

Abstract
Small cell lung cancer (SCLC) is an aggressive malignancy that accounts for 14% of all lung cancer diagnoses. Despite decades of active research, treatment options for SCLC are limited and resistance to the few Food and Drug Administration (FDA) approved therapies develops rapidly. With no approved targeted agents to date, new therapeutic strategies are desperately needed. SCLC is characterized by high mutation burden, ubiquitous loss of TP53 and RB1, mutually exclusive amplification of MYC family members, thereby, high genomic instability. Studies in the past few years have demonstrated the potential of targeting the DNA damage response (DDR) pathway as a promising therapeutic strategy for SCLC. Inhibitors targeting DDR proteins have shown promise in preclinical models, and are under clinical investigation as single agents and in combination with cytotoxic therapies. Recent efforts to expand the therapeutic arsenal toward SCLC have focused in part on immune checkpoint inhibitors, such as agents targeting the receptor-ligand pair programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1). Clinical trials have confirmed activity of these agents in extensive stage (ES)-SCLC. However, while several patients had dramatic responses, overall response rates to immune checkpoint blockade (ICB) remain poor. As a result, there is an urgent need to develop rational combination therapies to enhance response rates to immunotherapy in SCLC. Identification of predictive biomarkers for patient stratification, identifying effective combinations to overcome adaptive resistance to DDR-targeted therapies and identifying strategies to enhance response to immunotherapy are areas of active investigation in SCLC.
Keywords: DNA damage response (DDR), biomarkers, immunotherapy, small cell lung cancer (SCLC)
The “recalcitrant cancer”
With an estimated 250,000 cases worldwide annually, small cell lung cancer (SCLC) is the sixth major cause of cancer-related mortality (1-7). SCLC is the most aggressive form of lung cancer, characterized by rapid tumor growth and early metastatic spread (8,9). Currently, chemotherapy remains the cornerstone of treatment for both limited stage (LS) and extensive stage (ES)-SCLC, with a majority of patients obtaining an objective response with first-line treatment, as described by Farago and Keane (current standards for clinical management of SCLC) also in this issue. However, despite high rates of response initially, the duration of clinical response is extremely limited in most cases and almost all patients relapse—often within a few months (5,6,8,9). Recently, clinical trials have demonstrated that some SCLC patients can obtain durable responses to immunotherapy and this has led to the addition of nivolumab with or without ipilimumab to the NCCN treatment guidelines as the first non-chemotherapy option for relapsed SCLC (10,11). However, beyond this there are currently no targeted agents for the treatment of SCLC approved by the United States Food and Drug Administration (FDA).
In 2012, SCLC was recognized by US Congress as one of two “recalcitrant cancers” (the second being pancreatic cancer) based on its 5-year survival rate of <7%. The Recalcitrant Cancer Research Act led to new funding mechanisms and a renewed focus on translational research in SCLC intended to speed the pace of translational research and discovery for this disease (12). In the past several years, investigators have identified many new, highly promising drug targets through translational investigations of preclinical models and patient tissues (5,6). Among these are several drugs targeting the proteins involved in DNA damage response (DDR) such as poly-ADP-ribose polymerase (PARP), checkpoint kinase 1 (CHK1), ataxia telangiectasia and Rad3-related protein (ATR), and WEE1 (13). The finding that DDR targets are more highly expressed in SCLC (14)—together with data demonstrating preclinical activity of DDR inhibitors in SCLC models (14-19)—has had immediate translational implications, as several DDR inhibitors have been developed and are either approved for other cancer types (e.g., PARP inhibitors) or are in clinical trials (20). Only a small number of DDR inhibitor trials have included SCLC patients to date (Table 1). However, the available data suggest promising activity in a subset of SCLC patients. Moreover, candidate biomarkers are emerging that may aid in identifying subsets of SCLC with greatest vulnerability to specific DDR inhibitors.
Table 1. DDR-targeted therapy clinical trials in SCLC (list includes ongoing trials as of 12/2017).
| Trial | Treatment | Indication |
|---|---|---|
| PARP inhibitor trials in SCLC | ||
| NCT03227016 | Phase 1: veliparib alone; phase 2: veliparib + topotecan | Relapsed/refractory ES-SCLC |
| NCT02734004 | Phase 1/2: MEDI4736 (anti-PD-L1) in combination with olaparib | Advanced solid tumors including ES-SCLC cohort |
| NCT02289690 | Phase 2: carboplatin/etoposide +/− veliparib | Treatment-naïve ES-SCLC |
| NCT02769962 | Phase 1/2: CRLX101 (camptothecin nanoparticle) + olaparib | Relapsed/refractory ES-SCLC |
| NCT01642251 | Phase 1/2: cisplatin/etoposide +/− veliparib | Treatment-naïve ES-SCLC |
| NCT02498613 | Phase 2: olaparib + cediranib (anti-VEGFR TKI) | Advanced solid tumors including ES-SCLC cohort |
| NCT02446704 | Phase 1/2: olaparib + TMZ | Relapsed/refractory ES-SCLC |
| NCT03009682 | Phase 2: olaparib monotherapy | Relapsed/refractory ES-SCLC harboring HR mutations |
| NCT02511795 | Phase 1b: olaparib + AZD1775 (WEE1 inhibitor) | Advanced solid tumors including ES-SCLC cohort |
| ATR inhibitor trials including SCLC | ||
| NCT02487095 | Phase 1/2: topotecan + VX970 | Advanced small cell cancers |
| NCT02589522 | Phase 1: VX-970 + WBRT | Brain metastases from tumors |
| NCT02223923 | Phase 1: AZD6738 +/− RT | Advanced solid tumors |
| NCT02723864 | Phase 1: veliparib + VX-970 + cisplatin | Advanced solid tumors |
| NCT02595931 | Phase 1: VX-970 + irinotecan | Advanced solid tumors |
| NCT02157792 | Phase 1: VX-970 + chemotherapy | Advanced solid tumors |
| NCT03188965 | Phase 1: BAY1895344 monotherapy | Advanced solid tumors |
| CHK inhibitor trials including SCLC | ||
| NCT02735980 | Phase 2: prexasertib monotherapy | Relapsed/refractory ES-SCLC |
| NCT02797964 | Phase 1: SRA737 monotherapy | Advanced solid tumors |
| NCT02797977 | Phase 1: SRA737 + gemcitabine +/− cisplatin | Advanced solid tumors |
| NCT02873975 | Phase 2: prexasertib monotherapy | Advanced solid tumors with HR deficiency or replicative stress |
| NCT03057145 | Phase 1: prexasertib + olaparib | Advanced solid tumors |
| Wee1 inhibitor trials including SCLC | ||
| NCT02482311 | Phase 1: AZD1775 monotherapy | Advanced solid tumors including ES-SCLC cohort |
| NCT02511795 | Phase 1b: AZD1775 + olaparib | Advanced solid tumors including ES-SCLC cohort |
| NCT02593019 | Phase 2: AZD1775 monotherapy | Relapsed/refractory ES-SCLC |
| NCT02688907 | Phase 2: AZD1775 monotherapy | Relapsed/refractory ES-SCLC with MYC amplifications or CDKN2A + TP53 mutations |
| NCT02937818 | Phase 2: AZD1775 + carboplatin (arm) | Relapsed/refractory ES-SCLC |
| NCT01748825 | Phase 1: AZD1775 monotherapy | Advanced solid tumors |
| AURKA or AURKB inhibitor trials including SCLC | ||
| NCT03216343 | Phase 1: chiauranib | Relapsed/refractory ES-SCLC |
| NCT03092934 | Phase 1/2: AK-01 | Advanced solid tumors including ES-SCLC cohort |
| NCT02719691 | Phase 1b: MLN0128 (TORC1/2 inhibitor) + alisertib | Advanced solid tumors |
| NCT02134067 | Phase 1: TAS-119 + paclitaxel | Advanced solid tumors |
| NCT01118611 | Phase 1: GSK1070916A | Advanced solid tumors |
SCLC, small cell lung cancer; ES-SCLC, extensive stage-SCLC; DDR, DNA damage response; PARP, poly-ADP-ribose polymerase; PD-L1, programmed death-ligand 1; CEGFR, vascular endothelial growth factor; TKI, tyrosine kinase inhibitor; TMZ, temozolomide; WBRT, whole brain radiation therapy; RT, radiation therapy; AURKA/B, Aurora A/B.
In this review, we will discuss aspects of SCLC biology that may make it especially susceptible to DDR targeting approaches, preclinical and clinical data for several DDR inhibitors, and candidate biomarkers that may help identify patients who would benefit from DDR targeting. In recent years cancer treatment has been revolutionized with the clinical development of antibodies targeting immune checkpoints (21,22). In addition, there is active research to understand what drives immunogenicity in a cancer and the role of DDR pathways in this context has recently been in focus. Here, we will also discuss the mechanisms by which targeting the DDR pathways could regulate the antitumor immune response to immune checkpoint blockade (ICB) and describe clinical efforts to combine DDR targeted therapies and ICB in SCLC.
Genomic instability—a hallmark of SCLC
Cigarette smoking contributes significantly to the accumulation of DNA damage and is a major cause of SCLC (>95% of patients): in particular, the risk of developing SCLC increases with the number of cigarettes smoked each day and with the duration of smoking (23,24). A previous study exploring the mutational burden associated with tobacco smoking in SCLC performed deep sequencing on SCLC cell line, NCI-H209 (25) and identified a total of 22,910 somatic substitutions, including 134 in coding exons. Interestingly, even in this single cell line profiling study, they identified several distinctive point mutation patterns that reflected the effect of carcinogens present in cigarette smoke (25). They also observed signatures of the partially successful attempts of the cells’ surveillance machinery to repair DNA damage (25). Unsurprisingly, SCLC carries a high mutation burden and genomic instability (26).
On top of the mutational burden induced by tobacco exposure, almost all SCLC tumors have a loss of TP53 and RB1 and amplification of the MYC family of oncogenes (27,28). Defective TP53 and RB1, along with aberrant activation of the oncogene MYC, results in rapid proliferation and, consequently, replication stress in SCLC. In this background, SCLC cells become dependent on intact DDR pathways for survival. Any further perturbation to the DDR or cell cycle checkpoint regulation can potentially lead to mitotic catastrophe and cell death.
