

ABSTRACT
Pichia pastoris (syn. Komagataella phaffii) is one of the most common eukaryotic expression systems for heterologous protein production. Expression cassettes are typically integrated in the genome to obtain stable expression strains. In contrast to Saccharomyces cerevisiae, where short overhangs are sufficient to target highly specific integration, long overhangs are more efficient in P. pastoris and ectopic integration of foreign DNA can occur. Here, we aimed to elucidate the influence of ectopic integration by high-throughput screening of >700 transformants and whole-genome sequencing of 27 transformants. Different vector designs and linearization approaches were used to mimic the most common integration events targeted in P. pastoris. Fluorescence of an enhanced green fluorescent protein (eGFP) reporter protein was highly uniform among transformants when the expression cassettes were correctly integrated in the targeted locus. Surprisingly, most nonspecifically integrated transformants showed highly uniform expression that was comparable to specific integration, suggesting that nonspecific integration does not necessarily influence expression. However, a few clones (<10%) harboring ectopically integrated cassettes showed a greater variation spanning a 25-fold range, surpassing specifically integrated reference strains up to 6-fold. High-expression strains showed a correlation between increased gene copy numbers and high reporter protein fluorescence levels. Our results suggest that for comparing expression levels between strains, the integration locus can be neglected as long as a sufficient numbers of transformed strains are compared. For expression optimization of highly expressible proteins, increasing copy number appears to be the dominant positive influence rather than the integration locus, genomic rearrangements, deletions, or single-nucleotide polymorphisms (SNPs).
IMPORTANCE Yeasts are commonly used as biotechnological production hosts for proteins and metabolites. In the yeast Saccharomyces cerevisiae, expression cassettes carrying foreign genes integrate highly specifically at the targeted sites in the genome. In contrast, cassettes often integrate at random genomic positions in nonconventional yeasts, such as Pichia pastoris (syn. Komagataella phaffii). Hence, cells from the same transformation event often behave differently, with significant clonal variation necessitating the screening of large numbers of strains. The importance of this study is that we systematically investigated the influence of integration events in more than 700 strains. Our findings provide novel insight into clonal variation in P. pastoris and, thus, how to avoid pitfalls and obtain reliable results. The underlying mechanisms may also play a role in other yeasts and hence could be generally relevant for recombinant yeast protein production strains.
KEYWORDS: integration, Pichia pastoris, protein expression, genome analysis
INTRODUCTION
The yeast Pichia pastoris (syn. Komagataella phaffii) is widely used for heterologous protein production in both academia and industry (1–3). A recent review (4) suggested that P. pastoris is, after Escherichia coli, the second most commonly used expression system for single-protein expression. P. pastoris has several beneficial traits, such as suitability for high-cell-density bioreactor cultivation (5), the availability of exceptionally strong, tightly regulated promoters (6), and the capacity for secreting large amounts of heterologous proteins (1–3). Furthermore, P. pastoris is readily amenable to genetic modification, and it is relatively easy to integrate multiple copies of an expression cassette, thereby further boosting yields (7–9). Expression cassettes containing the gene of interest are typically integrated into the genome, resulting in stable expression strains (despite recent advances [10], plasmids have been used infrequently). In contrast to the model organism Saccharomyces cerevisiae, however, in P. pastoris it is more difficult to specifically integrate cassettes at a desired locus in the genome. In S. cerevisiae, short overhangs of ∼50 bp flanking the expression cassettes are sufficient to achieve close to 100% correct integration. In contrast, in P. pastoris even ∼1-kb-long overhangs may result in only <1 to 30% specific integration (11, 12). Hence, it appears that nonhomologous end joining (NHEJ) resulting in ectopic, nonspecific integration plays a stronger role in P. pastoris than in S. cerevisiae (where homologous recombination [HR] occurs nearly exclusively).
Over recent decades, relatively little attention has been paid to the effects of different integration events and whether they cause clonal variations that affect protein expression. Commonly, large numbers of transformant strains are screened using high-throughout (HTP) systems (13) resulting in a few highly producing “jackpot clones” (14). Although the term jackpot clone is sometimes used to refer to high-copy-number strains (9), other effects, such as the integration site and genome rearrangements, also may influence expression (15–19). The transcriptomes of P. pastoris strains secreting different amounts of human serum albumin were compared using microarrays, but the authors did not find clear regulatory patterns which could mechanistically explain the clonal variation observed (17).
With the advent of next-generation whole-genome sequencing (WGS), reference genomes (20–22) and refined sequencing and annotations (23–25) have become available for different P. pastoris wild-type strains. With further decreases in the cost of these technologies, systematic studies of the underlying mechanisms for clonal variation in P. pastoris transformants have become feasible. Recently, milestone papers by the group of Karl Friehs investigated the influence of integration events on protein expression (26, 27), providing insight into various mechanisms of multicopy integration (e.g., orientation of the cassettes [26]) and noncanonical integration events influencing growth phenotypes (27). While these integration events can have a profound influence on strain productivity and physiology, it remains partly unclear to what extent they occur, how they are influenced by expression cassette properties, and how they bias typical standard expression approaches in P. pastoris. For comparisons of different enzyme variants (e.g., from natural variants or protein engineering efforts) or promoter variants (e.g., to fine-tune expression [28–30]), it is necessary to perform multiple transformations for each clone expressing a different gene. Expression from the actual transformants used for comparisons may be biased by clonal variation to an unknown extent, possibly influencing the interpretation of the study if only a limited number of transformants are tested.
Here, we aimed to investigate how clonal variation in P. pastoris transformants affects protein expression levels and if this variation is influenced by commonly used vector designs and typical vector linearization strategies targeting different integration loci. Thus, we set up experiments mimicking commonly used P. pastoris protocols with regard to expression plasmids, how the plasmids are linearized to target integration, and amounts of DNA used. The most commonly used P. pastoris plasmids (11, 31, 32) are propagated in E. coli in circular form and linearized prior to transformation (as free DNA ends strongly increase the rate of integration into the genome [33–37]). As most standard plasmids contain the strong, methanol-inducible AOX1 promoter (PAOX1), enzymes cutting at the 5′ end of PAOX1 (typically BglII) or within the AOX1 promoter (commonly SacI) are used. In case the vector is linearized at the 5′ end of PAOX1, typically a second homologous sequence complementary to the downstream region 3′ of the AOX1 gene in the genome is provided. Correct integration by a double-crossover omega-type recombination event (ends-out configuration [38]) results in a replacement/deletion of the natural AOX1 gene with the heterologous expression cassette. Of note, deletion of the AOX1 gene results in a slow methanol utilization (mutS) phenotype that can be favorable for recombinant protein production (39) depending on the gene of interest (if already using a mutS strain, the ends can be designed to knock out any other gene). Alternatively, if linearization is performed within the AOX1 promoter, an ends-in (38)-type integration event is targeted, resulting in insertion of the expression cassette without deletions.
Here, we generated and screened >700 transformants expressing enhanced green fluorescent protein (eGFP) and analyzed 25 of these by WGS.
RESULTS
Effect of plasmid design, vector linearization, and type of integration event.
Three different vector/linearization approaches were tested (Fig. 1). (i) A vector containing an eGFP reporter gene under the control of PAOX1 with sequences homologous to the genome on both sides which, when linearized with SwaI, would target specific integration into the GUT1 (glycerol utilization 1) locus (the vector here is referred to as GUT1) (Fig. 1A). Gut1p is an essential enzyme in glycerol metabolism, hence gut1 knockout strains cannot grow on glycerol. However, growth on glucose and methanol is not impaired (11). (ii) The same vector was also linearized with SacI in the AOX1 promoter. Thus, insertion at the AOX1 promoter in the genome was targeted. (iii) Finally, we used an unmodified vector without the additional integration sequences targeting the GUT1 locus (Fig. 1B). This vector was intended as a control to more closely mimic standard expression strategies (termed STD). This control would show if the GUT1 targeting sequences influenced integration or expression of our eGFP reporter gene by the vector. This vector was also linearized with SacI, targeting the genomic AOX1 promoter.
FIG 1.

