

Abstract
The molecular development of diabetic encephalopathy remains ill-defined. Recently, we reported that elimination of inflammatory macrophages alleviated the progress and severity of diabetic encephalopathy. Here, we studied the underlying mechanism. Inflammatory macrophages were isolated from the brain of the mice that received i.p. injection of streptozotocin (STZ) to develop diabetes 6 weeks before, and showed enhanced autophagy activity, seemingly through augmentation of Beclin-1 levels. However, the increases in Beclin-1 levels did not result from enhanced gene transcription, but appeared to result from suppression of a Beclin-1-inhibitory microRNA, miR-384-5p. Overexpression of miR-384-5p in the inflammatory macrophages through an adeno-associated virus mediated gene transfer system significantly reduced inflammatory macrophages in the diabetic brain, resulting in attenuation of the STZ-induced decreases in brain malondialdehyde, catalase and superoxidase anion-positive cells, and the STZ-induced increases in brain nitric oxide. Thus, these data suggest that downregulation of miR-384-5p in the inflammatory macrophages may enhance macrophage autophagy and contribute to the development of diabetic encephalopathy, which may be suppressed by re-expression of miR-384-5p in macrophages.
Keywords: Diabetes, inflammatory macrophages, diabetic encephalopathy, miR-384-5p, autophagy, Beclin-1
Introduction
Diabetes is a metabolic disorder associated with increasing risks for central nervous system disorders [1]. Diabetic encephalopathy appears to be a complication that involves direct neuronal damage caused by hyperglycemia [2]. Diabetic encephalopathy received increasing attention recently, since it results in electrophysiological, structural, neurochemical as well as degenerative alterations to neuronal systems which seriously impair neural functions [3]. Indeed, diabetic encephalopathy is often associated with incidence of dementia, or even Alzheimer’s disease [4]. Oxidative stress and inflammation are responsible for diabetic encephalopathy and other neurodegenerative diseases [5-9], but the molecular development of diabetic encephalopathy remains ill-defined.
Brain inflammation due to pathogenesis of diabetes is an important mechanism of brain damage and the cause of diabetic encephalopathy [5-9]. Previous studies have demonstrated a role of activated and infiltrated macrophages in the progression of chronic central nervous system neurodegeneration [10]. Recently, we reported that elimination of inflammatory macrophages alleviated the progress and severity of diabetic encephalopathy, without affecting mouse blood glucose, serum insulin, glucose responses and beta cell mass [11]. However, the molecular regulation of brain inflammatory macrophages in diabetes is unknown.
Autophagy is a biological choice for the cells to survive nutrient deprival or harsh environment through recycling degraded cellular compartments [12]. During autophagy, autophagosomes engulf degraded cellular compartments, leading to conjugation of cytosolic microtubule-associated protein 1A/1B-light chain 3 (LC3-I) to phosphatidylethanolamine LC3-phosphatidylethanolamine conjugate (LC3-II). The ratio of LC3-II to LC3-I levels is thus a representative for the autophagic activity to be used commonly [12-14]. Many proteins coordinate the initiation and progression of autophagy, e.g. autophagy-associated protein 6 (Atg6, or Beclin-1), ATG7 and p62 [15]. Recent evidence showed that these proteins that regulate autophagy are sometimes controlled by a group of small non-coding RNA, termed microRNAs (miRNAs). Among all miRNAs, miR-384-5p was a specific one that was involved in the regulation of neural functions [16,17]. Very recently, we showed that miR-384-5p was a miRNA that targets Beclin-1 to control macrophage autophagy in the development of atherosclerosis [18]. However, the role of this regulatory axis in diabetic encephalopathy has not been examined. Here, we addressed this question.
Inflammatory macrophages were isolated from the brain of the mice that received i.p. injection of streptozotocin (STZ) to develop diabetes 6 weeks before, and showed enhanced autophagy activity, seemingly through augmentation of Beclin-1 levels. However, the increases in Beclin-1 levels did not result from enhanced gene transcription, but appeared to result from suppression of a Beclin-1-inhibitory microRNA, miR-384-5p. Overexpression of miR-384-5p in the inflammatory macrophages in vivo was achieved through an adeno-associated virus mediated gene transfer system, which significantly reduced inflammatory macrophages in the diabetic brain, resulting in attenuation of the STZ-induced decreases in brain malondialdehyde, catalase and superoxidase anion-positive cells, and the STZ-induced increases in brain nitric oxide.
