

Abstract
Trinucleotide repeat (TNR) expansions in the genome cause a number of degenerative diseases. A prominent TNR expansion involves the triplet CAG in the huntingtin (HTT) gene responsible for Huntington's disease (HD). Pathology is caused by protein and RNA generated from the TNR regions including small siRNA‐sized repeat fragments. An inverse correlation between the length of the repeats in HTT and cancer incidence has been reported for HD patients. We now show that siRNAs based on the CAG TNR are toxic to cancer cells by targeting genes that contain long reverse complementary TNRs in their open reading frames. Of the 60 siRNAs based on the different TNRs, the six members in the CAG/CUG family of related TNRs are the most toxic to both human and mouse cancer cells. siCAG/CUG TNR‐based siRNAs induce cell death in vitro in all tested cancer cell lines and slow down tumor growth in a preclinical mouse model of ovarian cancer with no signs of toxicity to the mice. We propose to explore TNR‐based siRNAs as a novel form of anticancer reagents.
Keywords: CAG repeats, cell death, RNAi, TLP nanoparticles, trinucleotide repeats
Subject Categories: Cancer, RNA Biology
Introduction
Trinucleotide repeat (TNR) expansions are the cause of a large number of degenerative disease syndromes characterized by amplification of DNA triplet motifs 1. They include spinocerebellar ataxias (SCAs), spinobulbar muscular atrophy (SBMA), myotonic dystrophy type 1 (DM1), and Huntington's disease (HD) 1, 2. HD is a dominantly inherited neurodegenerative disorder caused by expansion of CAG repeats in the huntingtin (HTT) gene. It has been shown that the resulting glutamine expansions (polyQ) in HTT are toxic to cells 3, 4 and that the length of the CAG amplifications determines severity and onset of the disease 2, 4. In addition to polyQ toxicity, repeat‐associated, non‐ATG (RAN) translation was discovered as another translation‐level pathogenic mechanism of CAG repeat‐containing mRNAs 5. More recent evidence, however, also points toward RNA playing a role in affecting cell viability by polytriplet repeats 1, 6. Indeed, many of the repeats in several TNR diseases are not located in open reading frames (ORFs) but in introns or untranslated regions (UTRs) 4. DM1 is the best‐characterized disease regarding RNA toxicity. The CUG repeats are in the 3′UTR of the dystrophia myotonica protein kinase (DMPK) gene, causing most of their toxicity by forming hairpin structures 7. These hairpins are believed to recruit a number of RNA‐binding proteins to nuclear RNA foci 8. Another mechanism by which CAG/CUG TNRs could be toxic at the RNA level is by interfering with cellular splicing. This has been shown for CUG in DM1 9 and CAG in HD 10.
Mounting evidence suggests the CAG TNR expansions are toxic at the RNA level. It was shown in Drosophila that the toxicity of the CAG repeat disease gene spinocerebellar ataxia type 3 (SCA3) protein ataxin‐3 is in large part caused by the trinucleotide repeat RNA and not by the polyQ protein 11. Replacing some of the glutamine coding CAG repeats with the other codon coding for glutamine, CAA, mitigated the toxicity despite similar polyQ protein expression levels. Direct toxicity of mRNA with extended CAG repeats was also demonstrated in mice 12. Finally, there is convincing evidence that CAG/CUG repeats can give rise to RNAi‐active small RNAs. In human neuronal cells, the expression of the CAG expanded exon 1 of HTT (above the threshold for complete penetrance which is > 40) 6 caused an increase in small CAG (sCAG) repeat‐derived RNAs of about 21 nt in length. Above a certain length, CAG/CUG repeats were found to be cleaved by Dicer, the enzyme that generates mature miRNAs from pre‐miRNAs before they are incorporated into the RNA‐induced silencing complex (RISC) 13. The CAG repeat‐derived fragments could bind to complementary transcripts and downregulate their expression via an RNAi‐based mechanism. In a mouse model of HD, treatment of the mice with a locked nucleic acid‐modified 20mer antisense oligonucleotide complementary to the CAG TNR (LNA‐CTG) which reduced the expression of sCAGs but not of HTT mRNA or protein reversed motor deficits 14. This study identified sCAG as a disease‐causing agent. Since sCAGs, isolated from HD human brains, when transfected reduced viability of neurons 6, these sequences might affect cell viability through RNAi by targeting genes that regulate cell survival.
We recently reported that si‐ and shRNAs derived from CD95, CD95L 15, and other genes in the human genome 16 kill cancer cells through RNAi by targeting a network of critical survival genes 15. DISE (death induced by survival gene elimination) was found to involve simultaneous activation of multiple cell death pathways, and cancer cells have a hard time developing resistance to this form of cell death 17. DISE was found to preferentially affect transformed cells 17. Because the length of the CAG repeats in different CAG repeat diseases has been inversely correlated with cancer incidence in various organs 18, 19, 20, 21, we were wondering whether RNAi‐active CAG‐based TNRs might be responsible for this phenomenon and whether they could be used to kill cancer cells.
We have now identified an entire family of TNR‐based siRNAs—which contains the CAG repeat that causes HD—to be at least 10 times more toxic to cancer cells than any tested DISE‐inducing si/shRNA. Our data suggest this super toxicity is caused by targeting multiple complementary TNR expansions present in the open reading frames (ORFs) of multiple genes, rather than in their 3′UTRs. As a proof of concept, we demonstrate that siCAG/CUG can be safely administered to mice to slow down the growth of xenografted ovarian cancer cells with no obvious toxicity to the animals. We are proposing to develop super toxic TNR expansion‐based siRNAs for cancer treatment.
Results
siCAG/CUG kills all cancer cells in vitro
CAG repeats are the defining factor in Huntington's disease, and their complement CTG is amplified in myotonic dystrophy type 1 (DM1) 1. We were interested in determining whether a 19mer duplex of CAG and CUG repeats (siCAG/CUG; Fig 1A) would affect the growth of cancer cells. When transfecting siCAG/CUG into various human (Fig 1B) and mouse (Fig 1C) cancer cell lines at 10 nM, all cancer cells stopped growing within hours of transfection and eventually most of the cells died with no outgrowth of recovering cells (Supplementary Movies EV1, EV2, EV3, EV4, EV5, EV6, EV7, EV8, EV9 and EV10; Appendix Fig S1A). All cancer cells transfected with siCAG/CUG showed morphological changes similar to the ones we observed in cells undergoing DISE (Appendix Fig S1B, 15, 17). We found that siCAG/CUG killed HCT116 cells even when transfected at 10 pM (Fig 1D). Compared to any other si‐ or shRNA we have tested, siCAG/CUG is ~10–100 times more toxic depending on the assay used. When monitoring cell viability (ATP content), the IC50 for siL3, the most toxic DISE‐inducing siRNA we have used, was determined to be 0.8 nM and for siCAG/CUG was 0.039 nM (Fig 1E).
Figure 1. An siRNA duplex comprised of CAG and CUG repeats is super toxic to various cancer cell lines of human and mouse origin.
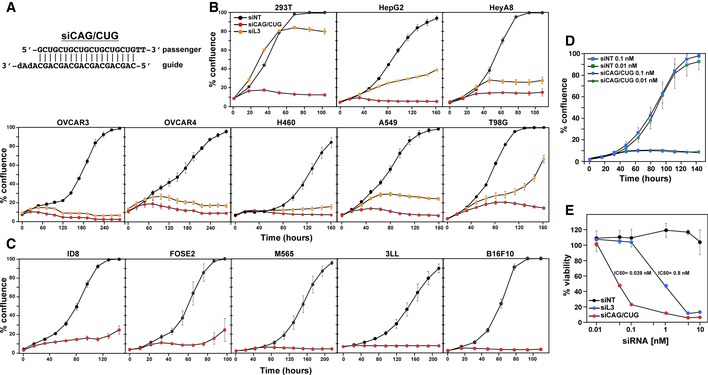
-
ASequence of the siCAG/CUG duplex. dA, deoxyadenosine.
-
B, CConfluences over time of human (B) and mouse (C) cell lines transfected with 10 nM of either siNT, siL3, or siCAG/CUG. M565, 3LL, and B16F10 cells were reverse‐transfected. Values are mean ± SEM. The 13 cell lines were tested between one and six times in each case with between three and six technical replicates.
-
DConfluence over time of HCT116 cells transfected with either siNT or siCAG/CUG at 0.1 or 0.01 nM. Values are mean ± SEM. n = 2 biological replicates, six technical replicates each.
-
EViability (ATP content) of HeyA8 cells transfected with different concentrations of siNT, siL3, or siCAG/CUG. Values are mean ± SD. n = 3 biological replicates, three technical replicates each.
Identification of the most toxic TNR‐based siRNAs
The siCAG/CUG repeat 19mer in all three frames showed roughly the same level of toxicity when transfected into HeyA8 cells (Appendix Fig S2). To test whether other TNR disease‐derived sequences were toxic to cancer cells when introduced as siRNAs, the repeats siGAA/UUC (GAA is amplified in Friedreich's ataxia 22) and siCGG/CCG [CGG found in fragile X tremor ataxia syndrome (FXTAS) and CCG found in fragile XE mental retardation (FRAXE) 1] were transfected into HeyA8 (ovarian) and A549 (lung) cancer cells (Fig 2A). In addition, siCGA/UCG was tested because it has the same base composition as the super toxic siCAG/CUG TNR. Interestingly, among the four tested TNR siRNA duplexes, two were super toxic to both cell lines, and two showed no toxicity. Most remarkable was the observation that siCGA/UCG was among the nontoxic repeats. This finding pointed at a sequence‐specific mechanism behind this phenomenon rather than a response of the cells to dsRNA of a specific base composition.
