

Abstract
Traumatic brain injury (TBI) is a common disease associated with a high rate of morbidity and mortality. Secondary brain injury following TBI triggers pathological, physiological, and biological reactions that lead to neurological dysfunctions. Alantolactone (ATL) is a well-known Chinese medicine that possesses strong anti-inflammatory properties, but its role in TBI remains poorly understood. The objective of this study was to evaluate the protective effect of ATL in a rat model of controlled cortical impact (CCI). We observed the neurological scores, brain water content, oxidative stress, neuroinflammation and apoptosis by performing an enzyme-linked immunosorbent assay, western blotting, quantitative real-time reverse transcription polymerase chain reaction (RT-qPCR), immunohistochemical (IHC) staining and other methods after CCI. The neurological scores, brain water content, levels of oxidative stress and inflammatory cytokines, and apoptosis index were markedly decreased following the ATL treatment in rats after TBI. Moreover, the antioxidant and anti-inflammatory effects of ATL in TBI may be partially mediated by inhibition of the NF-κB pathway and suppression of Cyclooxygenase 2 (COX-2). In addition, ATL attenuated TBI-induced neuronal apoptosis by suppressing the cytochrome c/caspase-dependent apoptotic pathway. Thus, ATL could exert neuroprotection in rats in a TBI model. Importantly, ATL has great potential in the clinical treatment of TBI.
Keywords: Alantolactone, traumatic brain injury, inflammation, apoptosis
Introduction
Traumatic brain injury (TBI) is currently the main cause of death and disability among young adults worldwide, causing great economic and psychological burdens on society and the patients’ families [1]. The pathological process of TBI includes primary and secondary injury [2]; the primary injury is the result of mechanical factors immediately after trauma. The brain damage and dysfunction observed after TBI are primarily attributed to the secondary injury [3]. The underlying mechanism of secondary injury in TBI mainly includes oxygen free radicals, neuroinflammation, brain edema formation and neuronal apoptosis [4,5]. Oxidative stress commonly occurs after acute injury, such as trauma and ischemic or hemorrhagic stroke [6-8]. However, the brain is rich with lipids and is susceptible to oxidative stress; therefore, the use of antioxidants is a potential targeted therapy for TBI [9-11].
The inflammatory response after TBI is as another major secondary injury process that increases the permeability of the blood-brain barrier (BBB), brain edema and intracranial pressure and eventually leads to delayed neuronal death and neurological dysfunction [12,13]. Interleukin (IL)-1β and IL-6, Prostaglandin E2 (PGE2), and necrosis factor-α (TNF-α) are crucial pro-inflammatory cytokines following TBI [14-17]. Targeting inflammation may serve as a promising treatment measure for TBI [18]. Unfortunately, most current research methods have failed to identify a single mechanism underlying secondary injury [19]. Therefore, new potential drugs that produce a combined therapeutic effect are urgently needed.
Alantolactone (ATL) is a sesquiterpene lactone compound that is isolated from the roots of Anula helenium and possesses a wide range of biological activities, including antibacterial, antifungal, hepatoprotective, antihelminthic, and antitumor activities. Muhammad Khan et al [20] found that ATL could exert antioxidant effects by inhibiting reactive oxygen species (ROS) and stabilizing Glutathione (GSH) in ATL antitumor studies. Cyclooxygenase (COX) catalyzes the first step in the production of prostaglandins by arachidonic acid, but reactive oxygen species are produced during this process [21]. In addition, COX-2, which is an inducible isoform, plays a key role in the regulation of PGE2 biosynthesis and is thought to play an important role in the inflammatory response after TBI [22]. In our previous studies, ATL could penetrate the BBB [23] and was more effective than celecoxib (a classical COX-2 inhibitor), which was demonstrated by both in vitro and in vivo studies. In addition, the precise effect of alantolactone on the NF-κB signaling pathway was demonstrated in our previous study [23]. Moreover, ATL has been shown to have a rapid onset without causing obvious damage to normal tissues and organs in vivo [24].
To the best of our knowledge, ATL has not been previously used for the treatment of TBI. In the current study, we examined whether ATL could have protective effects in TBI. Furthermore, the possible molecular mechanisms underlying the effect of ALT in TBI were found to be related to the COX-2 signaling pathway. Therefore, our results provide evidence supporting applications of ATL in TBI treatments.
Materials and methods
Plant materials and chemicals
Alantolactone (ATL, purity > 98%) was purchased from Tauto Biotech Co., LTD. (Shanghai, China). The chemical structure of alantolactone is shown in Figure 1. The malondialdehyde (MDA), superoxide dismutase (SOD), GSH, glutathione disulfide (GSSG), and Bradford protein assay kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Antibodies specific to COX-2, BAX, cleaved caspase-3, NF-κBp65 (p65), phospho-NF-κBp65 (p-p65), phospho-IκB-α (p-IκB-α), β-actin, and Lamin B1 and all secondary antibodies were purchased from Cell Signaling Technology (Cell Signaling Technology, Inc., USA). The primary antibodies for COX IV and cytochrome c were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO) unless otherwise specified. The remaining reagents were used according to the analyses performed.
Figure 1.
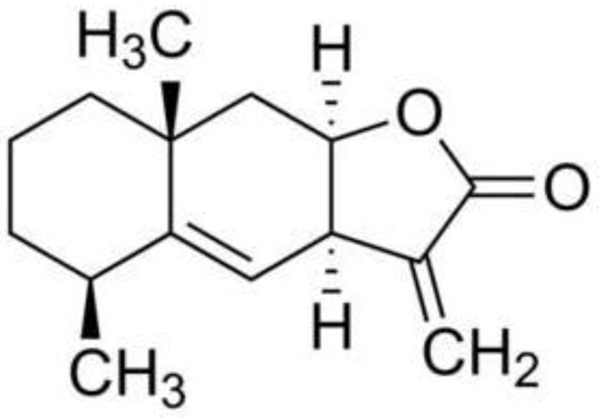
The chemical structure of Alantolactone. Molecular formula: C15H20O2. Molecular weight: 232.32 g/mol.
Animals and experimental grouping
Male Sprague-Dawley (SD) rats (pathogen-free grade males weighing 220-250 g) were purchased from the Animal Experimental Centre of Dalian Medical University, China (Certificate No. SCXK (Liao) 2008-0002). All animals had free access to sterilized food and water and were allowed to habituate for 7 days before the experiments. The study was approved by the Animal Research Ethics Committee of Dalian Medical University. All experimental procedures were performed according to the Guidance Suggestions for the Care and Use of Laboratory Animals published by the Ministry of Science and Technology of the People’s Republic of China.