Earlier studies have reported the vulnerability of TP53 defective cancer cells to DDR inhibitors (29,30). In the last few years data from proteomic and genomic studies of SCLC have also highlighted changes in the DDR pathways in SCLC. Proteomic analysis of 34 SCLC and 74 non-small cell lung cancer (NSCLC) cell lines demonstrated that SCLC had significantly higher levels of E2F1-regulated factors including DNA repair proteins such as PARP, ATR, CHK1/2, DNA-dependent protein kinase (DNA-PK), and ataxia telangiectasia-mutated (ATM) (14). Subsequent studies confirmed the overexpression of ATR, CHEK1 and WEE1 in SCLC patient tumors as compared to normal lung (17-19). A recent study by Doerr et al. further demonstrated by transcriptomic analysis of different subtypes of lung cancer that SCLC has a significantly higher expression of genes involved in the DDR (such as CHEK1, CDC25A, B and C) than other lung cancer subtypes (31). These new insights into the biology of SCLC represent an intriguing possibility of targeting DDR proteins (13,32) (Figure 1).
Figure 1.

Targeting DDR vulnerabilities of SCLC. Ubiquitous loss of TP53 and RB1 disrupts the G1-S cell cycle checkpoint SCLC making the cells dependent on G2-M cell cycle checkpoint for arrest upon DNA damage. MYC family amplifications and increased gene and protein expression of multiple DDR pathway molecules (indicated in green) suggest they may be effective targets in SCLC. DDR, DNA damage response; SCLC, small cell lung cancer; PARP, poly-ADP-ribose polymerase; ATM, ataxia telangiectasia-mutated; ATR, ataxia telangiectasia and Rad3-related protein.
Targeting DDR—an “Achilles’ heel” of SCLC?
Recent studies have demonstrated the therapeutic opportunities of targeting DDR pathways in highly aggressive cancers, including SCLC (14,15,17,18,33-37). The DDR network is a complex and dynamic system with almost 450 integral proteins. Five major DNA repair pathways are known: base excision repair (BER) to repair single-strand breaks (SSBs); homologous recombination repair (HRR) and non-homologous end-joining (NHEJ) to repair double-strand breaks (DSBs); mismatch repair (MMR) to repair replication errors, and nucleotide excision repair (NER) to repair bulky adducts (13,34-37). The different DDR pathways have built in redundancy to a certain extent that allows many of them to serve as a compensatory mechanism when other DDR pathways are compromised.
The transformation of a normal cell to a cancerous one requires, amongst other features, the acquisition of the ability of uncontrolled proliferation and failure of efficient and accurate DNA repair (38,39). The loss of robust DNA damage repair has important implications in tumor growth, cancer progression and therapeutic responses. Some common features of highly aggressive human malignancies include high replication stress, aberrant cell cycle control, and functional defects in one or more of the DNA repair pathways. The cellular stress is further enhanced with frequent overexpression and activation of oncogenes inducing proliferation such as MYC or CCNE1 or loss of tumor suppressor genes such as TP53 that further disrupt the balance of replication, transcription and cell cycle checkpoint control (40). As mentioned above, the mutational landscape of SCLC is complex and varied, but includes the ubiquitous functional inactivation of both TP53 and RB1 (26-28). Other notable abnormalities of SCLC are the frequent amplifications of the oncogenic transcription factors MYC and SOX2 in 27% of cases (26-28). Several studies have demonstrated the role of MYC as a vital regulator of cell growth and proliferation by enhancing the expression of genes controlling cell cycle (e.g., cyclins, CDKs, dNTP biosynthetic enzymes and replication factors), and repressing anti-proliferative genes (e.g., CDK inhibitors) (41,42). Also, MYC physically interacts with the pre-replicative complex and co-localizes with replication foci in early S phase regardless of transcription and hence drives cell cycle progression by directly controlling replication initiation (40,43). Hence, MYC overexpression and/or amplification affects replication initiation and potentially leads to premature origin firing and subsequent replication stress.
DNA-damaging chemotherapy in SCLC
By removing many of the mechanisms through which cells would normally halt the cell cycle to respond to errors in DNA replication or extrinsic DNA damage, cancer cells can proliferate at much higher rates. However, this characteristic also makes highly proliferative cancers more susceptible to the DNA damage induced by chemotherapy or radiation. This explains the initial high response rates seen in SCLC with chemotherapies that broadly target the process of cell division or DNA replication, including platinum agents (e.g., cisplatin, carboplatin) and topoisomerase I/II inhibitors (e.g., etoposide, irinotecan) (44,45).
Platinum-chemotherapy
Cisplatin and the cisplatin analog carboplatin have defined the backbone of SCLC chemotherapy for several decades (44,45). Platinum compounds bind to reactive centers on purine residues within DNA and generate cisplatin-purine cross-links to induce DNA damage, block cell division and, ultimately, promote apoptosis (46). Numerous studies have identified deficient DDR as a potential marker of platinum sensitivity and the upregulation of DDR machinery as a potential mechanism of platinum resistance (46).
Type II topoisomerase (Topo II) inhibitors
Etoposide is widely used in combination with platinum-chemotherapy in first-line treatment for SCLC (44,45). Etoposide targets Topo II, an enzyme class that plays a critical role during DNA replication (47). Specifically, Topo II cleaves double-stranded DNA to permit passage of intact helical DNA before ligating the cleavage site. Etoposide prevents this ligation event by stabilizing the complex formed by Topo II and the 5' cleaved ends of the DNA, resulting in stable, protein-linked double strand breaks in DNA and subsequent apoptosis (47).
Type I topoisomerase (Topo I) inhibitors
The Topo I inhibitors topotecan and irinotecan are commonly used in relapsed/refractory SCLC following resistance to platinum and Topo II targeting therapies (44,45). However, irinotecan is often also used (outside the United States) in the frontline setting in combination with platinum therapy. Topotecan is currently the only FDA-approved therapy for relapsed/refractory SCLC. The Topo I enzyme class is responsible for creating single strand breaks in DNA to relieve torsional strain created by twisting and supercoiling. Binding of topotecan or irinotecan to the Topo I-DNA complex prevents repair of these single strand nicks and, ultimately, to unrepaired double strand breaks and apoptosis (48).
DDR-targeted therapies in SCLC
In the last few years, targeted agents for DDR pathways have opened exciting new avenues for research and treatment in several cancers including SCLC. As mentioned above, DDR proteins were seen to be overexpressed in SCLC in vitro. Follow-on studies confirmed the higher gene expression of DDR pathway mediators such as PARP, CHEK1, ATM and ATR in SCLC patient tumors as compared to normal lung and/or NSCLC tumors (14,17-19,31,49). In addition, a high-throughput small molecule drug screen also identified DDR proteins, including CHK1 as a candidate therapeutic target in SCLC (50). Since then, several DNA repair inhibitors have been evaluated in SCLC preclinical models and clinical trials. Although DDR inhibitors have shown activity as monotherapy in SCLC models and in some patients (e.g., the PARP inhibitor talazoparib) (20), combinations with cytotoxic chemotherapies, other targeted agents (e.g., DDR inhibitor combinations), or possibly with immunotherapy are predicted to provide greater clinical response by increasing the percentage of patients likely to respond and potentially the duration of response.
Targeting PARP in SCLC treatment
PARPs are a family of nuclear protein enzymes involved in the DDR. Previous reports have extensively characterized the key role of PARP in DDR and repair of SSB through BER (51-54). More recently PARP’s role in homologous recombination (HR) and NHEJ was identified. PARP1 regulates the restart of replication forks after DNA damage repair by the attachment of ADP-ribose units to multiple proteins.
PARP inhibitors exert their effect by two main mechanisms first by trapping the enzyme to the SSBs by preventing the utilization of NAD+ (55); second, PARP1 inhibitors inhibit PARylation and therefore binding of PARP to DNA (55). The resulting PARP-DNA complexes lead to collapsing and stalling of replication forks and ultimately to the conversion of SSBs to DSBs leading to apoptosis (51-55). In cells with intact HR pathway, loss of PARP is not lethal. However, PARP targeting is highly effective in the context of synthetic lethality in models with existing deficiency in DNA repair, such as deleterious mutations in BRCA1/2. BRCA1 and 2 deficient cells are rendered defective in the ability to repair through HR and depend on error-prone NHEJ for DNA repair (56-60).
To date, three PARP inhibitors olaparib (AZD-2281/KU-0059436), niraparib (MK-4827) and rucaparib (AG-014699/PF-01367338) have been approved by the US FDA for the treatment of biomarker defined subsets of ovarian cancer (61). In SCLC, however, PARP inhibition is independent of BRCA1 status and likely depends more on intrinsically high levels of replication stress and genomic instability (14). In addition to the FDA-approved agents, PARP inhibitors that are currently undergoing active clinical investigation in SCLC also include CEP-9722, E7016/GPI-21016, talazoparib (BMN-673), and veliparib (ABT-888).
Although most PARP inhibitors are comparable in their enzymatic inhibition of PARP activity, they vary by the degree to which they induce direct cytotoxicity through PARP trapping. Among PARP inhibitors that are approved or in advanced stages of clinical trial testing, the lowest amount of trapping is seen with veliparib (making it more tolerable in combination with chemotherapy); moderate trapping with drugs such as olaparib, rucaparib, and niraparib; and the highest degree of trapping with talazoparib (formerly BMN-673) (55). Based on preclinical results described below, several clinical trials have been initiated to test the activity of these drugs in SCLC patients. Three main therapeutic approaches have been tested in SCLC patients to date: (I) single agent talazoparib in platinum-sensitive relapsed SCLC; (II) the combination of temozolomide (TMZ) with veliparib or olaparib in relapsed SCLC (NCT01638546, NCT02446704); and (III) the addition of veliparib to front-line platinum-etoposide therapy. None of these trials required biomarker testing as part of the eligibility criteria, although some exploratory biomarker analyses have been performed retrospectively. Available results from these clinical studies are outlined below.