Figure 2. Identification of the most toxic TNR‐based siRNAs.
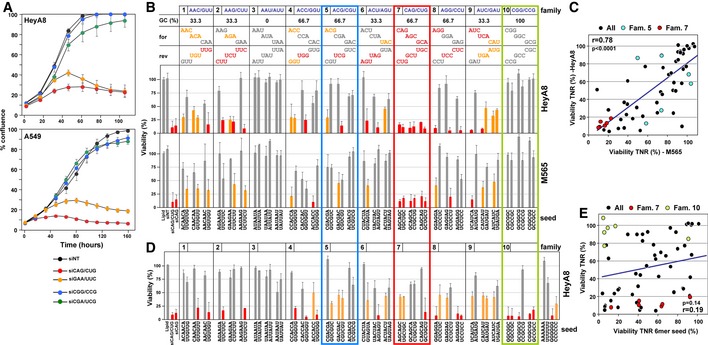
- Confluence over time of HeyA8 (top) or A549 (bottom) cells transfected with 1 nM of either siNT or the four TNR‐based siRNAs, siCAG/CUG, siCGG/CCG, siGAA/UUC, and siCGA/UCG. Values are mean ± SEM. n = 3/2 biological replicates (HeyA8/A549), eight technical replicates.
- Results of toxicity screens of 60 TNR‐based siRNAs in human HeyA8 (top) and mouse M565 (bottom) cells. Cells were reverse‐transfected in triplicate in 384‐well plates with 1 nM of each siRNA. In all cases, the complementary (sense) strand was inactivated by a 2′‐OMe modification in positions 1 and 2. Gray: no or less than 50% loss in viability; yellow: > 50% loss in viability; red: > 75% loss in viability. Family 7 is boxed in red as it is the only family in which all six duplexes were super toxic. Family 5 is boxed in blue. Its six members have the same GC content and the same nucleotide composition as the members of family 7. Family 10 is boxed in green. Each family contains three TNRs in the forward frame (for) and three TNRs that are the reverse complement (rev) of these TNRs. Values are mean ± SD. Three technical replicates.
- Correlation between the average of two viability screens performed in HeyA8 and two performed in M565 cells. The data points for the six members of TNR family 5 are labeled in blue those of family 7 are labeled in red. Pearson correlation and P‐value are given.
- Results of toxicity screen of 60 TNR‐based 6mer seeds in a nontoxic backbone siRNA (see Appendix Fig S5B) in HeyA8 cells. Values are mean ± SD. n = 3 technical replicates.
- Correlation between the average of two viability screens performed in HeyA8 with the 60 TNR‐based siRNAs and the screen performed with the 60 TNR‐based seeds. The data points for the six members of TNR family 7 are labeled in red and those of family 10 are labeled in green. Pearson correlation and P‐value are given.
To identify the most toxic TNR sequences in an unbiased screen, we designed a library of 19mer siRNAs based on the 60 possible TNRs (that contain more than one type of nucleotide). To reduce passenger strand loading and determine the toxicity of each repeat when loaded into the RISC as a guide strand, we replaced positions 1 and 2 of the passenger strand with 2′‐O‐methylated (OMe) nucleotides. To confirm the effect of the OMe modification, we modified the toxic CD95L‐derived siRNA siL3 in this way. While the siL3 duplex modified on the intended passenger strand (S‐OMe) was slightly more toxic to cells than unmodified siL3, likely reflecting a low level of passenger strand loading of siL3, siL3 modified neither on the antisense strand (AS‐OMe) nor on both strands (S/AS‐OMe) showed any toxicity (Appendix Fig S3).
All 60 TNRs were now synthesized with the sense strand carrying the OMe modification in positions 1 and 2, allowing us to determine the toxicity of each of the 60 antisense sequences. HeyA8 cells were transfected with 1 nM of each of the 60 TNRs, and viability was quantified 96 h after transfection. The 60 TNRs can be grouped into 10 families 23. Each family is comprised of three triplets shifted by one nucleotide plus its three complementary triplets. In total, 30 (50%) of the TNRs were not toxic, 11 (18%) were moderately toxic (> 50% loss of viability, shown in yellow), and 19 (31.7%) were super toxic (> 75% loss of viability, shown in red) to HeyA8 cells (Fig 2B, top panels). Among the nontoxic TNRs were all six members of family 3 (0% GC content) and all six members of family 10 (100% GC content). All other TNR families contained nontoxic and toxic TNRs. Interestingly, in some cases, just shifting the TNR sequence in the 19mer by one nucleotide resulted in opposite effects on viability (i.e., AGG and GGA in family 8). In other cases, members of a family showed toxicity of one strand but no toxicity of its complement (i.e., families 1, 2, 4, 5, 6, 8, and 9). This finding suggests a sequence‐specific and, in some cases, frame‐specific activity of the TNRs consistent with RNAi being involved. Due to the different base composition of targeted RNAs, the comparison of TNR families with the same GC content and base composition is most meaningful. Two families contain a balanced GC content of 66.7% and identical base composition: families 5 and 7. Remarkably, while family 5 contains toxic and nontoxic members, all six TNRs in family 7 were super toxic (boxed in red in Fig 2B). Family 7 stands out as it contains all permutations of both the CAG and the CUG repeats we identified as killing all cancer cells.
To determine how much of these activities was conserved between human and mouse cancer cells, the screen was repeated with the mouse liver cancer cell line M565 (Fig 2B, bottom panels). The results for the siRNAs in TNR families 1, 2, 4, 5, 8, and 9 were somewhat similar to the ones obtained with the human cell line, but also showed clear differences. This could be due to differences in tissue origin, cell line, or species. Three of the TNR families performed in an identical fashion between the two cell lines. Similar to HeyA8 cells, none of the 12 TNR‐derived siRNAs in families 3 or 10 showed any toxicity in M565 cells. Most strikingly, however, was the finding that again all six members of family 7, which contain both the CAG and the CUG repeat, were super toxic to the mouse cell line. Screens in both HeyA8 and M565 cells were repeated and results showed a high degree of congruence, especially in the results of family 7 (Appendix Fig S4). When the average of the screen in HeyA8 cells was plotted against the averages of the two screens in M565 cells, a significant correlation between the screens was found (Fig 2C) and, again, the six TNRs in family 7 were most consistently toxic. The data suggest the toxicity of this TNR family is conserved and it is independent of tissue, cell line, and species.
We recently reported that the 6mer seed sequence of siL3 was the main determinant of its toxicity 15. We therefore wondered how much of the toxicity of the super toxic TNRs was due to complete complementarity of the siRNA and how much was dependent on just the 6mer seed sequence. The data on siL3 were obtained by generating chimeric siRNA duplexes between a nontoxic control siRNA (siNT) and siL3 by replacing siL3 sequences from either end of the duplex with siNT sequences 15. To generate an artificial nontoxic siRNA backbone in which to test all 60 TNR 6mer seed sequences, we first replaced four positions in the center of siNT still identical to the same position in the siL3 sequence with the complementary nucleotides, thereby removing any identity between siNT and siL3 outside the seed, while maintaining GC content (Appendix Fig S5A). This siL3 seed siRNA (siL3 seed) was almost as toxic to HeyA8 cells as siL3, confirming that the 6mer seed determined a substantial part of the toxicity of siL3. We therefore used the modified siNT backbone to test all possible TNR‐derived 6mer seed sequences (Fig 2D, Appendix Fig S5B). While some TNR‐derived seeds were toxic to HeyA8 cells, there was only a moderate level of congruence between the screen with the entire TNR 19mers and one just with the 6mers in the modified siNT backbone (Fig 2E). Of the six super toxic TNRs in family 7, only one was also toxic in the 6mer screen (Fig 2D and E). Interestingly, most of the 6mers in family 10 were toxic although no toxicity was observed in the TNR screen (Fig 2B). We interpret this as the inability of these six TNRs with their 100% GC content to properly enter the RISC. Together, these data suggest 19mer TNR siRNAs are toxic to cancer cells by a mechanism distinct from the process of DISE which relies on just the seed sequences targeting the 3′UTRs of survival genes 15.
Super toxic TNR‐based siRNAs kill cancer cells through RNAi resulting in the loss of survival genes
To address the question whether the super toxic TNR‐based siRNAs killed cancer cells through RNAi, we first compared the toxicity of siCAG/CUG in HCT116 wild‐type and HCT116 Drosha−/− cells. DISE‐inducing si‐ and shRNAs kill Drosha−/− cells more efficiently than wild‐type cells 15. We had interpreted this as the RISC being more available in the absence of most cellular miRNAs, which rely on Drosha for processing. While siCAG/CUG was highly toxic to both cell lines at early time points, Drosha−/− cells were more sensitive to growth reduction induced by siCAG/CUG than their wild‐type counterparts (Appendix Fig S6, P = 0.038, according to polynomial fitting model). To directly test the requirement of AGO2 in the siCAG/CUG‐induced toxicity, we knocked down AGO2 in both HeyA8 and A549 cells (Fig 3A) and transfected the cells with either siNT or siCAG/CUG (Fig 3B). Removal of AGO2 from the cells almost completely prevented the toxicity of siCAG/CUG, confirming dependence on the RISC. A dependence on Ago2 for siCAG/CUG toxicity was confirmed in Ago1‐4 knockout mouse embryonic fibroblasts with re‐expressed AGO2 (Appendix Fig S7). These data indicated that siCAG/CUG was negatively affecting cells through canonical RNAi involving the RISC complex. To confirm this, we modified the siCAG/CUG siRNAs with the 2′‐O‐methylation to selectively block loading of either the siCAG‐ or the siCUG‐based strand into the RISC (Fig 3C). When the CAG‐based guide strand was modified (siCAG AS‐OMe), the toxicity of the siCAG/CUG duplex was severely reduced. It was not affected when the CUG repeat‐containing strand was 2′‐O‐methylated (siCAG S‐OMe), confirming that most of the toxicity of the siCAG/CUG repeat comes from the CAG repeat strand. siCAG/CUG did not have any toxicity when both strands were modified indicating most, if not all, of its toxicity requires RISC loading confirming that RNAi was responsible for cell death.