One hundred and fifty SD rats were randomly divided into the following five groups for the efficacy experiment, and each group was assessed at 2 time points (24 and 48 h): (a) Sham-operated control group (Sham): the rats only underwent a craniotomy, but their brain tissue was not damaged. After the operation, the rats received the same amount of vehicle by an intraperitoneal injection every 24 h (n = 30); (b) Sham-operated control + ATL group (Sham + ATL): After the operation, the rats received 20 mg/kg of ATL by an intraperitoneal injection every 24 h (n = 30); (c) TBI control group (Control): After TBI, the rats received the same amount of vehicle by an intraperitoneal injection every 24 h (n = 30); (d) 10 mg/kg of ATL treatment group: After TBI, the rats received 10 mg/kg of ATL by an intraperitoneal injection every 24 h (n = 30); (e) 20 mg/kg of ATL treatment group: After TBI, the rats received 20 mg/kg of ATL by an intraperitoneal injection every 24 h (n = 30).
TBI model
The TBI model used in the present study was based on the controlled cortical impact (CCI) model with some modifications as previously described by Brody DL, et al [25]. Briefly, the rats were anesthetized with 4% chloral hydrate and maintained at 37.0 ± 0.5°C on a thermal mat throughout the surgical procedure. CCI was produced in the rat using a PCI3000 PinPoint Precision Cortical Impactor (Hatteras Instruments, Cary, NC, USA). After exposing the skull through a central skin incision and removing the soft tissue with a cotton tip, a circular craniotomy approximately 4 mm in diameter was performed in the middle of the right parietal bone approximately 0.5 mm from the sagittal, coronal, and lambdoid sutures, while the dura remained intact. The CCI parameters were as follows: impact tip diameter, 3 mm; velocity, 2 m/s; compression time, 85 ms; and compression distance, 1 mm. Using these impact parameters, we established a moderate injury model. The impact tip was wiped with alcohol after each impact. Finally, we sutured the wounds, woke the rats, and placed the rats on a heating cushion to maintain normal body temperature for 30 to 60 min. The sham-operated rats underwent the same procedure without percussion.
Neurobehavioral score
The neurological score was assessed 24 h and 48 h after TBI using the neurological severity score (NSS) system. Two observers independently assessed the ability of each rat (N = 6) to perform 10 different tasks, and if the results were inconsistent, the results were obtained through discussion. Rats that failed to perform the tasks received one or no points (Table 1) [26]. The observers were blinded to all group information.
Table 1.
Neurological severity scores
| Items | Detailed description | Points (success/failure) |
|---|---|---|
| Exit circle | Ability to enter or exit a 30-cm circle (3 min) | 0/1 |
| Mono/hemiparesis | Paresis of contralateral upper or lower limb | 0/1 |
| Startle reflex | Innate reflex (flinching in response to a loud hand clap) | 0/1 |
| Seeking behavior | Physiological behavior as a sign of “interest” in the environment | 0/1 |
| Straight walk | Alertness/initiative/straight walking when placed on the floor | 0/1 |
| Round stick balance | Ability to balance on a round stick 5 mm in diameter for 10 s | 0/1 |
| Beam balance | Ability to balance on a beam 7 mm in width for 10 s | 0/1 |
| Beam walk (3 cm) | Ability to cross a beam (length × width, 30 × 3 cm) | 0/1 |
| Beam walk (2 cm) | Ability to cross a beam (length × width, 30 × 2 cm) | 0/1 |
| Beam walk (1 cm) | Ability to cross a beam (length × width, 30 × 1 cm) | 0/1 |
| Maximum score | 10 |
N = 6 per group.
Brain water content
The water content in the brain was measured using the wet-dry weight method as previously described [27]. In brief, the contusion cortex and surrounding injured brain tissues were rapidly harvested, quickly weighed to obtain the wet weight, dried in an incubator at 120°C for 24 h and reweighed (dry weight). The brain water content was calculated by the following formula: ratios = [(wet weight-dry weight)/wet weight] × 100%.
Tissue processing
For the western blot analysis and quantitative real-time reverse transcription polymerase chain reaction, the rats were rapidly killed 24 h post-TBI, and their ipsilateral cortexes were collected. The tissue was positioned directly over the center of the injury site and included both contusions and penumbra. The samples were immediately frozen in liquid nitrogen and stored at -80°C until use. For the immunohistochemistry analysis, the ipsilateral brain tissue was removed 24 h after TBI, immersed in 4% paraformaldehyde overnight and was measured by a fluorescence-activated cell sorter (FACS) using the Annexin V-FITC Apoptosis Detection Kit (Nanjing KeyGEN Biotech. CO., LTD.).
Malondialdehyde (MDA) and superoxide dismutase (SOD) assay
The samples were homogenized and centrifuged at 3,000 × g for 10 min. Subsequently, the supernatants were collected, and the MDA levels and SOD activity were analyzed. The level of MDA and SOD were measured using commercially available kits (Nanjing Jiancheng Biochemistry Co., Nanjing, China) according to the manufacturer’s instructions. The MDA level is represented in nmol/mg protein, and the SOD activity is expressed as U/mg protein.
Enzyme-linked immunosorbent assay (ELISA)
The total protein was determined using a using a BCA protein assay kit (Beyotime Biotechnology, China). The levels of inflammatory cytokines in the brain tissue were quantified using ELISA kits specific to the rat (IL-1β, IL-6, PGE2, and TNF-α from Shanghai Saimo Biotechnology, Shanghai, China) according to the manufacturer’s instructions. The inflammatory cytokine contents in the brain tissues are expressed as nanogram per gram protein.
Isolation of rat cortical neurons
To rapidly isolate cortical neurons from the injured side of the rat brain, we followed a previously described method with certain minor modifications [28-30]. Briefly, after TBI, the injured-side cortex was cut into fragments, and the cells were dissociated by incubation with 2 mg/mL papain in DMEM for 30 min at 37°C. To produce a purified population of cells, we used the immune adherence method. The cell suspensions were poured into anti-neural cell adhesion molecule (NCAM)-coated petri dishes (Millipore, USA) and placed on a shaker for 1 h; then, the adhered cells were collected. Trypan blue was used to exclude the non-viable cells.