Single agent activity of PARP inhibitors in SCLC preclinical models
In previous studies, we demonstrated single agent activity of PARP inhibitors, olaparib and rucaparib, in SCLC preclinical models (14). A subsequent investigation further demonstrated the single agent potential of another PARP inhibitor, talazoparib, in SCLC preclinical models (15). In the first PARP inhibitor trial to include SCLC patients, single agent talazoparib was tested in an expansion cohort of platinum-sensitive patients following relapse as part of the first-in-man phase 1 trial (20). Talazoparib is a highly potent PARP inhibitor that works through two mechanisms—inhibition of PARP enzymatic activity and direct cytotoxicity induced by trapping PARP-DNA complexes at the sites of single-strand DNA breaks (so-called “PARP trapping”). High response rates were observed in BRCA-mutated breast and ovarian cancer patients treated on this phase 1 study and clinical benefit from this drug in BRCA-mutated breast cancer patients was recently confirmed in a phase 3 trial (62). Patients in the SCLC cohort (n=23) were treated with 1.0 mg daily by mouth [the established maximum tolerated dose (MTD)]. Two patients (9%) had partial responses (PRs) (duration 12.0 and 15.3 weeks, respectively) and four patients (18%) had stable disease (SD) (lasting >16 weeks). Median progression free survival (PFS) for the overall SCLC cohort was 11.1 weeks (20). This is the only clinical trial to date of single agent PARPi (or of talazoparib) for relapsed SCLC patients.
Strategies of combining PARP inhibitors with chemotherapy in SCLC preclinical models
Several preclinical studies have also demonstrated the effect of PARP inhibitors alone or in combination regimens with standard chemotherapy in SCLC models. For example, we found that olaparib in combination with cisplatin and etoposide in multiple SCLC cell lines caused approximately 80% more cell killing than in control cells and the cell count was significantly lower than the cell count after treatment with olaparib alone (P<0.05 for both) (14). Furthermore, treatment of H69 cells with olaparib in combination with irinotecan, another chemotherapeutic commonly used in the treatment of SCLCs, also resulted in a greater decrease in tumor cell viability than either agent alone (P=0.03 olaparib/irinotecan vs. control; P=0.007 olaparib/irinotecan vs. irinotecan alone) (14). In a study investigating the efficacy of PARP inhibitor, veliparib, as a single agent and in combination with cisplatin, carboplatin, and etoposide, Owonikoko et al., demonstrated that veliparib potentiates standard cytotoxic agents and the effect is correlated with platinum sensitivity, DNA-PKcs expression and a 5-gene (GLS, UBEC2, HACL1, MSI2 and LOC100129585) expression profile in SCLC cell lines (63). In another study, Lok et al., demonstrated that PARP inhibition by talazoparib is synergistic in combination with DNA alkylator, TMZ in SCLC cell lines and patient-derived xenografts (PDX) models (64).
Based on phase 2 data showing activity of TMZ in relapsed SCLC (65) and preclinical data demonstrating activity of TMZ in combination with PARP inhibitors (64), two clinical trials have tested this combination in SCLC. In the first study, 100 patients with 1–2 prior lines of therapy were treated with TMZ with either veliparib or placebo (TMZ 150–200 mg/m2/day on days 1–5 and veliparib/placebo 40 mg po twice daily on days 1–7) (66). The patient population included 59% platinum-refractory patients and 33% 3rd line patients, which is notable since these are features known to be associated with lower response rates and shorter survival. Although the primary endpoint of improved 4-month PFS was not met in this trial, patients receiving the combination of TMZ/veliparib had an almost 3-fold higher response rate as compared to the TMZ/placebo arm (39% vs. 19%). Median overall survival (OS) was 8.2 months in TMZ/veliparib arm and 7.0 months in the TMZ/placebo arm (P=0.50). However, a significantly longer PFS and OS were observed in patients receiving TMZ/veliparib combination who had detectable schlafen 11 (SLFN11) by immunohistochemistry. The putative DNA/RNA helicase SLFN11 has been shown by others and us to be a candidate predictive marker of chemotherapy and PARP inhibitor sensitivity (49,64,67,68). A second TMZ-PARP inhibitor trial using olaparib is now ongoing. Data presented from the first 13 patients, though early, showed promising activity with a response rate of 48% and median PFS of 5.6 months (69).
In the frontline setting, the PARP inhibitor veliparib has been explored in combination with cisplatin-etoposide (ECOG-ACRIN 2511; NCT01642251) and with carboplatin-etoposide (NCT02289690). In the first trial, 128 eligible patients with ES-SCLC were randomized to four cycles of cisplatin plus etoposide (75 and 100 mg/m2, respectively) with either veliparib (100 mg po twice daily on days 1–7) or placebo. No maintenance veliparib was given in this trial. As of December 2016 (median follow up 18.5 months), the primary endpoint of PFS was 6.1 months in the cisplatin-etoposide plus veliparib arm versus 5.5 months in the cisplatin-etoposide-placebo arm (P=0.06), with an adjusted one-sided PFS HR of 0.63 (P=0.01). A trend toward improved OS was also observed in patients receiving chemotherapy in combination with veliparib, although this did not reach statistical significance (OS 10.3 vs. 8.9 months, P=0.17). Similarly, response rates were numerically higher in the veliparib arm (72% vs. 64%), but not significantly different (P=0.57). The most frequent adverse events in both groups were neutropenia (grade 3/4: 49% in veliparib arm; 32% placebo arm); leukopenia (grade 3/4: 19% in veliparib arm; 14% placebo arm); and anemia (grade 3/4: 19% in veliparib arm; 12% placebo arm). A randomized phase 1/2 study of carboplatin-etoposide with or without veliparib which incorporates a higher dose of veliparib with chemotherapy (recommended phase 2 dose 240 mg po BID days −2 to 12) as well as veliparib in the maintenance setting is ongoing (NCT2289690) (70). The finding from the cisplatin-etoposide +/− veliparib trial suggest that a biomarker selection strategy (such as SLFN11 or other candidate markers) will be needed to identify those patients likely to receive the greatest benefit from the addition of PARP inhibition to standard chemotherapy. In addition to these studies, there are now nine trials of PARP inhibitors at various stages for SCLC, including trials testing for the first time the combination of the PARP inhibitor olaparib with immunotherapy (MEDIOLA trial, NCT02734004) (Table 1).
Combining inhibition of PI3K and PARP in SCLC
Several studies have shown that combined inhibition of PARP and PI3K is a potentially effective treatment regimen for breast cancers with elevated activity of the PI3K pathway (71,72). Activation of the PI3K pathway is a common feature of many tumor types. PI3K pathway activation leads to the stimulation of cell growth, motility, survival, and metabolism, and also sensing of DDR (73,74). In a subsequent study, Cardnell et al., further showed that the proteins most significantly upregulated following treatment with the PARP inhibitors olaparib and rucaparib belonged to the PI3K/mTOR pathway (p-mTOR, p-AKT, and pS6) (P=0.02), and amongst the most significantly down-regulated proteins were LKB1 and its targets AMPK and TSC, which negatively regulate the PI3K pathway (P=0.042) (16). The study concluded that the observed increase in PI3K/mTOR pathway activation following PARP inhibition results from decreased ATP usage and a subsequent decrease in stress response signaling via LKB1. The data further suggested that combining PARP and PI3K inhibitors enhances the effect of either agent alone in in vitro and in vivo SCLC models (16). Although there is a strong preclinical rationale for combining inhibitors of PARP and PI3K, the therapeutic possibilities for this combination are less clear. The main concerns would be that of toxicity, patient selection and finally the resistance to targeted therapies as Juvekar et al.’s identification of a drug resistant “pushing margin” that continues to grow during treatment with a PI3K inhibitor (72). However, this combination certainly warrants consideration for clinical trial and may pave the way for wider use of PARP inhibitors in SCLC patients.
Targeting ATR in SCLC
Another DDR protein implicated in SCLC is ATR. The ATR-CHK1 axis is part of a complex signaling network that is activated upon genotoxic stress and DNA damage. In the presence of extended stretches of ssDNA that are coated with replication protein A (RPA), ATR gets recruited by interactions with ATRIP. Once activated, ATR phosphorylates and activates multiple targets including CHK1. Upon stimulation, ATR-CHK1 axis enforces halting of cell cycle progression at the G2-M phase until damage is repaired (75-78). Preclinical observations suggest that ATR inhibitors may be especially active in TP53 and/or ATM deficient models from other tumor types (79). Beyond its role in regulating p53-mediated apoptosis, ATM is also reported to be involved in DNA DSB repair, particularly through the HR pathway, with a less well-defined role in the NHEJ pathway. Hence, in ATM deficient tumors (up to ~8% of SCLC), DSB repair is dependent primarily on the ATR/CHK1 axis (19,78). Recent reports find activity of ATR inhibitors in in vitro and in vivo models of SCLC (31). Clinically, results from a phase 1, first-in-class trial with the ATR inhibitor VX-970 included a complete response (CR) in a colorectal cancer patient with ATM loss and a durable PR in a BRCA1 mutant ovarian cancer, further supporting a role for ATR inhibitors in DDR-deficient tumors. Several ATR inhibitors are now in clinical trial alone or in combination with chemotherapy, radiation and PARP inhibitors, including VX-970 and VX-803 (EMD Serono), AZD6738 (AstraZeneca) and BAY-1895344 (Bayer). Notably, several of these trials include SCLC patients (NCT02487095; NCT02589522), while others enroll across solid tumors. The clinical outcomes and biomarker analyses of these studies will guide future investigations into ATR inhibitors for SCLC.
Targeting CHK1 in SCLC
The combined loss of TP53 and RB1 in SCLC leads to defective cell cycle arrest at the G1/S phase and hence an enhanced dependency on the G2/M phase for repair upon DNA damage. We previously showed that SCLC cell lines have a median of 4.5-fold higher (range, 2–7 folds) CHK1 protein expression than NSCLC cell lines (P=0.001) (18). This was further validated in SCLC tumors which were shown to have 4.2-fold higher CHK1 expression at the mRNA levels (P<0.0001) vs. normal lung. Based on this, we tested the activity of a second generation CHK1 inhibitor, prexasertib (80), as a single agent or combined with chemotherapy in both platinum sensitive and resistant SCLC models. In a comprehensive panel of 39 human and murine SCLC cell lines, prexasertib showed remarkable single agent activity with half-maximal inhibitory concentration (IC50) of prexasertib <100 nM in 50% human SCLC cell lines tested, as well as activity in SCLC syngeneic, genetically-engineered mouse (GEM) and chemoresistant models. In addition, prexasertib potentiated the effect of cisplatin in chemoresistant and chemosensitive models, a finding that has potential clinical relevance. CHK1 inhibition also reversed PARP inhibitor resistance in preclinical models (18). Similar findings were reported recently in an independent study by Doerr et al., where transcriptome analysis showed a significant overexpression of genes involved in the DDR, specifically CHEK1, in SCLC compared to lung adenocarcinoma. They further showed that the mRNAs encoding the phosphatases CDC25A, B and C (substrates of checkpoint kinases) are expressed at significantly higher levels in SCLC, compared to lung adenocarcinomas (ADC) and squamous cell carcinomas (31). Following these observations, they demonstrated that CHK1 inhibitor, PF-477736, induced genotoxic damage and apoptosis in human and murine SCLC cell lines, but not in lung adenocarcinoma in vitro and in vivo models (31).
