

Abstract
Caseinolytic peptidase P (ClpP) is a mammalian quality control protease that is proposed to play an important role in the initiation of the mitochondrial unfolded protein response (UPR mt), a retrograde signaling response that helps to maintain mitochondrial protein homeostasis. Mitochondrial dysfunction is associated with the development of metabolic disorders, and to understand the effect of a defective UPR mt on metabolism, ClpP knockout (ClpP −/−) mice were analyzed. ClpP −/− mice fed ad libitum have reduced adiposity and paradoxically improved insulin sensitivity. Absence of ClpP increased whole‐body energy expenditure and markers of mitochondrial biogenesis are selectively up‐regulated in the white adipose tissue (WAT) of ClpP −/− mice. When challenged with a metabolic stress such as high‐fat diet, despite similar caloric intake, ClpP −/− mice are protected from diet‐induced obesity, glucose intolerance, insulin resistance, and hepatic steatosis. Our results show that absence of ClpP triggers compensatory responses in mice and suggest that ClpP might be dispensable for mammalian UPR mt initiation. Thus, we made an unexpected finding that deficiency of ClpP in mice is metabolically beneficial.
Keywords: adipose tissue, caseinolytic peptidase P, insulin sensitivity, mitochondria, obesity
Subject Categories: Metabolism; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Mitochondria are critical for the normal function of eukaryotic cells through production of ATP, maintenance of calcium homeostasis, and regulation of programmed cell death, and are also the major site for fatty acid oxidation 1. The mitochondrion has its own quality control system consisting of proteases and chaperones that helps to maintain protein homeostasis that in turn preserves mitochondrial integrity 2. The QC proteases present in the outer mitochondrial membrane (ubiquitin–proteasome system), inner mitochondrial membrane (PARL, OMA1, YME1L1, AFG3L2, and paraplegin), the intermembrane space (HtrA2), and mitochondrial matrix (Lon and ClpXP) help to maintain mitochondrial proteostasis through degradation of misfolded or damaged proteins 3. Failure of the QC system has been linked to various neurological diseases, aging, and metabolic disorders 4, 5. For example, muscle‐specific knockdown of PARL impairs insulin signaling and deficiency of OMA1 causes obesity and defective thermogenesis in mice 6, 7.
Caseinolytic peptidase P (ClpP) is a highly conserved QC protease from bacteria to humans. ClpP lacks ATPase activity and multimerizes with the mitochondrial chaperone and ATPase, ClpX to form the functional protease ClpXP. In Caenorhabditis elegans, ClpP plays a critical role in the activation of UPRmt, a retrograde signaling response that induces the expression of mitochondrial chaperones that helps to maintain mitochondrial proteostasis. The peptides generated through the proteolytic cleavage of unfolded proteins by ClpP initiate the UPRmt response in C. elegans 8, 9. In mammalian cells, accumulation of unfolded proteins in mitochondrial matrix results in the transcriptional up‐regulation of mitochondrial chaperones and ClpP 10, 11. While the role of ClpP in mammalian UPRmt is not clear, a potential involvement of ClpP in mitochondrial peptide release, similar to C. elegans, has been suggested 12. In addition to its proposed role in UPRmt, ClpP is involved in other mitochondrial functions; for example, knockdown of ClpP in muscle cells causes mitochondrial dysfunction and reduces cell proliferation 13, and ClpP is also involved in the regulation of mitochondrial protein synthesis through mitochondrial ribosome assembly 14. Recessive mutations in CLPP cause Perrault syndrome in humans, characterized by sensorineural deafness and ovarian failure 15.
Acquired obesity in humans is associated with an impaired UPRmt response in subcutaneous WAT (sWAT) suggesting a possible relationship between metabolic stress and UPRmt 16. Because ClpP is proposed to play an important role in UPRmt, we analyzed mice deficient in ClpP (ClpP −/− mice) to understand the role of UPRmt‐mediated proteostasis in metabolism. ClpP −/− mice recapitulate the phenotypes of Perrault syndrome in humans and are characterized by mild mitochondrial dysfunction, up‐regulation of mitochondrial chaperones, and accumulation of ClpX and mtDNA in various tissues 17. We hypothesized that a defective UPRmt response and mitochondrial dysfunction due to ClpP deficiency will cause insulin resistance in ClpP −/− mice. On the contrary, ClpP −/− mice fed ad libitum showed improved insulin sensitivity, reduced adiposity, and elevated mitochondrial respiration in WAT. When challenged with a metabolic stress such as high‐fat diet (HFD), ClpP −/− mice are protected from diet‐induced obesity, glucose intolerance, insulin resistance, and hepatic steatosis. Our findings suggest that compensatory responses due to ClpP deficiency could contribute to the paradoxical beneficial metabolic effects in ClpP −/− mice.