Materials and methods
Protocols and animals
All mouse experiments were approved by research committee of Shanghai Sixth People’s Hospital affiliated to Shanghai Jiaotong University. Male C57BL/6 mice of 10 weeks of age were purchased from Jackson Labs (Bar Harbor, ME, USA). Fasting blood glucose and serum insulin were measured as described before [11]. Streptozotocin (STZ) was i.p. injected at a dose of 150 mg/kg body weight to induce high blood glucose in 10 week-old male mice. Control mice (CTL) received injection of same volume of saline. For viral injection, the mice received 1010 adeno-associated viruses (AAVs) from the tail vein at the time of STZ.
Flow cytometry for macrophages
Mouse brain was dissociated into single cells by incubation in 0.02% Trypsin at 37°C for 50 minutes. The single cell fraction was incubated with PEcy7-conjugated F4/80 antibody (Becton-Dickinson Biosciences, San Jose, CA, USA), followed by flow cytometry based analysis and cell sorting. Primary mouse macrophages were cultured in DMEM media (Invitrogen, CA, Carlsbad, USA) suppled with 5% fetal bovine serum (FBS, Invitrogen) and 1% penicillin/streptomycin (Invitrogen).
AAV production
We used a pAAV-CAG-GFP plasmid (Clontech, Mountain View, CA, USA) as a backbone for generating AAVs. The miR-384-5p mimics, or null controls were purchased from RiboBio Co., Ltd. (Guangzhou, Guangdong, China), as has been described before [18]. The GFP coding sequence was replaced with miR-384-5p or null, while CAG promoter was replaced with CD68 promoter. To generate AAVs, HEK293T cells were co-transfected with 10 µg of the prepared plasmids and 5 µg each of packaging plasmids using Lipofectamine-2000 (Invitrogen). The viruses were purified using CsCl density centrifugation and the titration was determined by a quantitative densitometric dot-blot assay.
RT-qPCR
RNA isolation, reserve transcription and RT-qPCR were performed as described [18]. Data analysis was performed using the 2-ΔΔCt method. All primers were purchased from Qiagen (Hilden, Germany). Values of gene of interest were first normalized against housekeeping gene and then compared to the experimental control.
Western blotting
Protein isolation and Western blotting have been described before [18]. Primary antibodies for Western Blot are anti-F4/80 (Invitrogen) and anti-α-tubulin (1:1000; Cell Signaling, San Jose, CA, USA). Secondary antibody is HRP-conjugated anti-rat and anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). Western blot quantification was performed using NIH ImageJ software (Bethesda, MA, USA).
Immunohistochemistry and beta cell mass
Mouse pancreas and brain were dissected out and fixed in 4% paraformaldehyde for 6 hours, cyro-protected in 30% sucrose overnight, and then sectioned in 6 μM. Primary antibodies are guinea pig polyclonal anti-insulin (1:500; DAKO, Carpinteria, CA, USA) and rat polyclonal anti-F4/80 (1:200; Invitrogen). Secondary antibodies were HRP-conjugated antibodies for corresponding species (1:1000; Jackson ImmunoResearch Labs). Hematoxylin counterstaining was performed at the end of the staining. The quantification of beta cell mass was done as has been described before [11].
Measurement of nitrite/nitrate production
Measurement of nitrite/nitrate production was described before [11].
Measurement of thiobarbituric reactive substances (TBARS)
Measurement of thiobarbituric reactive substances (TBARS) was described before [11].
Measurement of catalase activity
Measurement of catalase activity was described before [11].
Histological identification of superoxide anion
Histological identification of superoxide anion was described before [11].
Bioinformatics and dual luciferase-reporter assay
MiRNAs targets were predicted as has been described before [18], using the algorithms TargetSan [19]. Luciferase reporter assay was described before [18], using miR-384-5p mimics, antisense for miR-384-5p (as-miR-384-5p) or null controls (RiboBio Co., Ltd., Guangzhou, Guangdong, China), a Beclin-1 mRNA 3’-UTR wildtype clone (wt) and Beclin-1 mRNA 3’-UTR with a site mutation at the miR-384-5p binding site (mut) (Creative Biogene, Shirley, NY, USA).
Statistical analyses
Data were analyzed using one-away ANOVA with a Bonferroni correction, followed by Fisher’ Exact Test for comparison of two groups (GraphPad Prism, GraphPad Software, Inc. La Jolla, CA, USA), shown as the mean ± S.D. The p value less than 0.05 was considered significant.