Together, these findings provide a strong rationale for targeting CHK1 and other G2/M checkpoint proteins such as WEE1. Previous phase I clinical trial data with the CHK1 inhibitor MK-8776 (Merck) showed PRs in only 2/30 evaluable patients, but SD in an additional 13/30. However, this was a solid tumor population that did not select for DDR deficiencies and included no SCLC patients. Additional data from a phase I trial of the CHK1 inhibitor prexasertib (Eli Lilly) in patients with advanced refractory squamous NSCLC, head and neck cancers and anal cancers reported similar response rates [PR in 4.4% (2/45); SD in 33% (15/45)] (81). Notably, these cancers also have a significantly high rate of TP53 mutation. Building on the preclinical and phase 1 results, prexasertib has now entered a phase II trial for SCLC patients with relapsed disease (NCT02735980). This trial is ongoing and will assess single agent activity in 116 patients with platinum sensitive or refractory SCLC. Additional CHK1 inhibitors in clinical investigation include GDC-0575 (Genentech; NCT01564251) and SRA737 (Sierra Oncology; NCT02797964 and NCT027979977). Studies in triple-negative breast cancers and head and neck cancers (in which TP53 mutations often occur) have demonstrated that CHK1 inhibitors can augment the effects of DNA-damaging treatments, findings that have led to trials investigating combinations with CHK1 inhibitors with platinum chemotherapy or PARP inhibitors (NCT02797977; NCT03057145).
Targeting WEE1 in SCLC
WEE1 tyrosine kinase is a crucial component of the G2-M cell cycle checkpoint that halts the entry of the cell into mitosis upon DNA damage. Upon successful completion of the G2/M phase, the cell’s entry into mitosis is governed by the phosphorylation status of CDK1, also known as CDC2, and its association with cyclin B. WEE1 acts as a negative regulator of entry into mitosis at the G2-M transition by preventing the entry of cyclin B complexed with CDK1 activated in the cytoplasm (82-84).
Analysis of WEE1 mRNA expression levels in 68 SCLC and 26 normal lung tissue samples showed that compared with normal lung tissue samples, SCLC samples had >4-fold higher WEE1 expression (P<0.0001) (17). Furthermore, profiling of 63 SCLC and 114 NSCLC cell lines showed significantly higher WEE1 protein expression in SCLC cell line samples [P<0.0001; fold change (FC) =1.66] (17). The study further showed that a significant subset (~70%) of SCLC cell lines were sensitive to the WEE1 inhibitor, AZD1775 (AstraZeneca). Using high-throughput proteomic profiling, we found that SCLC models with primary and acquired resistance to AZD1775 expressed high levels of receptor tyrosine kinase, AXL and phosphorylated S6 and resistance could be overcome with WEE1/AXL or WEE1/mTOR inhibitor combinations in cell lines and mouse models (17). This study further demonstrates that AXL-mediated activation of CHK1 could lead to redundancy and consequent resistance to WEE1 inhibition in SCLC (17).
Currently, AZD1775 is the only WEE1 inhibitor in clinical development (85-87). Early data from the trial showed that two of four patients with SCLC had a response to single-agent AZD1775 (88), although the final study results have not yet been published. Single agent (NCT02482311; NCT02593019) and combination studies with platinum chemotherapy (NCT02937818) and PARP inhibitors (NCT02511795) are being investigated for SCLC and other cancer types. These include a study specifically targeting SCLC with MYC amplifications (NCT02688907).
Targeting Aurora kinase (AURK) in SCLC
The AURKs belong to a family of highly conserved serine/threonine kinases that are important for faithful transition through mitosis. The function and expression of Aurora A is regulated by the cell cycle and the protein controls important mitotic events including centrosome maturation, chromosome alignment, chromosome segregation, and cytokinesis (89). Indeed, genetic inhibition of Aurora A (AURKA) has been shown to inhibit the proliferation of SCLC cells (90). A small molecule Aurora B (AURKB) inhibitor, barasertib (AZD1152) has also been shown to inhibit the growth of SCLC in vitro and in vivo (91). Recent reports have shown activity of AURK A targeting in SCLC with high MYC and NEUROD1 expression (92). Alisertib (MLN8237; Takeda) is a selective inhibitor of AURKA, with an IC50 of 1.2 nM against AURKA and 396.5 nM against AURKB (93). Multiple studies have demonstrated the preclinical activity of alisertib in a wide range of human tumor models including lung, prostate, ovarian, and lymphoma cells (94-96). A multicenter phase I/II study of alisertib in patients with various solid tumors (including SCLC), defined the recommended phase II dose and schedule and established the dose-limiting toxicities (97,98). The phase II expansion cohort of recurrent SCLC patients showed 21% objective response rate (ORR) with single agent alisertib treatment. In a subsequent randomized phase II study, alisertib (or placebo) was then combined with paclitaxel as second-line therapy for SCLC patients. In that study, a significant improvement in PFS was observed (101 vs. 66 days, P=0.038) with alisertib in the overall population. A trend toward improvement in ORR and OS was also observed, although this was not statistically significant. An exploratory analysis was then performed to determine if there was enhanced activity in patients whose cancer expressed cMYC protein. In 46 evaluable patients, those with cMYC positive tumors (defined as those with IHC intensity of 1−3+) who received the alisertib/paclitaxel combination had significantly improved PFS as compared to those with cMYC negative tumors (IHC intensity of 0). Specifically, the hazard ratio (HR) comparing alisertib/paclitaxel vs. paclitaxel alone in cMYC positive was 0.29 (0.12–0.72) vs. a HR in the cMyc-negative population of 11.8 (range, 1.52–91.2) (binary P=0.0006). Given the relatively small number of patients and retrospective nature of the analysis, a prospective study is needed to further validate the predictive value of cMYC in the clinic (99,100). Together, these clinical data suggest AURKA inhibition is a promising treatment strategy in SCLC (99). Additional inhibitors of AURKA and AURKB are currently in clinical development including chiauranib (Chipscreen Biosciences), AK-01 (AurKa Pharma), TAS-119 (Taiho Oncology) and GSK1070916A (GlaxoSmithKline) with several trials specifically focusing on SCLC (NCT03216343; NCT03092934).
The biomarker landscape in SCLC
Key to the success of any targeted agent is an appropriate patient selection strategy. By leveraging a large number of preclinical models, candidate biomarkers have recently been identified for several DDRi in SCLC. These include SLFN11 and epithelial-mesenchymal transition (EMT) as predictive markers of chemotherapy and PARP inhibitor sensitivity (49,64,67,68,101); MYC (and TTF1) as markers of CHK1 and AURK inhibitor sensitivity (18,92,102,103); and AXL/mTOR axis as the predictive marker of WEE1 inhibitor resistance (17). These findings suggest that biomarker profiling of patient samples could ultimately help guide treatment detections in the clinic, moving us towards the goal of personalized SCLC treatment (Figure 2).
Figure 2.
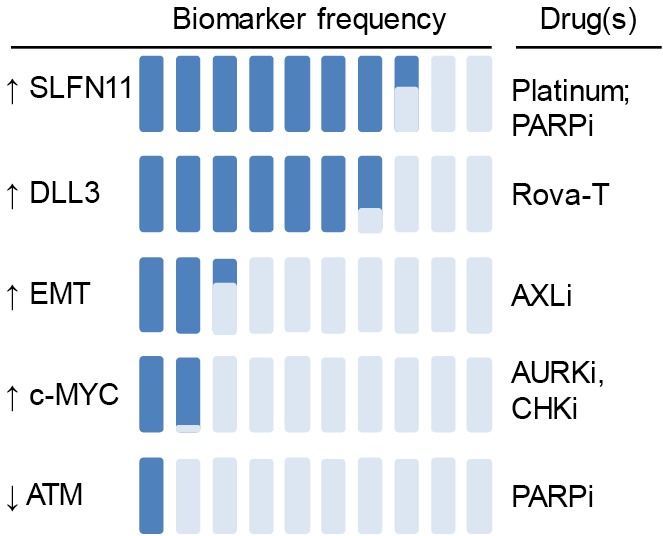
Landscape of predictive biomarkers in SCLC. Recent preclinical studies identified predictive biomarkers of response to DDR-targeted therapies in SCLC. SLFN11 predicts sensitivity to PARP inhibitors and chemotherapy; AXL and/or EMT predict resistance to PARP and WEE1 targeting; high ATM expression predicts resistance to PARP and ATR-targeted therapies; high MYC expression and amplification predicts sensitivity to AURK and CHK1 inhibitors; high expression of the inhibitory Notch ligand Delta-like protein 3 (DLL3) predicts response to Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate. Approximate proportions for each biomarker are estimated based on a combination of reported patient tumor and/or patient-derived xenograft expression and (where available) response rates in specific biomarker-selected populations [DLL3 (PMID: 27932068), MYC (PMID: 28490518), SLFN11/ATM/EMT (PMID: 28212573)]. For example, while >50% DLL3 expression was noted in in over two-thirds of evaluated patient samples, an objective response was observed in only 18% of evaluable patients, suggesting proportion of SCLC sensitive to DLL3 is less than that which expresses high DLL3 (PMID: 27932068). SCLC, small cell lung cancer; SLFN11, schlafen 11; PARP, poly-ADP-ribose polymerase; DDR, DNA damage response; EMT, epithelial-mesenchymal transition; ATM, ataxia telangiectasia-mutated; ATR, ataxia telangiectasia and Rad3-related protein; AURK, Aurora kinase; CHK1, checkpoint kinase 1; PDX, patient-derived xenograft.