Results
ClpP −/− mice have reduced adiposity and white adipocytes from ClpP −/− mice exhibit increased respiration
Gispert et al 17 reported that ClpP −/− mice have reduced gain in body weight compared to wild‐type (WT) littermates. In our cohort, at 5 months of age ClpP −/− male mice have 28% reduction in body weight compared to WT littermates or heterozygous ClpP +/− mice, despite increased food consumption by ClpP −/− mice (34% more compared to WT) (Fig 1A and B). ClpP −/− mice have 64% reduction in fat mass and 24% reduction in lean mass (Appendix Fig S1A). After normalizing to body weight, fat mass in ClpP −/− mice showed 38% reduction compared to WT, whereas lean mass showed a tendency to increase but did not reach statistical significance (Fig 1C). Thus, reduced body weight in ClpP −/− mice is attributable to reduced fat mass. Body weight, food intake, fat mass, and lean mass of ClpP +/− mice were similar to WT littermates (Fig 1A–C). ClpP −/− female mice (5‐month‐old) also showed a reduction in body weight and fat mass, comparable to male mice (Appendix Fig S1B and C). H&E staining of gonadal white adipose tissue (gWAT) showed smaller adipocytes (37% reduction in adipocyte area) in ClpP −/− mice compared to adipocytes in WT mice (Fig 1D). Transcript levels of adipocyte differentiation factors peroxisome proliferator‐activated receptor gamma (PPARγ), CCAAT/enhancer‐binding protein alpha (C/EBPα), and adipocyte protein 2 (aP2) in gWAT were similar in ClpP −/− and WT mice, suggesting that adipocyte differentiation is not impaired in ClpP −/− mice (Appendix Fig S1D).
Figure 1. ClpP −/− mice have reduced adiposity and elevated respiration in WAT .
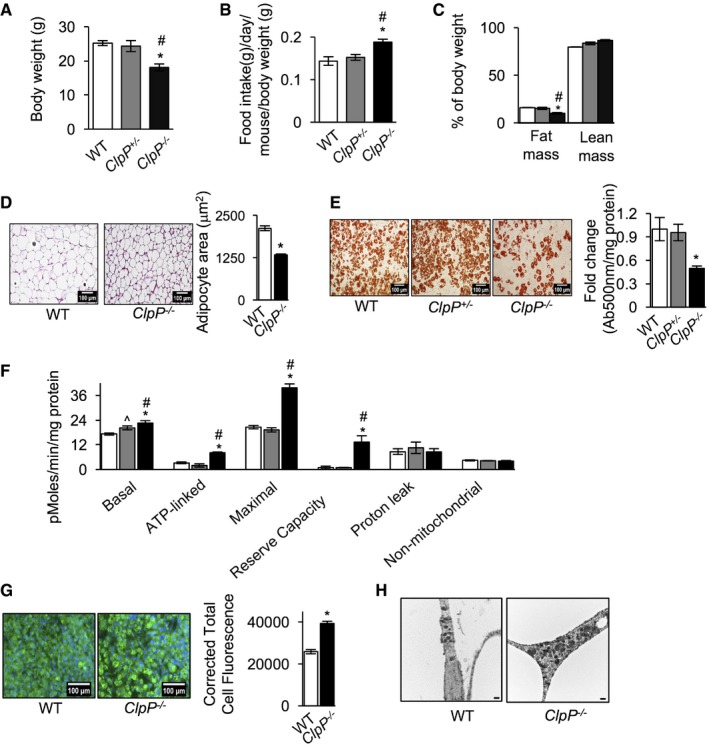
- Body weights of WT, ClpP +/−, and ClpP −/− male mice fed ad libitum at 5 months of age (n = 8–10).
- Food consumption of WT, ClpP +/−, and ClpP −/− mice fed ad libitum, normalized to body weight, at 5 months of age (n = 8–10).
- Fat mass and lean mass in WT, ClpP +/−, and ClpP −/− mice, assessed by quantitative magnetic resonance imaging and normalized to body weight age (n = 8–10).
- H&E staining of gWAT sections of WT and ClpP −/− mice (left panel, scale 100 μm, magnification 20×) and quantification of the average adipocyte area of gWAT sections (right panel) age (n = 3).
- Oil red O staining of differentiated adipocytes from WT, ClpP +/−, and ClpP −/− mice (left panel, scale 100 μm, magnification 20×). Quantification of total oil red O extracted from differentiated adipocytes (right panel) (n = 3). Data shown are mean ± SEM from three independent experiments.