Results
Enhanced macrophage autophagy is detected in the brains from STZ-treated mice
The beta-cell toxin streptozotocin (STZ) was i.p. injected at a dose of 150 mg/kg body weight to induce high blood glucose in C57BL/6 male at 10 week of age. The control mice (CTL) were injected with saline. Six weeks after STZ injection, the mouse brain was dissociated into single cells to sort F4/80+ macrophages (Figure 1A), as described before [11]. Since autophagy affects cell survival and death, we thus analyzed the autophagy levels in macrophages from the brains of these mice. We detected significant increases in LC3-II/I ratio in macrophages from STZ-treated mice, compared to CTL mice (Figure 1B, 1C, Supplementary Figure 1 upper panel). The alteration of autophagy activity in brain macrophages did not seem to result from changes in autophagy-associated factors ATG7 (Figure 1B, 1D), p62 (Figure 1B, 1E), but likely resulted from augmentation of Beclin-1 (Figure 1B, 1F). However, unlike protein, the mRNA levels of BECLIN-1 did not alter in brains from STZ-treated mice (Figure 1G), suggesting that STZ-diabetes may change the Beclin-1 protein at a post-transcriptional level.
Figure 1.
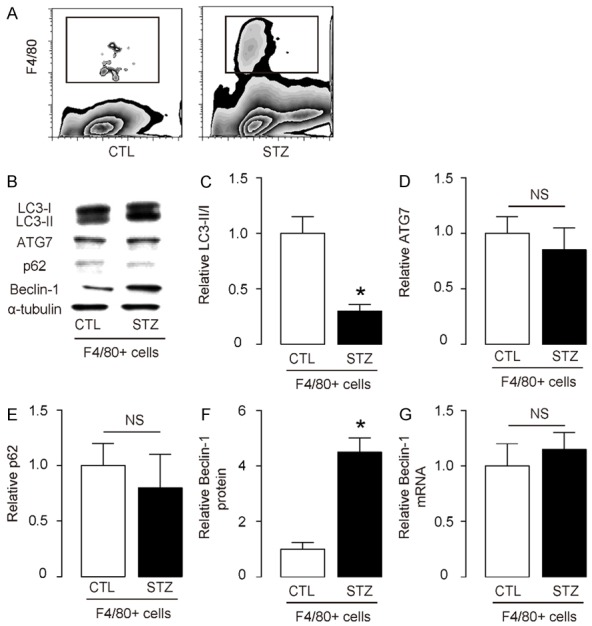
Enhanced macrophage autophagy is detected in the brains from STZ-treated mice. (A) The beta-cell toxin streptozotocin (STZ) was i.p. injected at a dose of 150 mg/kg body weight to induce high blood glucose in C57BL/6 male at 10 week of age. The control mice (CTL) were injected with saline. Six weeks after STZ injection, the mouse brain was dissociated into single cells to sort F4/80+ macrophages. (B) Representative Western blotting for LC3-II/I, ATG7, p62 and Belcin-1 (C-F) Quantification for LC3-II/I ratio (C), ATG7 (D), p62 (E) and Belcin-1 (F) blotting in macrophages from STZ-treated mice, compared to CTL mice. (G) RT-qPCR for Belcin-1 mRNA in macrophages from STZ-treated mice, compared to CTL mice. *p<0.05. NS: non-significant. N=10.
Enhanced macrophage autophagy in the brains from STZ-treated mice is likely due to reduced suppression of protein translation of Beclin-1 by miR-384-5p
Since we have previously reported that miR-384-5p was a miRNA that targets Beclin-1 to control macrophage autophagy in the development of atherosclerosis [18], here we examined whether this regulatory axis also works in the regulation of macrophages associated with diabetic encephalopathy. Indeed, miR-384-5p targets 3’-UTR of Beclin-1 mRNA at one binding site (Figure 2A). Moreover, miR-384-5p levels in brain macrophages significantly decreased after STZ-treated mice (Figure 2B). Modification of miR-384-5p levels was performed in macrophages, by transfection with miR-384-5p mimics, antisense for miR-384-5p (as-miR-384-5p) or null controls (null), which was confirmed by RT-qPCR (Figure 2C). Next, luciferase reporters containing the 3’-UTR of Beclin-1 mRNA or a mutated 3’-UTR of Beclin-1 mRNA in the miR-384-5p binding site were constructed (Figure 2A). We found that miR-384-5p markedly inhibited the luciferase activity of the vector containing the wild-type binding site, whereas the as-miR-384-5p increased the luciferase activity (Figure 2D). On the other hand, transfection of either miR-384-5p or as-miR-384-5p did not affect the luciferase activity of the reporter for 3’-UTR of Beclin-1 mRNA carrying the mutated miR-384-5p binding site (Figure 2E). Together, these data suggest that enhanced macrophage autophagy in the brains from STZ-treated mice is likely due to reduced suppression of protein translation of Beclin-1 by miR-384-5p.
