

Abstract
Gastric cancer is the fourth most common cancer and the second most frequent cause of cancer-associated mortality in the world. Previous studies have revealed that expression levels of microRNAs (miRNAs) are associated with the initiation and progression of several types of cancer, including gastric cancer. Previous studies have demonstrated that the abnormal expression of miRNA-136 may serve a function in the progression of several types of human cancer. However, the expression pattern of miR-136, its functions and underlying molecular mechanisms in gastric cancer remain unresolved. In the present study, it was revealed that the expression of miR-136 was aberrantly up regulated in gastric cancer tissues and cell lines. The suppression of miR-136 was able to inhibit proliferation and invasion in gastric cancer cell lines. Furthermore, phosphatase and tensin homolog (PTEN) was identified as a direct target gene of miR-136 in gastric cancer. PTEN was under expressed in gastric cancer tissues compared with non-tumor gastric tissues, and PTEN expression was negatively correlated with miR-136 expression. Furthermore, PTEN overexpression mimics the effects of miR-136 knockdown on gastric cancer cells. Additionally, miR-136 under expression decreased phospho-(p) AKT expression, but did not affect AKT expression in gastric cancer cells. In conclusion, the data of the present study suggest that miR-136 acts as an oncogene in gastric cancer via regulation of the PTEN/AKT/p-AKT signaling pathway and may potentially serve as a novel therapeutic target for the treatment of gastric cancer.
Keywords: gastric cancer, microRNA-136, proliferation, invasion, phosphatase and tensin homolog
Introduction
Gastric cancer is the fourth most common cancer (following lung cancer, breast cancer and colorectal cancer) and the second most frequent cause (following lung cancer) of cancer-associated mortality globally (1). It is estimated that there are ~850,000 newly diagnosed cases and 650,000 mortalities per year worldwide (2). Previous studies have demonstrated that multiple pathogenic factors contribute to the formation and progression of gastric cancer, including environmental factors, diet, infections and host genes, particularly the abnormal expression of oncogenes or tumor-suppressor genes (3,4). Despite developments in surgery, chemotherapy and radiation therapy, the prognosis of gastric cancer at advanced stages remains unsatisfactory, primarily due to tumor recurrence, local invasion and metastasis (5). The 5-year survival rate of patients with gastric cancer is <10% (6). Understanding the molecular mechanisms underlying the regulation of gastric cancer occurrence and development is necessary in order to identify novel and effective therapeutic methods to improve treatment outcome.
microRNAs (miRNAs) are small, endogenousnon-coding RNAs, which are between 19 and 22 bases in length (7). These oligonucleotides negatively regulate gene expression through perfect or imperfect base pairing with the 3′-untranslated regions (UTRs) of target genes thereby inducing degradation or translational repression of the target genes (8). Previously, miRNAs have been demonstrated to be functionally involved in the regulation of a series of physiological and pathological processes, including cell proliferation, apoptosis, differentiation and invasion (9). Alterations in the expression of miRNAs have been widely reported in almost all types of cancer, including hepatocellular carcinoma (10), cervical (11), thyroid (12), colorectal (13) and gastric cancer (14). Previous studies have demonstrated that expression levels of miRNAs are associated with cancer initiation and progression (15–17). The abnormal expression of miRNAs may suggest oncogenic or tumor suppressive functions of miRNAs in cancer, dependent on the tumor type and the target genes of the miRNA (18). Therefore, further studies on the regulatory mechanisms of miRNAs in cancer may provide an improved understanding of the oncogenes is network and potential therapeutic targets for cancer treatment.
Previous studies have reported that the abnormal expression of miR-136 has important roles in the progression of several types of human cancer (19–22). To the best of our knowledge, the expression pattern, precise function and underlying molecular mechanisms of miR-136 have not been resolved in gastric cancer thus far. In the present study, the expression, biological functions and underlying molecular mechanisms of miR-136 in gastric cancer were investigated.
Materials and methods
Tissue samples
The present study was approved by the Ethics Committee of The First Affiliated Hospital of Wenzhou Medical University (Wenzhou, China) and written informed consent was obtained from all patients for the use of tissue samples. Paired human gastric cancer tissues and adjacent non-tumorous gastric mucosae tissues were obtained from 26 gastric patients (18 males, 8 females; age range, 47–75 years; mean age, 62 years) who underwent gastrectomy at Department of Gastroenterology and Hepatology, The First Affiliated Hospital of Wenzhou Medical University between February 2014 and March 2015. Patients who were treated with chemotherapy or radiotherapy prior to surgery were excluded from the present study. All tissues were immediately frozen in liquid nitrogen and stored at −80°C.
Cell culture
Human gastric cancer cell lines (AGS, BGC-823, MGC-803, SGC-7901), normal human gastric epithelial GES-1 cells and 293T cells were obtained from the American Type Culture Collection (Manassas, VA, USA). All cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C under a humidified atmosphere containing 5% CO2.
Transfection
miR-136 inhibitor and negative control inhibitor (NC inhibitor) were synthesized by Shanghai GenePharma Co., Ltd., (Shanghai, China). Phosphatase and tensin homolog deleted on chromosome ten (PTEN) overexpression plasmid (pCDNA3.1-PTEN) and empty plasmid (pCDNA3.1) were obtained from Chinese Academy of Sciences (Changchun, China). MGC-803 and SGC-7901 cells were collected and seeded in six-well plates. Transfection was performed when the cell density reached between 30–50% confluence. Gastric cancer cells were transiently transfected with miR-136 inhibitor, NC inhibitor, pCDNA3.1-PTEN or pCDNA3.1 using Lipofectamine 2000® reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Following 48 h of transfection, reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed to determine transfection efficiency. Cell proliferation and Transwell cell invasion assays were conducted at 24 and 48 h post-transfection. Western blot analysis was performed at 72 h following transfection.
