.1111/j.1699-0463.1995.tb01130.x [DOI] [PubMed] [Google Scholar]
- Rigby K. M., DeLeo F. R. (2012). Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol. 34 237–259. 10.1007/s00281-011-0295-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers K. L., Fey P. D., Rupp M. E. (2009). Coagulase-negative staphylococcal infections. Infect. Dis. Clin. North Am. 23 73–98. 10.1016/j.idc.2008.10.001 [DOI] [PubMed] [Google Scholar]
- Rohde H., Burandt E. C., Siemssen N., Frommelt L., Burdelski C., Wurster S., et al. (2007). Polysaccharide intercellular adhesin or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials 28 1711–1720. 10.1016/j.biomaterials.2006.11.046 [DOI] [PubMed] [Google Scholar]
- Rohde H., Burdelski C., Bartscht K., Hussain M., Buck F., Horstkotte M. A., et al. (2005). Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol. Microbiol. 55 1883–1895. 10.1111/j.1365-2958.2005.04515.x [DOI] [PubMed] [Google Scholar]
- Rupp M. E., Ulphani J. S., Fey P. D., Bartscht K., Mack D. (1999a). Characterization of the importance of polysaccharide intercellular adhesin/hemagglutinin of Staphylococcus epidermidis in the pathogenesis of biomaterial-based infection in a mouse foreign body infection model. Infect. Immun. 67 2627–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp M. E., Ulphani J. S., Fey P. D., Mack D. (1999b). Characterization of Staphylococcus epidermidis polysaccharide intercellular adhesin/hemagglutinin in the pathogenesis of intravascular catheter-associated infection in a rat model. Infect. Immun. 67 2656–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schommer N. N., Christner M., Hentschke M., Ruckdeschel K., Aepfelbacher M., Rohde H. (2011). Staphylococcus epidermidis uses distinct mechanisms of biofilm formation to interfere with phagocytosis and activation of mouse macrophage-like cells 774A.1. Infect. Immun. 79 2267–2276. 10.1128/IAI.01142-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher-Perdreau F., Heilmann C., Peters G., Gotz F., Pulverer G. (1994). Comparative analysis of a biofilm-forming Staphylococcus epidermidis strain and its adhesion-positive, accumulation-negative mutant M7. FEMS Microbiol. Lett. 117 71–78. 10.1111/j.1574-6968.1994.tb06744.x [DOI] [PubMed] [Google Scholar]
- Spiliopoulou A. I., Kolonitsiou F., Krevvata M. I., Leontsinidis M., Wilkinson T. S., Mack D., et al. (2012). Bacterial adhesion, intracellular survival and cytokine induction upon stimulation of mononuclear cells with planktonic or biofilm phase Staphylococcus epidermidis. FEMS Microbiol. Lett. 330 56–65. 10.1111/j.1574-6968.2012.02533.x [DOI] [PubMed] [Google Scholar]
- Strunk T., Power Coombs M. R., Currie A. J., Richmond P., Golenbock D. T., Stoler-Barak L., et al. (2010). TLR2 mediates recognition of live Staphylococcus epidermidis and clearance of bacteremia. PLoS One 5:e10111. 10.1371/journal.pone.0010111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurlow L. R., Hanke M. L., Fritz T., Angle A., Aldrich A., Williams S. H., et al. (2011). Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 186 6585–6596. 10.4049/jimmunol.1002794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uckay I., Pittet D., Vaudaux P., Sax H., Lew D., Waldvogel F. (2009). Foreign body infections due to Staphylococcus epidermidis. Ann. Med. 41 109–119. 10.1080/07853890802337045 [DOI] [PubMed] [Google Scholar]
- Vuong C., Kocianova S., Voyich J. M., Yao Y., Fischer E. R., DeLeo F. R., et al. (2004a). A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J. Biol. Chem. 279 54881–54886. [DOI] [PubMed] [Google Scholar]
- Vuong C., Voyich J. M., Fischer E. R., Braughton K. R., Whitney A. R., DeLeo F. R., et al. (2004b). Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 6 269–275. [DOI] [PubMed] [Google Scholar]
- Vuong C., Otto M. (2002). Staphylococcus epidermidis infections. Microbes Infect. 4 481–489. 10.1016/S1286-4579(02)01563-0 [DOI] [PubMed] [Google Scholar]
- Wang A., Athan E., Pappas P. A., Fowler V. G., Jr., Olaison L., Paré C., et al. (2007). Contemporary clinical profile and outcome of prosthetic valve endocarditis. JAMA 297 1354–1361. 10.1001/jama.297.12.1354 [DOI] [PubMed] [Google Scholar]
- Wang R., Khan B. A., Cheung G. Y., Bach T. H., Jameson-Lee M., Kong K. F., et al. (2011). Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice. J. Clin. Invest. 121 238–248. 10.1172/JCI42520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y., Sturdevant D. E., Otto M. (2005). Genomewide analysis of gene expression in Staphylococcus epidermidis biofilms: insights into the pathophysiology of S. epidermidis biofilms and the role of phenol-soluble modulins in formation of biofilms. J. Infect. Dis. 191 289–298. 10.1086/426945 [DOI] [PubMed] [Google Scholar]
- Zhang Y. Q., Ren S. X., Li H. L., Wang Y. X., Fu G., Yang J., et al. (2003). Genome-based analysis of virulence genes in a non-biofilm-forming Staphylococcus epidermidis strain (ATCC 12228). Mol. Microbiol. 49 1577–1593. 10.1046/j.1365-2958.2003.03671.x [DOI] [PubMed] [Google Scholar]