Figure 2.
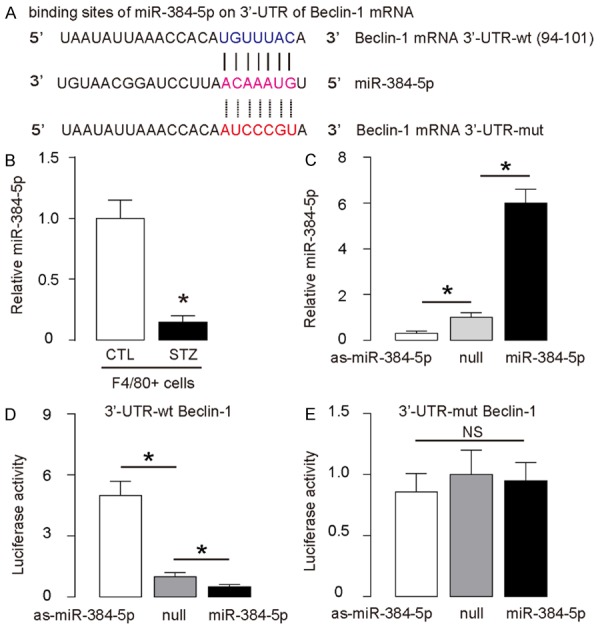
Enhanced macrophage autophagy in the brains from STZ-treated mice is likely due to reduced suppression of protein translation of Beclin-1 by miR-384-5p. (A) Bioinformatics show miR-384-5p binding sites on wildtype (wt) 3’-UTR of Beclin-1 mRNA and illustration of mutate (mut) 3’-UTR of Beclin-1 mRNA. (B) RT-qPCR for miR-384-5p levels in brain macrophages. (C) RT-qPCR for miR-384-5p levels in miR-384-5p-modified macrophages. (D, E) Luciferase reporters containing the 3’-UTR of Beclin-1 mRNA (D) or a mutated 3’-UTR of Beclin-1 mRNA in the miR-384-5p binding site (D), as well as miR-384-5p-modifiction plasmids were co-transfected into macrophages to analyze luciferase activity in a dual luciferase assay. *p<0.05. NS: non-significant. N=5.
Re-expression of miR-384-5p in macrophages does not alter diabetic state of STZ-treated mice
In order to figure out whether decreases in miR-384-5p in brain macrophages may be responsible for the enhanced macrophage autophagy and progress of diabetic encephalopathy, we generated AAVs carrying miR-384-5p or null under control of a CD68 promoter. CD68 is exclusively expressed by monocyte/macrophages, and thus gene therapy with AAVs using CD68 promoter specifically addressed macrophages. Four group of mice of 10 of each were included in this experiment. Group 1, the mice received saline only injection (CTL). Group 2, mice received STZ only (STZ). Group 3, mice received STZ and AAV-null (STZ+AAV-null). Group 4, mice received STZ and AAV-miR-384-5p (STZ+AAV-miR-384-5p). Mice were followed for 6 weeks. We found that STZ induced sustained hyperglycemia (Figure 3A), as well as significant and constant decreases in serum insulin (Figure 3B) in mice, regardless receiving AAVs or not. Moreover, STZ-treated mice had significantly reduced beta cell mass, which was also not affected by either AAVs (Figure 3C, 3D). Together, these data suggest that re-expression of miR-384-5p in macrophages does not alter diabetic state of STZ-treated mice.
Figure 3.