Figure 3. siCAG/CUG kills cancer cells through RNAi.
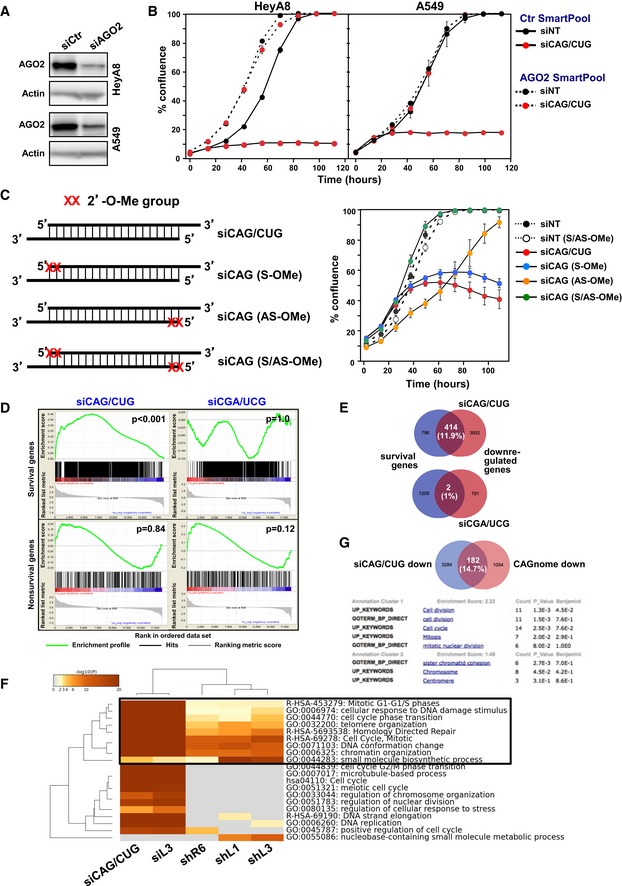
- Western blot analysis of HeyA8 and A549 cells treated with 25 nM of a control SmartPool (siCtr) or an AGO2 siRNA SmartPool for 48 h.
- Confluence over time of HeyA8 and A549 cells after transfection with 1 nM siNT or siCAG/CUG. Cells were first transfected with either a Ctr SmartPool or the AGO2 SmartPool and then, after 24 h, transfected with siNT or siCAG/CUG. Values are mean ± SEM. n = 3 biological replicates, three technical replicates each.
- Left: Sequences with positions of the 2′‐O‐methylation labeled in either the passenger/sense (S), the guide/antisense (AS), or both (A/AS) strands of siCAG/CUG. Right: Confluence over time of HeyA8 cells transfected with 10 nM of the four duplexes depicted on the left and two similarly modified duplexes derived from siNT. Values are mean ± SEM. n = 2 biological replicates, four to six technical replicates each.
- Gene set enrichment analysis for a group of 1,846 survival genes (top 2 panels) and 416 nonsurvival genes (bottom 2 panels) identified in a genome‐wide CRISPR lethality screen 45 after transfecting cells with either siCAG/CUG (left panels) or siCGA/UCG (right panels). Nontargeting siNT served as control. P‐values indicate the significance of enrichment. P‐values were calculated using the Kolmogorov–Smirnov test.
- Metascape analysis of 4 RNA‐Seq datasets of cells with introduced siL3, two CD95L‐derived shRNAs (shL1 and shL3), a CD95‐derived shRNA (shR6) ORF, previously described 15, and the downregulated genes in cells transfected with siCAG/CUG. The boxed GO term clusters were highly enriched in all five datasets.
- Top, Venn diagram comparing the 3,466 genes downregulated in the siCAG/CUG‐treated HeyA8 cells (> 1.5‐fold adj. P < 0.05) as determined by RNA‐Seq (siCAG/CUG down; see Fig 3D) and the 1,236 genes expression of which inversely correlate with the length of CAG repeats recently reported in HD patients (CAGnome down) 24. Bottom, DAVID gene ontology analysis of the overlapping 182 genes.
Source data are available online for this figure.
We recently reported that DISE‐inducing CD95L‐derived sh‐ and siRNAs kill cancer cells by targeting the 3′UTR of critical survival genes through canonical RNAi 15. To test whether the super toxic siCAG/CUG duplex also killed cancer cells through this mechanism, we transfected HeyA8 cells with siNT, siCAG/CUG, or the nontoxic siCGA/UCG and subjected the RNA 48 h after transfection to a RNA‐Seq analysis. Interestingly, in the cells transfected with siCAG/CUG, 3,466 genes were downregulated and 867 genes were upregulated (> 1.5‐fold, adjusted P‐value < 0.05; Dataset EV1). A DAVID gene ontology analysis of the upregulated genes did not reveal any evidence of an interferon response by the cells induced by the transfected siRNA (data not shown). In cells transfected with the nontoxic siCGA/UCG, only 194 genes were found to be downregulated and 420 genes upregulated.
Similar to cells undergoing DISE, when transfected with siCAG/CUG, ~1,800 critical survival genes but not ~400 nonsurvival control genes 15 were significantly enriched in the downregulated genes in cells transfected with siCAG/CUG but not with siCGA/UCG in a gene set enrichment analysis (Fig 3D). In fact, we detected a ~12‐fold increased percentage of survival genes compared to the nonsurvival genes among the downregulated RNAs in the siCAG/CUG‐treated cells (Fig 3E)—a higher difference than seen in cells treated with DISE‐inducing sh‐ or siRNAs (data not shown). A Metascape gene ontology analysis comparing the downregulated genes in cells treated with either CD95‐ or CD95L‐derived si‐ or shRNAs with the data from the siCAG/CUG‐treated cells showed a strong overlap in the GO terms including cell cycle, response to DNA damage, mitosis, and chromatin organization, suggesting that cells died through a mechanism similar to DISE (Fig 3F and Appendix Fig S8). When the RNA‐Seq data of cells treated with siCGA/UCG were included in the analysis, not a single GO cluster overlapped with all data sets (data not shown).
Interestingly, a large genome‐wide comparison of lymphoblastoid cell lines from 107 HD patients reported an inverse correlation between CAG repeat length and downregulated genes. Biological pathways that were significantly affected were ribosomal process, energy metabolism, and cell death pathways 24 all consistent with reduced cell viability. We compared the genes that were reported to be negatively and significantly correlated with the length of the CAG repeats in these patients (1,236 genes, according to Pearson correlation) with the 3,466 genes downregulated in the HeyA8 cells transfected with siCAG/CUG. Of the 1,236 genes downregulated in the patients, 182 (14.7%) were also downregulated in the siCAG/CUG‐treated HeyA8 cells (Fig 3G, top panel). In a DAVID gene ontology analysis with these 182 genes, the two most significantly enriched clusters were consistent with genes playing a role in cell division and mitosis, consistent with a major effect of siCAG/CUG on mitosis (Fig 3G, bottom panel). In summary, these data suggest that the toxicity of the CAG repeat‐based siRNA may involve loss of survival genes and that this form of cell death could be related to the TNR activities seen in patients with extended CAG repeats.
Super toxic TNR‐derived siRNAs kill cells by targeting TNR sequences present in the ORF of genes complementary to the toxic siRNA guide strand
To determine which genes and what part of the mRNAs could be targeted by toxic TNR‐derived siRNAs, we subjected ranked lists of downregulated genes of cells treated with either siCAG/CUG or siCGA/UCG to a Sylamer analysis 25. This method detects enrichment of seed matches in mRNAs that are complementary to the seed of the introduced siRNAs. When the seed length was set to 6nts, we detected a minor enrichment of the 6mer TGCTGC in the 3′UTRs of the downregulated genes in the cells treated with siCAG/CUG (Appendix Fig S9A, left panel). TGCTGC is the expected seed match (positions 2–7) of the siCAG 19mer guide strand. No significant seed match enrichment was found in cells treated with siCGA/UCG or when the ORFs of these genes were analyzed (Appendix Fig S9A, right panel and B). However, when we analyzed the ORFs of cells treated with siCAG/CUG and even when setting the seed length to the maximum of 10 nts, we found a very profound enrichment of two 10 nt sequences (Bonferroni‐adjusted P‐value ~10−50) that corresponded to positions 1–10 and 2–11, respectively, of the targeting siCAG 19mer (Fig 4A). No such enrichment was found when the 3′UTRs of the genes were used for the analysis (Appendix Fig S9C). These data suggest that in contrast to DISE‐inducing si/shRNAs, siCAG/CUG killed cancer cells by targeting long repeat sequences located mainly in ORFs. Consistent with this conclusion, genes containing either of the two targeted 10mers in their ORFs were very strongly enriched among the downregulated genes in siCAG/CUG‐treated cells, while only a weak enrichment was found when the 3′UTRs were analyzed (Fig 4B).
Figure 4. The toxicity of the TNR‐based siRNAs correlates with the presence of long complementary repeats in the ORFs of genes.