Setup of the study using integration events targeted by different plasmid designs and linearization. (A) A reporter plasmid bearing sequences homologous to the GUT1 locus (GUT1 5′ and 3′) is linearized with SwaI or SacI. Linearization with SwaI results in overhangs suitable for an omega/ends-out-type recombination event (38) via double crossover at the GUT1 locus in the genome. Correct integration will result in a replacement of the GUT1 gene with the heterologous expression cassette (i.e., Δgut1). Linearization of the same vector with SacI targets a recombination event via the ends-in (38) at the AOX1 promoter in the genome. (B) A SacI integration event was also performed with a control vector lacking GUT1 integration sequences. The reporter plasmid bears an enhanced GFP (eGFP) gene under the control of the AOX1 promoter (pAOX1) and the AOX1 transcription terminator (AOX1TT). The zeocin resistance (ZeoR) cassette consists of an ILV5 promoter for expression in P. pastoris, an EM72 promoter for expression in E. coli, the Sh ble gene, and a terminator (details not shown). The gray sequence between pUC ORI and GUT1 3′UTR/PAOX1 is a remnant present in typical pPpT4-derived vectors (11). Panels A and B are not drawn on the same scale (although elements within panels A or B are at correct relative scale).
Our integration plasmids (Fig. 1) were based on phleomycin D1 (zeocin) antibiotic selection, as auxotrophic markers had resulted in mixed cultures in previous studies (26). We linearized the expression plasmids with the respective restriction enzymes and gel purified the bands (to avoid carryover of uncut vector or foreign DNA present [26, 27]). P. pastoris cells were transformed in triplicate with each plasmid to test variability and potential influence of the transformation events. Additionally, we intended to perform whole-genome sequencing of a subset of strains, and we aimed to select transformants from different transformation events. It is possible that cells duplicate during the regeneration phase after the transformation. Hence, clones showing similar expression levels arising from the same transformation event theoretically could be identical. By performing the transformations in triplicate, we ensured that at least three different clones showing similar expression were available for genome sequencing.
For each transformation, one 96-well deep-well plate (DWP) was screened (inoculated with 84 transformants and 12 controls), resulting in a total of 755 transformants screened (see Fig. S1 in the supplemental material for the complete screening data).
Summarized results for the three replicate transformation events with the three vector designs and linearization strategies are shown in Fig. 2A and B. In Fig. 2A, eGFP expression values for each transformant are sorted from lowest to highest, resulting in expression landscapes as typically used in the literature to present such results (40–43). For statistical interpretation, the same results are shown as boxplots in Fig. 2B. In a box plot, 50% of the data points lie within the box, with box limits indicating the 25th and 75th percentiles. The whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, representing approximately ±2.7 standard deviations. Hence, according to a normal distribution, 99.3% of the data were expected to be within the whiskers (44), and outliers are represented by dots. Transformation results from all plasmids/linearization approaches appear to follow a normal distribution, except for outliers (GUT1-SwaI, 7; GUT1-SacI, 5; STD-SacI, 12), which accounted for 3, 2, and 5% of the sample population, respectively (n = 252, 252, and 251) (Fig. 2B). Hence, the percentage of outliers was higher than expected from a normal distribution (0.7%). The expression range of the outliers also varied more strongly. Comparing the highest-expressing clone with the lowest one gives ranges of 28 × 103, 57 × 103, and 97 × 103 relative fluorescence units [RFU] per unit of optical density at 600 nm [OD600] for GUT1-SwaI, GUT1-SacI, and STD-SacI, respectively (using a cutoff of <500 RFU/unit of OD600 to discriminate nonexpressing clones). Hence, the range of expression levels for transformants with the SacI-linearized plasmids was 2.0 (GUT1)- and 3.4 (STD)-fold larger than that of transformants with the SwaI-linearized plasmid.
FIG 2.

Screening of 755 P. pastoris transformants indicates that plasmid design, vector linearization, and the type of integration event (specific/nonspecific) mostly influences the expression range of outliers but not the population distribution. (A) Vectors providing GUT1 integration sequences and a standard vector design were linearized with SwaI and/or SacI targeting the integration events depicted in Fig. 1. Cells were pregrown on glucose for 60 h and subsequently induced with methanol for 48 h. eGFP reporter fluorescence normalized to cell growth (OD600) is shown. Results of landscapes typical for work with P. pastoris (40–43) are shown. Each bar represents a transformant (n = 252, 252, and 251 sample points). (B) The data from panel A are shown as a boxplot (59). Center lines show the medians, and box limits indicate the 25th and 75th percentiles as determined by R software. Whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, and outliers are represented by dots. n = 252, 252, and 251 sample points. (C) Expression landscapes of the vector providing GUT1 integration sequences linearized with SwaI sorted by specific/nonspecific integration. Hence, the first third of the data from panel A is shown in a rearranged fashion. Transformants were replica plated in glycerol-containing medium after growth on glucose for 60 h to test for specific/nonspecific integration. Note that the GUT1-SacI and STD-SacI integrating vectors cannot be tested for correct integration in this way. (D) The same data from panel C are shown as a boxplot (59). Center lines show the medians, and box limits indicate the 25th and 75th percentiles as determined by R software. Whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, and outliers are represented by dots; the width of the boxes is proportional to the square root of the sample size. n = 158 and 94 sample points.
In Fig. 2A and B, results for all plasmids/linearizations are plotted independently of the integration event. For the GUT1-SwaI linearized plasmid, it was possible to determine the integration event by checking for abolished growth on glycerol (Fig. 1A; results are shown in Fig. 2C and D). Sixty-three percent of transformants showed specific integration by knocking out the GUT1 gene (158 versus 94 transformants), and no clear differences were noted between separate transformation replicates (Table S2). Separate expression landscapes (Fig. 2C) and box plots (Fig. 2D) are shown for specific and nonspecific integration events. Transformants from both integration types showed a similar median-and-whiskers spread. However, the number of outliers was different. Specific integration showed only one outlier, whereas in the case of nonspecific integration seven outliers were found. These outliers accounted for 0.6 and 7% of the sample for specific and nonspecific integration, respectively (n = 158 and 94) (Fig. 2D). In addition, the expression range was 1.3-fold larger for nonspecific integration than specific expression (expression ranges, 21 × 103 and 28 × 103 RFU/OD600, again using a cutoff of <500 RFU/OD600 to discriminate between nonexpressing clones).
Rescreening of biological replicates shows that the clones span a 25-fold expression range.
The results shown in Fig. 2 are single biological measurements (e.g., each colony arising from the transformation event was only measured once). While this summarized data can be used to draw conclusions about the whole sample size (Fig. 2B and D), the biological variation of specific transformants is unknown (e.g., highly expressing clones could show a larger variation than average or low expressers). Therefore, we rescreened 44 selected transformants in 4-fold biological replicates (Fig. 3 and Fig. S3). Colonies from transformation plates may represent mixed populations (although unlikely, two cells may end up in close proximity on the plate and appear after growth as one colony). Hence, we restreaked all transformants and used only separated single colonies for the rescreening (Fig. 3A).
FIG 3.

Strains selected for whole-genome sequencing span a 25-fold expression range. (A) Workflow from screening to whole-genome sequencing (WGS). Forty-four transformants from the screening pool of 755 transformants were used for dilution streaking and rescreened in biological 4-fold replicates. Twenty-seven strains eventually were used for WGS. The image of the dilution streaking is taken from the Public Health Image Library (identifier 7925), CDC/James Gathany. (B) eGFP fluorescence measurements of the strains selected for WGS span a 25-fold expression range, and the highest-expressing strain surpasses average clones by more than 6-fold. Cells were pregrown on glucose for 60 h and subsequently induced with methanol for 48 h. eGFP reporter fluorescence normalized to cell growth (OD600) is shown. Mean values and standard deviations of biological 4-fold replicates are shown. Here, 25 strains transformed with eGFP plasmids are shown, and the parental strain (mutS) and the mutS strain transformed without DNA were sequenced. Identifiers refer to internal strain collection numbers assigned at QUT.
The transformants were selected based on different criteria: (i) outliers showing higher or lower reporter fluorescence than expected from the normally distributed landscape (Fig. 2) and (ii) clones showing average expression, arising from either specific or nonspecific integration. We selected these transformants from each vector/linearization combination with the aim of investigating how similar expression is obtained despite different integration sites. In general, the initial screening results were reproduced in the rescreening (see Table S3 for a list of transformants tested, brief comments on their selection, and extended discussion of the results).