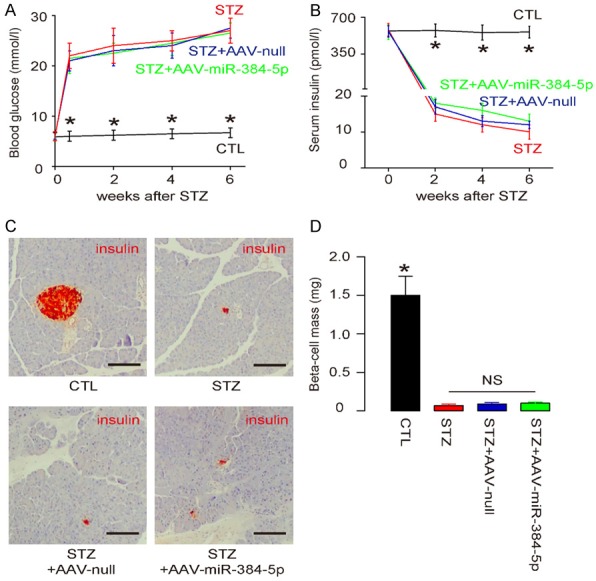
Re-expression of miR-384-5p in macrophages does not alter diabetic state of STZ-treated mice. Four group of mice of 10 of each were included in this experiment. Group 1, the mice received saline only injection (CTL). Group 2, mice received STZ only (STZ). Group 3, mice received STZ and AAV-null (STZ+AAV-null). Group 4, mice received STZ and AAV-miR-384-5p (STZ+AAV-miR-384-5p). All AAVs used CD68 promoter. Mice were followed for 6 weeks. A. Fasting blood glucose. B. Serum insulin. C. Immunostaining for insulin. D. Beta cell mass. *p<0.05. NS: non-significant. N=10. Scale bars are 100 μm.
Re-expression of miR-384-5p in macrophages significantly reduces inflammatory macrophages in the brain
Next, we analyzed the effects of Re-expression of miR-384-5p on macrophages. We found that Re-expression of miR-384-5p significantly reduced the F4/80 protein (Figure 4A, Supplementary Figure 1 lower panel) and F4/80 mRNA (Figure 4B) in the brains of STZ-treated mice, which was also supported by immunohistochemistry for F4/80 in these mice, shown by representative images (Figure 4C), and by quantification (Figure 4D). Hence, re-expression of miR-384-5p in macrophages significantly reduces inflammatory macrophages in the brain.
Figure 4.

Re-expression of miR-384-5p in macrophages significantly reduces inflammatory macrophages in the brain. (A, B) Re-expression of miR-384-5p significantly reduced the F4/80 protein (A) and F4/80 mRNA (B) in the brains of STZ-treated mice. (C, D) Immunohistochemistry for F4/80 was shown by representative images (C), and by quantification (D). *p<0.05. NS: non-significant. N=10. Scale bars are 50 μm.
Re-expression of miR-384-5p in macrophages alleviates brain degradation in STZ-treated mice
Finally, we analyzed the effects of re-expression of miR-384-5p in macrophages on brain degradation markers, brain malondialdehyde (MDA), catalase, superoxidase anion-positive cells and nitric oxide (NO). We found that re-expression of miR-384-5p in macrophages attenuated the STZ-induced decreases in brain MDA (Figure 5A), catalase (Figure 5B) and superoxidase anion-positive cells (Figure 5C), as well as the STZ-induced increases in brain NO (Figure 5D). These data suggest that re-expression of miR-384-5p in macrophages alleviates brain degradation in STZ-treated mice.
Figure 5.
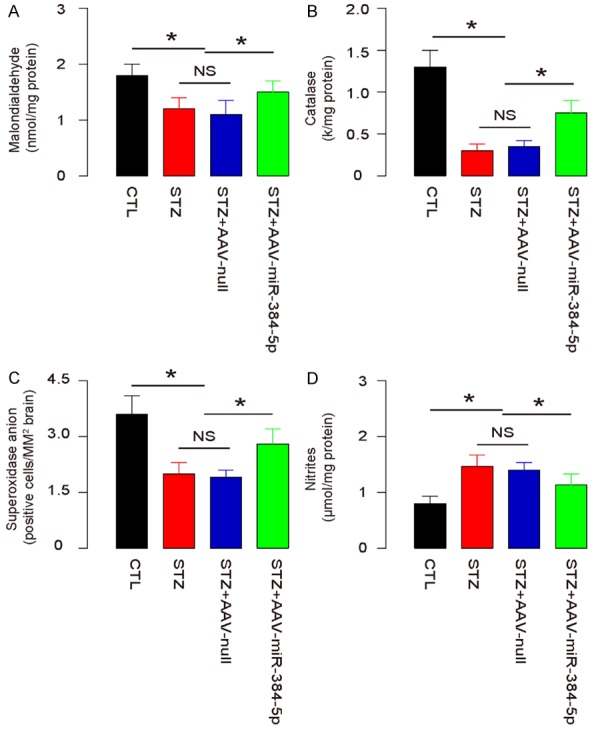
Re-expression of miR-384-5p in macrophages alleviates brain degradation in STZ-treated mice. (A-D) Measurement of brain MDA (A), catalase (B), superoxidase anion-positive cells (C), and NO (D). *p<0.05. NS: non-significant. N=10.