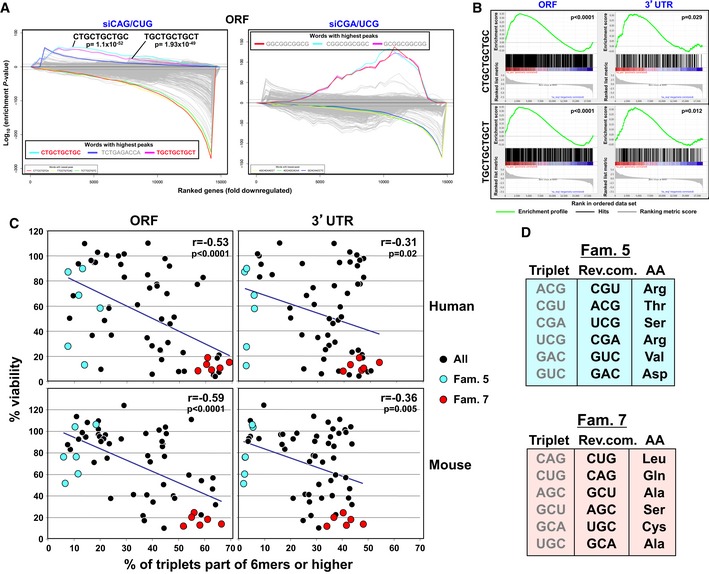
- Ten nucleotide Sylamer analysis of the ORFs of genes deregulated in HeyA8 cells transfected with either siCAG/CUG (left) or siCGA/UCG (right) ordered from most downregulated to most upregulated. The three most highly enriched sequences are shown. The 10 nucleotide motifs in bold in the siCAG/CUG analysis correspond to the reverse complement of positions 1–10 and 2–11 in the 19mer siCAG strand. Bonferroni‐adjusted P‐values are shown. Sequences without a P‐value were either not significantly enriched or not at the top of the ranked list.
- Gene set enrichment analysis for genes containing the CTGCTGCTGC (top panels) or TGCTGCTGCT (bottom panels) sequences identified as enriched in the downregulated genes (see A) in cells transfected with siCAG/CUG when compared to cells transfected with siNT. The analyses were performed with either the ORF (left panels) or the 3′UTR (right panels) of the downregulated genes. P‐values indicate the significance of enrichment. P‐values were calculated using the Kolmogorov–Smirnov test.
- Correlation between the average of the two viability screens performed in HeyA8 (top panels) or M565 (bottom panels) cells with the percentage of TNRs that are part of 6mer or higher longer repeat sequences in either the ORF (left panels) or 3′UTR (right panel) of the genes. The data points for the six members of TNR family 5 are labeled in blue and those of family 7 are labeled in red. Pearson correlations and P‐values are given.
- The amino acids that are coded by the targeted triplets in families 5 and 7 Rev.com., reverse component.
Now, knowing that the toxicity of the siCAG/CUG correlated with the presence of targeted trinucleotide repeats found in the ORFs of genes, we wondered whether the toxicity across all 60 TNR‐derived siRNAs correlated with the presence of higher ordered reverse complementary TNRs in ORFs that could be targeted by the TNR siRNAs and whether this was conserved between human and mouse cells. When counting all 60 single triplets in both the ORFs and 3′UTRs of all human and mouse genes, they were found to be slightly more abundant in the ORF of genes (Appendix Fig S10A). When separating into individual triplets of the 10 families, most triplets are found at similar frequencies in both the ORFs and 3′UTRs in humans and in mice (Appendix Fig S10B, top panel in each set). This situation changed when we focused on targeted (reverse complements of the targeting TNRs) repeats. We plotted the results for 6mers (as this is the minimum sequence required for RNAi‐based targeting), 10mers (the maximum seed length allowed by Sylamer), and 19mers (the length of the siRNAs used). In all cases, with longer sequences, the preference for certain triplets became clearer (Appendix Fig S10B, bottom three panels in each set). When analyzing the 19mers, in both human and mouse, the most abundant TNRs are members of the super toxic family 7 and, barely, any 19mers were found in the related family 5. Genes containing the 19mer TNR (or longer) in their ORF targeted by siCAG were enriched in the most downregulated genes in cells transfected with siCAG/CUG (Appendix Fig S10C). We selected five of the six most highly expressed and downregulated genes from the RNA‐Seq analysis for validation (Dataset EV2 and Appendix Fig S11). HeyA8 cells were transfected with siRNAs at 1 nM, and the mRNAs levels were quantified by real‐time PCR 10, 20, or 40 h after transfection. When transfecting siCAG/CUG, all 5 CUG repeat‐containing mRNAs were downregulated as early as 10 h with maximal downregulation at 40 h. Specificity of the targeting was established by transfecting the cells with either siCAG or siCUG (in which the passenger strand was disabled by adding the 2′‐O‐methylation). Only the siCAG‐based siRNAs were active in silencing the CUG TNR‐containing genes. These data strongly suggested that the toxicity of TNR‐based siRNAs in general might be explained by the presence of extended reverse complementary repeat sequences present preferentially in the ORF of targeted genes. This was confirmed by plotting the viability of cells treated with any of the 60 TNR siRNAs and the number of targeted TNR sequences of 6 nts or longer (Fig 4C). The highest and most significant correlation was found for both human and mouse ORFs. Remarkably, while 80–90% of triplets targeted by the six members of the CGA‐containing family 5 are present as singular events (blue dots), between 50 and 70% of the triplets targeted by members of the CAG‐containing family 7 are found to be part of 6mers or higher ordered TNRs (red dots). Interestingly, the TNRs targeted by the six toxic members of family 7 code for five different amino acids (Fig 4D). As all the six members of family 7 are equally toxic to cancer cells, this suggests that this involves targeting long repeat elements (i.e., TNRs), rather than a requirement for poly‐homo amino acid coding stretches. This view is supported by an analysis of species conservation (Appendix Fig S12). Of the genes that contain targeted 19mers, only seven of the 99 genes found in either the mouse or the human genome overlapped and the genes that are targeted do not have shared functions (Appendix Fig S12B). In summary, our data provide evidence that TNR‐based siRNAs are toxic to cells by targeting a number of genes that contain high‐order trinucleotide repeats that are reverse complementary to the targeting TNR. The resulting cell death has features of what we recently described as DISE, with the main difference that DISE is the result of a miRNA‐like targeting of short seed matches in the 3′UTRs of survival genes, whereas the TNR‐induced cell death is an on‐target effect affecting a larger number of genes that contain targeted sequences in their ORF. This now provides an explanation for why the most toxic TNR‐based siRNAs are much more toxic than DISE‐inducing siRNAs. Intriguingly, the CAG repeats found in HD are part of the most toxic family of TNRs and their reverse complementary 19mers that can serve as targets are the most abundant TNR sequences in the ORFs of both human and mouse genomes.
Super toxic CAG/CUG TNR‐based siRNAs slow down tumor growth in vivo with no toxicity to normal tissues
We were wondering whether the super toxic TNR‐based siRNAs could be used for cancer therapy. We decided to deliver the siRNAs to cancer cells in vivo using templated lipoprotein (TLP) nanoparticles 26. Before using the TLP particles loaded with the siCAG/CUG duplex (siCAG/CUG‐TLP) in vivo, we tested their effects on tumor cells in vitro. They killed HeyA8 cells more efficiently than siL3‐TLPs (Fig 5A) and also slowed down growth of the tested human or mouse cancer cell lines (Fig 5B and data not shown). They also killed neurospheres derived from patients with glioblastoma (Fig 5C). To test the activity of the siCAG/CUG‐TLPs in vivo, we treated orthotopically xenografted HeyA8 ovarian cancer cells in mice. Mice injected with 100,000 tumor cells were i.p. injected with nanoparticles five times a week for 2 weeks (Fig 5D). After the tenth treatment, mice were split into two groups: One group continued to receive treatment in the third week and the second group did not. This was done to determine whether large established tumors would still respond to the treatment. The large tumors in treatment group 1 still benefited from the effect of siCAG/CUG in the third week of treatment (Fig 5E, left panel). In contrast, some tumors in the mice in treatment group 2 grew out rapidly, while others showed persistent growth reduction (Fig 5E, right panel). These results suggest that established tumors respond to the siCAG/CUG treatment. This was confirmed in another experiment in which 106 HeyA8 cells were injected and mice were first treated three times a week and then switched to daily treatment 19 days after tumor cell injection (data not shown).
Figure 5. Killing cancer cells using siCAG/CUG coupled to TLP nanoparticles both in vitro and in vivo .