Based on the rescreening results, we selected a diverse set of 25 strains for whole-genome sequencing (Fig. 3 and Table 1). Two control strains were also included (the untransformed parental strain [mutS] and the parental strain electroporated without DNA to account for possible influences from the transformation event itself [empty; QTV19]). Thus, a total of 27 strains were sequenced. We selected average transformants from each plasmid/linearization combination; for GUT1 SwaI we included specifically and nonspecifically integrated transformants. Transformants showing large standard deviations in the rescreening (Table S3) were omitted. Furthermore, we also included low expressers (with one clone, QTV84, showing no detectable expression at all) and high expressers. The highest- and lowest-expressing clones selected for sequencing spanned a 25-fold range, and notably the highest-expressing strain surpassed average expression more than 6-fold (Fig. 3).
TABLE 1.
Details of the 27 P. pastoris strains analyzed by next-generation sequencingf
| No. | Identifier | Vector | Linearization | Integration | Group | Replicate | SNPs and indelsc | Estimated CNd |
Integration locus | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SAMtools | TPM | Rounded | |||||||||
| 1 | wt/mutSa | Control | NA | NA | NA | NA | 37e (15 in exons) | NA | NA | NA | NA |
| 2 | QTV19b | Control | NA | NA | NA | NA | 27e (11 in exons) | NA | NA | NA | NA |
| 3 | QTV76 | GUT1 vector | SwaI | Specific | Avg | 1 | 1 (intergenic) | 0.6058 | 0.6768 | 1 | GUT1 |
| 4 | QTV77 | GUT1 vector | SwaI | Specific | Avg | 2 | 2 (intergenic) | 0.6608 | 0.5824 | 1 | GUT1 |
| 5 | QTV78 | GUT1 vector | SwaI | Specific | Avg | 3 | 5 (intergenic, one mitochondrial intergenic) | 0.7201 | 0.6951 | 1 | GUT1 |
| 6 | QTV79 | GUT1 vector | SwaI | Nonspecific | Avg | 1 | 3 (2 in exons, 1 intergenic) | 0.7010 | 0.5707 | 1 | Chromosome 2: 3,438,50 bp in ACIB2EUKG769578 |
| 7 | QTV80 | GUT1 vector | SwaI | Nonspecific | Avg | 2 | 3 (1 in exon, 2 intergenic) | 0.8522 | 0.7346 | 1 | Chromosome 2: 1,536,800 bp between two genes |
| 8 | QTV81 | GUT1 vector | SwaI | Nonspecific | Avg | 3 | 2 (intergenic) | 0.9243 | 0.8395 | 1 | Chromosome 2: 1,472,800 bp, no annotation |
| 9 | QTV82 | GUT1 vector | SwaI | Nonspecific | High | 1 | 2 (1 in exon, 1 intergenic) | 1.8054 | 1.7745 | 2 | Chromosome 4: 6,794,00 bp near ACIB2EUKG772429 |
| 10 | QTV83 | GUT1 vector | SwaI | Nonspecific | High | 2 | 2 (1 intergenic, 1 mitochondrial intergenic) | 1.4162 | 1.3572 | 1 | Chromosome 1: 1,994,650 bp, ACIB2EUKG768896 |
| 11 | QTV84 | GUT1 vector | SwaI | Nonspecific | Low/no | 1 | 23 (intergenic) | 1.2110 | 1.1716 | 1 | ∼69-kbp deletion at the end of chromosome 4: 1,752,000–1,821,000 bp; 17 genes deleted |
| 12 | QTV85 | GUT1 vector | SwaI | Low/no | 2 | 1 (exon) | 0.7803 | 0.6157 | 1 | Chromosome 1: 1,579,000 bp, ∼550 bp deleted between ACIB2EUKG768656 and ACIB2EUKG768657 | |
| 13 | QTV86 | GUT1 vector | SacI | Avg (lower) | 1 | 2 (1 intergenic, 1 mitochondrial intergenic) | 1.0203 | 0.7384 | 1 | Chromosome 4: 2,380,00 bp in AOX1 promoter | |
| 14 | QTV87 | GUT1 vector | SacI | Avg (lower) | 2 | 2 (intergenic) | 0.8369 | 0.7783 | 1 | Not found | |
| 15 | QTV88 | GUT1 vector | SacI | Avg (lower) | 3 | 3 (1 in exon, 2 intergenic) | 0.8240 | 0.7028 | 1 | Not found | |
| 16 | QTV89 | GUT1 vector | SacI | Avg (higher) | 1 | 3 (intergenic) | 0.8657 | 0.7019 | 1 | Not found | |
| 17 | QTV90 | GUT1 vector | SacI | Avg (higher) | 2 | 10 (1 intergenic, others in same gene) | 0.8637 | 0.7428 | 1 | Not found | |
| 18 | QTV91 | GUT1 vector | SacI | Avg (higher) | 3 | 1 (intergenic) | 0.8371 | 0.7704 | 1 | Not found | |
| 19 | QTV92 | GUT1 vector | SacI | High | 1 | 6 (1 in exon, 5 intergenic) | 1.8785 | 2.0084 | 2 | Not found | |
| 20 | QTV93 | GUT1 vector | SacI | High | 2 | 4 (3 intergenic, 1 mitochondrial intergenic) | 2.0182 | 2.1124 | 2 | Not found | |
| 21 | QTV94 | GUT1 vector | SacI | High | 3 | 2 (1 in an exon, 1 intergenic) | 2.2047 | 2.3392 | 2 | Not found | |
| 22 | QTV95 | STD T4 | SacI | High | 1 | 1 (intergenic) | 2.3142 | 2.3378 | 2 | Not found | |
| 23 | QTV96 | STD T4 | SacI | High | 2 | 1 (intergenic) | 4.6788 | 4.7903 | 5 | Not found | |
| 24 | QTV97 | STD T4 | SacI | High | 3 | 2 (intergenic) | 4.9057 | 4.9626 | 5 | Not found | |
| 25 | QTV98 | STD T4 | SacI | Avg | 1 | 1 (intergenic) | 0.8635 | 0.8346 | 1 | Not found | |
| 26 | QTV99 | STD T4 | SacI | Avg | 2 | 1 (intergenic) | 0.8234 | 0.7851 | 1 | Not found | |
| 27 | QTV100 | STD T4 | SacI | Avg | 3 | 10 (2 in mitochondrial genes, rest intergenic) | 1.3011 | 1.3548 | 1 | Not found | |
Unmodified parental strain (mutS).
Empty transformation (without DNA) of the unmodified parental/reference strain wt/mutS (to check if electroporation procedure for transformation of DNA has a negative effect).
Number of unique variants with a quality score of >20 using bcfvariant tools (i.e., unique compared to the mutS reference strain).
See Table S7 in the supplemental material for details on the copy number calculation methods SAMtools (BAM stats) and TPM.
Relative to the PacBio reference sequence (23).
NA, not applicable.
Whole-genome sequencing of 27 strains. (i) Mapping to reference and integration sites.
Approximately 3 million to 16 million high-quality 150-bp paired-end reads were obtained for the 27 strains (Table S4). The reads were mapped to the newest reference sequence of the P. pastoris CBS7435 strain (9,381,467 bp) (23), the mitochondrial genome FR839632.1 (35,683 bp), and the corresponding plasmid (GUT1 or STD). To accurately map the reads to chromosome 4, the reference sequence was modified to remove the AOX1 gene and an additional sequence, introduced when it was deleted, was added. On average, 92.32% of paired reads mapped to the genomes with average coverage across the four chromosomes, mitochondrial genome, and plasmid ranging from approximately 40 to 250 times (Table S4). The mapping quality was high and error rate low (Table S4). When the reads were mapped against the unmodified reference sequence of the wild-type strain, it was clear that the AOX1 gene was deleted in all strains as expected from using a mutS parental strain.
We used the mapping data to look for plasmid insertion sites and any other large changes to the sequence due to the transformation events. We used a BLAST read-walking method similar to that described in Chambers et al. (45), starting with the cut ends of the plasmids. We then looked for gaps or mismapping in the regions identified. Due to the presence of endogenous P. pastoris sequences on the plasmid, some of the BLAST results were ambiguous. In total, we could clearly identify the integration site in 44% of the strains (11 out of 25) (Table 1), as the genome sequence was disrupted. As expected, the GUT1 gene on chromosome 4 was deleted in QTV76, QTV77, and QTV78. Insertion sites could also be found for the other strains transformed with the GUT1 plasmid cut with SwaI (Table 1; see Figure S5 for examples). Most were between genes (Table 1), and there was no obvious similarity in the sequence around the insertion site. Closer examination of the reads bordering these putative insertion sites usually showed that they contained parts of the GUT1 3′ or 5′ sequence and then the corresponding genomic sequence. Only one potential insertion site could be clearly observed with a SacI-cut plasmid (strain QTV86); this was in the AOX1 promoter, as expected.