Mitochondrial membrane potential (MMP) measurement
JC-1 probe (Invitrogen, CA, USA) was employed to measure mitochondrial depolarization in isolated neurons. Briefly, the cells were cultured in six-well plates after the indicated treatments, incubated with an equal volume of JC-1 staining solution (5 μg/ml) at 37°C for 20 min and rinsed twice with PBS. The mitochondrial membrane potentials were monitored by determining the relative amounts of dual emissions from mitochondrial JC-1 monomers or aggregates using FACS Accuri C6 (Genetimes Technology Inc.). Mitochondrial depolarization is indicated by a decrease in the red/green fluorescence intensity ratio.
Western blot analysis
The total and nuclear proteins were extracted from the ipsilateral cerebral cortical tissue using the Total and Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, China) according to the manufacturer’s instructions. The mitochondrial and cytoplasmic proteins were extracted from the remaining ipsilateral tissue using the Tissue Mitochondria Isolation Kit for Tissue Protocols (Beyotime Biotechnology, China). These proteins were separated by electrophoresis on a 7.5-12% SDS-PAGE gel and probed with specific antibodies. The protein bands were detected by enhanced chemiluminescence. The protein concentrations were determined using a BCA protein assay kit (Beyotime Biotechnology, China). Similar experiments were performed at least three times.
Quantitative real-time reverse transcription polymerase chain reaction (RT-qPCR)
The total RNA was extracted from brain tissue using TRIzol reagent according to the manufacturer’s protocol (TaKaRa Bio, Dalian, China). cDNA was reverse-transcribed using the PrimeScript RT Reagent Kit (TaKaRa Bio, Dalian, China) according to the manufacturer’s instructions. The Q-PCR reaction was performed following the manufacturer’s protocol (TaKaRa Bio, Dalian, China), and the amplification was performed using a Mx3005P Real-Time PCR System (Agilent, CA, USA). The relative mRNA expression of each gene was normalized to the β-actin RNA levels. The primers were synthesized by Invitrogen (Shanghai, China). The primers for COX-2 were as follows: 5’-ACCAACGCTGCCACAACT-3 and 5’-GGTTGGAACAGCAAGGATTT-3’; the primers for β-actin were as follows: 5’-GGCACCCAGCACAATGAA-3’ and 5’-TAGAAGCATTTGCGGTGG-3’.
Immunohistochemical staining
The brain tissue samples were fixed with 10% neutral formalin and embedded in paraffin. For the immunohistochemical examination, the tissue sections (4 mm) were incubated with the COX-2 (1:50) and p-p65 NF-κB (1:50) antibodies and examined under a light microscope. The images were examined under a Leica DM 4000B microscope equipped with a digital camera.
Statistical analysis
Student’s t-tests were performed to compare the values of the test and control samples in vitro and in vivo. All experiments were repeated three times. The data are represented as the mean ± standard deviation (SD). SPSS 17.0 software was used for all statistical analysis. ##P < 0.01, *P < 0.05 and **P < 0.01.
Results
ATL improved neurological function and alleviated cerebral edema after TBI
To evaluate whether ATL exerts a neuroprotection effect after TBI, we examined the NSS scores and brain water content 24 h and 48 h after TBI as shown in Figure 2. First, no differences were found in the NSS scores and brain water content between the sham and sham + ATL groups. However, the NSS scores and brain water content in the TBI control group were obviously higher than those in the sham group at the two time point (##P < 0.01). Furthermore, the ATL treatment significantly alleviated the neurological deficits and brain edema in a dose-dependent manner compared with those seen in the control group, (*P < 0.05, **P < 0.01). Thus, ATL might have a potential protective effect in rats and, hence, could be used in the subsequent trials.
Figure 2.
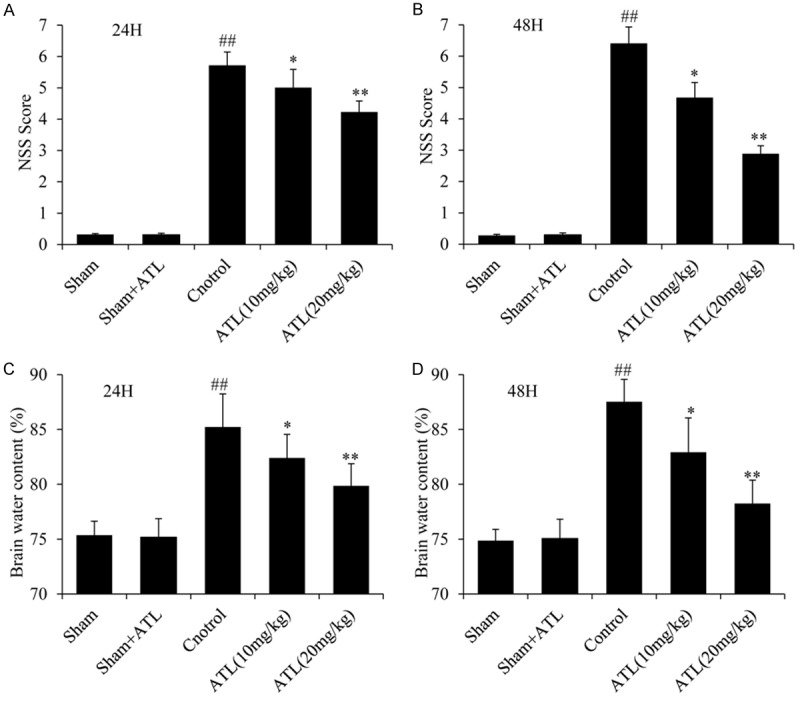
ATL decreases the NSS after TBI in rats and reduces the water content in the injured tissue at 24 h and 48 h. A and B: NSS Score. C and D: Brain water content. ##P < 0.01, vs. sham group. *P < 0.05, **P < 0.01, vs. control group.
ATL attenuated oxidative stress caused by TBI
To assess whether the neuroprotective effect of ATL was mediated by oxidative stress-related signaling pathways, the indicators of lipid peroxidation and antioxidant enzyme activity, including MDA level, SOD activity, GSH level, GSSG level and GSH/GSSG ratio, were measured 24 h after TBI as shown in Figure 3A, 3B) and Table 2. The TBI control group had a higher level of MDA and GSSG than the sham group (##P < 0.01). However, the levels of MDA and GSSG in the ATL treatment groups were significantly lower than those in the TBI control group (*P < 0.05, **P < 0.01). The activity of SOD, GSH and GSH/GSSH in the TBI control group was significantly lower after TBI than that after the sham injury (##P < 0.01), whereas the ATL treatment groups showed an obvious up-regulation of SOD, GSH and GSH/GSSH activity in a dose-dependent manner (*P < 0.05, **P < 0.01). Importantly, ATL had no effects on the sham group.
Figure 3.