The rise of SLFN11
The Schlafen (SLFN) family (from the word schlafen, which in German means sleeping) includes several mouse and human genes (104-108). Multiple reports in the past year have highlighted the role of SLFN11 as a critical determinant of response to chemotherapy and PARP inhibitors in SCLC in vitro and in vivo models. Polley et al., screened 63 human SCLC lines and three NSCLC lines for response to 103 FDA-approved oncology agents and 423 investigational agents (68). They found that high SLFN11 expression was associated with sensitivity to the PARP-1 inhibitors (R =−0.42) (68). This finding was further substantiated in two subsequent papers. Lok et al., demonstrated that SLFN11 expression correlates with response multiple PARP inhibitors and loss of SLFN11 conferred resistance to PARP inhibition in SCLC cell line and animal models (64). Independently, using a high-throughput, integrated proteomic, transcriptomic, and genomic analysis of SCLC PDXs and profiled cell lines, Allison Stewart et al., demonstrated that high levels of SLFN11 (as well as high E-cadherin and low ATM) predicted sensitivity to PARP inhibition and chemotherapy (49). They further showed that SLFN11 expression was downregulated after treatment with cisplatin or PARP inhibitors, and knockdown of SLFN11 reduced in vitro sensitivity and drug-induced DNA damage (49). SLFN11 expression is regulated by multiple mechanisms (105) including methylation, which is often regulated by the catalytic subunit of the Polycomb repressive complex 2, enhancer of zeste homologue 2 (EZH2), discussed below.
EZH2 has been previously shown to be overexpressed in many tumors, including SCLC (14,109). Of its more important cellular functions, EZH2 plays a crucial role in methylation via chromatin modification and activation of DNA methyltransferases (110-112). Its other functions include suppression of apoptosis, increased cell proliferation and activation of ASCL1 expression (via suppression of TGF-β signaling) (113). Expression of the EZH2 gene is under the direct control of the E2F family of transcription factors, including E2F1 (110-112). Interestingly, a study published in 2017 demonstrated that overexpression of EZH2 downregulates SLFN11 via histone modification and methylation thereby making the SCLC resistant to chemotherapy (101). Furthermore, cisplatin and EZH2 inhibitor combination was successful in overcoming drug resistance in both chemo-sensitive and resistant SCLC models. Furthermore, EZH2 inhibition was found to prevent SLFN11 loss and subsequent resistance to chemotherapy and PARP targeting (101), suggesting an intriguing possibility of combining EZH2 inhibitors with standard chemotherapy in the clinic for SCLC patients.
Role of MYC in DDR inhibitor response
After TP53 and RB1, MYC family amplifications are the next most common form of genetic alterations. Amplifications and/or overexpression of MYC family genes (MYC, MYCL and MYCN) are mutually exclusive and are observed in about 20% of tumors and cell lines (27,28,114-117). MYC amplification is a known driver of genomic instability and replication stress. In SCLC, higher MYC expression in the mouse lung promotes an aggressive phenotype, highly metastatic tumors and rapid development of chemotherapy resistance similar to human SCLC (92,118). However, inhibitors of MYC have yet to be translated into clinically viable therapies.
Studies from our group and others have demonstrated the potential of targeting MYC indirectly as a synthetic lethal strategy in SCLC. A recent study from our group demonstrated that higher expression of and/or MYC amplification predicted sensitivity to CHK1 targeting in SCLC models (18). We further showed that knockdown of MYC made SCLC cells more resistant to CHK1 inhibitors, indicating the functional role of MYC in DDR inhibitor response (18). Using model-based clustering, Cardnell et al., found two major proteomic subtypes of SCLC characterized by either high thyroid transcription factor-1 (TTF1)/low cMYC protein expression or high cMYC/low TTF1 (102). The study further demonstrated that protein levels of TTF1 and cMYC predict response to targeted therapies including AURK, BCL2, and HSP90 inhibitors (102). Combined genomic and chemical vulnerability analysis in a panel of 60 SCLC cell lines demonstrated that sensitivity to structurally diverse AURK inhibitors VX680, MLN8237, PHA680632, and ZM447439 was significantly enriched in MYC amplified cells (P=0.004 MLN8237; P=0.003 PHA680632; P=0.01 VX680; P=0.01 ZM447439) (103). In an elegant study using SCLC GEM models, Mollaoglu et al., showed that SCLC with high MYC expression is vulnerable to AURK inhibition and chemotherapy and the combination causes tumor suppression and significant survival benefit (92). Mollaoglu et al., (92) and our data (18) demonstrate that MYC expression is observed in a larger cohort of SCLC patients than MYC amplification alone and some patients have high MYC expression even without amplification. The data thus suggests that MYC expression rather than MYC amplification alone may identify additional MYC “positive” patients and therefore may be a more sensitive biomarker to identify patients who may respond to certain DDR inhibitors. SCLC patients with MYC amplifications are being enrolled on an active clinical trial with the WEE1 inhibitor AZD1775 (NCT02688907). Trial designs such as this along with detailed mechanistic studies are warranted to establish the role of MYC in different SCLC subtypes.
Role of EMT and AXL in SCLC
EMT has emerged as a key regulator of metastasis, resistance to targeted therapy, chemotherapy, and radiation; as well as an emerging role in immune escape (119,120). Allison Stewart et al., recently demonstrated that in SCLC preclinical models, resistance to PARP inhibitors is frequently associated with EMT and low expression of CDH1 (E-cadherin). High levels of SLFN11 and E-cadherin and low ATM were significantly associated with in vitro sensitivity to cisplatin and topoisomerase1/2 inhibitors in SCLC. Interestingly, SCLC with loss of E-cadherin and high EMT signature scores displayed alterations in expression of the miR200 family and key neuroendocrine and SCLC genes (e.g., NEUROD1, ASCL1, ALDH1A1, MYCL) (49). In a 76-gene EMT signature in NSCLC, the mesenchymal phenotype was strongly correlated with the expression of the receptor tyrosine kinase, AXL (121).
AXL, activated by a complex interaction between its ligand growth arrest-specific protein 6 (Gas6) and phosphatidylserine, regulates various vital cellular processes, including proliferation, survival, motility, and immunologic response (122-124). AXL has been suggested to promote both intrinsic and acquired resistance to chemotherapeutic, immunotherapeutic and molecularly targeted agents in both solid and hematologic malignancies, including acute myeloid leukemia, NSCLC, gastric and colorectal ADC, and breast and prostate cancers (121,125,126). Thus, several AXL inhibitors are currently in preclinical and clinical development, but thus far, AXL’s role in SCLC had not been well studied. A recent study from our group demonstrated that higher basal expression of AXL is associated with primary resistance to WEE1 targeting (17). Moreover, AXL expression is upregulated upon acquired resistance to WEE1 inhibitor, AZD1775 and co-inhibition of AXL overcomes resistance in SCLC models (17). Further exploration is needed to determine if AXL contributes functionally to additional DDR inhibitor resistance as it is frequently overexpressed in tumors that have undergone EMT.
Future perspective—DDR-immune system crosstalk and its therapeutic implications in SCLC
Earlier studies showed that defects in the DDR network and function could lead to changes in the immune microenvironment of tumors (127). Therapeutically, IL-2 and intravesicular Bacillus Calmette-Guerin (BCG) have long track-records of being used to activate the host immune system for multiple cancer types including melanoma, renal cell carcinoma and bladder cancer (128-130). In the past several years, many cancer immunotherapy regimens have been developed that use therapeutic antibodies against inhibitory signaling molecules on tumor and immune cells (131,132). Common targets of these monoclonal antibodies include the immune checkpoints PD-1, PD-L1, and cytotoxic T-lymphocyte associated protein 4 (CTLA4) (132-137). Since the first FDA approval of ipilimumab (anti-CTLA4) for melanoma, several such ICB agents have been approved for patients with advanced cancers, including NSCLC (138-140).
Though very promising, ICBs vary in their activity across different cancer types and hence there is an active effort to develop therapeutic strategies including novel drug combinations that may enhance the anti-tumor efficacy of ICBs in cancers with relatively low response rates to immunotherapy. Several studies have now reported high mutational burden as a potential predictor of effective immunotherapy response (141) however, despite having one of the highest mutational burdens, SCLCs (26), the clinical efficacy of checkpoint inhibitors in this disease seems to be far less than as expected. Antibodies targeting PD-1 or PD-L1 are being actively evaluated in clinical trials for SCLC alone and in combination with other therapeutic agents or radiation (142). Recently, nivolumab with or without ipilimumab was added to the National Comprehensive Cancer Network guidelines as a treatment option for relapsed SCLC (11). However, only a minority of SCLC patients respond to anti-PD-1 monotherapy (~10%) or the anti-PD1/anti-CTLA4 combination (~23%) (10,143). Notably, SCLC has relatively immunosuppressed phenotypes with low levels of infiltrating T cells and reduced antigen presentation (144-146). Furthermore, PD-L1 levels appear to be lower in SCLC than in NSCLC (147), which is paradoxical given the high overall mutation burden in SCLC (25) and might at least partially explain the poor outcomes from anti-PD-1/PD-L1 trials (Table 2).