There were no obvious large rearrangements in any of the strains, but there was a large deletion of approximately 69 kbp coding for 17 genes at the end of chromosome 4 in strain QTV84, which had no eGFP expression (see Table S6 for a full list). The other lower-expressing line, QTV85, also has an approximately 550-bp deletion that could affect the function and/or expression of ACIB2EUKG768656 or ACIB2EUKG768657 on chromosome 1, which encode phosphatidylethanolamine N-methyltransferase and a P-loop containing nucleoside triphosphate hydrolase, respectively.
Effect of CN variations, SNPs, and insertions and deletions (indels).
To estimate plasmid copy number (CN), we calculated the ratio of reads mapped to the chromosomes compared to reads mapped to the plasmid, assuming that coverage is even across all sequences (rounded values are summarized in Table 1, and raw data/calculations are provided in Data Set S7).The CNs were calculated using the read-mapping data from SAMtools (46). The coverage (number of reads mapped to the chromosome divided by its length) should be roughly equivalent to one copy (as there is only one copy of each chromosome). Hence, a relatively higher coverage of the plasmid is attributable to more than one copy of the plasmid. We also used Salmon (47), normally applied to RNA sequencing data, to calculate transcripts per million (TPM) and used these values to infer CN (see Materials and Methods and Data Set S7 for details) for GFP, the zeocin resistance gene, and the pUC-origin sequence, all genetic elements on the vector that can be unambiguously distinguished from genomic P. pastoris endogenous sequences. The CNs for eGFP, zeocin, and pUC were averaged (see Data Set S7 for the raw data and calculations) and were in excellent agreement with the values obtained from the SAMtools BAM stats coverage data (R2 = 0.99; Data Set S7B). This CN determination method yielded highly reproducible results. For the independently generated strains QTV76 to QTV78 (specifically integrated in the GUT1 locus and showing uniform expression), the calculated CNs varied by only ca. 10% (0.61% ± 0.07%). The expected CN of 1 was slightly underestimated, but variation to this extent was also noted with quantitative PCR (qPCR) (48) CN determinations in P. pastoris (43, 49, 50). Also, relative conclusions on the CN differences between the strains were not influenced.
Interestingly, CNs correlated well (R2 = 0.82) with the expression level (Fig. 4, reporter protein fluorescence), suggesting that this is the main factor contributing to the striking difference in expression levels we observed. The two highest-expressing lines were estimated to contain four or five copies of the plasmid compared to one copy in most lines.
FIG 4.
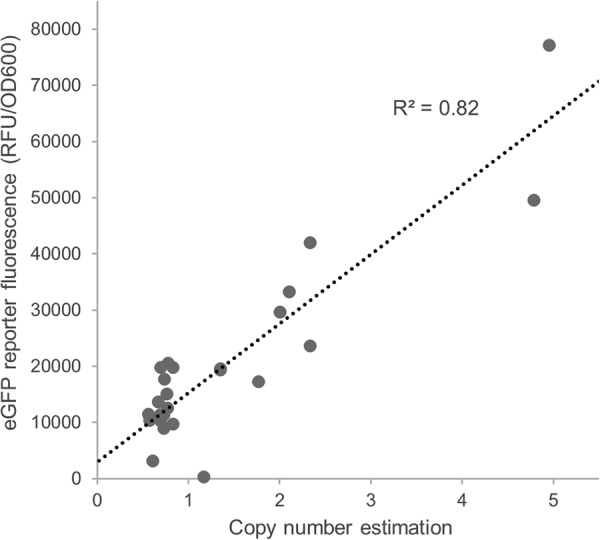
Copy numbers correlate with measured eGFP reporter protein fluorescence. Copy numbers (summarized in Table 1; raw data and calculation are shown in Data Set S7), and eGFP reporter protein fluorescence measurements (normalized by OD600, as obtained from the rescreening and shown in Fig. 3) were correlated. The raw data of the unrounded copy numbers are shown (Data Set S7).
We also analyzed the sequencing data for SNPs and indels. We used bcfvariant tools to identify SNPs and indels between the 27 lines and the CBS7435 reference (results are summarized in Table 1; see Data Set S8 for details on each strain). We first analyzed the two control strains: (i) the unmodified parental strain and (ii) strain QTV19, which was electroporated without plasmid DNA (to check for detrimental effects of the transformation event itself) (Table 1). In the mutS control we found 37 changes, including SNPS (12) and indels (25), spread across all four chromosomes and the mitochondrial genomes. Indels were between 1 and 21 bp in length, with insertions being most prevalent (15 insertions between 1 and 10 bp in length and 9 deletions between 11 and 21 bp; see Data Set S8 for the exact changes). We also found 27 SNPs and indels in QTV19 on chromosomes 1, 3, and 4 and the mitochondrial genomes.
We next examined the occurrence of SNPs and indels in the 25 transformants (Table 1 and Data Set S8). The number of unique variants in the transformed strains ranged from 1 to 23 (Table 1 and Data Set S8), including changes in the mitochondrial genome. Interestingly, unlike CN, the number and type of variants varied in the three transformants that were replicates (QTV76 to QTV78). However, there was a very poor correlation (R2 = 0.08) between the number of SNPs/indels and reporter protein fluorescence (Fig. S9), indicating that unlike CNs (Fig. 4), the detectable SNPs do not appear to have a prevalent influence on expression. The lowest-expressing strain, QTV84, did have the highest number of variations, but these were artifacts related to the large deletion on chromosome 4.
DISCUSSION
Effect of plasmid design, vector linearization, and type of integration event.
The high-throughput screening of >700 P. pastoris transformants in this study found highly uniform eGFP reporter protein fluorescence among the vast majority of transformants (>90%) and, surprisingly, for most nonspecifically integrated transformants (Fig. 2D). However, the number of outliers increased upon nonspecific integration, showing a greater variation spanning a 25-fold range, surpassing specifically integrated reference strains up to 6-fold.
Interestingly, linearization of the same plasmid (GUT1) with two different restriction endonucleases yielded different expression medians (Fig. 2B). These differences may only partly be explained by specific versus nonspecific integration (Table 1), as both specifically and nonspecifically integrated GUT1 SwaI clones (Fig. 2D) showed lower reporter fluorescence than the SacI-linearized plasmid. A possible explanation is an influence of the GUT1 5′ region on the expression of PAOX1: upon SacI linearization the GUT1 5′ region stays attached to PAOX1, whereas it is separated upon SwaI cleavage (Fig. 1A and extended discussion in Supplement S10 in the supplemental material). Hence, the GUT1 5′ region may have a repressing effect on PAOX1. Similarly, in S. cerevisiae the integration site (and thereby possibly the upstream region) was shown to have a pronounced effect on expression (51).
Varying the plasmid design by omitting the GUT1 5′ and 3′ regions for targeted integration (Fig. 1B, STD design) did not have a clear effect compared to the GUT1-containing design (Fig. 2B). The STD design yielded outliers with higher reporter protein fluorescence (and copy numbers [Table 1]) than the GUT1 design. However, these rare events (<5%) are difficult to interpolate to general conclusions, as they may appear stochastically, and the occurrence of three higher-expressing clones in the STD design cannot be generalized.
While we performed these experiments with the GUT1 locus and the AOX1 promoter region, in principle, any locus could have been used for this experiment. We chose GUT1, as it was reported to be well suited for HTP screening (12). Notably, we obtained higher specific integration rates in the GUT1 locus by homologous recombination than those reported in previous studies (12), despite using identical 5′ and 3′ homologous regions and expression cassettes based on a similar vector family. Although hard to rationalize, this difference may be related to the insertion of longer sequences (PAOX1, eGFP, and terminator expression cassette) between the GUT1 5′ and 3′ regions in our study, as the previous knockout cassette contained only a resistance marker (12, 31). Elucidating the exact mechanisms will require further studies.