Discussion
Diabetic encephalopathy is one major complication of severe diabetes, likely resulting from hyperglycemic and inflammatory damages [3,20-22]. Although great efforts have been made to understand the molecular mechanisms underlying development of diabetic encephalopathy [5-9], our current knowledge on this prevalent disease is quite limited.
Brain inflammatory events secondary to diabetes contribute significantly to the development of diabetic encephalopathy. Most interestingly, recent evidence points to a critical role of macrophages in the progress of chronic central nervous system neurodegeneration [10]. Recently, we used a mouse STZ-diabetes model to figure out that the overall function of inflammatory macrophages in the diabetic brain appears to be negative [11]. Elimination of brain inflammatory macrophages not only significantly inhibited the STZ-induced decreases in brain MDA, catalase and superoxidase anion-positive cells, but also significantly attenuated STZ-induced increases in NO [11]. However, we did not give answers to the regulation of brain macrophages in the pathological state of diabetes.
In the current study, we took advantage of another finding in our study on atherosclerosis [18], in which we demonstrated the regulatory axis of miR-384-5p/Belcin-1 in the regulation of macrophage autophagy. Here, it seemed that this regulatory axis worked in an opposite way, since macrophage autophagy appeared to play a malignant role in development of diabetic encephalopathy but a beneficial role against development of atherosclerosis. Nevertheless, interference with this target appeared to be effective in both models.
Here, we used an AVV system to alter gene expression in macrophages. A CD68 promoter was chosen not only due to its specificity in macrophages, but also due to its relative small size to be well resembled into an AVV vector. Compared to CD68 promoter, F4/80 promoter region is too big to be fit into an AVV. We used both STZ and STZ with control AAV as controls, since we would like to exclude the possibility that the infection itself may alter macrophage characteristics [23], although our final results did not support this possibility.
The next question may be the exact mechanism leading to the suppression of miR-384-5p in macrophages in the diabetic state. Further experiments may be applied to answer this question, which may further improve our understanding of the molecular pathology of diabetic encephalopathy.
To summarize, here we showed evidence for miR-384-5p as a novel target to experimentally control the development of diabetic encephalopathy.
Acknowledgements
This work was supported by internal funding from Shanghai Sixth People’s Hospital affiliated to Shanghai Jiaotong University.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Zhao L, Zheng Z, Huang P. Diabetes mellitus and the risk of glioma: a meta-analysis. Oncotarget. 2016;7:4483–4489. doi: 10.18632/oncotarget.6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z, Huang Y, Cheng Y, Tan Y, Wu F, Wu J, Shi H, Zhang H, Yu X, Gao H, Lin L, Cai J, Zhang J, Li X, Cai L, Xiao J. Endoplasmic reticulum stress-induced neuronal inflammatory response and apoptosis likely plays a key role in the development of diabetic encephalopathy. Oncotarget. 2016;7:78455–78472. doi: 10.18632/oncotarget.12925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong S, Li G, Zheng D, Wu J, Sun D, Yang F, Yu X, Li T, Sun A, Liu J, Zhong X, Xu C, Lu F, Zhang W. A novel role for the calcium sensing receptor in rat diabetic encephalopathy. Cell Physiol Biochem. 2015;35:38–50. doi: 10.1159/000369673. [DOI] [PubMed] [Google Scholar]
- 4.Ashraghi MR, Pagano G, Polychronis S, Niccolini F, Politis M. Parkinson’s disease, diabetes and cognitive impairment. Recent Pat Endocr Metab Immune Drug Discov. 2016;10:11–21. doi: 10.2174/1872214810999160628105549. [DOI] [PubMed] [Google Scholar]