RNA preparation and RT-qPCR
Total RNA was extracted from the tissue samples and cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) as per the manufacturer's protocol. The concentration and purity of total RNA was examined using a Nanodrop 2000 (Thermo Fisher Scientific, Inc.). To determine the level of miR-136 expression, reverse transcription was performed using a TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). qPCR was carried out on an ABI7500 Real-time PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.) using a TaqMan MicroRNA PCR kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) to detect the expression levels of miR-136. The cycling conditions were as follows: 50°C for 2 min, 95°C for 10 min, and then 40 cycles of denaturation at 95°C for 15 sec; and annealing/extension at 60°C for 60 sec. The expression level of miR-136 was normalized against the endogenous U6 small nuclear RNA (U6 snRNA). For quantification of PTEN mRNA, cDNA was synthesized using Moloney Murine Leukemia Virus Reverse Transcription system (Promega Corporation, Madison, WI, USA). qPCR was performed using SYBR Premix Ex Taq (TaKaRa Biotechnology Co., Ltd., Dalian, China), and the results were quantified using GAPDH as an internal reference. The cycling conditions for qPCR were as follows: 5 min at 95°C, followed by 40 cycles of 95°C for 30 sec and 65°C for 45 sec. The primers were designed as follows: miR-136, 5′-ACUCCAUUUGUUUUGAUGAUGGA-3′ (forward) and 5′-UCCAUCAUCAAAACAAAUGGAGU-3′ (reverse); U6, 5′-GCTTCGGCAGCACATATACTAAAAT-3′ (forward) and 5′-CGCTTCACGAATTTGCGTGTCAT-3′ (reverse); PTEN, 5′-TGGCGGAACTTGCAATCCTCAGT-3′ (forward) and 5′-TCCCGTCGTGTGGGTCCTGA-3′ (reverse); and GAPDH, 5′-GCCTTCCGTGTCCCCACTGC-3′ (forward) and 5′-CAATGCCAGCCCCAGCGTCA-3′ (reverse). Relative gene expression was calculated using the 2−ΔΔCq method (23).
Cell proliferation assay
Cell proliferation was assessed at day 1, 2 and 3 using Cell Counting Kit-8 assay (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan). At 24 h post-transfection, the transfected cells were harvested, seeded in 96-well plates (3×103 cells/well) and incubated at 37°C in a humidified atmosphere containing 5% CO2. At 1, 2 and 3 days 10 µl CCK-8 solution was added to each well, and the cells were incubated at 37°C for an additional 2 h. The absorbance at 450 nm was detected on a micro plate reader (Model 550; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Each assay was performed in triplicate.
Transwell cell invasion assay
Cell invasion was assayed using Transwell chambers (pore size, 8 µm; EMD Millipore, Billerica, MA, USA) coated with 30 µl Matrigel (BD Biosciences, San Jose, CA, USA). At 48 h post-transfection, the transfected cells were harvested, and 5×104 cells per well were seeded in DMEM medium (600 µl) containing 20% FBS was added to the lower Transwell chamber. After incubating for 48 h, non-invaded cells were removed with cotton swabs. The cells from the lower chamber were fixed with 100% methanol at room temperature for 15 min and then stained with 0.5% crystal violet at room temperature for 15 min. The images of the cells were captured, and the cells were counted using an inverted microscope (Olympus, Tokyo, Japan) at ×200 magnification. Each assay was performed in triplicate and repeated 3 times.
Bioinformatic analysis
The potential target genes of miR-136 were analyzed using microRNA (www.microrna.org/microrna/home.do) and TargetScan (www.targetscan.org) databases.
Luciferase reporter assay
For the luciferase reporter assay, luciferase reporter plasmids, including pGL3-PTEN-3′UTR wild-type (WT) and pGL3-PTEN-3′UTR with mutated 3′UTR (Mut), were synthesized by Shanghai GenePharma Co., Ltd. 293T cells were collected and seeded in 24-well plates. Transfection was performed when cells reached between 30–50% confluence. 293T cells were transfected with miR-136 inhibitor, NC inhibitor, and pGL3-PTEN-3′UTR Wt or pGL3-PTEN-3′UTR Mut, using Lipofectamine 2000 reagents. After incubation for 48 h, the transfected cells were harvested, washed with PBS and then luciferase activity was determined using the Dual-Luciferase Reporter Assay system (Promega Corporation), according to the manufacturer's protocol. Each assay was performed in triplicate and replicated three times.