- Graphical representation of cellular bioenergetics in differentiated primary adipocyte cultures from WT, ClpP +/−, and ClpP −/− mice was measured using the Seahorse Bioscience XF24 Extracellular Flux Analyzer mitostress assay, normalized to protein concentration per well. Data represent mean ± SEM from three independent experiments.
- Immunostaining of differentiated adipocytes from WT and ClpP −/− mice using Tom20 antibody (left panel, scale 100 μm, magnification 20×) and quantification of fluorescent intensity (right panel) (n = 3).
- Electron micrographs of gWAT from WT and ClpP −/− mice (n = 3). Scale 500 nm. Magnification 5,000×.
Differentiated adipocytes from ClpP −/− mice accumulated 50% less triglycerides than adipocytes from WT or ClpP +/− mice (Fig 1E). Basal respiration (30% increase), ATP‐linked respiration (93% increase), maximal respiration (127% increase), and spare respiratory capacity (1,319% increase) were increased in ClpP −/− mice compared to WT or ClpP +/− adipocytes (Fig 1F). Levels of non‐mitochondrial respiration and proton leak were similar in WT, ClpP +/−, and ClpP −/− adipocytes. For ClpP +/− adipocytes, only basal respiration showed a 17% increase compared to WT adipocytes, whereas other parameters were comparable to WT mice. Similarly, in vitro knockdown of ClpP (90% reduction in ClpP protein) in 3T3‐L1 cells also increased respiration, compared to control cells (Appendix Fig S1E and F). Thus, ClpP deficiency increased respiration in adipocytes both in vitro and in vivo.
An increased number of mitochondria per cell or the same number of highly active mitochondria per cell can contribute to high mitochondrial respiration. Immunostaining of differentiated adipocytes using an antibody for mitochondrial outer membrane protein Tom20 showed more intense staining in ClpP −/− adipocytes than WT adipocytes, suggesting there is an increase in mitochondrial content in ClpP −/− adipocytes (Fig 1G). In agreement with this, mitochondrial content in gWAT of ClpP −/− mice was higher (and the mitochondria appeared larger) than WT mice, as assessed by electron microscopy (Fig 1H). Thus, elevated mitochondrial content could contribute to elevated respiration and reduced fat mass in ClpP −/− mice.
Markers of mitochondrial biogenesis, mitochondrial chaperones, and mitochondrial fission/fusion regulator OPA1 are elevated in gWAT of ClpP −/− mice
Peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α) is a key transcription factor that regulates mitochondrial biogenesis. In gWAT of ClpP −/− mice, expression of PGC‐1α is up‐regulated 3.3‐fold compared to WT mice (Fig 2A). Protein expression of transcription factor A, mitochondrial (Tfam) (mitochondrial DNA transcription factor, 3.3‐fold), and voltage‐dependent anion channel (VDAC) (a highly conserved outer mitochondrial membrane protein, 2.8‐fold) is also elevated in ClpP −/− mice of gWAT (Fig 2A). Protein expression of PGC‐1α and Tfam was comparable in gWAT of ClpP +/− mice and WT mice; however, expression of VDAC was twofold higher in ClpP +/− mice compared to WT mice (Fig 2A). We also found a significant increase in the protein expression of electron transport chain (ETC) subunits: ATP Synthase, H+ Transporting, Mitochondrial F1 Complex, Alpha Subunit 1 (ATP5a1, 2.4‐fold); ATP5ab (2.5‐fold); Succinate Dehydrogenase Complex Flavoprotein Subunit A (SDHA, 3.4‐fold); SDHAB (2.4‐fold); SDHC (2.2‐fold); and Ubiquinol‐Cytochrome C Reductase Core Protein II (UQCRC2, 2.6‐fold) in gWAT of ClpP −/− mice, compared to WT mice (Fig 2B and Table EV1). However, expression of ETC subunits in WAT of ClpP +/− mice was similar to WT mice (Fig 2B). In addition, protein expression of citrate synthase was increased twofold (Appendix Fig S2A and Table EV1) and mitochondrial DNA (mtDNA) content was increased fourfold in the WAT of ClpP −/− mice (Fig 2C). Taken together, mitochondrial biogenesis markers are increased in gWAT of ClpP −/− mice suggesting an increase in mitochondrial mass, compared to WT or ClpP +/− mice. PGC‐1α expression is also elevated (2.7‐fold) in subcutaneous WAT (sWAT) of ClpP −/− mice, but not in brown adipose tissue (BAT), heart, or skeletal muscle (Fig 2D). We also looked for a potential signal that might drive PGC‐1α expression in gWAT. Increased levels of reactive oxygen species (ROS) are linked to activation of PGC‐1α expression in skeletal muscle 18, 19, 20. In humans, oxidative stress induced by short‐term exercise increases PGC‐1a expression in skeletal muscle 21. Based on this, we also tested whether ROS levels are altered in gWAT of ClpP −/− mice. Levels of 4‐Hydroxynonenal (4‐HNE) were used as a marker of oxidative stress 22 and Western blotting of gWAT showed increased 4‐HNE levels (1.3‐fold) in ClpP −/− mice compared to WT mice, suggesting increased oxidative stress in gWAT (Fig 2E). Elevated levels of H2O2 are shown to induce PGC‐1α expression through activation of AMPK in skeletal muscle 18. Assessing AMP‐activated protein kinase (AMPK) activation in gWAT of ClpP −/− mice showed that the ratio of phospho‐AMPK/AMPK is increased in ClpP −/− mice compared to WT mice, suggesting increased AMPK activation (Fig 2F). Thus, increase in ROS and AMPK activation could contribute to the increased expression of PGC‐1α in gWAT of ClpP −/− mice.