The amount of DNA transformed typically influences the outcome of transformation events in P. pastoris, as large amounts of DNA (several micrograms) give higher chances of multicopy integrations. We aimed to investigate specific/nonspecific integration, hence we transformed small amounts of DNA to avoid the pronounced occurrence of multicopy integrations. Previous studies with reporter plasmids from the pPpT4 family (28–30) showed that 1 μg of linearized vector DNA resulted in an even expression landscape, i.e., similarly expressing clones. Hence, we also used amounts equivalent to 1 μg of the reference constructs from references 28–30. Due to the long integration sequences, the GUT1-targeting vector is considerably larger (6,480 bp) than the reference constructs, so 1.5 μg was transformed to reach similar numbers of vector molecules. We obtained even expression landscapes, yet outliers and multicopy integration did occur (Fig. 3) and could not be completely avoided. If larger amounts of linearized plasmid were transformed (i.e., several micrograms), the transformant population distribution would likely be even more skewed by outliers and highly expressing multicopy strains. Hence, we assume that our results represent a rather conservative estimate of the influence of outliers, copy number, and integration events in P. pastoris.
Whole-genome sequencing.
High-coverage (Table S4) whole-genome sequencing of 27 transformants (Table 1) provided insights into the effect of CNs, helped to identify the integration locus, and allowed comparisons of SNPs and indels. Copy number estimations pinpointed CN variations as the major influence on reporter protein fluorescence in our setting (Fig. 4), as supported by more detailed studies on multicopy integration orientation (26).
It has previously been noted that genetic changes can occur in transformed strains from the same transformation event in P. pastoris (15). Viader-Salvadó and colleagues noted altered amplified fragment length polymorphism (AFLP) patterns between transformants (15), yet it has remained unclear which genetic changes caused these altered patterns. We reasoned that newly introduced single-nucleotide polymorphisms (SNPs) or short indels may have caused these pattern changes and could affect recombinant protein expression. Fifteen (out of 37 in total) and 11 (out of 27 in total) of the changes in the parental mutS and QTV19 strain (Table 1 and Fig. S8), respectively, were in exons of annotated genes. Similarly, Sturmberger et al. (23) found 24 differences (SNPs and indels) between CBS7435 and paired-end Illumina HiSeq reads of BioGrammatics' strains BG08 and BG10, stating that “Only small clonal variations occur between these closely related P. pastoris strains, even after storage at different sites for many years, indicating defined molecular manipulation can be precise with relatively little clonal drift.” Many of the indels are in repetitive regions and thus could reflect the differences in short Illumina read (used in this study) and long PacBio read (used for obtaining the reference sequence [23]) sequencing technologies (52) to correctly sequence repetitive and GC-rich regions. Even though these mutations obtained quality scores of >20, which primarily indicates that all of the reads that mapped to that region had the same change in it compared to the reference, the change could still also represent technical bias arising from the sequencing method used. Regardless, there was no overall correlation between the unique variants in the transformants and their expression levels (Fig. S9).
We could unambiguously map the integration sites in 44% (11 out of 25) of the strains (Table 1). In most cases the integration site did not have a clear influence on reporter protein fluorescence (Fig. 3), yet we did notice in one strain (QTV84) a large deletion of 69 kbp on chromosome 4. This strain showed impaired growth, likely attributable to the 17 genes deleted, which included five plasma membrane transporters (including two sugar transporters and an iron transporter), one S. cerevisiae Gal4-like transcription factor, two glycoside hydrolases, and a putative cell wall flocculin, among others (Table S6). Such rare integration events have been reported in the past yet were mostly attributed to the integration of E. coli sequences (27), whereas the large deletion observed in our case suggests a different mechanism. Only one potential insertion site could be clearly observed with a SacI-cut plasmid (strain QTV86); this was in the AOX1 promoter, as expected. It appears possible that in several lines, the cassette inserted seamlessly into the AOX1 promoter region, but this could not be observed by a disruption in the mapping. For some transformant strains, such as QTV96 and QTV97, the BLAST readwalking also pointed to a region on chromosome 3, but no disruptions were apparent and this appeared to be an artifact, since a 466-bp sequence on chromosome 3 in gene ACIB2EUKG771569 is almost identical to the AOX1 terminator sequence. The number of apparently specifically integrated transformants at the genomic AOX1 promoter appeared higher than expected from a literature report (11); linearization within PAOX1 using SacI results in uneven and partly relatively short overhangs (729 and 205 bp) that are thought to be suboptimal for integration via HR (11) (Fig. 1A and B, bottom).
In the transformants, where the integration locus could not be clearly identified (Table 1), limitations with the capabilities of the sequencing technology and the plasmid architecture were apparent. Since the plasmids used also contain P. pastoris endogenous sequences (e.g., the AOX1 promoter/terminator, the promoter/terminator of the zeocin resistance cassette, and GUT1 5′ and 3′ sequences, if applicable), it was somewhat difficult to discriminate between reads mapping to endogenous sequences in the genome and reads arising from the inserted plasmid. The 150-bp paired-end reads used did unambiguously cover the foreign/unique eGFP, pUC, and zeocin resistance elements, but as these sequences were embedded between endogenous sequences, it was difficult to clearly infer the genomic integration locus by assembling the reads into longer contigs. Multicopy integration in some transformants (Table 1) may have further complicated the mapping and/or assembly. In future studies these issues could be avoided by following different approaches. (i) Longer reads covering the transitions between foreign and endogenous vector elements would allow unambiguous alignment of all reads and assembly of the integration site. Therefore, single-molecule real-time sequencing (SMRT) (53, 54) with read lengths of, on average, >10 kbp would simplify analysis if available. (ii) Mapping issues could be avoided by omitting (or reducing) the use of P. pastoris endogenous sequences at the 5′ and 3′ ends of the expression cassette used. This approach will not be feasible for all elements, as integration sequences need to be identical for targeted integration. However, regarding promoter or terminator sequences, synthetic promoters or heterologous terminators established in P. pastoris could be used. Alternatively, it would also be possible to add synthetic barcode sequences in the vector to facilitate identification.
Integration studies in P. pastoris and other yeasts.
A considerable decrease in the cost of next-generation sequencing and WGS over the last decade (54) has permitted comparisons of interclonal variation rather than sequencing only a single strain. Recently, Karl Friehs' group thoroughly investigated integration event-induced changes in recombinant protein productivity in P. pastoris, focusing on the nature of multicopy integration events and clones with abnormal colony morphology in two studies by Schwarzhans et al. (26, 27). While these studies provide insights into P. pastoris clonal variation, our alternative study design focused on different aspects and overcame some experimental limitations. For instance, Schwarzhans et al. noticed contamination in their sequencing efforts, possibly arising from mixed cultures due to the use of an auxotrophic HIS marker and the lack of dilution plating (26). Our integration plasmids (Fig. 1) were based on phleomycin D1 (zeocin) antibiotic selection, and transformants were carefully restreaked and rescreened (Fig. 3). Thus, we could accurately assess clonal variation and pinpoint the expression spread. In the clones that we sequenced, we did not observe the cointegration of DNA from the E. coli plasmid host, leading to impaired growth as reported by Schwarzhans et al. (27). We did, however, observe the loss of a large fragment of approximately 69 kbp in one of our strains with a growth defect (QTV84), suggesting deletions as an additional possible cause for clonal variation in P. pastoris.
Other notable differences in the study design by Schwarzhans et al. (26, 27) and our work are the vector design/linearization (Fig. 1), DNA amount transformed, and the authors changing the terminator of their expression plasmid to avoid recombination with the endogenous AOX1 terminator. We did not notice such recombination events in the combination of our vector and transformation setup, suggesting that such vector modifications are not imperative in every experimental context.
Our study suggests that nonspecific integration does not necessarily affect expression across the population of transformants but rather that the number of high- and low-expression outliers is influenced (Fig. 2D). This notion has interesting implications for comparing P. pastoris expression strains arising from different transformation events. If enzyme or promoter variants are being compared for expression levels, picking outliers from a population may strongly bias the results, leading to an unclear assessment of the effect of the enzyme or promoter variations. To rule out these effects, two strategies appear feasible: (i) confirming specific integration (by demonstrating growth phenotypes, by colony PCR, or by using site-specific molecular “landing pad” recombination systems [55]) or (ii) screening a sufficient number of transformants to obtain a representative population distribution and thus avoid outliers.