Protein extraction and western blot analysis
For western blot analysis, the proteins were isolated using radioimmunoprecipitation assay lysis buffer (50 mM Tris-HCl, pH 8.0; 10 mM NaCl; 1 mM DTT; 2 mM EDTA; 1 mM phenylmethylsulfonylfluoride; 5 mg/ml leupeptin; 2 mg/ml aprotinin; 2 mg/ml pepstatin; 1% NP-40 and 0.1% SDS). The concentration of protein was examined using a bicinchoninic acid protein assay kit (Beyotime Institute of Biotechnology, Haimen, China), according to the manufacturer's protocol. Equal quantities of protein (30 µg) were separated using SDS-PAGE (10% gel) and then transferred onto polyvinylidene difluoride membranes (EMD Millipore). The membranes were blocked with 5% skimmed milk in Tris-buffered saline with Tween-20 (TBST) at room temperature for 1 h and incubated overnight at 4°C with primary antibodies for detection of PTEN (1:1,000; catalog no. sc-7974; Santa Cruz Biotechnology, CA, US), AKT (1:1,000; catalog no. sc-8312; Santa Cruz Biotechnology), phospho-(p)-AKT (1:1,000; catalog no. sc-514032; Santa Cruz Biotechnology)and GAPDH (1:1,000; catalog no. sc-166574; Santa Cruz Biotechnology). Following washing four times with TBST, the membranes were further incubated with a corresponding horseradish peroxidase-conjugated secondary antibody (1:5,000; cat. no. sc-2004 for AKT and sc-2005 for PTEN, p-AKT, GAPDH; Santa-Cruz Biotechnology) at room temperature for 2 h. Finally, the immunoreactive bands were visualized using an Enhanced Chemiluminescence Immunoblot Detection system (Pierce Biotechnology, Inc., Rockford, IL, USA) and analyzed using Quantity One software (version 4.62; Bio-Rad Laboratories, Inc.). The relative protein expression was normalized to GAPDH expression.
Statistical analysis
The data are expressed as the mean ± standard deviation or presented as box plots. Statistical analysis was performed using SPSS software (version 17.0; SPSS, Inc., Chicago, IL). The difference between the groups was compared with Student's t-test or a one-way analysis of variance with a Student-Newman-Keulspost-hoc test. Spearman's correlation analysis was used to evaluate the correlation between miR-136 and PTEN mRNA expression in gastric cancer tissues. P<0.05 was considered to indicate a statistically significant difference.
Results
Expression level of miR-136 in clinical gastric cancer tissues and cell lines
To determine the expression pattern of miR-136 in gastric cancer, miR-136 expression in gastric cancer tissues and adjacent non-tumorous gastric mucosae tissues was detected using RT-qPCR. The results revealed that miR-136 was up regulated in gastric cancer tissues compared with adjacent non-tumorous gastric mucosae tissues (P<0.05; Fig. 1A). Additionally, miR-136 expression was evaluated in four gastric cancer cell lines and normal human gastric epithelial GES-1 cells. As presented in Fig. 1B, the expression levels of miR-136 was increased in the gastric cancer cell lines compared with GES-1 cells (P<0.05).
Figure 1.
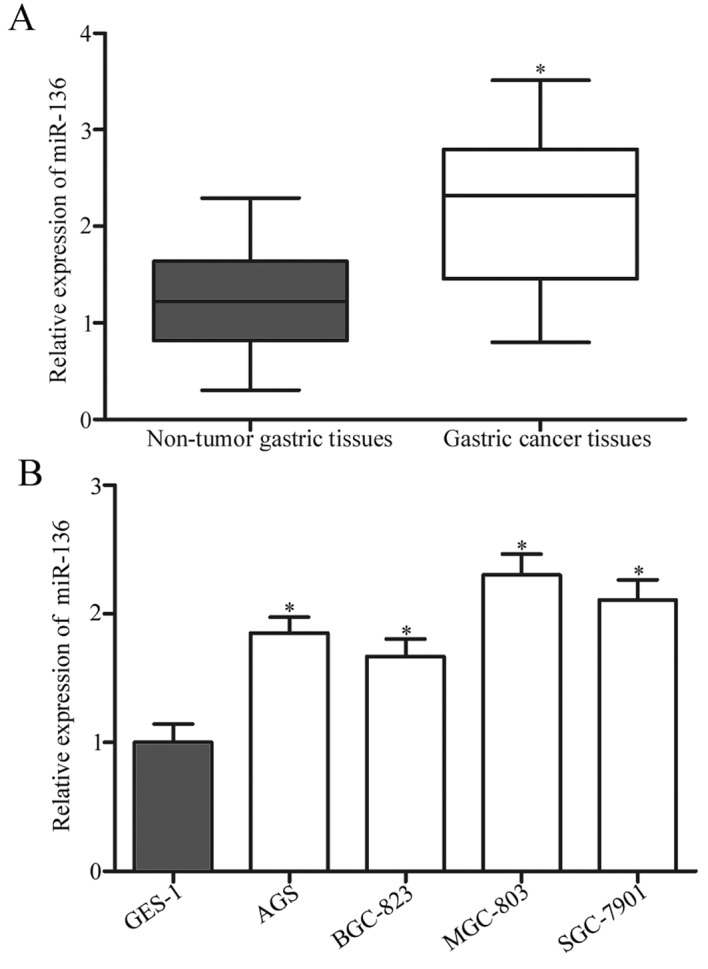
Reverse transcription-quantitative polymerase chain reaction analysis of miR-136 expression in gastric cancer tissues and cell lines. (A) miR-136 expression was determined in 26 paired gastric cancer tissues and matched adjacent non-tumorous gastric mucosae tissues. The data were presented as box plots. The top of the box indicates upper quartile, and the bottom of the box indicates lower quartile. The central line in the box indicates median, and the whiskers indicate the range. *P<0.05 vs. non-tumor tissues (B) Relative expression of miR-136 in four gastric cancer cell lines (AGS, BGC-823, MGC-803 and SGC-7901) and normal human gastric epithelial GES-1 cells. Data are presented as the mean ± standard deviation. *P<0.05 vs. GES-1. miR, microRNA.
miR-136 under expression inhibits proliferation and invasion of gastric cancer cells
Given the up regulation of miR-136 in gastric cancer, it was hypothesized that miR-136 may act as an oncogene in gastric cancer. To confirm this, miR-136 inhibitor was introduced into MGC-803 and SGC-7901 cells, and the transfection efficiency was assessed using RT-qPCR (P<0.05; Fig. 2A). The proliferation of transfected cells was determined using CCK-8 method. As presented in Fig. 2B, miR-136 suppression inhibited the proliferation of MGC-803 and SGC-7901 cells compared with cells that were transfected with NC inhibitor (P<0.05). A Transwell cell invasion assay was used to investigate the effect of miR-136 under expression on gastric cancer cell invasion. The downregulation of miR-136 suppressed the invasion of MGC-803 and SGC-7901 cells compared with the NC inhibitor group (P<0.05; Fig. 2C). These results indicated that miR-136 may act as an oncogene in gastric cancer.