Figure 2. Markers of mitochondrial biogenesis, mitochondrial chaperones, and mitochondrial fission/fusion regulator OPA1 are elevated in gWAT of ClpP −/− mice.
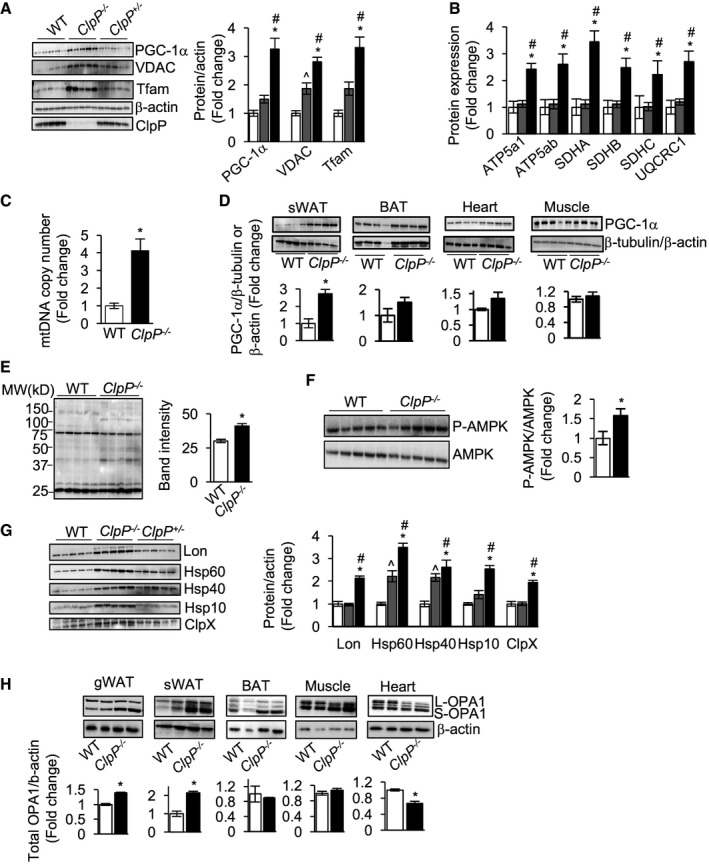
- Left panel: immunoblots of gWAT extracts from WT, ClpP +/−, and ClpP −/− mice for PGC‐1α, Tfam, VDAC, β‐actin, and ClpP (n = 5). Right panel: graphical representation of quantified blots normalized to β‐actin.
- Quantification of protein levels of electron transport chain (ETC) subunits ATP5a1, ATP5ab, SDHA, SDHAB, SDHC, and UQCRC2 in gWAT of WT, ClpP +/−, and ClpP −/− mice obtained by mass spectrometry (n = 5).
- Quantification of mtDNA/nDNA content in gWAT from WT and ClpP −/− mice (n = 6–8).
- Top panels: immunoblots of sWAT, BAT, heart, and muscle extracts from WT and ClpP −/− mice for PGC‐1α and β‐tubulin (Western blot shows representative examples for n = 4, and quantification is based on n = 6). Bottom panels: graphical representation of quantified blots normalized to β‐tubulin.
- Left panel: immunoblots of gWAT extracts (30 μg/lane) from WT and ClpP −/− mice for 4‐HNE. Right panel: Quantification of band intensity of the entire line represented graphically (n = 5).
- Top panel: immunoblots of gWAT extracts from WT and ClpP −/− mice for P‐AMPK and AMPK (n = 5). Right panel: graphical representation of the ratio of P‐AMPK to AMPK.