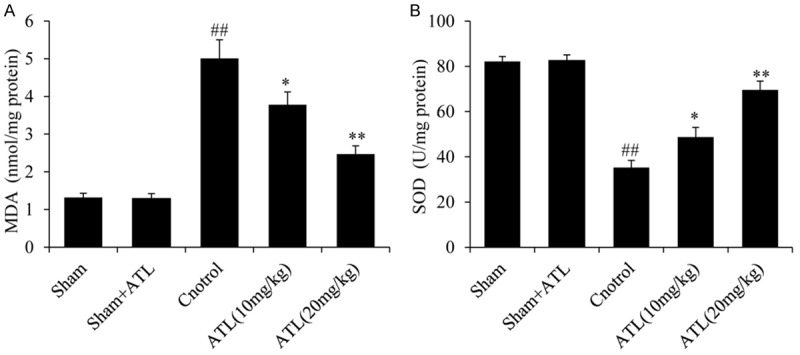
ATL reduces oxidative stress in brain tissue following TBI. A: The level of MDA in the cortex 24 h post-injury. B: The activities of SOD in the cortex 24 h post-injury. ##P < 0.01, vs. sham group. *P < 0.05, **P < 0.01, vs. control group.
Table 2.
Reduced and oxidized glutathione levels in TBI rats (24 h post-injury)
| Group | GSH (µmol/L) | GSSG (µmol/L) | GSH/GSSG |
|---|---|---|---|
| Sham | 99.35 ± 5.32 | 25.76 ± 1.87 | 3.86 ± 0.15 |
| Sham + ATL | 99.42 ± 4.98 | 25.16 ± 2.13 | 3.92 ± 0.19 |
| Control | 58.82 ± 4.17## | 54.51 ± 4.26## | 1.08 ± 0.24## |
| ATL (10 mg/kg) | 70.59 ± 6.19* | 45.82 ± 3.25* | 1.54 ± 0.32* |
| ATL (20 mg/kg) | 87.49 ± 7.13** | 32.93 ± 2.86** | 2.66 ± 0.39** |
Each value represents the mean ± SD (n = 6).
P < 0.01 compared to the sham group.
P < 0.05 compared to the control group;
P < 0.01 compared to the control group.
ATL induced significant reductions in multiplex cytokine levels after TBI
Inflammatory processes are considered major components of the secondary injury cascade following TBI [31]. As shown in Figure 4, the expression levels of the pro-inflammatory cytokines (i.e., IL-1β, IL-6, PGE2 and TNF-α) were low in the brains of rats in the sham group. The levels of those cytokines were markedly increased following TBI compared with those in the sham group (##P < 0.01); however, the administration of ATL significantly decreased the cytokine expression levels compared with those in the TBI control group (*P < 0.05, **P < 0.01). The above-mentioned indicators did not significantly differ between the sham and sham + ATL groups.
Figure 4.
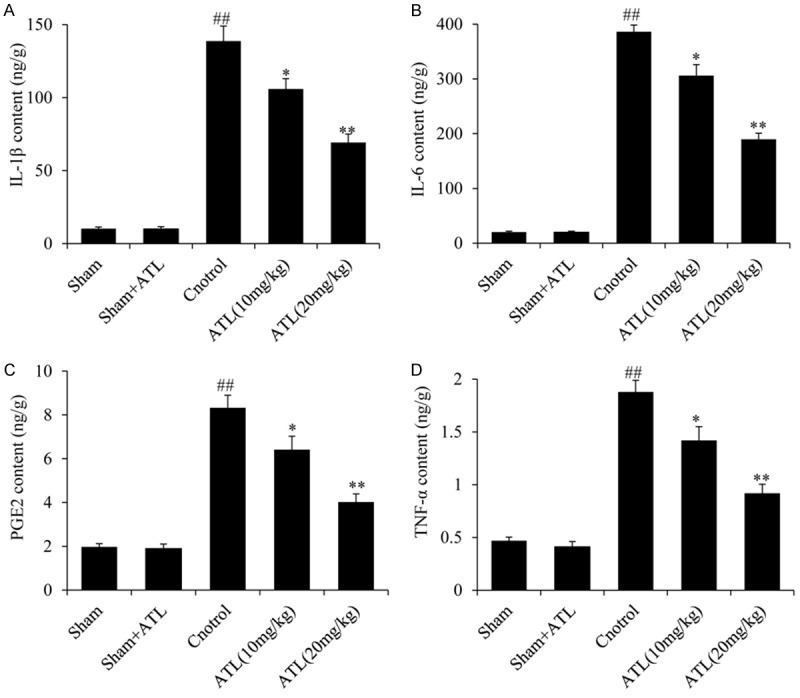
ATL ameliorates TBI-induced inflammation. ELISA analysis of the expression levels of pro-inflammatory genes, including (A) IL-1β, (B) IL-6, (C) PGE2 and (D) TNF-α 24 h after TBI. ##P < 0.01, vs. sham group. *P < 0.05, **P < 0.01, vs. control group.
ATL inhibited the COX-2 signaling pathway in TBI
As mentioned above, ATL can significantly inhibit the expression of PGE2 and other inflammatory factors in TBI, and COX-2 is an inducible enzyme responsible for prostaglandin production at sites of inflammation [32,33]. We immediately detected the COX-2 protein and molecular levels in TBI (Figure 5). As shown in Figure 5A, 5B and 5D, according to the western blot and IHC analyses, the expression levels of COX-2 were low in the sham group. Following TBI, the expression levels of COX-2 in the cortex of rats in the TBI control group were significantly higher than those in the sham group (##P < 0.01). The expression levels of COX-2 were markedly lower in the ATL treatment group than those in the TBI control group (**P < 0.01). Furthermore, the RT-qPCR results were consistent. As shown in Figure 5C, the treatment with ATL significantly down-regulated the COX-2 mRNA levels in the ATL treatment group (**P < 0.01).
Figure 5.