The fact that many jackpot clones showing high expression were obtained only by extensive screening in the past (for examples, see references 7, 14, and 56) suggests that copy number variation, genomic integration sites, and even genomic deletions and rearrangements (27) altogether have unique effects for different proteins of interest. This notion may explain why P. pastoris has become such a successful heterologous protein expression system and represents one of its key assets: even without mechanistic understanding of the expression requirements of a little-studied or unknown protein, highly expressing jackpot clones can be obtained by large-scale screenings and beneficial events arising stochastically from the clonal variation in P. pastoris. Disentangling the exact mechanisms will require larger-scale studies focusing on a multitude of proteins. However, screening thousands of transformants of multiple proteins is highly laborious. Hence, a promising approach would be to collect high-expressing strains reported in the literature and to sequence them as a community effort.
MATERIALS AND METHODS
Strains and materials.
The strain used in this study was the P. pastoris BG11 strain (derivative of P. pastoris BG10 strain, Δaox1; methanol utilization slow) from ATUM, Inc. (Newark, CA). The NEB 5′-alpha competent Escherichia coli strain (New England BioLabs) was used for cloning. Small-scale cultivations were performed using the DWP and induction protocols reported previously (13).
Reporter constructs.
The vector used for cloning was pD912 from ATUM (formerly DNA2.0), which is based on the pPpT4S vector family (11). An eGFP reporter gene was cloned into pD912 by linearizing it with EcoRI and NotI. The eGFP gene then was amplified with the primers pAOX1-EcoRI-eGFP-Gib (5′-GAGAAGATCAAAAAACAACTAATTATTGAAAGAATTCCGAAACGATGGCTAGCAAAGGAGAAGAACTTTTCAC-3′) and AOX1TT-NotI-eGFP-Gib (5′-CAAATGGCATTCTGACATCCTCTTGAGCGGCCGCTTACTTGTACAATTCATCCATGCCATGTG-3′) and cloned into the vector backbone by Gibson assembly (57). The insert was sequenced using primers seqAOX1TT-120.0.143-rev (5′-CGAGATAGGCTGATCAGGAGCAAG-3′) and seq-pAOX1-fwd-778.0.801 (5′-TAAACAGAAGGAAGCTGCCCTGTC-3′). This plasmid is referred to as the standard (STD) vector in this study. For the GUT1 integration vector (GUT1), GUT1 5′ and 3′ homologous sequences reported by Weninger et al. (12) were cloned into the STD vector, which was first linearized with SwaI. The 5′ GUT1 region was amplified using the primers GUT1-3prime-GUT1-5prime-Gib (5′-CACGATACGAACGTTGTTTCCTCTTATATTTAAATCTAGGTCATCCTACAGCAAACACC-3′) and GUT1-5prime-pAOX1-Gib (5′-GTTTCATTCAACCTTTCGTCTTTGGATGTTTATAGTAGATATATCTGTGGTATAGTGTGAAAAAGTAGAAG-3′). The 3′ region was amplified with the primers Element2-GUT1-3prime-Gib (5′-AGATCGGGAACACTGAAAAATACACAGTTATTATTCAGAGCAGCTGTAATTATATTATCATGTTAGGTCA-3′) and GUT1-5prime-GUT1-3prime-Gib (5′-GTGTTTGCTGTAGGATGACCTAGATTTAAATATAAGAGGAAACAACGTTCGTATCGTGA-3′). These primers contain overhangs for Gibson assembly with the 5′ and 3′ vector regions and between the 5′ and 3′ GUT1 sequences. The vector backbone and 5′ and 3′ GUT1 regions were linked by Gibson assembly of the three fragments in equimolar ratios. The inserted sequences were verified by sequencing with the primers seq-pAOX1-94.0.117-rev (5′-GCAACGGTCTGCTGCTAGTGTATC-3′), seq-GUT1-3prime-481.0.504-fwd (5′-TATGTGACAGCTCTGGCAGCGTTG-3′), and seqElement5-41.0.64-fwd (5′-CTGCCTGAAATCTCCATCGCCTAC-3′).
Transformations, screening, and eGFP measurements.
P. pastoris cells were transformed following a condensed standard protocol (58). The amounts of DNA used were adjusted depending on the vector size (see Results). After the transformation, screenings and rescreenings of the indicated numbers of transformants (Fig. 2 and 3) were performed as outlined previously (13, 28, 30). Correct integration in the GUT1 locus (leading to glycerol auxotrophy) was performed as outlined previously (12). Notably, we did not stamp the cells on glycerol- or glucose-containing plates but rather inoculated them in liquid media in deep-well plates containing buffered minimal media (BM; 1.34% yeast nitrogen base, 4 × 10−5% biotin, 200 mM potassium phosphate buffer [pH 6.0], and either 1% glucose [in the form of dextrose; BMD] or 1% glycerol [BMG]) as the carbon source. The liquid media were inoculated with 1 μl from pregrown liquid cultures in DWPs (containing BMD medium) that were initially inoculated from solid transformation plates.
For eGFP fluorescence measurements, the cultures were diluted 20-fold (10 μl + 190 μl double-distilled H2O), and eGFP fluorescence was normalized to biomass (using OD600 measurements) to account for pipetting errors. eGFP fluorescence (excitation/emission wavelengths of 488/507 nm) was measured using a FLUOstar plate reader (BMG Labtech, Ortenberg, Germany) using ex/em 485-12/520 filters (gain setting, 1,300). Absorption at 600 nm was measured using a SpectraMax plus 384 plate reader (Molecular Devices, Germany). After subtracting background fluorescence/absorbance from diluted media, fluorescence was normalized to the OD600. Box plots depicting the results were generated with BoxPlotR (59).
Isolation of genomic DNA, library preparations, and sequencing.
Genomic DNA (gDNA) was isolated using the Isolate II genomic DNA kit (Bioline, Pty Ltd., Alexandria, Australia) by following the standard protocol, with minor modifications. The strains were grown in 5 ml YPD (1%, wt/vol, yeast extract, 2%, wt/vol, peptone, 2% glucose) medium overnight in 50-ml plastic tubes. Subsequently, 0.75 ml of the cultures was harvested by centrifugation at 4,000 × g for 5 min and washed once with 1 ml 10 mM EDTA, pH 8. Cell pellets were resuspended in 600 μl sorbitol buffer (1.2 M sorbitol, 10 mM CaCl2, 0.1 M Tris-HCl, pH 7.5, 35 mM β-mercaptoethanol) with 100 U lyticase (L2524; Sigma-Aldrich, Castle Hill, NSW, Australia), incubated for 30 min, and then centrifuged at 2,000 × g for 10 min. Pellets were resuspended in 180 μl lysis buffer GL, 25 μl proteinase K (provided in the kit), and 10 μl RNase A (20 mg/ml; 12091-039; Invitrogen PureLink; Thermo Fisher Scientific Pty Ltd., Scoresby, VIC, Australia). Spin column purification steps then were performed according to the manual. Fifty nanograms of gDNA from each strain was prepared for sequencing using the Nextera DNA library preparation kit (Illumina, San Diego, CA) according to the manufacturer's instructions. Indexed libraries then were sequenced on the Illumina MiSeq system at the Central Analytical Research Facility at Queensland University of Technology (QUT) by following the manufacturer's instructions.
Sequence analysis.
The paired reads were mapped to the P. pastoris genome (23) (with the appropriate plasmid) using Bowtie2 2.2.9 (60) with default settings. The resulting SAM file was compressed and sorted using SAMtools 1.3.1 (46). BAM QC analysis mapping statistics were generated using Qualimap 2.2.1. Mapping was viewed using the Integrative Genomics Viewer (IGV) from the Broad Institute (61, 62).
Variant detection (SNP).
Sequence variants were detected using Bcftools 1.3.1 (63), with the ploidy set to 1, and filtered with a minimum quality score of 20. In addition, variants that existed in the wild-type (mutS) strain were also removed from the other strains. However, we did retain changes at the same position if the change was different in a specific individual strain. The effect of the variant was determined using snpEff 4.3p with a custom-built database.
Copy number estimations.
To estimate integration cassette copy numbers in the transformants, two approaches were taken. (i) The average coverage of reads mapped to the plasmid was compared to the average coverage of reads mapped to each chromosome using the mapping stats (BAM stats) calculated by SAMtools. (ii) The genomic reads were quasimapped to a quasitranscriptome using Salmon 0.8.2 (47). Salmon is a tool for quantifying the expression of transcripts using transcriptome sequencing (RNA-seq) data. A set of quasitranscripts was built for the genome and the eGFP, zeocin resistance, and pUC-origin sequences from the plasmids added. Transcripts then were quantified with Salmon and tximport (64), and the edgeR (65) package (R 3.3.3 software) was used to import and normalize the counts across the data sets in R. This gave a transcripts per million (TPM) value for each gene which should, assuming an even mapping distribution across the genome, be equivalent to one copy for most genes and then can be compared against the levels of the plasmid genes to see if they are present at a higher copy number. More details about how we calculated copy number are given in Data Set S7 in the supplemental material.