- Left panel: immunoblots of gWAT extracts from WT, ClpP +/−, and ClpP −/− mice for Lon, Hsp60, Hsp40, Hsp10, ClpX, and ClpP (n = 5). Right panel: graphical representation of quantified blots normalized to β‐actin.
- Western blots showing protein expression of OPA1 isoforms and β‐actin in gWAT, sWAT, BAT, muscle and heart of WT, and ClpP −/− mice (top panel) (Western blot shows representative examples for n = 2, and quantification is based on n = 5). Quantification of the blots normalized to β‐actin is shown as bar graphs (bottom panel).
Loss of ClpP was reported to induce the expression of mitochondrial chaperones and the mitochondrial protease Lon in testis, heart, liver, and brain of ClpP −/− mice 17. In gWAT of ClpP −/− mice, protein expression of Lon protease (2.1‐fold) and mitochondrial chaperones [Heat shock protein 60 (Hsp60, 3.5‐fold), Hsp40 (2.6‐fold), Hsp10 (2.5‐fold), and ClpX (1.9‐fold)] was significantly elevated compared to WT mice (Fig 2G). However, this increase in the expression of mitochondrial chaperones in gWAT could be attributed to the increase in mitochondrial number. In ClpP +/− mice, protein levels of Lon, Hsp10, and ClpX were similar to WT mice, whereas protein expression of Hsp60 (2.2‐fold) and Hsp40 (2.2‐fold) was significantly elevated (Fig 2G). Similarly, sWAT of ClpP −/− mice showed a strong induction of Lon and mitochondrial chaperones and BAT showed increased expression of Lon protease and Hsp40 compared to WT mice (Appendix Fig S2B).
Previously, we found that knockdown of ClpP (70% reduction compared control cells) in C2C12 muscle cells increased the expression of mitochondrial fission protein dynamin‐related protein1, Drp1 13. In gWAT of ClpP −/− mice, we did not see any change in the expression of fusion protein mitofusin 2 (Mfn2) or fission proteins Drp1 or Fis1 (data not shown). However, protein expression of mitochondrial fission–fusion regulator Optic Atrophy 1 (OPA1, total) was increased in the gWAT (17%) and sWAT (114%) of ClpP −/− mice (Fig 2H). OPA1 exists in multiple long forms (L‐OPA1) and short forms (S‐OPA1) and processing of OPA1 to L‐OPA1 and S‐OPA1 balances mitochondrial fission/fusion and the antibody we used detected five different isoforms, as previously reported 23, 24. In BAT and skeletal muscle of ClpP −/− mice, total OPA1 was similar to WT mice, whereas in heart, total OPA1 was reduced by 34% (Fig 2H). Thus, PGC‐1α and OPA1 showed a tissue‐specific difference in their expression and WAT depots showed higher expression, compared to other tissues.
Pink1 and Parkin are known initiators of mitophagy 25. Therefore, we quantified the expression of mitophagy markers PINK1 and Parkin in gWAT to test whether activation of mitophagy contributes to the recovery of healthy mitochondria in the absence of ClpP. Surprisingly, expression of PINK1 and Parkin is decreased in gWAT of ClpP −/− mice, suggesting that mitophagy is not activated in ClpP −/− mice (Appendix Fig S2C).
Absence of ClpP increases whole‐body energy expenditure and mitochondrial uncoupling and alters expression of metabolic enzymes in gWAT of ClpP −/− mice
ClpP −/− mice exhibited 37 and 12% increases in oxygen consumption during dark and light phases, respectively, when normalized to total body mass (Fig 3A). When normalized to lean body mass, ClpP −/− mice showed a 15% increase in oxygen consumption during dark phase and this difference was not statistically significant (Fig 3B). Similarly, energy expenditure (EE) normalized to total body mass was 40 and 34% higher for ClpP −/− mice during dark and light phases, respectively (Fig 3C). Normalizing EE to lean body mass also reduced the increased EE in ClpP −/− mice to 10%, which did not reach statistical significance (Fig 3D). Furthermore, WT and ClpP −/− mice had a similar respiratory exchange ratio (RER) and cage activity levels (Fig 3E and F). Metabolically, ClpP +/− mice were similar to WT mice except for significantly reduced RER during the light phase (Fig 3E). Thus, the finding that higher oxygen consumption and EE rates of ClpP −/− mice were reduced when normalized to lean rather than total body mass is consistent with increased adipose tissue metabolism in ClpP −/− mice.
Figure 3. Absence of ClpP increases whole‐body energy expenditure and mitochondrial uncoupling and alters expression of metabolic enzymes in gWAT .