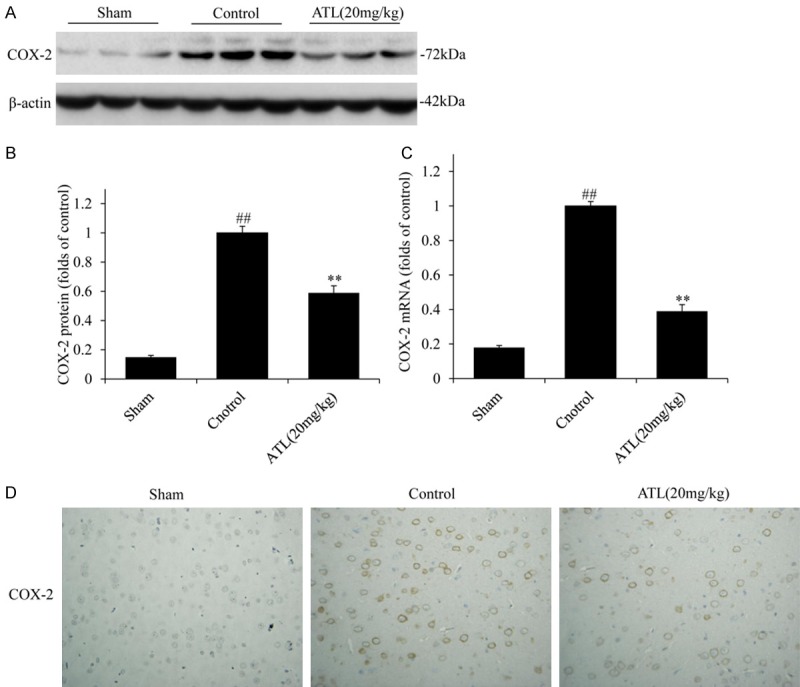
ATL inhibits COX-2 expression in brain tissue 24 h after TBI. A and B: COX-2 expression by western blot assays. C: mRNA expression by RT-qPCR. D: COX-2 protein expression by immunohistochemical analysis (Original magnification, 400 ×). ##P < 0.01, vs. sham group. **P < 0.01, vs. control group.
ATL inhibited the activation of the NF-κB signaling pathway and its translocation into the nucleus after TBI
The above-mentioned results demonstrate that ATL effectively suppressed the COX-2 gene expression after TBI; however, COX-2 gene expression is strictly and specifically controlled by the binding of many transcription factors to the corresponding sites of its promoters, such as NF-κB [34,35]. The activation of the NF-κB signaling pathway is closely related to the overexpression of COX-2 [36,37]. As shown in Figure 6, according to the western blot and IHC analyses, the expression levels of p-p65 and p-IκB-α in the TBI control group were higher than those in sham group. However, these protein contents were obviously decreased in the ATL treatment group compared with those in the TBI control group (**P < 0.01). Then, we extracted the nuclear protein and performed a western blot analysis (Figure 6E and 6F); the expression levels of NF-κB and p65 were significantly up-regulated in the TBI control group. Meanwhile, the protein content of p65 was markedly decreased in the ATL treatment group compared with that in the TBI control group (**P < 0.01).
Figure 6.
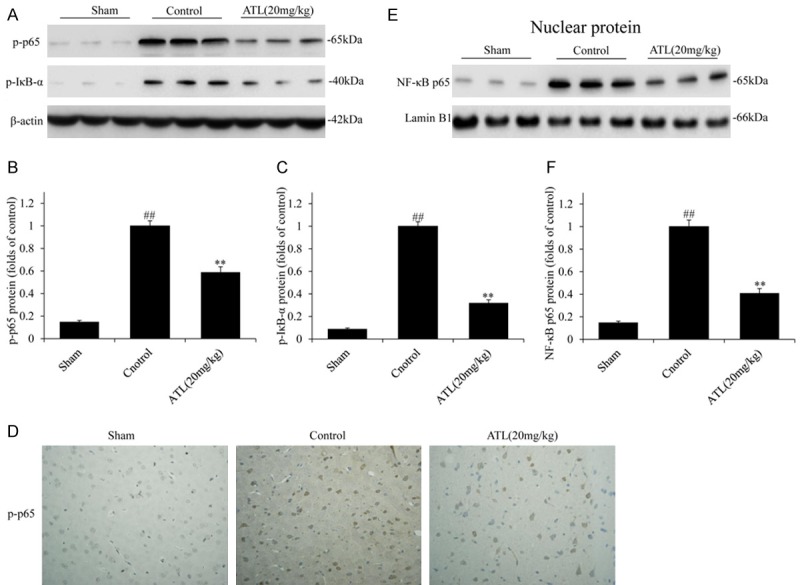
ATL inhibits the activation of the NF-κB signaling pathway and its translocation into the nucleus 24 h after TBI. A-C: The total protein expression of p-p65 and p-IκB-α by WB assays. D: The protein expression of p-p65 and p-IκB-α by immunohistochemical analysis (Original magnification, 400 ×). E and F: The nuclear protein expression of NF-κB p65 by WB assays. ##P < 0.01, vs. sham group. **P < 0.01, vs. control group.
ATL attenuated neuronal apoptosis in brain tissue after TBI
To determine whether ATL could inhibit neuronal apoptosis after TBI, we first detected the apoptosis-related proteins by performing a western blot analysis. As shown in Figure 7A-C, the protein levels of pro-apoptotic Bax and cleaved caspase-3 were markedly increased in the TBI control group compared with those in the sham group. However, these protein levels were significantly reduced after the treatment with ATL. Subsequently, we detected the changes in the mitochondrial membrane potential (Figure 7D). The MMP was obviously reduced in the TBI control group and significantly increased following the treatment with ATL. In addition, we extracted the cytosolic and mitochondrial proteins and performed a western blot analysis (Figure 7E-H). Relative to the sham groups, the cytosolic and mitochondrial cyt c levels increased and decreased, respectively, after TBI, while the cytosolic and mitochondrial cyt c levels markedly decreased and increased, respectively, after the treatment with ATL in a dose-dependent manner. Thus, following TBI, ATL could attenuate neuronal apoptosis by inhibiting cyt c release in the mitochondrion and suppressing caspase activation in the cytoplasm.
Figure 7.