BLAST readwalking.
A BLAST-based readwalking approach adapted from that described in Chambers et al. (45) was used to locate plasmid insertion sites in the genome. Reads were mapped to the cut end of the relevant plasmid using BLASTN 2.3.0 (66). Reads that mapped to the end of the plasmid with an overhang of between 50 and 100 bases were retained. These reads then were used to query a database of all reads. Again, reads with the overhang were kept and reads that had not been used in earlier rounds were then used to query the read database again for up to 10 rounds. All of the reads selected by this process then were mapped back to the genome.
Supplementary Material
ACKNOWLEDGMENTS
The data reported in this paper were obtained at the Central Analytical Research Facility operated by the Institute for Future Environments (QUT). Access to CARF is supported by generous funding from the Science and Engineering Faculty (QUT). We acknowledge the Queensland node of Metabolomics Australia (MA) at The University of Queensland, an NCRIS initiative under Bioplatforms Australia, Pty. Ltd. T.V. was supported by an Endeavor Research Fellowship by the Australian Government Department of Education and Training.
We thank Kevin Dudley, Vincent Chand, and Sahana Manoli from the Central Analytical Research Facility at QUT for excellent support and technical assistance. We also thank Lukas Sturmberger for providing the published (23), refined P. pastoris genome sequence. We also thank Knut Madden and Tom Chappell from Biogrammmatics, Inc., for providing sequence information used for generating the Δaox1 strain used in this study (BG11). We also thank Lars K. Nielsen, Michele Bruschi, and Carl Davies for fruitful discussions.
Regarding author contributions, T.V. and R.S. conceived the study. T.V. performed molecular cloning and wet laboratory experiments. R.W.P. performed computational analyses of sequencing data. L.G. performed data analysis. T.V., L.G., R.W.P., and R.S. interpreted the results. T.V and L.G. wrote the manuscript. R.S. supervised the research.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02712-17.
REFERENCES
- 1.Ahmad M, Hirz M, Pichler H, Schwab H. 2014. Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol 98:5301–5317. doi: 10.1007/s00253-014-5732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gasser B, Prielhofer R, Marx H, Maurer M, Nocon J, Steiger M, Puxbaum V, Sauer M, Mattanovich D. 2013. Pichia pastoris: protein production host and model organism for biomedical research. Fut Microbiol 8:191–208. doi: 10.2217/fmb.12.133. [DOI] [PubMed] [Google Scholar]
- 3.Vogl T, Hartner FS, Glieder A. 2013. New opportunities by synthetic biology for biopharmaceutical production in Pichia pastoris. Curr Opin Biotechnol 24:1094–1101. doi: 10.1016/j.copbio.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bill RM. 2014. Playing catch-up with Escherichia coli: using yeast to increase success rates in recombinant protein production experiments. Front Microbiol 5:85. doi: 10.3389/fmicb.2014.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jahic M, Veide A, Charoenrat T, Teeri T, Enfors S-O. 2006. Process technology for production and recovery of heterologous proteins with Pichia pastoris. Biotechnol Prog 22:1465–1473. doi: 10.1021/bp060171t. [DOI] [PubMed] [Google Scholar]
- 6.Vogl T, Glieder A. 2013. Regulation of Pichia pastoris promoters and its consequences for protein production. N Biotechnol 30:385–404. doi: 10.1016/j.nbt.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 7.Aw R, Polizzi KM. 2013. Can too many copies spoil the broth? Microb Cell Fact 12:128. doi: 10.1186/1475-2859-12-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aw R, Polizzi KM. 2016. Liquid PTVA: a faster and cheaper alternative for generating multi-copy clones in Pichia pastoris. Microb Cell Fact 15:29. doi: 10.1186/s12934-016-0432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sunga AJ, Tolstorukov I, Cregg JM. 2008. Posttransformational vector amplification in the yeast Pichia pastoris. FEMS Yeast Res 8:870–876. doi: 10.1111/j.1567-1364.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 10.Liachko I, Youngblood RA, Keich U, Dunham MJ. 2013. High-resolution mapping, characterization, and optimization of autonomously replicating sequences in yeast. Genome Res 23:698–704. doi: 10.1101/gr.144659.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Näätsaari L, Mistlberger B, Ruth C, Hajek T, Hartner FS, Glieder A. 2012. Deletion of the Pichia pastoris KU70 homologue facilitates platform strain generation for gene expression and synthetic biology. PLoS One 7:e39720. doi: 10.1371/journal.pone.0039720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weninger A, Hatzl A-M, Schmid C, Vogl T, Glieder A. 2016. Combinatorial optimization of CRISPR/Cas9 expression enables precision genome engineering in the methylotrophic yeast Pichia pastoris. J Biotechnol 235:139–149. doi: 10.1016/j.jbiotec.2016.03.027. [DOI] [PubMed] [Google Scholar]
- 13.Weis R, Luiten R, Skranc W, Schwab H, Wubbolts M, Glieder A. 2004. Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res 5:179–189. doi: 10.1016/j.femsyr.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 14.Brooks CL, Morrison M, Lemieux JM. 2013. Rapid expression screening of eukaryotic membrane proteins in Pichia pastoris. Protein Sci 22:425–433. doi: 10.1002/pro.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viader-Salvadó JM, Cab-Barrera EL, Galán-Wong LJ, Guerrero-Olazarán M. 2006. Genotyping of recombinant Pichia pastoris strains. Cell Mol Biol Lett 11:348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panagiotou V. 2010. Clonal selection and characterization of epigenetic variation in Pichia pastoris. Ph.D. dissertation, Massachusetts Institute of Technology, Boston, MA: http://hdl.handle.net/1721.1/59881. [Google Scholar]
- 17.Aw R, Barton GR, Leak DJ. 2017. Insights into the prevalence and underlying causes of clonal variation through transcriptomic analysis in Pichia pastoris. Appl Microbiol Biotechnol 101:5045–5058. doi: 10.1007/s00253-017-8317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Love KR, Politano TJ, Panagiotou V, Jiang B, Stadheim TA, Love JC. 2012. Systematic single-cell analysis of Pichia pastoris reveals secretory capacity limits productivity. PLoS One 7:e37915. doi: 10.1371/journal.pone.0037915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love KR, Panagiotou V, Jiang B, Stadheim TA, Love JC. 2010. Integrated single-cell analysis shows Pichia pastoris secretes protein stochastically. Biotechnol Bioeng 106:319–325. [DOI] [PubMed] [Google Scholar]
- 20.De Schutter K, Lin Y-C, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, Rouzé P, Van de Peer Y, Callewaert N. 2009. Genome sequence of the recombinant protein production host Pichia pastoris. Nat Biotechnol 27:561–566. doi: 10.1038/nbt.1544. [DOI] [PubMed] [Google Scholar]
- 21.Küberl A, Schneider J, Thallinger GG, Anderl I, Wibberg D, Hajek T, Jaenicke S, Brinkrolf K, Goesmann A, Szczepanowski R, Pühler A, Schwab H, Glieder A, Pichler H. 2011. High-quality genome sequence of Pichia pastoris CBS7435. J Biotechnol 154:312–320. doi: 10.1016/j.jbiotec.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Mattanovich D, Graf A, Stadlmann J, Dragosits M, Redl A, Maurer M, Kleinheinz M, Sauer M, Altmann F, Gasser B. 2009. Genome, secretome and glucose transport highlight unique features of the protein production host Pichia pastoris. Microb Cell Fact 8:29. doi: 10.1186/1475-2859-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sturmberger L, Chappell T, Geier M, Krainer F, Day KJ, Vide U, Trstenjak S, Schiefer A, Richardson T, Soriaga L, Darnhofer B, Birner-Gruenberger R, Glick BS, Tolstorukov I, Cregg J, Madden K, Glieder A. 2016. Refined Pichia pastoris reference genome sequence. J Biotechnol 235:121–131. doi: 10.1016/j.jbiotec.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love KR, Shah KA, Whittaker CA, Wu J, Bartlett MC, Ma D, Leeson RL, Priest M, Borowsky J, Young SK, Love JC. 2016. Comparative genomics and transcriptomics of Pichia pastoris. BMC Genomics 17:550. doi: 10.1186/s12864-016-2876-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valli M, Tatto NE, Peymann A, Gruber C, Landes N, Ekker H, Thallinger GG, Mattanovich D, Gasser B, Graf AB. 2016. Curation of the genome annotation of Pichia pastoris (Komagataella phaffii) CBS7435 from gene level to protein function. FEMS Yeast Res 16:fow051. doi: 10.1093/femsyr/fow051. [DOI] [PubMed] [Google Scholar]