Table 2. Reported immunotherapy trials in SCLC.
| Reference | Trial details |
|---|---|
| Antonia SJ et al. (CheckMate 032) (10) | |
| Phase 1/2: LS or ES-SCLC patients with progression after prior platinum therapy | |
| Patients received nivolumab (nivo) 3 mg/kg q2w (every 2 weeks) until PD or toxicity or nivo/ipilimumab (ipi) (1+1 mg/kg, or 1+3 mg/kg, or 3+1 mg/kg) q3w (every 3 weeks) ×4 cycles, followed by nivo 3 mg/kg q2w until PD or toxicity | |
| ORR was 10% (10/98) in nivo alone, 33% (1/3) for nivo 1 + ipi 1, 23% (14/61) for nivo 1 + ipi 3 and 19% (10/54) for nivo 3 + ipi 1 (21% or 25/118 total for nivo + ipi) | |
| PD-L1 expression ≥1% in 17% and ≥5% in 5% of patients analyzed using 28-8 (Dako) antibody | |
| Responses were irrespective of PD-L1 expression | |
| Antonia SJ et al. (CheckMate 032 exploratory analysis) (148) | |
| Evaluable patients from CheckMate 032 were divided into low/medium/high TMB (<143, 143–247, and >247 missense mutations, respectively) | |
| Breakdown into three tertiles compared favorably to Carbone DP et al. (149) CheckMate 026 data for NSCLC | |
| Improved ORR, 1-year PFS and 1-year OS for both nivo alone and nivo/ipi with high TMB | |
| Low/medium/high ORRs: 4.8%, 6.8%, 21.3% (nivo alone) and 22.2%, 16.0%, 46.2% (nivo + ipi); 1-year PFS: not-calculable, 3.1%, 21.2% (nivo alone) and 6.2%, 8%, 30% (nivo + ipi); 1-year OS: 22.1%, 26%, 35.2% (nivo alone) and 23.4%, 19.6%, 62.4% (nivo + ipi) | |
| Ott PA et al. (KEYNOTE-028) (150) | |
| Pre-treated ES-SCLC with PD-L1 expression ≥1% were treated with pembrolizumab (pembro) 10 mg/kg q2w for 24 months or until PD or toxicity | |
| Forty-six of 145 (31.7%) evaluated patients had PD-L1 expression ≥1% using 22C3 (Merck) antibody | |
| Twenty-four patients total treated, 1 CR, 7 PRs (ORR of 33% or 8/24) | |
| Gadgeel SM et al. (151) | |
| Phase II study of pembro 200 mg flat-dose q3w for maximum of 2 years following 4–6 cycles of platinum/etoposide with PR or SD (pembro maintenance) | |
| PD-L1 staining only positive in 1/35 patients; maintenance pembro failed to improve PFS compared to historical controls | |
| Reck M et al. (152) | |
| Phase II study featuring 1:1:1 randomization between carbo/taxol + placebo + ipi (concurrent) or + placebo followed by + ipi (phased); no improvement in PFS or OS | |
PD, progressive disease; ORR, objective response rate; TMB, tumor mutational burden; PFS, progression-free survival; OS, overall survival; PR, partial response; SD, stable disease; SCLC, small cell lung cancer; LS-SCLC, limited stage SCLC; ES-SCLC, extensive stage SCLC; PD-L1, programmed death-ligand 1; NSCLC, non-small cell lung cancer; CR, complete response.
In other cancer types, defects in the DDR pathway have been associated with enhanced response to ICB (153,154). For example, somatic alterations in DDR genes such as POLE, POLD1 and MSH2 have been linked with durable benefit to ICB response in patients, suggesting that defects in DDR network and changes in the genomic landscape (such as higher mutation burden and increased levels of predicted neo-antigens) may contribute to enhanced ICB response (141). Recently, our group and others found that co-targeting DDR proteins such as PARP and CHK1 can increase expression of PD-L1 and anti-tumor immune response of anti-PD-L1 in SCLC and breast cancer models (155,156). These findings suggest that DDR targeting through pharmacologic inhibition could potentially enhance the anti-tumor immune response to ICB in SCLC and other malignancies (157-159).
Conclusions
Proteomic, genomic and transcriptomic profiling have helped identify novel, promising DDR targets for SCLC like PARP, CHK1, WEE1 and ATR. Drugs targeting these proteins are currently in various phases of clinical development. Identification of predictive biomarkers for DDR-targeted therapies and identifying optimal combination regimens to overcome adaptive resistance to DDR-targeted therapies are areas of active investigation in SCLC. The importance of liquid biopsies and blood-based biomarker identification strategies are additional areas of interest in this disease. Immuno-oncology and the use of ICB in SCLC treatment is an area of intensive ongoing investigation. Optimizing strategies to activate the immune system would be one of most interesting future areas of research in this disease. The results from the ongoing DDR-targeted clinical trials would indicate whether we can finally expand and improve the therapeutic options for this recalcitrant disease for which the treatment options have remained stagnant for over three decades.
Acknowledgements
The authors would like to thank Dr. Emily Roarty for editing the manuscript.
Funding: This work was supported by NIH/NCI 1-R01-CA207295 (LA Byers); NIH-U01 (JV Heymach/J Sage/LA Byers); Uniting Against Lung Cancer (LA Byers); Lung Cancer Research Foundation Research Grant (T Sen); LUNGevity (LA Byers & DL Gibbons); The University of Texas MD Anderson Cancer Center Small Cell Lung Cancer Working Group and Abell Hangar Foundation Distinguished Professor Endowment (BS Glisson); Rexanna’s Foundation for Fighting Lung Cancer (LA Byers & DL Gibbons); Sidney Kimmel Scholar Award (LA Byers); by Free to Breathe (formerly National Lung Cancer Research Partnership), North Carolina Lung Cancer Partnership (LA Byers); MD Anderson Physician Scientist Award (LA Byers & DL Gibbons); R. Lee Clark Fellow Award, made possible by the Jeanne F. Shelby Scholarship Fund (LA Byers); National Cancer Institute Cancer Clinical Investigator Team Leadership Award (P30 CA016672) (LA Byers); The University of Texas Lung Cancer Moon Shot Program and the MD Anderson Cancer Center Support Grant (P30CA01667); Lung Cancer Research Foundation (LCRF) (T Sen).
Footnotes
Conflicts of Interest: LA Byers has served as an advisory board member and/or consultant to AstraZeneca, Medivation, and AbbVie.
References
- 1.Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2095-128. 10.1016/S0140-6736(12)61728-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nicholson AG, Chansky K, Crowley J, et al. The International Association for the Study of Lung Cancer Lung Cancer Staging Project: Proposals for the Revision of the Clinical and Pathologic Staging of Small Cell Lung Cancer in the Forthcoming Eighth Edition of the TNM Classification for Lung Cancer. J Thorac Oncol 2016;11:300-11. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin 2007;57:43-66. 10.3322/canjclin.57.1.43 [DOI] [PubMed] [Google Scholar]
- 4.Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 2006;24:4539-44. 10.1200/JCO.2005.04.4859 [DOI] [PubMed] [Google Scholar]
- 5.Sabari JK, Lok BH, Laird JH, et al. Unravelling the biology of SCLC: implications for therapy. Nat Rev Clin Oncol 2017;14:549-61. 10.1038/nrclinonc.2017.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nature Reviews Cancer 2017;17:725. 10.1038/nrc.2017.87 [DOI] [PubMed] [Google Scholar]
- 7.Society AC. Cancer Facts & Figures 2016. American Cancer Society, 2016. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2016.html
- 8.William WN, Jr, Glisson BS. Novel strategies for the treatment of small-cell lung carcinoma. Nat Rev Clin Oncol 2011;8:611-9. 10.1038/nrclinonc.2011.90 [DOI] [PubMed] [Google Scholar]
- 9.Byers LA, Rudin CM. Small cell lung cancer: where do we go from here? Cancer 2015;121:664-72. 10.1002/cncr.29098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonia SJ, Lopez-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883-95. 10.1016/S1470-2045(16)30098-5 [DOI] [PubMed] [Google Scholar]
- 11.Network NCC. National Comprehensive Cancer Network Guidelines: Small Cell Lung Cancer Version 2.2017. Available online: https://www.tri-kobe.org/nccn/guideline/lung/english/small.pdf
- 12.Recalcitrant Cancer Research Act of 2012. Available online: https://deainfo.nci.nih.gov/advisory/ctac/0716/4-SCLprogressReport_Jul%202016.pdf
- 13.Foy V, Schenk MW, Baker K, et al. Targeting DNA damage in SCLC. Lung Cancer 2017;114:12-22. 10.1016/j.lungcan.2017.10.006 [DOI] [PubMed] [Google Scholar]
- 14.Byers LA, Wang J, Nilsson MB, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov 2012;2:798-811. 10.1158/2159-8290.CD-12-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardnell RJ, Feng Y, Diao L, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res 2013;19:6322-8. 10.1158/1078-0432.CCR-13-1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cardnell RJ, Feng Y, Mukherjee S, et al. Activation of the PI3K/mTOR Pathway following PARP Inhibition in Small Cell Lung Cancer. PLoS One 2016;11:e0152584. 10.1371/journal.pone.0152584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen T, Tong P, Diao L, et al. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin Cancer Res 2017;23:6239-53. 10.1158/1078-0432.CCR-17-1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen T, Tong P, Stewart CA, et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res 2017;77:3870-84. 10.1158/0008-5472.CAN-16-3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sen T, Tong P, Wang J, et al. Proteomic profiling identifies ATM expression level as a predictive biomarker to ATR and PARP inhibition in small cell lung cancer (SCLC). Cancer Research 2016;76: abstract LB132.