Figure 2.
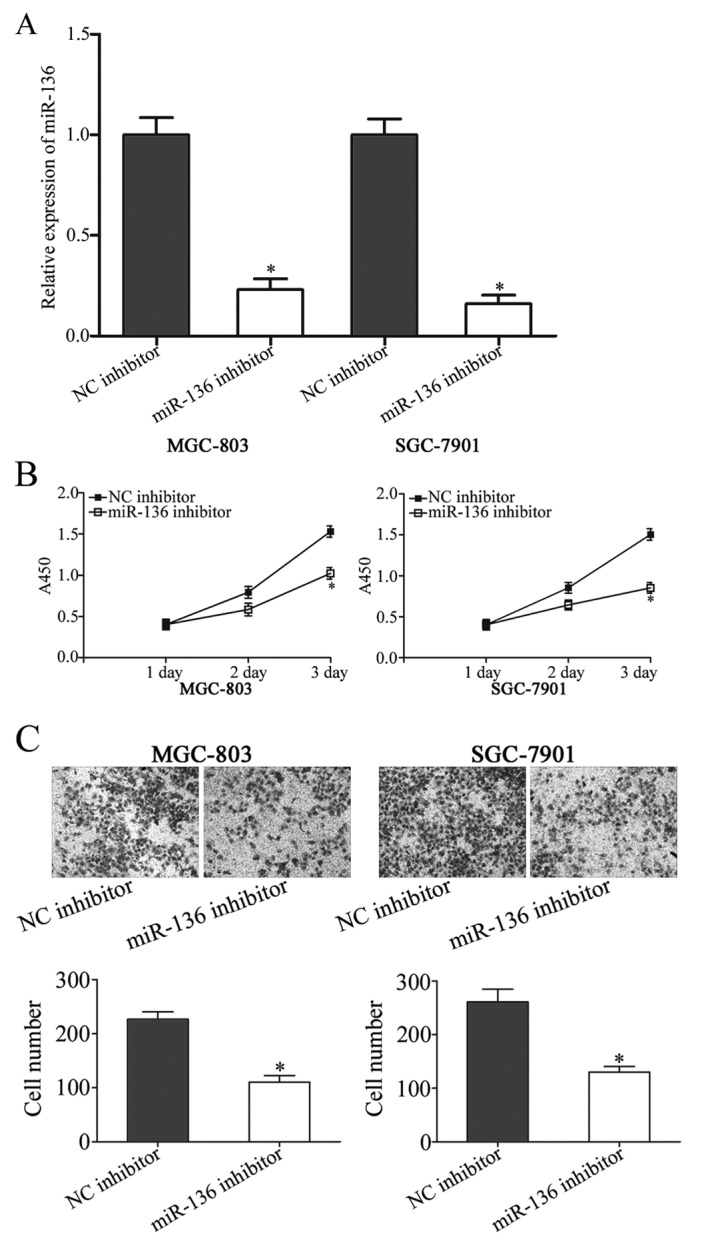
miR-136 knockdown inhibits proliferation and invasion of gastric cancer cells. (A) The expression level of miR-136 in MGC-803 and SGC-7901 cells that were treated with miR-136 inhibitor or NC inhibitor. (B) A cell proliferation assay was performed at 1, 2 and 3 days following the plating of transfected cells. (C) A Transwell cell invasion assay of MGC-803 and SGC-7901 cells that were transfected with miR-136 inhibitor or NC inhibitor. Magnification, ×200. Data are presented as the mean ± standard deviation. *P<0.05 vs. NC inhibitor. miR, microRNA; NC, negative control.
miR-136 directly targets and regulates PTEN expression in gastric cancer
The potentialtarget gene of miR-136 in gastric cancer was investigated. Bioinformatic analysis was used to predict potential target genes of miR-136. Among the potential candidates, PTEN was selected as the focus of the present study (Fig. 3A). A low expression of PTEN was previously demonstrated that in gastric cancer tissues, and PTEN expression was associated with the formation and progression of gastric cancer (24–27). A luciferase reporter assay was performed to evaluate whether PTEN is a direct target gene of miR-136. 293T cells were co-transfected with miR-136 inhibitor or NC inhibitor, and pGL3-PTEN-3′UTR Wt or pGL3-PTEN-3′UTR Mut, followed by determination of luciferase activity. As presented in Fig. 3B, miR-136 under expression increased the luciferase activity of pGL3-PTEN-3′UTR Wt compared with that in cells co-transfected with pGL3-PTEN-3′UTR Wt and NC inhibitor(Fig. 3B; P<0.05), whereas there was no alteration in luciferase activity in cells that were transfected with pGL3-PTEN-3′UTR Mut and miR-136 inhibitor compared with that in cells co-transfected with NC inhibitor.
Figure 3.