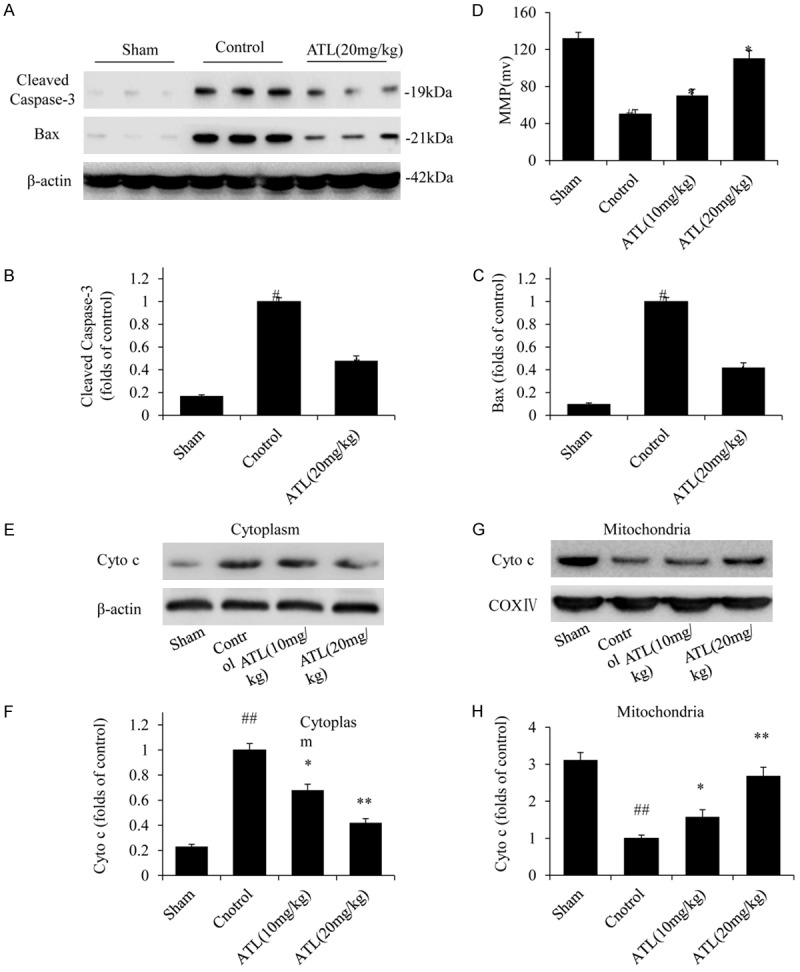
ATL attenuates TBI-induced neuronal apoptosis 24 h post-injury. A-C: The protein expression of cleaved caspase-3 and Bax by western blot assays. D: MMP. E and F: The cytoplasmic protein expression of cyt c by western blot assays. G and H: The mitochondrial protein expression of cyt c by western blot assays. ##P < 0.01, vs. sham group. *P < 0.05, **P < 0.01, vs. control group.
Discussion
Secondary brain injury after TBI occurs in a series of pathological processes, and oxidative stress, the inflammatory response and cell apoptosis are crucial mechanisms that mediate the subsequent histopathology and neurobehavioral deficits [38,39]. In our study, we used a rat model to evaluate the protective effects of ATL after TBI and the potential molecular mechanisms and obtained the following results: (a) the administration of ATL improved the neurological function and alleviated cerebral edema after TBI; (b) ATL inhibited TBI-induced oxidative stress, which was represented by the level of MDA and GSH and the activity of SOD; (c) ATL suppressed the production of pro-inflammatory cytokines after TBI by inhibiting the NF-κB/COX-2 signaling pathway; and (d) ATL attenuated neuronal apoptosis after TBI by suppressing the cytochrome c/caspase-dependent apoptotic pathway.
The brain is particularly vulnerable to oxidative stress attacks after TBI, and brain tissue produces more toxicants than other organs [40]. Due to oxidative stress following TBI, peroxidized enzymes become activated, destroying membrane phospholipids and mitochondrial functions, and several cellular components, such as RNA, DNAs, lipids, carbohydrates, and proteins, are harmed. Ultimately, neuronal cells and brain tissue suffer irreversible damage [41]. To inhibit the oxidative stress response after TBI, key enzymes related to oxidative stress have become the focus of current research studies. SOD and GSH are the most important antioxidants that prevent the brain from oxidative injury. SOD can catalyze the conversion of superoxide to hydrogen peroxide [42], and GSH is among the smallest antioxidants in body fluids [43]. MDA, which is a product of lipid peroxidation, is an important marker of oxidative damage. The content of MDA in the tissue increases as the level of oxidative stress increases [44]. Following oxidative stress, GSH can combine with free radicals and convert to GSSG. The GSH/GSSG ratio is an effective indicator used to monitor antioxidant activity. In the present study, ATL significantly improved the SOD activities, GSH level, and GSH/GSSG ratio in brain tissues after TBI. Meanwhile, ATL evidently diminished the MDA and GSSG levels.
The inflammatory responses induced following TBI are important therapeutic targets for reducing tissue damage following trauma [45]. IL-1β, IL-6, PGE2 and TNF-α are key pro-inflammatory cytokines after acute injuries, and these cytokines were up-regulated at the early stage, which initiated a continuous regional inflammatory response. In our study, ATL could markedly reduce the levels of these pro-inflammatory cytokines during the early period of TBI.
COX-2 expression is strictly and specifically controlled by the binding of transfactors to the corresponding sites of its promoters. However, the transcription factor NF-κB is involved in the regulation of a variety of inflammatory mediators, which is a cis-acting element of the COX-2 transcriptional regulatory sequence. NF-κB consists of homo- and heterodimers of different Rel family proteins (p65, RelB, c-Rel, p52, and p50) and is sequestered by binding to the inhibitory subunit I-κB in most non-stimulated cells [46]. The NF-κB pathway is mainly activated by the degradation of the phosphorylation of the inhibitory protein IκB, thereby liberating free NF-κB dimers to translocate to the nucleus [47]. Thus, activated NF-κB could bind to the COX-2 promoter region and initiate COX-2 transcription. In the present study, ATL significantly suppressed the expression of total p-p65, p-IκB-α, and nuclear p65. Thus, ATL suppressed COX-2 expression by inhibiting the activation of the NF-κB pathway.
Apoptosis is an important pathophysiological component during secondary damage in TBI [48,49]. Apoptosis of neurons and oligodendrocytes in brain tissues causes neural functional deficits [50,51]. Caspase-3 is activated during apoptosis in many types of central nervous system (CNS) cells, and its activation appears to be an important apoptotic event in the CNS [52,53]. The Bcl-2 family proteins are the key step in the regulation of apoptosis in the nervous system. The pro-apoptotic Bax modulates intrinsic apoptotic cell death [54,55]. In our study, ATL significantly inhibited the expression of cleaved caspase-3 and Bax after TBI. To further determine the possible mechanism underlying the apoptosis inhibition effect of ATL in TBI, we first detected whether the mitochondrial membrane was damaged; then, the increased release of cytochrome c from the mitochondria to the cytoplasm was observed, and increased apoptosis via activating caspase signaling cascades was confirmed. ATL inhibited the decrease in the mitochondrial membrane potential, and the cytosolic and mitochondrial cyt c levels were markedly decreased and increased, respectively, after the treatment with ATL, in a dose-dependent manner. Therefore, ATL attenuated TBI-induced neuronal apoptosis by inhibiting the mitochondrial apoptotic pathway.