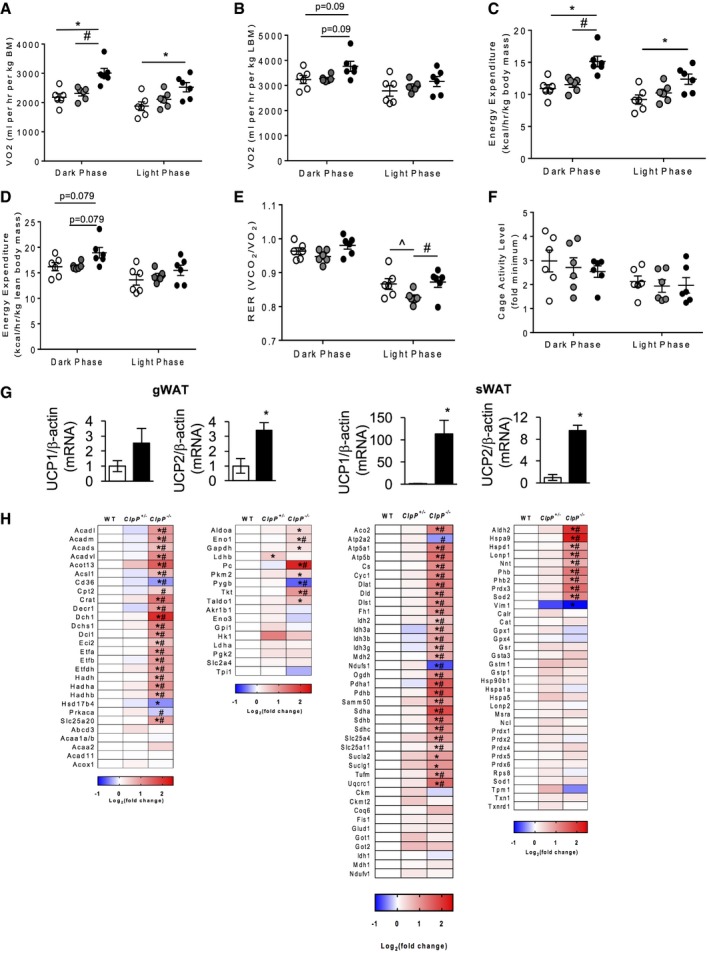
-
A–FOxygen consumption rate (OCR) normalized to body weight (A), OCR normalized to lean body mass (B), EE normalized to body weight (C), EE normalized to lean body mass (D), RER (E), and cage activity (F) (n = 6).
-
GTranscript levels of UCP1 and UCP2 in gWAT and sWAT of WT and ClpP −/− mice (n = 6–8).
-
HHeatmaps showing changes in the expression of protein in fatty acid metabolism (first panel), glucose metabolism (second panel), TCA cycle, electron transport chain (ETC), and other mitochondrial proteins (third panel) and stress response (detoxification/antioxidant enzymes, chaperones, heat shock proteins, and proteases) (forth panel) in gWAT of WT, ClpP +/−, and ClpP −/− mice (n = 5). Average value of WT was used to normalize values of ClpP +/− and ClpP −/− mice. The darker the red indicates the greater the increase in expression, and the darker the blue indicates the greater the decrease in expression.
Transcript level of uncoupling protein 1 (UCP1) was significantly increased in sWAT, not in gWAT, of ClpP −/− mice compared to WT mice (Fig 3G). UCP2 was significantly elevated in gWAT and sWAT of ClpP −/− mice compared to WT mice (Fig 3G), whereas UCP3 levels were similar in WT and ClpP −/− mice WAT depots (data not shown). Beige fat or “brown‐like” fat present in WAT is known to increase energy expenditure 26, 27. Therefore, we tested markers of browning/beiging in sWAT and found that in addition to UCP1, transcript levels of PGC‐1α (11.6‐fold), cell death‐inducing DFFA‐like effector A (CIDEA, 11.4‐fold), and cytochrome c oxidase subunit 8b (Cox8b, 15.8‐fold) were elevated in sWAT of ClpP −/− mice; however, levels of PR/SET Domain 16 (Prdm16) were similar in ClpP −/− and WT mice (Appendix Fig S3A). Western blotting to assess protein expression of UCP1 in BAT showed no significant change in UCP1 expression (Appendix Fig S3B). Thus, increased uncoupling in WAT depots as well as “browning” of sWAT could contribute to increased energy expenditure in ClpP −/− mice.
A targeted quantitative proteomic approach was employed for a detailed study of the changes in protein expression of mitochondrial metabolic enzymes in gWAT of WT, ClpP +/−, and ClpP −/− mice. Protein expression of mitochondrial fatty acid oxidation enzymes, and enzymes/proteins involved glucose metabolism, tricarboxylic acid (TCA) cycle, and ETC, and antioxidants are altered in gWAT of ClpP −/− mice, compared to WT mice (Fig 3H and Table EV1). Thus, absence of ClpP altered expression of metabolic enzymes in gWAT, and the increase in mitochondrial biogenesis might partly contribute to this increase.