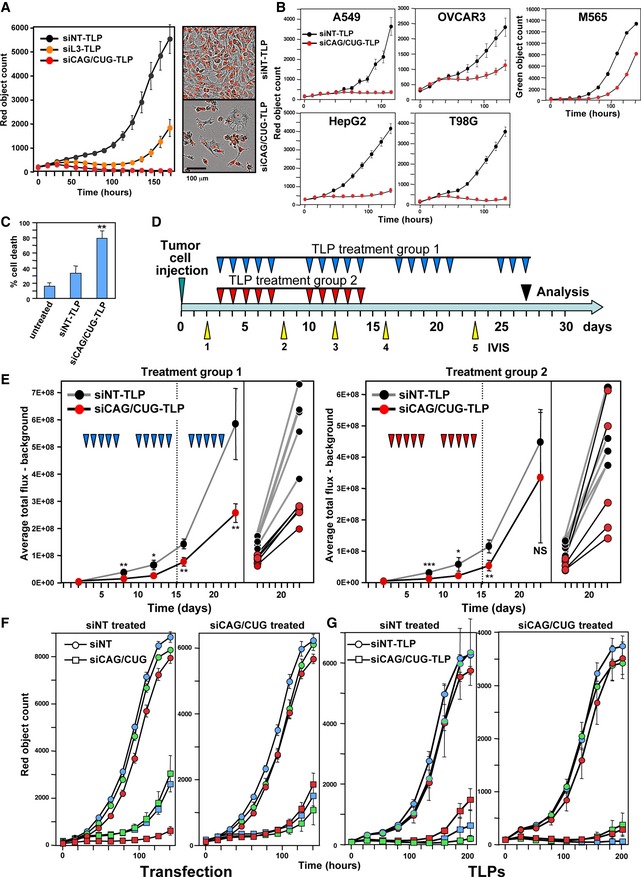
-
ALeft: Confluency over time of HeyA8 (NucRed) cells treated with 10 nM of either siNT‐TLP, siL3‐TLP, or siCAG/CUG‐TLP. Right: Phase and red fluorescence image of HeyA8 (NucRed) cells 90 h after transfection with 15 nM of TLPs. Values are mean ± SEM. n = 3 technical replicates.
-
BConfluency over time of different human and mouse cancer cell lines treated with either 10 nM (OVCAR3, HepG2, M565) or 20 nM (A549, T98G) of TLPs. Values are mean ± SEM. n = 2 biological replicates, six technical replicates each.
-
CPercent cell death (Trypan blue counting) of GIC‐20 neurospheres derived from a patient with glioblastoma 6 days after adding the TLPs (30 nM). Values are mean ± SD. n = 3 technical replicates. **P < 0.01 (Student's t‐test, two‐tailed distribution, two‐sample equal variance).
-
DTreatment scheme.
-
ETumor growth over time based on small animal imaging of 105 HeyA8‐NucRed‐Luc‐neo cells injected i.p. into NSG mice treated with either siNT‐TLPs or siCAG/CUG‐TLPs. Treatment group 1 received 18 injections over 4 weeks, and treatment group 2 received 10 injections over 3 weeks. The bioluminescence signal of IVIS #4 and #5 for individual mice is shown (right panel). The experiment represents one of two similar experiments. Values are mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.0001; NS, not significant (Student's t‐test, two‐tailed distribution, two‐sample equal variance).
-
F, GChange in red object count (growth) of tumor cells from three mice of the siNT‐TLP and the siCAG/CUG‐TLP treatment group 1 either after transfection with 1 nM siNT or siCAG/CUG (F) or after incubation with 7.5 nM siNT‐TLP or siCAG/CUG‐TLP (G). A total of 1,000 cells per well were plated. Values are mean ± SEM. n = 3, eight technical replicates.
To determine whether siCAG/CUG was detrimental to mice, mice in treatment group 1 were treated a few more times with the siRNA and were analyzed just when the control‐treated mice were moribund at around day 27. We did not see any signs of toxicity in any of the mice. They were feeding well (not shown), did not lose weight (Appendix Fig S13A), had normal liver histology (Appendix Fig S13B), and showed no increase in liver enzymes in the serum (Appendix Fig S13C). These data demonstrated that super toxic CAG/CUG TNR‐based siRNAs delayed tumor growth in vivo 5–6 days with no gross toxicity to normal cells and that they could be safely administered using TLP nanoparticles.
To determine whether tumor cells acquired resistance to the treatment, we tested tumors from three mice in treatment group 1 ex vivo. Three tumors of mice treated with siNT‐TLP and three tumors of mice treated with siCAG/CUG‐TLP were transfected with the same siRNAs in vitro a day after tumor isolation (Fig 5F). In parallel, these tumor cells were also treated with the nanoparticles again (Fig 5G). In all cases, the tumors from the mice that had received the toxic siRNA were as sensitive to the toxic effects of siCAG/CUG in vitro as were the tumors from mice treated with siNT suggesting that cancer cells do not become resistant to cell death induced by the toxic TNR‐based siRNAs, at least not in the classical sense observed after targeted therapy, and that preferentially tumor cells were responding as there were no signs of toxicity in the mice.
Discussion
The siRNA we used for cancer treatment was a duplex between the basic TNR module found in HD (CAG) and the fully complementary strand CUG found in myotonic dystrophy (DM1). In a screen of all TNR‐derived siRNAs, both CAG and CUG were part of a family that contains six members all of which were highly toxic to cancer cells of both human and mouse origin. This hybrid duplex between the two disease molecules was also recently tested in a well‐established Drosophila model of DM1 27. Expression of the two transcripts led to the generation of Dicer‐2 (dcr‐2) and ago2‐dependent 21‐nt TNR‐derived siRNAs, resulting in high toxicity to the cells. In a separate study, it was shown that the expression of these complementary repeat RNAs leads to dcr‐2‐dependent neurodegeneration 28. These results suggest that the co‐expression of CAG and CUG repeat‐derived sequences may dramatically enhance toxicity in human repeat expansion diseases in which antisense transcription occurs. Antisense transcription was reported to occur in SCA8 in two genes encompassing the repeats are expressed: ATXN8 (CAG repeat), on the sense strand, and ATXN8OS (CTG repeat), on the antisense strand 29.
One could argue that a cancer therapy based on delivering siCAG/CUG could be detrimental to patients as TNR expansion patients suffer from various pathologies. However, similar to many other genes with amplified CAG repeats, HTT is ubiquitously expressed throughout the body with somewhat higher expression in the brain and in testis 30. The disease is characterized by neurodegeneration affecting the cerebral cortex and neuropathology in the striatum, but it also affects other tissues 31. So, if sCAGs are produced in multiple tissues, the effects on most normal tissues seem to be moderate. Even in the brain, although sCAGs are detrimental to HD patients long term, most patients do not have major symptoms before the age of 40 4, 31. Short‐term exposure to toxic sCAGs for cancer therapy, as suggested by our mouse experiments, may not have a dramatic effect on normal tissues but may be enough to kill cancer cells. If a CAG‐based siRNA were to produce side effects particularly in the brain, it may be possible to protect the brain through local administration of neuroprotecting LNA‐CTGs as described 14.
What could be the mechanism of the relative resistance of normal versus tumor cells to the toxic siRNAs? Our recent data suggested that miRNAs inhibit DISE and in fact may protect normal cells from it 15. Both Drosha and Dicer k.o. HCT116 cells were found to be hypersensitive to a DISE‐inducing siRNA. This is entirely consistent with reported activities of CAG repeats which may also act through RNAi. Pathogenic Ataxin‐3 with amplified CAG repeats showed strongly enhanced toxicity in HeLa cells after knockdown of Dicer 32. In addition, it was previously shown in Drosophila that impairing miRNA processing dramatically enhanced neurodegeneration caused by the CAG repeat gene Ataxin‐3. Two fly mutants were tested, one with a deficiency in dcr‐1, the Drosophila Dicer ortholog that is required for miRNA biogenesis, and another with a deficiency in R3D1, a gene required for dcr‐1 to function 33. The authors concluded that “miRNA pathways normally play a protective role in polyQ‐induced neurodegeneration”. In light of our data, it is possible that miRNAs might actually protect cells from the toxic effects of TNR‐based siRNAs.
While HD is the best‐known disease caused by CAG repeats, one of the two diseases first discovered to be caused by TNR repeat expansions is the neurodegenerative disorder SBMA/Kennedy disease 34, wherein the pathogenic CAG repeat is found in exon 1b of the androgen receptor (AR). If patients with amplified CAG repeats produce sCAGs, which according to our work may be detrimental to cancer cells, one would have to expect that such patients have a reduced cancer incidence due to toxic siRNA expression in many tissues. Indeed, reduced cancer incidence was reported for HD and SBMA patient populations in Sweden 18 and HD patients in France 19, Denmark 20, and England 21.
Possibly, the clearest connection to cancer has been reported for the CAG repeats in the AR gene and prostate cancer (PCa). The CAG repeat length in the AR has been inversely linked to PCa. While longer repeats (> 20 CAGs) confer a protective effect among the PCa patients 45 years or older 35, shorter CAG repeats have been shown to result in a twofold increased cancer risk 36, a more aggressive disease, and a high risk of distant metastases 37, 38, 39. Shortening of CAG repeat length was found in in situ lesions of PCa and its possible precursors 40, suggesting that PCa avoids longer CAG repeats. This is consistent with our finding of super toxicity of CAG‐based siRNAs.
There are two observations that suggest the targeted TNRs present in the ORFs of certain genes are not there because these proteins require stretches of the same amino acid for their function which would presumably be conserved between human and mouse: First, all six members of the TNR family 7 were super toxic targeting six different reverse complementary TNRs that code for five different amino acids (Fig 4D), and second, the genes with the longest repeats with complete complementarity to the most toxic siCAG/CUG of 19 nts showed little overlap between human and mouse (Appendix Fig S12B) and the different targeted genes do not share similar functions (Appendix Fig S12C). Interestingly, both the AR and the HTT genes contain some of the longest CAG repeats in the human genome; however, those are not found in the mouse orthologs at the same positions in the ORF.
So, if there was no pressure to maintain these TNRs in specific genes but rather anywhere in the genome, could there be an evolutionary link between TNRs and cancer? A hint may come from the way the repeat expansion is generated. It is believed that among other mechanisms, DNA replication slippage and/or defective base excision repair causes expansion of TNRs 41. Therefore, CAG repeats could be part of a mechanism used during evolution to maintain genome integrity and, in the context of multicellular organisms, to prevent cancer formation by producing toxic siRNAs. This would occur whenever too many mutations start accumulating in cells, one property all cancers have in common.