In conclusion, ATL could have a potential neuroprotective effect in TBI and should be further explored for suitability in clinical applications for the treatment of TBI.
Acknowledgements
This work is supported by grants from National Natural Science Foundation of China (Nos. 81372714, 81672480), Liaoning Provincial Natural Science Foundation of China (No. 201602244), Distinguished Professor Project of Liaoning Province, Special Grant for Translational Medicine, Dalian Medical University (No. 2015002), Traditional Chinese Medicine Foundation of Dalian (17Z1007).
Disclosure of conflict of interest
None.
References
- 1.Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–353. [PubMed] [Google Scholar]
- 2.Dixon DJ. Closed head injury--an inflammatory disease? Phys Med Rehabil Clin N Am. 2017;28:215–225. doi: 10.1016/j.pmr.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Wei J, Xiao GM. The neuroprotective effects of progesterone on traumatic brain injury: current status and future prospects. Acta Pharmacol Sin. 2013;34:1485–90. doi: 10.1038/aps.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury-an inflammatory disease? Brain Res Brain Res Rev. 2005;48:388–399. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 6.Xiong XY, Wang J, Qian ZM, Yang QW. Iron and intracerebral hemorrhage: from mechanism to translation. Transl Stroke Res. 2014;5:429–441. doi: 10.1007/s12975-013-0317-7. [DOI] [PubMed] [Google Scholar]
- 7.Seifert HA, Pennypacker KR. Molecular and cellular immune responses to ischemic brain injury. Transl Stroke Res. 2014;5:543–553. doi: 10.1007/s12975-014-0349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasegawa Y, Suzuki H, Uekawa K, Kawano T, Kim-Mitsuyama S. Characteristics of cerebrovascular injury in the hyperacute phase after induced severe subarachnoid hemorrhage. Transl Stroke Res. 2015;6:458–466. doi: 10.1007/s12975-015-0423-9. [DOI] [PubMed] [Google Scholar]
- 9.Miyamoto K, Ohtaki H, Dohi K, Tsumuraya T, Song D, Kiriyama K, Satoh K, Shimizu A, Aruga T, Shioda S. Therapeutic time window for edaravone treatment of traumatic brain injury in mice. Biomed Res Int. 2013;2013:379206. doi: 10.1155/2013/379206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodrigo R, Fernández-Gajardo R, Gutiérrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12:698–714. doi: 10.2174/1871527311312050015. [DOI] [PubMed] [Google Scholar]
- 11.Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Israelsson C, Bengtsson H, Kylberg A, Kullander K, Lewén A, Hillered L, Ebendal T. Distinct cellular patterns of upregulated chemokine expression supporting a prominent inflammatory role in traumatic brain injury. J Neurotrauma. 2008;25:959–974. doi: 10.1089/neu.2008.0562. [DOI] [PubMed] [Google Scholar]
- 14.Kamm K, Vanderkolk W, Lawrence C, Jonker M, Davis AT. The effect of traumatic brain injury upon the concentration and expression of interleukin-1β and interleukin-10 in the rat. J Trauma. 2006;60:152–157. doi: 10.1097/01.ta.0000196345.81169.a1. [DOI] [PubMed] [Google Scholar]
- 15.Aibiki M, Maekawa S, Ogura S, Kinoshita Y, Kawai N, Yokono S. Effect of moderate hypothermia on systemic and internal jugular plasma IL-6 levels after traumatic brain injury in humans. J Neurotrauma. 1999;16:225–232. doi: 10.1089/neu.1999.16.225. [DOI] [PubMed] [Google Scholar]
- 16.Hang CH, Shi JX, Li JS, Li WQ, Wu W. Expressions of intestinal NF-kappaB, TNF-α, and IL-6 following traumatic brain injury in rats. J Surg Res. 2005;123:188–193. doi: 10.1016/j.jss.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Allan SM, Rothwell NJ. Inflammation in central nervous system injury. Philos Trans R Soc Lond B Biol Sci. 2003;358:1669–1677. doi: 10.1098/rstb.2003.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neruol. 2015;72:355–362. doi: 10.1001/jamaneurol.2014.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun M, Zhao Y, Gu Y, Zhang Y. Protective effects of taurine against closed head injury in rats. J Neurotrauma. 2015;32:66–74. doi: 10.1089/neu.2012.2432. [DOI] [PubMed] [Google Scholar]
- 20.Khan M, Yi F, Rasul A, Li T, Wang N, Gao H, Gao R, Ma T. Alantolactone induces apoptosis in glioblastoma cells via GSH depletion, ROS generation, and mitochondrial dysfunction. IUBMB Life. 2012;64:783–94. doi: 10.1002/iub.1068. [DOI] [PubMed] [Google Scholar]
- 21.Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, Panikashvili D, Shohami E, Jansen SA, Narayan RK, Strauss KI. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the Rat. J Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Yu Z, Wang C, Cheng W, Tian X, Huo X, Wang Y, Sun C, Feng L, Xing J, Lan Y, Sun D, Hou Q, Zhang B, Ma X, Zhang B. Alantolactone, a natural sesquiterpene lactone, has potent antitumor activity against glioblastoma by targeting IKKβ kinase activity and interrupting NF-κB/COX-2-mediated signaling cascades. J Exp Clin Cancer Res. 2017;36:93. doi: 10.1186/s13046-017-0563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JY, Kim SB, Chun J, Song KH, Kim YS, Chung SJ, Cho HJ, Yoon IS, Kim DD. High body clearance and low oral bioavailability of alantolactone, isolated from Inula helenium, in rats: extensive hepatic metabolism and low stability in gastrointestinal fluids. Biopharm Drug Dispos. 2016;37:156–167. doi: 10.1002/bdd.2005. [DOI] [PubMed] [Google Scholar]
- 25.Brody DL, Mac Donald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, Kim E, Schwetye KE, Holtzman DM, Bayly PV. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma. 2007;24:657–673. doi: 10.1089/neu.2006.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. 2009;4:1328–1337. doi: 10.1038/nprot.2009.148. [DOI] [PubMed] [Google Scholar]
- 27.Roof RL, Duvdevani R, Heyburn JW, Stein DG. Progesterone rapidly decreases brain edema: treatment delayed up to 24 hours is still effective. Exp Neurol. 1996;138:246–251. doi: 10.1006/exnr.1996.0063. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Song R, Chen Y, Zhao M, Zhao KS. Polydatin-a new mitochondria protector for acute severe hemorrhagic shock treatment. Expert Opin Investig Drugs. 2013;22:169–179. doi: 10.1517/13543784.2013.748033. [DOI] [PubMed] [Google Scholar]