PTEN is a direct target of miR-136. (A) Binding sequences for miR-136 in the 3′UTR of PTEN, and the mutations in the 3′UTR of PTEN are presented. (B) A luciferase reporter assay was performed in 293T cells at 48 h after co-transfection with miR-136 inhibitor or NC inhibitor, and pGL3-PTEN-3′UTR Wt or pGL3-PTEN-3′UTR Mut. The treatment with a miR-136 inhibitor was able to increase luciferase activity in cells that were transfected with pGL3-PTEN-3′UTR Wt. By contrast, miR-136 inhibitor did not affect the luciferase activity in in cells that were transfected with pGL3-PTEN-3′UTR Mut. *P<0.05 vs. NC inhibitor. (C) RT-qPCR analysis of PTEN mRNA in MGC-803 and SGC-7901 cells following transfection with miR-136 inhibitor or NC inhibitor. *P<0.05 vs. NC inhibitor. (D) Western blotting was used to detect PTEN protein expression in MGC-803 and SGC-7901 cells following transfection with miR-136 inhibitor or NC inhibitor. (E) The relative PTEN mRNA expression levels were determined using RT-qPCR in gastric cancer tissues and adjacent non-tumorous gastric mucosae tissues. The data are presented as box plots. The top of the box indicates the upper quartile, and the bottom indicates the lower quartile. The central line in the box indicates median, and the whiskers indicate the range. *P<0.05 vs. non-tumorous gastric mucosae tissues. (F) Analysis of correlation between miR-136 and PTEN mRNA expression in gastric cancer tissues. r=−0.6035, P=0.0011. The data are presented as the mean ± standard deviation. PTEN, phosphatase and tensin homolog deleted on chromosome ten; miR-136, microRNA-136; NC inhibitor, negative control inhibitor; 3′UTR, 3′-untranslated regions; Wt, wild-type; Mut, mutant; RT-qPCR, reverse transcription-quantitative polymerase chain reaction.
To further investigate the association between miR-136 and PTEN, RT-qPCR and western blotting were utilized to analyze PTEN mRNA and protein expression in MGC-803 and SGC-7901 cells that were treated with miR-136 inhibitor or NC inhibitor. The results indicated that PTEN mRNA (P<0.05; Fig. 3C) and protein (P<0.05; Fig. 3D) expression were markedly up regulated in MGC-803 and SGC-7901 cells that were transfected with miR-136 inhibitor compared with cells that were transfected with NC inhibitor. Furthermore, the PTEN mRNA expression in gastric cancer tissues was investigated and its possible correlation with miR-136 expression was evaluated. As presented in Fig. 3E and F, PTEN mRNA was markedly downregulated in gastric cancer tissues compared with non-tumor tissues (P<0.05), and an inverse correlation between PTEN mRNA expression and miR-136 expression in gastric cancer tissues was detected (r=−0.6035, P=0.0011). These data support the hypothesis that miR-136 directly targets and regulates PTEN expression in gastric cancer.
PTEN overexpression mimics the effects of miR-136 under expression on proliferation and invasion gastric cancer cells
To confirm whether miR-136-dependent oncogenic effects on the biological behaviors of gastric cancer cells were mediated by PTEN, the functions of PTEN in gastric cancer were investigated. pCDNA3.1-PTEN was utilized to increase PTEN expression in MGC-803 and SGC-7901 cells. Western blotting revealed that PTEN was markedly up regulated in MGC-803 and SGC-7901 cells following transfection with pCDNA3.1-PTEN compared with cells transfected with pCDNA3 (P<0.05; Fig. 4A). As presented in Fig. 4B and C, the up regulation of PTEN was able to inhibit cell proliferation (P<0.05) and invasion (P<0.05) in MGC-803 and SGC-7901 cells, which is similar to the effects of miR-136 inhibitor (Fig. 2B). These results further demonstrated thatmiR-136knockdown was able to inhibit cell proliferation and invasion in gastric cancer by targeting PTEN.
Figure 4.

Up regulation of PTEN mimics the effects of miR-136 under expression in gastric cancer cell proliferation and invasion. (A) Western blot analysis of PTEN expression in MGC-803 and SGC-7901 cells that were treated with pcDNA3.1-PTEN or pcDNA3.1. (B) Cell proliferation assay analysis of cell proliferation following pcDNA3.1-PTEN or pcDNA3.1 transfection in MGC-803 and SGC-7901 cells. (C) Transwell cell invasion assay was performed to detect the effects on cell invasion following pcDNA3.1-PTEN or pcDNA3.1 transfection in MGC-803 and SGC-7901 cells. The data are presented as the mean ± standard deviation. *P<0.05 vs. pcDNA3.1. PTEN, phosphatase and tensin homolog.
miR-136 is involved in the PTEN/AKT/p-AKT signaling pathway
PTEN is a negative regulator of the PI3K/AKT signaling pathway; therefore AKT and p-AKT expression levels were evaluated in MGC-803 and SGC-7901 cells following transfection with miR-136 inhibitor or NC inhibitor. The results indicated that miR-136 under expression decreased p-AKT expression in MGC-803 and SGC-7901 cells (P<0.05), however AKT expression remained unaffected by the miR-136 under expression (Fig. 5). These results suggested that miR-136 is an onco-miRNA that exerts its phenotypic effects partly through the PTEN/AKT/p-AKT signaling pathway in gastric cancer.
Figure 5.