While the treatment with siCAG/CUG requires optimization, our data on the toxicity of CAG TNR‐based siRNAs for cancer cells but not normal cells when administered in vivo and the reported decreased incidence rate for different types of cancer in patients with CAG expansions suggest that TNR‐based siRNAs may be useful for cancer therapy.
Materials and Methods
Cell lines and tissue culture
All cells were grown in an atmosphere of 5% carbon dioxide (CO2) at 37°C. Unless indicated otherwise, base media were supplemented with 10% heat‐inactivated fetal calf serum (FBS; Sigma‐Aldrich) and 1% penicillin/streptomycin and L‐glutamine (Mediatech Inc.). Cells were dissociated with 0.25% (w/v) trypsin–0.53 mM EDTA solution (Mediatech Inc.). The following cell lines were cultured in supplemented RPMI1640 medium (Mediatech Inc.): ovarian cancer cell lines HeyA8 (RRID:CVCL_8878), OVCAR3 and OVCAR4 (both from Tumor Biology Core, Northwestern University), and lung cancer cells A549 (ATCC CRM‐CCL‐185) and H460 (ATCC HTB‐177). The GBM cell line T98G (ATCC CRL1690) was cultured in Eagle's minimum essential medium (EMEM; ATCC). Melanoma B16F10 cells (ATCC CRL‐6475) and 293T cells (RRID:CVCL_0063) were cultured in DMEM (Cellgro). HepG2 (ATCC HB‐80645) was cultured in EMEM (ATCC). ID8, a mouse ovarian cancer cell line, was cultured in DMEM supplemented with 4% FBS, and 10 mg/l insulin, 5.5 mg/l transferrin, and 6.7 μg/ml selenium (ITS, Mediatech, Inc., 1:100 diluted). 3LL Lewis lung cancer cells (ATCC CRL‐1642) were cultured in DMEM. FOSE2 cells are spontaneously immortalized ovarian surface epithelial cells, and M565 cells are from a spontaneously formed liver cancer in a female and male mouse, respectively, both isolated from mice carrying a floxed Fas allele 17. Both were cultured in DMEM/F12 (Gibco #11330), 1% insulin–transferrin–selenium (ITS, Gibco 51300‐044). M565 cells were dissociated with Accutase detachment reagent (Fisher Sci.). HCT116 Drosha−/− were generated by Dr. Narry Kim 42. HCT116 parental (cat#HC19023, RRID:CVCL_0291) and the Drosha−/− clone (clone #40, cat#HC19020) were purchased from Korean Collection for Type Cultures (KCTC). All HCT116 cells were cultured in McCoy's medium (ATCC, cat#30‐2007). Mouse Ago1‐4 k.o. embryonic stem cells inducibly expressing human FLAG‐HA‐AGO2 were described in 43. CELLSTAR tissue culture dishes (Greiner Bio‐One, cat#664160, cat#639160) were coated with 0.1% gelatin solution (Sigma, cat#ES‐006‐B) for 10–30 min before use. Cells were cultured in DMEM (Gibco, cat#12430054) supplemented with 15% fetal bovine serum (Sigma, cat#F2442), 1% NEAA solution (HyClone, cat#SH3023801), 1% GlutaMAX 100x (Gibco, cat#35050061), 0.0007% 2‐mercaptoethanol (Fisher, cat#BP176100), and 106 units/l LIF (Sigma, #ESG1107). The cell culture media were refreshed daily. FLAG‐HA‐AGO2 is under the control of a TRE‐tight (TT) doxycycline (Dox)‐inducible promoter. 100 ng/ml doxycycline (Sigma, cat#D9891) was added to the media in order to induce moderate level of Ago2 expression to maintain normal cell growth. To deplete Ago2 expression in cells, doxycycline was withdrawn from media for 4 days. To induce wild‐type level of hAgo2 expression, 2.5 μg/ml doxycycline was added to the media. The human GBM‐derived neurosphere cell line GIC‐20 (infected with pLV‐Tomato‐IRES‐Luciferase) was obtained from Dr. Alexander Stegh. Cells were grown as neurospheres in DMEM/F12 50:50 with L‐glutamine (Corning), supplemented with 1% PenStrep, B27 (Invitrogen), N2 (Invitrogen), human epidermal growth factor (hEGF; Shenandoah Biotech), fibroblast growth factor (FGF; Shenandoah Biotech), leukemia inhibitory factor (LIF; Shenandoah Biotech), and GlutaMAX (Life Technologies). HeyA8 xenografted tumor nodules were dissected from mice, cut, washed in sterile PBS, and dissociated with 0.25% (w/v) trypsin–0.53 mM EDTA solution for 20 min at 37°C. The digestion was stopped by adding full RPMI‐1640 medium. After centrifugation, the trypsin solution mix was removed, and the tumor cells were resuspended in fresh full medium and strained through 70‐micron cell strainer. The tumor cell suspension was plated over night on 10‐cm tissue culture dishes. The following day, cells were harvested, counted, and plated on 96‐well plates for further experiments. Cells were transfected with siRNAs after cells had adhered or incubated with siRNA‐TLPs and then plated.
Western blot analysis and reagents
Primary antibodies for Western blot used were anti‐β‐actin antibody (Santa Cruz #sc‐47778, RRID:AB_626632), anti‐human AGO2 (Abcam #AB186733, RRID:AB_2713978). Secondary antibodies for Western blot used were goat anti‐rabbit; IgG‐HRP (Southern Biotech #SB‐4030‐05, RRID:AB_2687483). Reagents used were propidium iodide (Sigma‐Aldrich #P4864), puromycin (Sigma‐Aldrich #P9620), and Lipofectamine RNAiMAX (ThermoFisher Scientific #13778150). Western blot analysis was performed as recently described 15.
Transfection with short oligonucleotides
For transfection of cancer cells with siRNAs, RNAiMAX was used at a concentration optimized for each cell line, following the instructions of the vendor. Cell lines were either transfected after cells had adhered (forward transfection), or during plating (reverse transfection). For an IncuCyte experiment, cells were typically plated in 200 μl antibiotic free medium, and 50 μl transfection mix with RNAiMAX and siRNAs were added. During growth curve acquisitions, the medium was not exchanged to avoid perturbations. All individual siRNA oligonucleotides were ordered from integrated DNA technologies (IDT). Individual RNA oligos were ordered for the sense and antisense oligo; the sense strand had 2 Ts added to the 3′ end; antisense strand had 2 deoxy As at the 3′ end. When indicated, the first two positions at the 5′‐end were 2′‐O‐methylated. Sense and antisense oligos were mixed with nuclease‐free Duplex buffer (IDT, Cat. No# 11‐01‐03‐01; 100 mM potassium acetate, 30 mM HEPES, pH 7.5) to 20 μM (working solution), heated up for 2 min at 94°C, and then, the oligos were allowed to cool down to room temperature for 30 min. siRNA solutions were aliquoted and stored at −80°C. The cells were transfected with siRNAs at a final concentration of 0.01–10 nM. The following siRNA sequences were used: siNT (siNT#2): UGGUUUACAUGUUGUGUGA (nontargeting in mammalian cells), siNT1: UGGUUUACAUGUCGACUAA (nontargeting in mammalian cells), siL3: GCCCUUCAAUUACCCAUAU (human CD95L exon 1), and siNT/siL3: UGGUUUACAUGUCCCAUAA; siNT seed: UGGUAAACUAGUUGUCUGA and siL3 seed: UGGUAAACUAGUCCCAUAA. All TNR‐based 19mer siRNAs were designed as follows: The TNR‐based siRNA was named according to its antisense/guide strand: 2 nt 3′ overhangs were added as described above. All TNR‐based siRNAs were fully complementary 19mers. For instance, the siCAG/CUG sequences are as follows: S: CAGCAGCAGCAGCAGCAGCdAdA. and AS: GCUGCUGCUGCUGCUGCUGTT. siRNA duplexes used in the screens are shown in Dataset EV3. In all siRNAs used in screens, the sense/passenger strand was disabled by 2′‐O‐methylation in positions 1 and 2 of the sense strand.
For transfecting Ago1‐4 k.o. mouse ESC, cells were cultured without doxycycline for 3 days. Half of the cells were then cultured for one more day without doxycycline before transfection. The other half was cultured in media containing 2.5 μg/ml of doxycycline for 1 day to induce WT‐level AGO2 expression before transfection. For IncuCyte experiments, the ESCs were transfected with either 5 nM siNT or siCAG/CUG using reverse transfection method in a 96‐well plate coated with 0.1% gelatin. A total of 5,000 cells/well and 0.2 μl RNAiMAX/well were used. One day after transfection, 100 μl of media (with or without 2.5 μg/ml doxycycline) was added to each well. After that, media were refreshed every 2~3 days until the cells grew confluent. For flow cytometry experiments, cells were transfected with either 5 nM unlabeled siNT or siNT labeled with Cy5 on the 5′ end of the antisense strand using reverse transfection method in a 12‐well plate coated with gelatin in triplicates. A total of 300,000 cells/well and 1 μl RNAiMAX/well were used. Flow cytometry measurements were conducted 24 h after transfection. For AGO2 knockdown experiment, 100,000 cells/well HeyA8 or 200,000 cells/well A549 cells were reverse‐transfected in six‐well plate with either nontargeting (Dharmacon, cat#D‐001810‐10‐05) or an AGO2 targeting siRNA SMARTpool (Dharmacon, cat#L004639‐00‐005) at 25 nM. 1 μl RNAiMAX per well was used for HeyA8 cells, and 6 μl RNAiMAX per well was used for A549 cells. Twenty‐four hours after transfection with the SMARTpools, cells were reversed‐transfected in a 96‐well plate with either siNT or siCAG/CUG at 1 nM and monitored in the IncuCyte. 0.1 μl/well RNAiMAX was used for HeyA8 cells, and 0.6 μl/well RNAiMAX was used for A549 cells.