- 20.de Bono J, Ramanathan RK, Mina L, et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov 2017;7:620-9. 10.1158/2159-8290.CD-16-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khalil DN, Smith EL, Brentjens RJ, et al. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 2016;13:273-90. 10.1038/nrclinonc.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348:56-61. 10.1126/science.aaa8172 [DOI] [PubMed] [Google Scholar]
- 23.Varghese AM, Zakowski MF, Yu HA, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol 2014;9:892-6. 10.1097/JTO.0000000000000142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pesch B, Kendzia B, Gustavsson P, et al. Cigarette smoking and lung cancer--relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int J Cancer 2012;131:1210-9. 10.1002/ijc.27339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pleasance ED, Stephens PJ, O'Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463:184-90. 10.1038/nature08629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415-21. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47-53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 2012;44:1111-6. 10.1038/ng.2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma CX, Cai S, Li S, et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest 2012;122:1541-52. 10.1172/JCI58765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med 2011;17:88-96. 10.1016/j.molmed.2010.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doerr F, George J, Schmitt A, et al. Targeting a non-oncogene addiction to the ATR/CHK1 axis for the treatment of small cell lung cancer. Sci Rep 2017;7:15511. 10.1038/s41598-017-15840-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pietanza MC, Byers LA, Minna JD, et al. Small cell lung cancer: will recent progress lead to improved outcomes? Clin Cancer Res 2015;21:2244-55. 10.1158/1078-0432.CCR-14-2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown JS, O'Carrigan B, Jackson SP, et al. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov 2017;7:20-37. 10.1158/2159-8290.CD-16-0860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Gent DC, Kanaar R. Exploiting DNA repair defects for novel cancer therapies. Mol Biol Cell 2016;27:2145-8. 10.1091/mbc.E15-10-0698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012;481:287. 10.1038/nature10760 [DOI] [PubMed] [Google Scholar]
- 36.O'Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell 2015;60:547-60. 10.1016/j.molcel.2015.10.040 [DOI] [PubMed] [Google Scholar]
- 37.Pearl LH, Schierz AC, Ward SE, et al. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 2015;15:166-80. 10.1038/nrc3891 [DOI] [PubMed] [Google Scholar]
- 38.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70. 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- 40.Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer 2015;15:276-89. 10.1038/nrc3916 [DOI] [PubMed] [Google Scholar]
- 41.Dominguez-Sola D, Gautier J. MYC and the control of DNA replication. Cold Spring Harb Perspect Med 2014;4. pii: a014423. 10.1101/cshperspect.a014423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srinivasan SV, Dominguez-Sola D, Wang LC, et al. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep 2013;3:1629-39. 10.1016/j.celrep.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dominguez-Sola D, Ying CY, Grandori C, et al. Non-transcriptional control of DNA replication by c-Myc. Nature 2007;448:445-51. 10.1038/nature05953 [DOI] [PubMed] [Google Scholar]
- 44.Chan BA, Coward JI. Chemotherapy advances in small-cell lung cancer. J Thorac Dis 2013;5 Suppl 5:S565-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crivellari G, Monfardini S, Stragliotto S, et al. Increasing chemotherapy in small-cell lung cancer: from dose intensity and density to megadoses. Oncologist 2007;12:79-89. 10.1634/theoncologist.12-1-79 [DOI] [PubMed] [Google Scholar]
- 46.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007;7:573-84. 10.1038/nrc2167 [DOI] [PubMed] [Google Scholar]
- 47.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 2009;9:338-50. 10.1038/nrc2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bodley AL, Liu LF. Topoisomerases as Novel Targets for Cancer Chemotherapy. Bio/Technology 1988;6:1315. [Google Scholar]
- 49.Allison Stewart C, Tong P, Cardnell RJ, et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017;8:28575-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christensen CL, Kwiatkowski N, Abraham BJ, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014;26:909-22. 10.1016/j.ccell.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drew Y, Plummer R. PARP inhibitors in cancer therapy: two modes of attack on the cancer cell widening the clinical applications. Drug Resist Updat 2009;12:153-6. 10.1016/j.drup.2009.10.001 [DOI] [PubMed] [Google Scholar]
- 52.Lin KY, Kraus WL. PARP Inhibitors for Cancer Therapy. Cell 2017;169:183. 10.1016/j.cell.2017.03.034 [DOI] [PubMed] [Google Scholar]
- 53.Rouleau M, Patel A, Hendzel MJ, et al. PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010;10:293-301. 10.1038/nrc2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vyas S, Chesarone-Cataldo M, Todorova T, et al. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat Commun 2013;4:2240. 10.1038/ncomms3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012;72:5588-99. 10.1158/0008-5472.CAN-12-2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913-7. 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- 57.Dziadkowiec KN, Gasiorowska E, Nowak-Markwitz E, et al. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Prz Menopauzalny 2016;15:215-9. 10.5114/pm.2016.65667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evers B, Helleday T, Jonkers J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol Sci 2010;31:372-80. 10.1016/j.tips.2010.06.001 [DOI] [PubMed] [Google Scholar]
- 59.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917-21. 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- 60.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 2011;5:387-93. 10.1016/j.molonc.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim G, Ison G, McKee AE, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res 2015;21:4257-61. 10.1158/1078-0432.CCR-15-0887 [DOI] [PubMed] [Google Scholar]
- 62.Litton JK, Blum JL, Im YH, et al. A phase 3, open-label, randomized, parallel, 2-arm international study of the oral PARP inhibitor talazoparib (BMN 673) in BRCA mutation subjects with locally advanced and/or metastatic breast cancer (EMBRACA). J Clin Oncol 2015;33:TPS1107. [Google Scholar]
- 63.Owonikoko TK, Zhang G, Deng X, et al. Poly (ADP) ribose polymerase enzyme inhibitor, veliparib, potentiates chemotherapy and radiation in vitro and in vivo in small cell lung cancer. Cancer Med 2014;3:1579-94. 10.1002/cam4.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lok BH, Gardner EE, Schneeberger VE, et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin Cancer Res 2017;23:523-35. 10.1158/1078-0432.CCR-16-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pietanza MC, Kadota K, Huberman K, et al. Phase II trial of temozolomide in patients with relapsed sensitive or refractory small cell lung cancer, with assessment of methylguanine-DNA methyltransferase as a potential biomarker. Clin Cancer Res 2012;18:1138-45. 10.1158/1078-0432.CCR-11-2059 [DOI] [PubMed] [Google Scholar]
- 66.Byers LA, Krug L, Waqar S, et al. MA11.07 Improved Small Cell Lung Cancer (SCLC) Response Rates with Veliparib and Temozolomide: Results from a Phase II Trial. J Thorac Oncol 2016;12:S406-7. 10.1016/j.jtho.2016.11.466 [DOI] [Google Scholar]
- 67.Murai J, Feng Y, Yu GK, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016;7:76534-50. 10.18632/oncotarget.12266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Polley E, Kunkel M, Evans D, et al. Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. J Natl Cancer Inst 2016;108(10). 10.1093/jnci/djw122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farago AF, Drapkin BJ, Charles A, et al. Abstract CT048: Phase 1/2 study of olaparib tablets and temozolomide in patients with small cell lung cancer (SCLC) following failure of prior chemotherapy. Cancer Research 2017;77:CT048.-CT. 10.1158/1538-7445.AM2017-CT048 [DOI] [Google Scholar]
- 70.Atrafi F, Groen HJM, Byers LA, et al. Phase 1/2 study of veliparib (V) combined with carboplatin (Cb) and etoposide (E) in patients (pts) with extensive-stage disease (ED) small cell lung cancer (SCLC) and other solid tumors: Phase 1 results. J Clin Oncol 2017;35:8530. [Google Scholar]
- 71.Ibrahim YH, Garcia-Garcia C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov 2012;2:1036-47. 10.1158/2159-8290.CD-11-0348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Juvekar A, Burga LN, Hu H, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov 2012;2:1048-63. 10.1158/2159-8290.CD-11-0336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010;28:1075-83. 10.1200/JCO.2009.25.3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A 2010;107:7491-6. 10.1073/pnas.0914242107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J 2011;436:527-36. 10.1042/BJ20102162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qiu Z, Oleinick NL, Zhang J. ATR/CHK1 inhibitors and cancer therapy. Radiother Oncol 2017. [Epub ahead of print]. 10.1016/j.radonc.2017.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rundle S, Bradbury A, Drew Y, et al. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers (Basel) 2017;9. pii: E41. 10.3390/cancers9050041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith J, Tho LM, Xu N, et al. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010;108:73-112. 10.1016/B978-0-12-380888-2.00003-0 [DOI] [PubMed] [Google Scholar]
- 79.Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011;7:428-30. 10.1038/nchembio.573 [DOI] [PubMed] [Google Scholar]
- 80.King C, Diaz HB, McNeely S, et al. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther 2015;14:2004-13. 10.1158/1535-7163.MCT-14-1037 [DOI] [PubMed] [Google Scholar]
- 81.Hong D, Infante J, Janku F, et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol 2016;34:1764-71. 10.1200/JCO.2015.64.5788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 2009;185:193-202. 10.1083/jcb.200812045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J 1995;14:1878-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McGowan CH, Russell P. Cell cycle regulation of human WEE1. EMBO J 1995;14:2166-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Do K, Wilsker D, Ji J, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol 2015;33:3409-15. 10.1200/JCO.2014.60.4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hamilton EP, Wang JS, Falchook G, et al. A phase Ib study of AZD1775 and olaparib combination in patients with refractory solid tumors. J Clin Oncol 2016;34:5562. [Google Scholar]
- 87.Leijen S, van Geel RM, Pavlick AC, et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol 2016;34:4371-80. 10.1200/JCO.2016.67.5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bauer TM, Jones SF, Greenlees C, et al. Abstract CT013: A phase Ib, open-label, multicenter study to assess the safety, tolerability, pharmacokinetics, and antitumor activity of AZD1775 monotherapy in patients with advanced solid tumors: initial findings. Cancer Research 2016;76:CT013.-CT. 10.1158/1538-7445.AM2016-CT013 [DOI] [Google Scholar]
- 89.Nikonova AS, Astsaturov I, Serebriiskii IG, et al. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci 2013;70:661-87. 10.1007/s00018-012-1073-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu Y, Liu Y, Jiang J, et al. Knocking down the expression of Aurora-A gene inhibits cell proliferation and induces G2/M phase arrest in human small cell lung cancer cells. Oncol Rep 2014;32:243-9. 10.3892/or.2014.3194 [DOI] [PubMed] [Google Scholar]
- 91.Helfrich BA, Kim J, Gao D, et al. Barasertib (AZD1152), a Small Molecule Aurora B Inhibitor, Inhibits the Growth of SCLC Cell Lines In Vitro and In Vivo. Mol Cancer Ther 2016;15:2314-22. 10.1158/1535-7163.MCT-16-0298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mollaoglu G, Guthrie MR, Bohm S, et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017;31:270-85. 10.1016/j.ccell.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Manfredi MG, Ecsedy JA, Chakravarty A, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res 2011;17:7614-24. 10.1158/1078-0432.CCR-11-1536 [DOI] [PubMed] [Google Scholar]