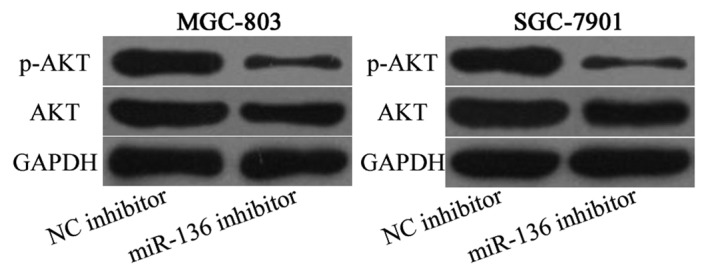
miR-136 knockdown decreases p-AKT expression, but not AKT expression in MGC-803 and SGC-7901 cells. AKT, protein kinase B. p-, phosphorylated; miR-136, microRNA-136; NC, negative control.
Discussion
miRNAs, a new group of regulatory molecules, have been reported to be abnormally expressed in various types of human cancer and associated with the pathogenesis of tumorigenesis and tumor development (28,29). Therefore, it is of great value to investigate the function of miRNAs in gastric cancer formation and progression, and to examine the underlying molecular mechanisms of their involvement. In the present study, it was revealed that miR-136 was up regulated in gastric cancer tissues and cell lines compared with adjacent non-tumorous gastric mucosae tissues and normal human gastric epithelial GES-1 cells. Importantly, the silencing of miR-136 was able to suppress the proliferation and invasion of gastric cancer cells in vitro. Furthermore, it was indicated that the effects of miR-136 under expression in gastric cancer were mediated through the PTEN/AKT/p-AKT signaling pathway. To the best of our knowledge, the present study is the first to investigate the expression, functions and mechanisms of miR-136 in gastric cancer, and also the first study to identify PTEN as a direct and functional target of miR-136.
The deregulation of miR-136 is observed in multiple types of human cancer, suggesting that miR-136 may serve a function in carcinogenesis and progression. For example, Yan et al (19) reported that miR-136 was under expressed in triple-negative breast cancer and negatively associated with the World Health Organization tumor grades (30). Ectopic expression of miR-136 suppressed migration, invasion and epithelial-to-mesenchymal transition in breast cancer cells (19). A study by Zhao et al (31) identified that miR-136 expression level was decreased in patients with primary platinum-resistant ovarian cancer tissues compared with that in platinum-sensitive ovarian cancer tissues. Additionally, miR-136 was identified as a tumor suppressor and miR-136 under expression in ovarian cancer improved chemo resistance partly via the inhibition of apoptosis and promotion of the repair of cisplatin-induced DNA damage (31). Yang et al (20) revealed that miR-136 was downregulated in glioma tissues. The up regulation of miR-136 promoted apoptosis in glioma cells. These findings suggested that miR-136 serves tumor suppressive functions in these types of cancer. Conversely, in non-small cell lung cancer, miR-136 was up regulated and markedly associated with tumor type and differentiation (22). The downregulation of miR-136 expression repressed anchorage-dependent and anchorage-independent proliferation in non-small cell lung cancer (22). These studies draw conflicting conclusions that miR-136 may act as an oncogene in certain types of cancer and tumor suppressor in others. These differences in the function of miR-136 may be explained by the imperfect complementarity of the interactions between miRNAs and target genes (32).
The molecular mechanism by which miR-136 acts as an oncogene in gastric cancer was also studied. Previous studies have validated several targets of miR-136, including RAS protein activator like 2 in breast cancer (19), E2F transcription factor 1, metadherin and B-cell lymphoma 2 in glioma (20,21) and protein phosphatase 2 regulatory subunit B-α in lung cancer (22). Bioinformatic analysis was performed and PTEN was identified as a potential target of miR-136. A subsequent luciferase reporter assay revealed that miR-136 under expression increased the luciferase activity controlled by the wild-type PTEN 3′UTR construct. However, this effect was diminished when the 3′UTR of the PTEN sequence was mutated. Additionally, the knockdown of miR-136 increased PTEN mRNA and protein levels, and decreased p-AKT protein expression in gastric cancer cells. Furthermore, PTEN mRNA was downregulated in gastric cancer tissues and inversely correlated with miR-136 expression. Moreover, it was revealed that the restoration of PTEN expression was able to mimic proliferation and invasion-suppressive effects induced by miR-136 suppression in gastric cancer cells. Collectively, these results demonstrated that miR-136 may partially exert its phenotypic effects through the PTEN/AKT/p-AKT signaling pathway in gastric cancer.
PTEN is one of the most common tumor suppressors and is frequently demonstrated to be decreased or mutated in numerous types of cancer, including cervical cancer (33), colorectal cancer (34), glioma (35), bladder cancer (36) and prostate cancer (37). It has been well established that PTEN may serve functions in a number of biological processes, including cell proliferation, the cell cycle, apoptosis, migration, invasion, metastasis, metabolism, differentiation, transcription and translation through negative regulation of the PI3K/AKT signaling pathway (38–42). Li et al (24) reported that in gastric cancer, PTEN expression is low in tumor tissues and was associated with advanced clinical stage and poor prognosis of patients with gastric cancer. Zheng et al (43) identified that decreased PTEN expression was significantly associated with depth of invasion, lymphatic invasion, lymph node metastasis, liver metastasis and Union Internationale Contre le Cancer staging of gastric cancer (44). Functional experiments indicated tumor suppressive functions for PTEN in cell apoptosis, cell cycle arrest, proliferation, invasion and metastasis in gastric cancer cells (25–27). Consistent with previous data, the present study confirmed the low expression of PTEN in gastric cancer tissues compared with non-tumor gastric tissuesand identified the tumor suppressive functions of PTEN during gastric cancer progression. Therefore, PTEN is a potential target for anti-cancer therapy.
In conclusion, the present study revealed that miR-136 exhibits high expression in gastric cancer tissues and cell lines. Suppression of miR-136 may be able to inhibit proliferation and invasion of gastric cancercellvia the regulation of the PTEN/AKT/p-AKT signaling pathway. The present study may provide a novel target for the treatment of gastric cancer.