- 26.Schwarzhans J-P, Wibberg D, Winkler A, Luttermann T, Kalinowski J, Friehs K. 2016. Integration event induced changes in recombinant protein productivity in Pichia pastoris discovered by whole genome sequencing and derived vector optimization. Microb Cell Fact 15:84. doi: 10.1186/s12934-016-0486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwarzhans J-P, Wibberg D, Winkler A, Luttermann T, Kalinowski J, Friehs K. 2016. Non-canonical integration events in Pichia pastoris encountered during standard transformation analysed with genome sequencing. Sci Rep 6:38952. doi: 10.1038/srep38952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vogl T, Sturmberger L, Kickenweiz T, Wasmayer R, Schmid C, Hatzl AM, Gerstmann MA, Pitzer J, Wagner M, Thallinger GG, Geier M, Glieder A. 2016. A toolbox of diverse promoters related to methanol utilization: functionally verified parts for heterologous pathway expression in Pichia pastoris. ACS Synth Biol 5:172–186. doi: 10.1021/acssynbio.5b00199. [DOI] [PubMed] [Google Scholar]
- 29.Portela RMC, Vogl T, Kniely C, Fischer JE, Oliveira R, Glieder A. 2017. Synthetic core promoters as universal parts for fine-tuning expression in different yeast species. ACS Synth Biol 6:471–484. doi: 10.1021/acssynbio.6b00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogl T, Ruth C, Pitzer J, Kickenweiz T, Glieder A. 2014. Synthetic core promoters for Pichia pastoris. ACS Synth Biol 3:188–191. doi: 10.1021/sb400091p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogl T, Ahmad M, Krainer FW, Schwab H, Glieder A. 2015. Restriction site free cloning (RSFC) plasmid family for seamless, sequence independent cloning in Pichia pastoris. Microb Cell Fact 14:103. doi: 10.1186/s12934-015-0293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Invitrogen-Life Technologies. 2014. Pichia expression kit user guide, revision A.0. Publication number MAN0000012Invitrogen-Life Technologies, Waltham, MA. [Google Scholar]
- 33.Smih F, Rouet P, Romanienko PJ, Jasin M. 1995. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res 23:5012–5019. doi: 10.1093/nar/23.24.5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rouet P, Smih F, Jasin M. 1994. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol 14:8096–8106. doi: 10.1128/MCB.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caldecott KW. 2008. Single-strand break repair and genetic disease. Nat Rev Genet 9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 36.Storici F, Durham CL, Gordenin DA, Resnick MA. 2003. Chromosomal site-specific double-strand breaks are efficiently targeted for repair by oligonucleotides in yeast. Proc Natl Acad Sci U S A 100:14994–14999. doi: 10.1073/pnas.2036296100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weninger A, Killinger M, Vogl T. 2016. Key methods for synthetic biology: genome engineering and DNA assembly, p 101–141. In Glieder A, Kubicek CP, Mattanovich D, Wiltschi B, Sauer M (ed), Synthetic biology. Springer International Publishing, Cham, Switzerland. [Google Scholar]
- 38.Hastings PJ, McGill C, Shafer B, Strathern JN. 1993. Ends-in vs. ends-out recombination in yeast. Genetics 135:973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krainer FW, Dietzsch C, Hajek T, Herwig C, Spadiut O, Glieder A. 2012. Recombinant protein expression in Pichia pastoris strains with an engineered methanol utilization pathway. Microb Cell Fact 11:22. doi: 10.1186/1475-2859-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krainer FW, Gerstmann MA, Darnhofer B, Birner-Gruenberger R, Glieder A. 2016. Biotechnological advances towards an enhanced peroxidase production in Pichia pastoris. J Biotechnol 233:181–189. doi: 10.1016/j.jbiotec.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Krainer FW, Capone S, Jäger M, Vogl T, Gerstmann M, Glieder A, Herwig C, Spadiut O. 2015. Optimizing cofactor availability for the production of recombinant heme peroxidase in Pichia pastoris. Microb Cell Fact 14:4. doi: 10.1186/s12934-014-0187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Pscheidt B, Avi M, Gaisberger R, Hartner FS, Schuster C, Skranc W, Gruber K, Glieder A. 2008. Laboratory evolved biocatalysts for stereoselective syntheses of substituted benzaldehyde cyanohydrins. Chembiochem 9:58–61. doi: 10.1002/cbic.200700514. [DOI] [PubMed] [Google Scholar]
- 43.Mellitzer A, Weis R, Glieder A, Flicker K. 2012. Expression of lignocellulolytic enzymes in Pichia pastoris. Microb Cell Fact 11:61. doi: 10.1186/1475-2859-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krzywinski M, Altman N. 2014. Points of significance 05: visualizing samples with box plots. Nat Methods 11:119–120. doi: 10.1038/nmeth.2813. [DOI] [PubMed] [Google Scholar]
- 45.Chambers K, Lowe RG, Howlett BJ, Zander M, Batley J, Van de Wouw AP, Elliott CE. 2014. Next-generation genome sequencing can be used to rapidly characterise sequences flanking T-DNA insertions in random insertional mutants of Leptosphaeria maculans. Fungal Biol Biotechnol 1:10. doi: 10.1186/s40694-014-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 49.Abad S, Kitz K, Hörmann A, Schreiner U, Hartner FS, Glieder A. 2010. Real-time PCR-based determination of gene copy numbers in Pichia pastoris. Biotechnol J 5:413–420. doi: 10.1002/biot.200900233. [DOI] [PubMed] [Google Scholar]
- 50.Mellitzer A, Ruth C, Gustafsson C, Welch M, Birner-Grünberger R, Weis R, Purkarthofer T, Glieder A. 2014. Synergistic modular promoter and gene optimization to push cellulase secretion by Pichia pastoris beyond existing benchmarks. J Biotechnol 191:187–195. doi: 10.1016/j.jbiotec.2014.08.035. [DOI] [PubMed] [Google Scholar]
- 51.Flagfeldt DB, Siewers V, Huang L, Nielsen J. 2009. Characterization of chromosomal integration sites for heterologous gene expression in Saccharomyces cerevisiae. Yeast 26:545–551. doi: 10.1002/yea.1705. [DOI] [PubMed] [Google Scholar]
- 52.Rhoads A, Au KF. 2015. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13:278–289. doi: 10.1016/j.gpb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 54.Mardis ER. 2017. DNA sequencing technologies: 2006-2016. Nat Protoc 12:213–218. doi: 10.1038/nprot.2016.182. [DOI] [PubMed] [Google Scholar]
- 55.Perez-Pinera P, Han N, Cleto S, Cao J, Purcell O, Shah KA, Lee K, Ram R, Lu TK. 2016. Synthetic biology and microbioreactor platforms for programmable production of biologics at the point-of-care. Nat Commun 7:12211. doi: 10.1038/ncomms12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hasslacher M, Schall M, Hayn M, Bona R, Rumbold K, Lückl J, Griengl H, Kohlwein SD, Schwab H. 1997. High-level intracellular expression of hydroxynitrile lyase from the tropical rubber tree Hevea brasiliensis in microbial hosts. Protein Expr Purif 11:61–71. doi: 10.1006/prep.1997.0765. [DOI] [PubMed] [Google Scholar]
- 57.Gibson DG, Young L, Chuang R, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 58.Lin-Cereghino J, Wong WW, Xiong S, Giang W, Luong LT, Vu J, Johnson SD, Lin-Cereghino GP. 2005. Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. Biotechniques 38:44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spitzer M, Wildenhain J, Rappsilber J, Tyers M. 2014. BoxPlotR: a web tool for generation of box plots. Nat Methods 11:121–122. doi: 10.1038/nmeth.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soneson C, Love MI, Robinson MD. 2015. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.