- 94.Bavetsias V, Linardopoulos S. Aurora Kinase Inhibitors: Current Status and Outlook. Front Oncol 2015;5:278. 10.3389/fonc.2015.00278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hilton JF, Shapiro GI. Aurora Kinase Inhibition As an Anticancer Strategy. J Clin Oncol 2014;32:57-9. 10.1200/JCO.2013.50.7988 [DOI] [PubMed] [Google Scholar]
- 96.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nature Reviews Cancer 2004;4:927. 10.1038/nrc1502 [DOI] [PubMed] [Google Scholar]
- 97.Melichar B, Adenis A, Lockhart AC, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol 2015;16:395-405. 10.1016/S1470-2045(15)70051-3 [DOI] [PubMed] [Google Scholar]
- 98.Niu H, Manfredi M, Ecsedy JA. Scientific Rationale Supporting the Clinical Development Strategy for the Investigational Aurora A Kinase Inhibitor Alisertib in Cancer. Front Oncol 2015;5:189. 10.3389/fonc.2015.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Owonikoko T, Nackaerts K, Csoszi T, et al. OA05.05 Randomized Phase 2 Study: Alisertib (MLN8237) or Placebo + Paclitaxel as Second-Line Therapy for Small-Cell Lung Cancer (SCLC). J Thorac Oncol 2017;12:S261-2. 10.1016/j.jtho.2016.11.253 [DOI] [Google Scholar]
- 100.Owonikoko TK, Nackaerts K, Csoszi T, et al. Randomized phase 2 study of investigational aurora A kinase (AAK) inhibitor alisertib (MLN8237) + paclitaxel (P) vs placebo + P as second line therapy for small-cell lung cancer (SCLC). Ann Oncol 2016;27:493-6. 10.1093/annonc/mdw389.01 [DOI] [Google Scholar]
- 101.Gardner EE, Lok BH, Schneeberger VE, et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017;31:286-99. 10.1016/j.ccell.2017.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cardnell RJ, Li L, Sen T, et al. Protein expression of TTF1 and cMYC define distinct molecular subgroups of small cell lung cancer with unique vulnerabilities to aurora kinase inhibition, DLL3 targeting, and other targeted therapies. Oncotarget 2017;8:73419-32. 10.18632/oncotarget.20621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sos ML, Dietlein F, Peifer M, et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A 2012;109:17034-9. 10.1073/pnas.1207310109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Berns K, Berns A. Awakening of "Schlafen11" to Tackle Chemotherapy Resistance in SCLC. Cancer Cell 2017;31:169-71. 10.1016/j.ccell.2017.01.013 [DOI] [PubMed] [Google Scholar]
- 105.Mavrommatis E, Fish EN, Platanias LC. The schlafen family of proteins and their regulation by interferons. J Interferon Cytokine Res 2013;33:206-10. 10.1089/jir.2012.0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mu Y, Lou J, Srivastava M, et al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep 2016;17:94-109. 10.15252/embr.201540964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang SW, Bilke S, Cao L, et al. SLFN11 Is a Transcriptional Target of EWS-FLI1 and a Determinant of Drug Response in Ewing Sarcoma. Clin Cancer Res 2015;21:4184-93. 10.1158/1078-0432.CCR-14-2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zoppoli G, Regairaz M, Leo E, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A 2012;109:15030-5. 10.1073/pnas.1205943109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Poirier JT, Gardner EE, Connis N, et al. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 2015;34:5869-78. 10.1038/onc.2015.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bracken AP, Pasini D, Capra M, et al. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 2003;22:5323-35. 10.1093/emboj/cdg542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer 2012;106:243-7. 10.1038/bjc.2011.551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Coe BP, Thu KL, Aviel-Ronen S, et al. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS One 2013;8:e71670. 10.1371/journal.pone.0071670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Murai F, Koinuma D, Shinozaki-Ushiku A, et al. EZH2 promotes progression of small cell lung cancer by suppressing the TGF-beta-Smad-ASCL1 pathway. Cell Discov 2015;1:15026. 10.1038/celldisc.2015.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jahchan NS, Lim JS, Bola B, et al. Identification and Targeting of Long-Term Tumor-Propagating Cells in Small Cell Lung Cancer. Cell Rep 2016;16:644-56. 10.1016/j.celrep.2016.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim YH, Girard L, Giacomini CP, et al. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 2006;25:130-8. 10.1038/sj.onc.1208997 [DOI] [PubMed] [Google Scholar]
- 116.Sato M, Shames DS, Gazdar AF, et al. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol 2007;2:327-43. 10.1097/01.JTO.0000263718.69320.4c [DOI] [PubMed] [Google Scholar]
- 117.Peifer M, Fernandez-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012;44:1104-10. 10.1038/ng.2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brägelmann J, Böhm S, Guthrie MR, et al. Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle 2017;16:1489-98. 10.1080/15384101.2017.1339849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Davis FM, Stewart TA, Thompson EW, et al. Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci 2014;35:479-88. 10.1016/j.tips.2014.06.006 [DOI] [PubMed] [Google Scholar]
- 120.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002;2:442-54. 10.1038/nrc822 [DOI] [PubMed] [Google Scholar]
- 121.Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013;19:279-90. 10.1158/1078-0432.CCR-12-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer 2017;116:415-23. 10.1038/bjc.2016.428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rankin EB, Giaccia AJ. The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers (Basel) 2016;8. pii: E103. 10.3390/cancers8110103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wu X, Liu X, Koul S, et al. AXL kinase as a novel target for cancer therapy. Oncotarget 2014;5:9546-63. 10.18632/oncotarget.2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Balaji K, Vijayaraghavan S, Diao L, et al. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol Cancer Res 2017;15:45-58. 10.1158/1541-7786.MCR-16-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Levin PA, Brekken RA, Byers LA, et al. Axl Receptor Axis: A New Therapeutic Target in Lung Cancer. J Thorac Oncol 2016;11:1357-62. 10.1016/j.jtho.2016.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004;22:329-60. 10.1146/annurev.immunol.22.012703.104803 [DOI] [PubMed] [Google Scholar]
- 128.Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999;17:2105-16. 10.1200/JCO.1999.17.7.2105 [DOI] [PubMed] [Google Scholar]
- 129.Klapper JA, Downey SG, Smith FO, et al. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer 2008;113:293-301. 10.1002/cncr.23552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sylvester RJ, van der Meijden AP, Witjes JA, et al. Bacillus calmette-guerin versus chemotherapy for the intravesical treatment of patients with carcinoma in situ of the bladder: a meta-analysis of the published results of randomized clinical trials. J Urol 2005;174:86-91; discussion 91-2. 10.1097/01.ju.0000162059.64886.1c [DOI] [PubMed] [Google Scholar]
- 131.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-9. 10.1038/nature10673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015;27:450-61. 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28:3167-75. 10.1200/JCO.2009.26.7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65. 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23. 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517-26. 10.1056/NEJMoa1104621 [DOI] [PubMed] [Google Scholar]
- 137.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909-20. 10.1016/S0140-6736(16)00561-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ferris RL, Blumenschein G, Jr, Fayette J, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med 2016;375:1856-67. 10.1056/NEJMoa1602252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab vs. Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016;375:1823-33. 10.1056/NEJMoa1606774 [DOI] [PubMed] [Google Scholar]
- 140.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320-30. 10.1056/NEJMoa1412082 [DOI] [PubMed] [Google Scholar]
- 141.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Reck M, Heigener D, Reinmuth N. Immunotherapy for small-cell lung cancer: emerging evidence. Future Oncol 2016;12:931-43. 10.2217/fon-2015-0012 [DOI] [PubMed] [Google Scholar]
- 143.Ott P, Felip E, Hiret S, et al. OA05.01 Pembrolizumab in Patients with Extensive-Stage Small Cell Lung Cancer: Updated Survival Results from KEYNOTE-028. J Thorac Oncol;12:S259 10.1016/j.jtho.2016.11.250 [DOI] [Google Scholar]
- 144.Cheng Y, Li H, Liu Y, et al. Distribution and clinical significance of CTLA4, PD-1 and PD-L1 in peripheral blood of patients with small-cell lung cancer. J Clin Oncol 2015;33:7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ishii H, Azuma K, Kawahara A, et al. Significance of programmed cell death-ligand 1 expression and its association with survival in patients with small cell lung cancer. J Thorac Oncol 2015;10:426-30. 10.1097/JTO.0000000000000414 [DOI] [PubMed] [Google Scholar]
- 146.Yamane H, Isozaki H, Takeyama M, et al. Programmed cell death protein 1 and programmed death-ligand 1 are expressed on the surface of some small-cell lung cancer lines. Am J Cancer Res 2015;5:1553-7. [PMC free article] [PubMed] [Google Scholar]
- 147.Schultheis AM, Scheel AH, Ozretic L, et al. PD-L1 expression in small cell neuroendocrine carcinomas. Eur J Cancer 2015;51:421-6. 10.1016/j.ejca.2014.12.006 [DOI] [PubMed] [Google Scholar]
- 148.Impact of tumor mutation burden on the efficacy of nivolumab or nivolumab + ipilimumab in small cell lung cancer: an exploratory analysis of Checkmate 032. Available online: http://healthdocbox.com/Cancer/71670941-Iaslc-18th-world-conference-on-lung-cancer.html.
- 149.Carbone DP, Reck M, Paz-Ares L, et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N Engl J Med 2017;376:2415-26. 10.1056/NEJMoa1613493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ott PA, Elez E, Hiret S, et al. Pembrolizumab in Patients With Extensive-Stage Small-Cell Lung Cancer: Results From the Phase Ib KEYNOTE-028 Study. J Clin Oncol 2017;35:3823-9. 10.1200/JCO.2017.72.5069 [DOI] [PubMed] [Google Scholar]
- 151.Gadgeel SM, Ventimiglia J, Kalemkerian GP, et al. Phase II study of maintenance pembrolizumab (pembro) in extensive stage small cell lung cancer (ES-SCLC) patients (pts). J Clin Oncol 2017;15:8504. [Google Scholar]
- 152.Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol 2013;24:75-83. 10.1093/annonc/mds213 [DOI] [PubMed] [Google Scholar]
- 153.Mouw KW, Goldberg MS, Konstantinopoulos PA, et al. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov 2017;7:675-93. 10.1158/2159-8290.CD-17-0226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sato H, Niimi A, Yasuhara T, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 2017;8:1751. 10.1038/s41467-017-01883-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sen T, Chen L, Leticia Rodriguez B, et al. Abstract B72: Combining immune checkpoint inhibition and DNA damage repair (DDR) targeted therapy in small cell lung cancer (SCLC). Available online: http://cancerimmunolres.aacrjournals.org/content/5/3_Supplement/B72
- 156.Jiao S, Xia W, Yamaguchi H, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res 2017;23:3711-20. 10.1158/1078-0432.CCR-16-3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hartlova A, Erttmann SF, Raffi FA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015;42:332-43. 10.1016/j.immuni.2015.01.012 [DOI] [PubMed] [Google Scholar]
- 158.Shen YJ, Le Bert N, Chitre AA, et al. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep 2015;11:460-73. 10.1016/j.celrep.2015.03.041 [DOI] [PubMed] [Google Scholar]
- 159.Yu Q, Katlinskaya YV, Carbone CJ, et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep 2015;11:785-97. 10.1016/j.celrep.2015.03.069 [DOI] [PMC free article] [PubMed] [Google Scholar]