Acknowledgements
The present study is supported by a grant from the National Natural Science Foundation of China (FHY2014029).
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Yang L. Incidence and mortality of gastric cancer in China. World J Gastroenterol. 2006;12:17–20. doi: 10.3748/wjg.v12.i1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo M, Yan W. Epigenetics of gastric cancer. Methods Mol Biol. 2015;1238:783–799. doi: 10.1007/978-1-4939-1804-1_41. [DOI] [PubMed] [Google Scholar]
- 4.Ueda T, Volinia S, Okumura H, Shimizu M, Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, et al. Relation between microRNA expression and progression and prognosis of gastric cancer: A microRNA expression analysis. Lancet Oncol. 2010;11:136–146. doi: 10.1016/S1470-2045(09)70343-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terry MB, Gaudet MM, Gammon MD. The epidemiology of gastric cancer. Semin Radiat Oncol. 2002;12:111–127. doi: 10.1053/srao.30814. [DOI] [PubMed] [Google Scholar]
- 6.Orditura M, Galizia G, Sforza V, Gambardella V, Fabozzi A, Laterza MM, Andreozzi F, Ventriglia J, Savastano B, Mabilia A, et al. Treatment of gastric cancer. World J Gastroenterol. 2014;20:1635–1649. doi: 10.3748/wjg.v20.i7.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammond SM. MicroRNAs as oncogenes. Curr Opin Genet Dev. 2006;16:4–9. doi: 10.1016/j.gde.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 9.Macfarlane LA, Murphy PR. MicroRNA: Biogenesis, Function and Role in Cancer. Curr Genomics. 2010;11:537–561. doi: 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu G, Zheng K, Xia S, Wang Y, Meng X, Qin X, Cheng Y. MicroRNA-655-3p functions as a tumor suppressor by regulating ADAM10 and β-catenin pathway in Hepatocellular Carcinoma. J Exp Clin Cancer Res. 2016;35:89. doi: 10.1186/s13046-016-0368-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen XF, Liu Y. MicroRNA-744 inhibited cervical cancer growth and progression through apoptosis induction by regulating Bcl-2. Biomed Pharmacother. 2016;81:379–387. doi: 10.1016/j.biopha.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 12.Sheng W, Chen Y, Gong Y, Dong T, Zhang B, Gao W. miR-148a inhibits self-renewal of thyroid cancer stem cells via repressing INO80 expression. Oncol Rep. 2016;36:3387–3396. doi: 10.3892/or.2016.5203. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Yu P. miR-300 promotes proliferation and EMT-mediated colorectal cancer migration and invasion by targeting p53. Oncol Rep. 2016;36:3225–3232. doi: 10.3892/or.2016.5193. [DOI] [PubMed] [Google Scholar]
- 14.Duan J, Zhang H, Qu Y, Deng T, Huang D, Liu R, Zhang L, Bai M, Zhou L, Ying G, et al. Onco-miR-130 promotes cell proliferation and migration by targeting TGFβR2 in gastric cancer. Oncotarget. 2016;7:44522–44533. doi: 10.18632/oncotarget.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farooqi AA, Rehman ZU, Muntane J. Antisense therapeutics in oncology: Current status. Onco Targets Ther. 2014;7:2035–2042. doi: 10.2147/OTT.S49652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 17.Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, Liu CG, Bhatt D, Taccioli C, Croce CM. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297:1901–1908. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- 18.Qu Y, Zhang H, Duan J, Liu R, Deng T, Bai M, Huang D, Li H, Ning T, Zhang L, et al. MiR-17-5p regulates cell proliferation and migration by targeting transforming growth factor-beta receptor 2 in gastric cancer. Oncotarget. 2016;7:33286–33296. doi: 10.18632/oncotarget.8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan M, Li X, Tong D, Han C, Zhao R, He Y, Jin X. miR-136 suppresses tumor invasion and metastasis by targeting RASAL2 in triple-negative breast cancer. Oncol Rep. 2016;36:65–71. doi: 10.3892/or.2016.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Wu J, Guan H, Cai J, Fang L, Li J, Li M. MiR-136 promotes apoptosis of glioma cells by targeting AEG-1 and Bcl-2. FEBS Lett. 2012;586:3608–3612. doi: 10.1016/j.febslet.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 21.Chen W, Yang Y, Chen B, Lu P, Zhan L, Yu Q, Cao K, Li Q. MiR-136 targets E2F1 to reverse cisplatin chemosensitivity in glioma cells. J Neurooncol. 2014;120:43–53. doi: 10.1007/s11060-014-1535-x. [DOI] [PubMed] [Google Scholar]
- 22.Shen S, Yue H, Li Y, Qin J, Li K, Liu Y, Wang J. Upregulation of miR-136 in human non-small cell lung cancer cells promotes Erk1/2 activation by targeting PPP2R2A. Tumour Biol. 2014;35:631–640. doi: 10.1007/s13277-013-1087-2. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Cui J, Zhang CH, Yang DJ, Chen JH, Zan WH, Li B, Li Z, He YL. High-expression of DJ-1 and loss of PTEN associated with tumor metastasis and correlated with poor prognosis of gastric carcinoma. Int J Med Sci. 2013;10:1689–1697. doi: 10.7150/ijms.7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng T, Meng X, Wang J, Chen X, Yin D, Liang Y, Song X, Pan S, Jiang H, Liu L. PTEN- and p53-mediated apoptosis and cell cycle arrest by FTY720 in gastric cancer cells and nude mice. J Cell Biochem. 2010;111:218–228. doi: 10.1002/jcb.22691. [DOI] [PubMed] [Google Scholar]