ClpP −/− mice have improved insulin sensitivity
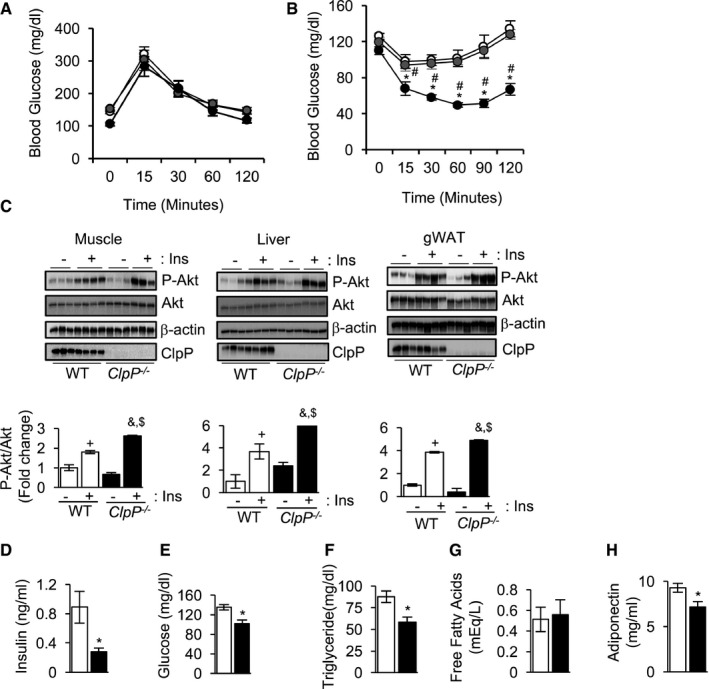
Mitochondrial dysfunction is associated with the development of insulin resistance 28. To understand the effect of ClpP deficiency on glucose metabolism, glucose clearance was measured by glucose tolerance test (GTT) and was found to be similar in WT, ClpP +/−, and ClpP −/− mice (Fig 4A). However, ClpP −/− mice exhibited improved insulin sensitivity compared to WT or ClpP +/− mice when subjected to insulin tolerance test (ITT) (Fig 4B). Improved insulin sensitivity suggests enhanced insulin‐stimulated Akt activation to enable faster glucose uptake. Consistent with this, insulin‐stimulated Akt phosphorylation was significantly elevated in skeletal muscle (45%), liver (62%), and gWAT (27%) of ClpP −/− mice compared to WT mice (Fig 4C). Circulating level of insulin and glucose was reduced by 68 and 44%, respectively, in ClpP −/− mice compared to WT mice, further supporting improved insulin sensitivity in ClpP −/− mice (Fig 4D and E). Circulating triglycerides were also significantly reduced (33%) in ClpP −/− mice, whereas free fatty acid levels were similar in ClpP −/− mice and WT mice (Fig 4F and G). Surprisingly, circulating level of the insulin‐sensitizing adipokine, adiponectin was significantly lower (23%) in ClpP −/− mice compared to WT (Fig 4H).
Figure 4. ClpP −/− mice exhibit improved insulin sensitivity.
-
A, BGlucose tolerance test (A) and insulin tolerance test (B) of WT, ClpP +/−, and ClpP −/− mice fed ad libitum (n = 6–8).
-
CWestern blots showing expression P‐Akt (T308), Akt, β‐actin, and ClpP in WT and ClpP −/− mice muscle (first panel), liver (second panel), and gWAT (third panel) injected with PBS (−Ins) or insulin (+Ins) (top panels) (n = 3–4). Quantification of P‐Akt/Akt is shown in bottom panels.
-
D–HLevels of circulating insulin (D), glucose (E), triglyceride (F), free fatty acids (G), and adiponectin (H) in WT and ClpP −/− mice in fed state (n = 6–8).
ClpP −/− mice are resistant to diet‐induced obesity and are protected from HFD‐induced glucose intolerance and insulin resistance
To understand the effect of ClpP deficiency during metabolic stress, WT, ClpP +/−, and ClpP −/− mice were fed defined diets containing low fat (10% of calories from fat, LFD) or high fat (60% of calories from fat, HFD) for 10 weeks. Weight gain for WT, ClpP +/−, and ClpP −/− mice at 10 weeks on a LFD was 26, 26, and 0.7%, respectively, and weight gain for HFD‐fed mice was 83, 65, and 12%, respectively (Fig 5A and Appendix Fig S4A). This difference in body weight between WT and ClpP −/− mice with HFD feeding is regardless of any significant change in food intake (Appendix Fig S4B).