Total RNA isolation and RNA‐Seq analysis
HeyA8 cells were transfected in six wells with siNT or either siCAG/CUG or siCGA/UCG oligonucleotides at 1 nM. The transfection mix was removed after 9 h. Total RNA was isolated 48 h after transfection using the miRNeasy Mini Kit (Qiagen, Cat.No. 74004) following the manufacturer's instructions. An on‐column digestion step using the RNase‐free DNase Set (Qiagen, Cat.No.: 79254) was included. NGS RNA‐Seq library construction and sequencing was performed by the University of Chicago Genomics Facility. The quality and quantity of RNA samples was assessed using an Agilent bio‐analyzer. RNA‐Seq libraries were generated using Illumina Stranded TotalRNA TruSeq kits according to the Illumina provided protocol, and sequencing was performed using the Illumina HiSEQ4000 according to Illumina provided protocols and reagents. The resulting paired‐end reads were aligned to the hg38 assembly of the human genome with Tophat2. HTseq was used to associate the aligned reads with genes, and EdgeR was used to identify genes significantly differentially expressed between treatments, all as recently described 15. The accession number for the RNA‐Seq and expression data reported in this work are GSE104552.
Real‐time PCR
Real‐time PCR was performed as described recently 15 using the following primers: GAPDH (Hs00266705_g1), RPL14 (Hs03004339_g1), LRRC59 (Hs00372611_m1), CNPY3 (Hs01047697_m1), CTSA (Hs00264902_m1), and LRP8 (Hs00182998_m1) (Thermo Scientific).
Monitoring growth over time and quantification of cell death
To monitor cell growth over time, cells were seeded between 125 and 4,000 per well in a 96‐well plate in triplicates. The plate was then scanned using the IncuCyte ZOOM live‐cell imaging system (Essen BioScience). Images were captured at regular intervals, at the indicated time points, using a 10× objective. Cell confluence was calculated using the IncuCyte ZOOM software (version 2015A). IC50 values for siL3 and siCAG/CUG were determined using GraphPad Prism 6 software (by logarithm‐normalized sigmoidal dose curve fitting). Quantification of DNA fragmentation (subG1 DNA) was done as previously described 15.
siRNA screens and cell viability assay
HeyA8 or M565 cells were expanded and frozen down at the same passage. One week before transfection, cells were thawed and cultured in RPMI1640 medium, 10% FBS and 1% pen/strep. Cells were split three times during the week and each time seeded at 4 × 106 cells total in one T75 flask. On the day of the transfection, RNA duplexes were first diluted with Opti‐MEM to make 30 μl solution of 10 nM (for the duplexes with the 6mer seeds) or 1 nM (for the TNR‐based duplexes) as final concentration in a 384‐well plate by Multidrop Combi. Lipofectamine RNAiMAX (Invitrogen) was diluted in Opti‐MEM (6 μl lipid + 994 μl of Opti‐MEM for HeyA8 and 15.2 μl lipid + 984.8 μl of Opti‐MEM for M565 cells). After incubating at room temperature for 5–10 min, 30 μl of the diluted lipid was dispensed into each well of the plate that contains RNA duplexes. The mixture was pipetted up and down three times by PerkinElmer EP3, incubated at room temperature for at least 20 min, and then, the mixture was mixed again by PerkinElmer EP3. 15 μl of the mixture was then transferred into wells of three new plates (triplicates) using the PerkinElmer EP3. 50 μl with 320 HeyA8 or 820 M565 cells was then added to each well containing the duplex and lipid mix, which results in a final volume of 65 μl. Plates were left at room temperature for 30 min and then moved to a 37°C incubator. Ninety‐six hours post‐transfection, cell viability was assayed using CellTiter‐Glo (Promega) quantifying cellular ATP content. 35 μl medium was removed from each well, and 30 μl CellTiter‐Glo cell viability reagent was added. The plates were shaken for 5 min and incubated at room temperature for 15 min. Luminescence was then read on the BioTek Synergy Neo2.
Treatment of xenografted ovarian cancer cells in vivo with templated lipoprotein particles (TLP) loaded with siRNAs
Synthesis of TLPs and production of siRNA‐TLPs were done exactly as recently described 44. 105 HeyA8 cells [infected with a luciferase lentivirus and a NucRed lentivirus (NucLight Red Lentivirus, EF‐1α, Puro, Essen Bioscience)] were injected i.p. into 6‐week‐old female NSG mice 44 following the Northwestern University Institutional Animal Care and Use Committee (IACUC)‐approved protocol. The growth of tumor cells in the mice over time was monitored noninvasively using the IVIS® Spectrum in vivo imaging system as recently described 44. Each mouse of a treatment group was injected with 150 μl of either siNT‐TLP or siCAG/CUG‐TLP (1 μM stock).
Data analyses
To determine the number of triplets, 6mer, 10mer, or 19mer repeat sequences in the ORFs or 3′UTR of human or mouse genes, all ORF and 3′UTRs were extracted from the Homo sapiens (GRCh38.p7) or Mus musculus (GRCm38.p5) gene dataset of the Ensembl database using the Ensembl Biomart data mining tool. For each gene, only the longest deposited ORF or 3′UTR was considered. Custom perl scripts were used to identify whether each 3′UTR or ORF contained an identical match to a particular triplet, 6mer, 10mer, or 19mer.
GSEA was performed using the GSEA v.3.0 software from the Broad Institute (http://software.broadinstitute.org/gsea); 1,000 permutations were used. Two lists of 1,846 survival and 418 nonsurvival genes were used as recently described 15, 45. They were set as custom gene sets to determine enrichment of survival genes versus the nonsurvival control genes in downregulated genes from the RNA‐Seq data. Log(Fold downregulation) was used as the ranking metric. Nominal P‐values below 0.05 were considered significantly enriched. The GO enrichment analysis shown was performed using all genes that after alignment and normalization were found to be at least 1.5‐fold downregulated with adjusted P‐values of < 0.05, using the software available on www.Metascape.org and default running parameters. The datasets of HeyA8 cells with introduced siL3, shL1, shL3, or shR6 were recently described 15.
Sylamer analyses 25 were performed using the RNA‐Seq datasets from the HeyA8 cells transfected with siNT, siCAG/CUG, or siCGA/UCG as recently described 15. The analyses were performed using default settings. Enriched 6 or 10mer motifs were analyzed using either the longest ORFs or the 3′UTRs sequences used in [15]. Sylamer (version 12‐342) was run with the Markov correction parameter set to 4. DAVID gene ontology analysis was performed using the tool at https://david.ncifcrf.gov/home.jsp and default settings.
Statistical analyses
Two‐way analyses of variances (ANOVAs) were performed using the Stata 14 software to compare growth curves. One‐tailed Student's t‐test was performed in the software package R to compare tumor load between treatment groups. Wilcoxon rank‐sum test was performed in R to compare IVIS signal between treatment groups. The effects of treatment on wild‐type versus Drosha−/− cells were statistically assessed by fitting regression models that included linear and quadratic terms for value over time; main effects for treatment and cell type; and two‐ and three‐way interactions for treatment, cell type, and time. The three‐way interaction on the polynomial terms with treatment and cell type was evaluated for statistical significance since this represents the difference in treatment effects over the course of the experiment for the varying cell types. All statistical analyses were conducted in Stata 14 (RRID:SCR_012763) or R 3.3.1 in Rstudio (RRID:SCR_000432) except for Pearson correlation analyses, which were performed using StatPlus 6.2.2.
Author contributions
MEP and AEM designed and supervised the project; AEM, WP, QG, MP, CL, and BB performed research and analyzed the data; ETB provided bioinformatics support and analyzed the data; SC performed the siRNA screens; KMM and CST provided the siRNA‐TLP particles; MEP wrote the manuscript; and all authors reviewed and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Source Data for Supplementary Figure S7
Review Process File
Source Data for Figure 3
Acknowledgements
We are grateful to Dr. Evangelos Kiskinis for advice on trinucleotide repeat expansion diseases and Sarah Fazal for performing siRNA screens. We would like to thank Johannes Peter for help in creating the movies. The mouse Ago1‐4 k.o. embryonic stem cells were a kind gift from Dr. Markus Hafner. This work was funded by training grants T32CA070085 (to M.P.) and T32CA009560 (to W.P. and K.M.M), and R35CA197450 (to M.E.P.). C.S.T. would like to thank the Department of Defense/Air Force Office of Scientific Research (FA95501310192) for grant funding, and grant funding from the National Institutes of Health/National Cancer Institute (R01CA167041). K.M.M acknowledges the Ryan Family, the Malkin Family, the Driskill Family, Chicago Baseball Charities Cancer Fellowship, and The Northwestern University Feinberg School of Medicine Developmental Therapeutic Institute for financial and fellowship support.
EMBO Reports (2018) 19: e45336
Contributor Information
Andrea E Murmann, Email: a-murmann@northwestern.edu.
Marcus E Peter, Email: m-peter@northwestern.edu.