- 5.Zhou X, Zhu Q, Han X, Chen R, Liu Y, Fan H, Yin X. Quantitative-profiling of neurotransmitter abnormalities in the disease progression of experimental diabetic encephalopathy rat. Can J Physiol Pharmacol. 2015;93:1007–1013. doi: 10.1139/cjpp-2015-0118. [DOI] [PubMed] [Google Scholar]
- 6.Liu YW, Zhu X, Zhang L, Lu Q, Zhang F, Guo H, Yin XX. Cerebroprotective effects of ibuprofen on diabetic encephalopathy in rats. Pharmacol Biochem Behav. 2014;117:128–136. doi: 10.1016/j.pbb.2013.11.027. [DOI] [PubMed] [Google Scholar]
- 7.Wang CF, Li DQ, Xue HY, Hu B. Oral supplementation of catalpol ameliorates diabetic encephalopathy in rats. Brain Res. 2010;1307:158–165. doi: 10.1016/j.brainres.2009.10.034. [DOI] [PubMed] [Google Scholar]
- 8.Sima AA, Zhang W, Kreipke CW, Rafols JA, Hoffman WH. Inflammation in diabetic encephalopathy is prevented by C-peptide. Rev Diabet Stud. 2009;6:37–42. doi: 10.1900/RDS.2009.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhad A, Chopra K. Curcumin attenuates diabetic encephalopathy in rats: behavioral and biochemical evidences. Eur J Pharmacol. 2007;576:34–42. doi: 10.1016/j.ejphar.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Kierdorf K, Wang Y, Neumann H. Immune-mediated CNS damage. Results Probl Cell Differ. 2010;51:173–196. doi: 10.1007/400_2008_15. [DOI] [PubMed] [Google Scholar]
- 11.Wang B, Miao Y, Zhao Z, Zhong Y. Inflammatory macrophages promotes development of diabetic encephalopathy. Cell Physiol Biochem. 2015;36:1142–1150. doi: 10.1159/000430285. [DOI] [PubMed] [Google Scholar]
- 12.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu QH, Yu D, Hu Z, Liu X, Yang Y, Luo Y, Zhu J, Li Z. miR-26a and miR-384-5p are required for LTP maintenance and spine enlargement. Nat Commun. 2015;6:6789. doi: 10.1038/ncomms7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogata K, Sumida K, Miyata K, Kushida M, Kuwamura M, Yamate J. Circulating miR-9* and miR-384-5p as potential indicators for trimethyltin-induced neurotoxicity. Toxicol Pathol. 2015;43:198–208. doi: 10.1177/0192623314530533. [DOI] [PubMed] [Google Scholar]
- 18.Wang B, Zhong Y, Huang D, Li J. Macrophage autophagy regulated by miR-384-5p-mediated control of Beclin-1 plays a role in the development of atherosclerosis. Am J Transl Res. 2016;8:606–614. [PMC free article] [PubMed] [Google Scholar]
- 19.Coronnello C, Benos PV. ComiR: combinatorial microRNA target prediction tool. Nucleic Acids Res. 2013;41:W159–164. doi: 10.1093/nar/gkt379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samarghandian S, Azimi-Nezhad M, Samini F. Ameliorative effect of saffron aqueous extract on hyperglycemia, hyperlipidemia, and oxidative stress on diabetic encephalopathy in streptozotocin induced experimental diabetes mellitus. Biomed Res Int. 2014;2014:920857. doi: 10.1155/2014/920857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamboj SS, Chopra K, Sandhir R. Neuroprotective effect of N-acetylcysteine in the development of diabetic encephalopathy in streptozotocin-induced diabetes. Metab Brain Dis. 2008;23:427–443. doi: 10.1007/s11011-008-9104-7. [DOI] [PubMed] [Google Scholar]
- 22.Reske-Nielsen E, Lundbaek K. Diabetic Encephalopathy. Diffuse and focal lesions of the brain in long-term diabetes. Acta Neurol Scand Suppl. 1963;39(SUPPL4):273–290. [PubMed] [Google Scholar]
- 23.Xiao X, Guo P, Prasadan K, Shiota C, Peirish L, Fischbach S, Song Z, Gaffar I, Wiersch J, El-Gohary Y, Husain SZ, Gittes GK. Pancreatic cell tracing, lineage tagging and targeted genetic manipulations in multiple cell types using pancreatic ductal infusion of adeno-associated viral vectors and/or cell-tagging dyes. Nat Protoc. 2014;9:2719–2724. doi: 10.1038/nprot.2014.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.