- 29.Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protoc. 2007;2:1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- 30.Ray B, Bailey JA, Sarkar S, Lahiri DK. Molecular and immunocytochemical characterization of primary neuronal cultures from adult rat brain: differential expression of neuronal and glial protein markers. J Neurosci Methods. 2009;184:294–302. doi: 10.1016/j.jneumeth.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Israelsson C, Bengtsson H, Kylberg A, Kullander K, Lewén A, Hillered L, Ebendal T. Distinct cellular patterns of upregulated chemokine expression supporting a prominent inflammatory role in traumatic brain injury. J Neurotrauma. 2008;25:959–974. doi: 10.1089/neu.2008.0562. [DOI] [PubMed] [Google Scholar]
- 32.Chen R, Zhang J, Fan N, Teng ZQ, Wu Y, Yang H, Tang YP, Sun H, Song Y, Chen C. Δ9-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell. 2013;155:1154–1165. doi: 10.1016/j.cell.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 34.Chung MH, Kim DH, Na HK, Kim JH, Kim HN, Haegeman G, Surh YJ. Genistein inhibits phorbol ester-induced NF-κB transcriptional activity and COX-2 expression by blocking the phosphorylation of p65/RelA in human mammary epithelial cells. Mutat Res. 2014;768:74–83. doi: 10.1016/j.mrfmmm.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 35.Jung KT, Lim KJ. Curcumin, COX-2, and Protein p300/CBP. Korean J Pain. 2014;27:365. doi: 10.3344/kjp.2014.27.4.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.García-Rivera D, Delgado R, Bougarne N, Haegeman G, Berghe WV. Gallic acidindanone and mangiferin xanthone are strong determinants of immunosuppressive anti-tumour effects of Mangifera indica L. bark in MDA-MB231 breast cancer cells. Cancer Lett. 2011;305:21–31. doi: 10.1016/j.canlet.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Kim HN, Kim DH, Kim EH, Lee MH, Kundu JK, Na HK, Cha YN, Surh YJ. Sulforaphane inhibits phorbol ester-stimulated IKK-NF-κB signaling and COX-2 expression inhuman mammary epithelial cells by targeting NF-κB activating kinase and ERK. Cancer Lett. 2014;35:41–49. doi: 10.1016/j.canlet.2014.03.037. [DOI] [PubMed] [Google Scholar]
- 38.Sharp DJ, Scott G, Leech R. Network dysfunction after traumatic brain injury. Nat Rev Neurol. 2014;10:156–166. doi: 10.1038/nrneurol.2014.15. [DOI] [PubMed] [Google Scholar]
- 39.Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta. 2012;1822:675–84. doi: 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann GL, Gradilone SA, Marinelli RA. Aquaporin water channels in central nervous system. Curr Neurovasc Res. 2004;1:293–303. doi: 10.2174/1567202043362081. [DOI] [PubMed] [Google Scholar]
- 41.Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One. 2012;7:e34504. doi: 10.1371/journal.pone.0034504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D, Yuan X, Liu T, Liu L, Hu Y, Wang Z, Zheng Q. Neuroprotective activity of lavender oil on transient focal cerebral ischemia in mice. Molecules. 2012;17:9803–9817. doi: 10.3390/molecules17089803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khalaj L, Nejad SC, Mohammadi M, Zadeh SS, Pour MH, Ahmadiani A, Khodagholi F, Ashabi G, Alamdary SZ, Samami E. Gemfibrozil pretreatment proved protection against acute restraint stress-induced changes in the male rats’ hippocampus. Brain Res. 2013;1527:117–130. doi: 10.1016/j.brainres.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 44.Wang QS, Xie KQ, Zhang CL, Zhu YJ, Zhang LP, Guo X, Yu SF. Allyl chloride induced time dependent changes of lipid peroxidation in rat nerve tissue. Neurochem Res. 2005;30:1387–1395. doi: 10.1007/s11064-005-8391-1. [DOI] [PubMed] [Google Scholar]
- 45.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 46.Niederberger E, Geisslinger G. The IKK-NF-kappaB pathway: a source for novel molecular drug targets in pain therapy? FASEB J. 2008;22:3432–3442. doi: 10.1096/fj.08-109355. [DOI] [PubMed] [Google Scholar]
- 47.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappa B inactivation. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 48.Ng I, Yeo TT, Tang WY, Soong R, Ng PY, Smith DR. Apoptosis occurs after cerebral contusions in humans. Neurosurgery. 2000;46:949–956. doi: 10.1097/00006123-200004000-00034. [DOI] [PubMed] [Google Scholar]
- 49.Yang SY, Xue L. Human neuronal apoptosis secondary to traumatic brain injury and the regulative role of apoptosis-related genes. Chin J Traumatol. 2004;7:159–164. [PubMed] [Google Scholar]
- 50.Yang XF, Liu WG, Shen H, Gong JB, Yu J, Hu WW, Lü ST, Zheng XJ, Fu WM. Correlation of cell apoptosis with brain edema and elevated intracranial pressure in traumatic brain injury. Chin J Traumatol. 2005;8:96–100. [PubMed] [Google Scholar]
- 51.Dressler J, Hanisch U, Kuhlisch E, Geiger KD. Neuronal and glial apoptosis in human traumatic brain injury. Int J legal Med. 2007;121:365–375. doi: 10.1007/s00414-006-0126-6. [DOI] [PubMed] [Google Scholar]
- 52.Sullivan PG, Keller JN, Bussen WL, Scheff SW. Cytochrome c release and caspase activation after traumatic brain injury. Brain Res. 2002;949:88–96. doi: 10.1016/s0006-8993(02)02968-2. [DOI] [PubMed] [Google Scholar]
- 53.Eldadah BA, Faden AI. Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma. 2000;17:811–829. doi: 10.1089/neu.2000.17.811. [DOI] [PubMed] [Google Scholar]
- 54.Luo C, Lu Y, Jiang J, Zhu C. Changes of bcl-x(L) and bax mRNA expression following traumatic brain injury in rats. Chin J Traumatol. 2002;5:299–302. [PubMed] [Google Scholar]
- 55.Luo C, Zhu C, Jiang J, Lu Y, Zhang G, Yuan G, Cai R, Ye T. Alterations of bcl-2, bcl-x and bax protein expressions in area CA-3 of rat hippocampus following fluid percussion brain injury. Chin J Traumatol. 1999;2:101–104. [PubMed] [Google Scholar]