References
- 1. Nalavade R, Griesche N, Ryan DP, Hildebrand S, Krauss S (2013) Mechanisms of RNA‐induced toxicity in CAG repeat disorders. Cell Death Dis 4: e752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gatchel JR, Zoghbi HY (2005) Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 6: 743–755 [DOI] [PubMed] [Google Scholar]
- 3. Ross CA (2002) Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron 35: 819–822 [DOI] [PubMed] [Google Scholar]
- 4. Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30: 575–621 [DOI] [PubMed] [Google Scholar]
- 5. Cleary JD, Ranum LP (2017) New developments in RAN translation: insights from multiple diseases. Curr Opin Genet Dev 44: 125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banez‐Coronel M, Porta S, Kagerbauer B, Mateu‐Huertas E, Pantano L, Ferrer I, Guzman M, Estivill X, Marti E (2012) A pathogenic mechanism in Huntington's disease involves small CAG‐repeated RNAs with neurotoxic activity. PLoS Genet 8: e1002481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Napierala M, Krzyzosiak WJ (1997) CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J Biol Chem 272: 31079–31085 [DOI] [PubMed] [Google Scholar]
- 8. Wojciechowska M, Krzyzosiak WJ (2011) Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet 20: 3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA (2005) Colocalization of muscleblind with RNA foci is separable from mis‐regulation of alternative splicing in myotonic dystrophy. J Cell Sci 118: 2923–2933 [DOI] [PubMed] [Google Scholar]
- 10. Lin L, Park JW, Ramachandran S, Zhang Y, Tseng YT, Shen S, Waldvogel HJ, Curtis MA, Faull RL, Troncoso JC et al (2016) Transcriptome sequencing reveals aberrant alternative splicing in Huntington's disease. Hum Mol Genet 25: 3454–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li LB, Yu Z, Teng X, Bonini NM (2008) RNA toxicity is a component of ataxin‐3 degeneration in Drosophila . Nature 453: 1107–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hsu RJ, Hsiao KM, Lin MJ, Li CY, Wang LC, Chen LK, Pan H (2011) Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS One 6: e16417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M, Krzyzosiak WJ (2007) Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell 25: 575–586 [DOI] [PubMed] [Google Scholar]
- 14. Rue L, Banez‐Coronel M, Creus‐Muncunill J, Giralt A, Alcala‐Vida R, Mentxaka G, Kagerbauer B, Zomeno‐Abellan MT, Aranda Z, Venturi V et al (2016) Targeting CAG repeat RNAs reduces Huntington's disease phenotype independently of huntingtin levels. J Clin Invest 126: 4319–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Putzbach W, Gao QQ, Patel M, van Dongen S, Haluck‐Kangas A, Sarshad AA, Bartom E, Kim KY, Scholtens DM, Hafner M et al (2017) Many si/shRNAs can kill cancer cells by targeting multiple survival genes through an off‐target mechanism. eLife 6: e29702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patel M, Peter ME (2017) Identification of DISE‐inducing shRNAs by monitoring cellular responses. Cell Cycle https://doi.org/10.1080/15384101.2017.1383576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hadji A, Ceppi P, Murmann AE, Brockway S, Pattanayak A, Bhinder B, Hau A, De Chant S, Parimi V, Kolesza P et al (2014) Death induced by CD95 or CD95 ligand elimination. Cell Rep 10: 208–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ji J, Sundquist K, Sundquist J (2012) Cancer incidence in patients with polyglutamine diseases: a population‐based study in Sweden. Lancet Oncol 13: 642–648 [DOI] [PubMed] [Google Scholar]
- 19. Coarelli G, Diallo A, Thion MS, Rinaldi D, Calvas F, Boukbiza OL, Tataru A, Charles P, Tranchant C, Marelli C et al (2017) Low cancer prevalence in polyglutamine expansion diseases. Neurology 88: 1114–1119 [DOI] [PubMed] [Google Scholar]
- 20. Sorensen SA, Fenger K, Olsen JH (1999) Significantly lower incidence of cancer among patients with Huntington disease: an apoptotic effect of an expanded polyglutamine tract? Cancer 86: 1342–1346 [PubMed] [Google Scholar]
- 21. Turner MR, Goldacre R, Goldacre MJ (2013) Reduced cancer incidence in Huntington's disease: record linkage study clue to an evolutionary trade‐off? Clin Genet 83: 588–590 [DOI] [PubMed] [Google Scholar]
- 22. Clark RM, Dalgliesh GL, Endres D, Gomez M, Taylor J, Bidichandani SI (2004) Expansion of GAA triplet repeats in the human genome: unique origin of the FRDA mutation at the center of an Alu. Genomics 83: 373–383 [DOI] [PubMed] [Google Scholar]
- 23. Kozlowski P, de Mezer M, Krzyzosiak WJ (2010) Trinucleotide repeats in human genome and exome. Nucleic Acids Res 38: 4027–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee JM, Galkina EI, Levantovsky RM, Fossale E, Anne Anderson M, Gillis T, Srinidhi Mysore J, Coser KR, Shioda T, Zhang B et al (2013) Dominant effects of the Huntington's disease HTT CAG repeat length are captured in gene‐expression data sets by a continuous analysis mathematical modeling strategy. Hum Mol Genet 22: 3227–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Dongen S, Abreu‐Goodger C, Enright AJ (2008) Detecting microRNA binding and siRNA off‐target effects from expression data. Nat Methods 5: 1023–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McMahon KM, Plebanek MP, Thaxton CS (2016) Properties of native high‐density lipoproteins inspire synthesis of actively targeted in vivo siRNA delivery vehicles. Adv Funct Mater 26: 7824–7835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu Z, Teng X, Bonini NM (2011) Triplet repeat‐derived siRNAs enhance RNA‐mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet 7: e1001340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lawlor KT, O'Keefe LV, Samaraweera SE, van Eyk CL, McLeod CJ, Maloney CA, Dang TH, Suter CM, Richards RI (2011) Double‐stranded RNA is pathogenic in Drosophila models of expanded repeat neurodegenerative diseases. Hum Mol Genet 20: 3757–3768 [DOI] [PubMed] [Google Scholar]
- 29. Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ et al (2006) Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 38: 758–769 [DOI] [PubMed] [Google Scholar]
- 30. Sharp AH, Loev SJ, Schilling G, Li SH, Li XJ, Bao J, Wagster MV, Kotzuk JA, Steiner JP, Lo A et al (1995) Widespread expression of Huntington's disease gene (IT15) protein product. Neuron 14: 1065–1074 [DOI] [PubMed] [Google Scholar]
- 31. Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57: 369–384 [DOI] [PubMed] [Google Scholar]
- 32. Bilen J, Liu N, Burnett BG, Pittman RN, Bonini NM (2006) MicroRNA pathways modulate polyglutamine‐induced neurodegeneration. Mol Cell 24: 157–163 [DOI] [PubMed] [Google Scholar]
- 33. Jiang F, Ye X, Liu X, Fincher L, McKearin D, Liu Q (2005) Dicer‐1 and R3D1‐L catalyze microRNA maturation in Drosophila . Genes Dev 19: 1674–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutations in X‐linked spinal and bulbar muscular atrophy. Nature 352: 77–79 [DOI] [PubMed] [Google Scholar]
- 35. Gu M, Dong X, Zhang X, Niu W (2012) The CAG repeat polymorphism of androgen receptor gene and prostate cancer: a meta‐analysis. Mol Biol Rep 39: 2615–2624 [DOI] [PubMed] [Google Scholar]
- 36. Ingles SA, Ross RK, Yu MC, Irvine RA, La Pera G, Haile RW, Coetzee GA (1997) Association of prostate cancer risk with genetic polymorphisms in vitamin D receptor and androgen receptor. J Natl Cancer Inst 89: 166–170 [DOI] [PubMed] [Google Scholar]
- 37. Giovannucci E, Stampfer MJ, Krithivas K, Brown M, Dahl D, Brufsky A, Talcott J, Hennekens CH, Kantoff PW (1997) The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci USA 94: 3320–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Irvine RA, Yu MC, Ross RK, Coetzee GA (1995) The CAG and GGC microsatellites of the androgen receptor gene are in linkage disequilibrium in men with prostate cancer. Cancer Res 55: 1937–1940 [PubMed] [Google Scholar]
- 39. Hakimi JM, Schoenberg MP, Rondinelli RH, Piantadosi S, Barrack ER (1997) Androgen receptor variants with short glutamine or glycine repeats may identify unique subpopulations of men with prostate cancer. Clin Cancer Res 3: 1599–1608 [PubMed] [Google Scholar]
- 40. Tsujimoto Y, Takakuwa T, Takayama H, Nishimura K, Okuyama A, Aozasa K, Nonomura N (2004) In situ shortening of CAG repeat length within the androgen receptor gene in prostatic cancer and its possible precursors. Prostate 58: 283–290 [DOI] [PubMed] [Google Scholar]
- 41. Liu Y, Wilson SH (2012) DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci 37: 162–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim YK, Kim B, Kim VN (2016) Re‐evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc Natl Acad Sci USA 113: E1881–E1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zamudio JR, Kelly TJ, Sharp PA (2014) Argonaute‐bound small RNAs from promoter‐proximal RNA polymerase II. Cell 156: 920–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murmann AE, McMahon KM, Halluck‐Kangas A, Ravindran N, Patel M, Law C, Brockway S, Wei JJ, Thaxton CS, Peter ME (2017) Induction of DISE in ovarian cancer cells in vivo . Oncotarget 8: 84643–84658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM (2015) Identification and characterization of essential genes in the human genome. Science 350: 1096–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A et al (2015) Gene essentiality and synthetic lethality in haploid human cells. Science 350: 1092–1096 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Source Data for Supplementary Figure S7
Review Process File
Source Data for Figure 3