- 26.Zhang LL, Liu J, Lei S, Zhang J, Zhou W, Yu HG. PTEN inhibits the invasion and metastasis of gastric cancer via downregulation of FAK expression. Cell Signal. 2014;26:1011–1020. doi: 10.1016/j.cellsig.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 27.He RF, Hu ZL, Wen JF. Biological implication of PTEN gene expression in human gastric cancer and related molecular mechanisms. Zhonghua Bing Li Xue Za Zhi. 2007;36:324–328. (In Chinese) [PubMed] [Google Scholar]
- 28.Sachdeva M, Mo YY. miR-145-mediated suppression of cell growth, invasion and metastasis. Am J Transl Res. 2010;2:170–180. [PMC free article] [PubMed] [Google Scholar]
- 29.Wu D, Niu X, Pan H, Zhou Y, Zhang Z, Qu P, Zhou J. Tumor-suppressing effects of microRNA-429 in human renal cell carcinoma via the downregulation of Sp1. Oncol Lett. 2016;12:2906–2911. doi: 10.3892/ol.2016.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christgen M, Länger F, Kreipe H. Histological grading of breast cancer. Pathologe. 2016;37:328–336. doi: 10.1007/s00292-016-0182-8. (In German) [DOI] [PubMed] [Google Scholar]
- 31.Zhao H, Liu S, Wang G, Wu X, Ding Y, Guo G, Jiang J, Cui S. Expression of miR-136 is associated with the primary cisplatin resistance of human epithelial ovarian cancer. Oncol Rep. 2015;33:591–598. doi: 10.3892/or.2014.3640. [DOI] [PubMed] [Google Scholar]
- 32.Yu Z, Ni L, Chen D, Zhang Q, Su Z, Wang Y, Yu W, Wu X, Ye J, Yang S, et al. Identification of miR-7 as an oncogene in renal cell carcinoma. J Mol Histol. 2013;44:669–677. doi: 10.1007/s10735-013-9516-5. [DOI] [PubMed] [Google Scholar]
- 33.Loures LF, Candido EB, Vidigal PV, Seabra MA, Marco LA, Silva-Filho AL. PTEN expression in patients with carcinoma of the cervix and its association with p53, Ki-67 and CD31. Rev Bras Ginecol Obstet. 2014;36:205–210. doi: 10.1590/S0100-7203201400050004. [DOI] [PubMed] [Google Scholar]
- 34.de Araujo WM, Robbs BK, Bastos LG, de Souza WF, Vidal FC, Viola JP, Morgado-Diaz JA. PTEN overexpression cooperates with lithium to reduce the malignancy and to increase cell death by apoptosis via PI3K/Akt suppression in colorectal cancer cells. J Cell Biochem. 2016;117:458–469. doi: 10.1002/jcb.25294. [DOI] [PubMed] [Google Scholar]
- 35.Zhou YH, Tan F, Hess KR, Yung WK. The expression of PAX6, PTEN, vascular endothelial growth factor, and epidermal growth factor receptor in gliomas: Relationship to tumor grade and survival. Clin Cancer Res. 2003;9:3369–3375. [PubMed] [Google Scholar]
- 36.Koksal IT, Yasar D, Dirice E, Usta MF, Karauzum S, Luleci G, Baykara M, Sanlioglu S. Differential PTEN protein expression profiles in superficial versus invasive bladder cancers. Urol Int. 2005;75:102–106. doi: 10.1159/000085933. [DOI] [PubMed] [Google Scholar]
- 37.Mithal P, Allott E, Gerber L, Reid J, Welbourn W, Tikishvili E, Park J, Younus A, Sangale Z, Lanchbury JS, et al. PTEN loss in biopsy tissue predicts poor clinical outcomes in prostate cancer. Int J Urol. 2014;21:1209–1214. doi: 10.1111/iju.12571. [DOI] [PubMed] [Google Scholar]
- 38.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 39.Mukhopadhyay UK, Mooney P, Jia L, Eves R, Raptis L, Mak AS. Doubles game: Src-Stat3 versus p53-PTEN in cellular migration and invasion. Mol Cell Biol. 2010;30:4980–4995. doi: 10.1128/MCB.00004-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waite KA, Eng C. Protean PTEN: Form and function. Am J Hum Genet. 2002;70:829–844. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nogueira C, Kim KH, Sung H, Paraiso KH, Dannenberg JH, Bosenberg M, Chin L, Kim M. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene. 2010;29:6222–6232. doi: 10.1038/onc.2010.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee H, Choi SK, Ro JY. Overexpression of DJ-1 and HSP90alpha, and loss of PTEN associated with invasive urothelial carcinoma of urinary bladder: Possible prognostic markers. Oncol Lett. 2012;3:507–512. doi: 10.3892/ol.2011.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng H, Takahashi H, Murai Y, Cui Z, Nomoto K, Tsuneyama K, Takano Y. Low expression of FHIT and PTEN correlates with malignancy of gastric carcinomas: Tissue-array findings. Appl Immunohistochem Mol Morphol. 2007;15:432–440. doi: 10.1097/01.pai.0000213127.96590.2d. [DOI] [PubMed] [Google Scholar]
- 44.Ahn HS, Kim SH, Kodera Y, Yang HK. Gastric cancer staging with radiologic imaging modalities and UICC staging system. Dig Surg. 2013;30:142–149. doi: 10.1159/000350881. [DOI] [PubMed] [Google Scholar]