Figure 5. ClpP −/− mice are resistant to diet‐induced obesity and are protected from HFD‐induced glucose intolerance and insulin resistance.
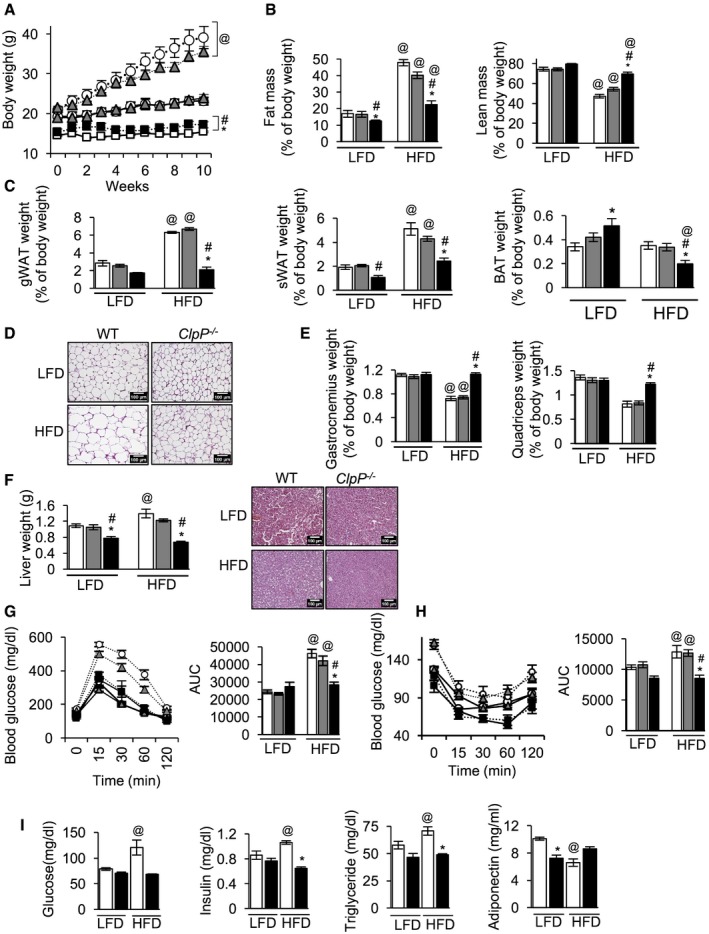
- Change in body weights of WT, ClpP +/−, and ClpP −/− mice fed low fat diet (LFD, solid line) or high‐fat diet (HFD, dotted line) (n = 8–10). WT—white circle, ClpP +/−—gray triangle, ClpP −/−—black square.
- Quantitative magnetic resonance analysis showing fat mass (left panel) and lean mass (right panel), normalized to body weight (n = 8–10).
- Weights of different fat depots, normalized to body weight (gWAT—gonadal white adipose tissue, sWAT—subcutaneous WAT, and BAT—brown adipose tissue) (n = 8–10).
- H&E staining of gWAT sections of WT and ClpP −/− mice fed a LFD or HFD. Scale bar 100 μm, magnification 20×.
- Tissue weight of gastronenius muscle (left) and quadriceps muscle (right) normalized to body weight (n = 8–10).
- Liver weight of WT, ClpP +/−, and ClpP −/− mice fed LFD or HFD (left panel, n = 8–10) and H&E staining of liver sections of WT and ClpP −/− mice fed a LFD or HFD. Scale bar 100 μm, magnification 20×.
- Glucose tolerance test of WT, ClpP +/−, and ClpP −/− mice fed LFD or HFD (left panel) (n = 8–10). Area under the curve is represented graphically (right panel).
- Insulin tolerance test of WT, ClpP −/−, and ClpP +/− mice fed LFD or HFD (left panel) (n = 8–10). Graphical representation of area under the curve (right panel).
- Levels of circulating glucose (first panel) and insulin (second panel) in LFD‐ or HFD‐fed WT and ClpP −/− mice, fasted for 16 h, and levels of circulating triglycerdies (third panel) and adiponectin (fourth panel) in LFD‐ or HFD‐fed WT and ClpP −/− mice in fed state (n = 8–10).
WT, ClpP +/−, and ClpP −/− mice after 10 weeks on HFD showed 184, 143, and 79% increase in fat mass and 41, 26, and 12% decrease in lean mass as a percentage of body weight, respectively (Fig 5B, left panel). gWAT mass as a percentage of body weight in WT and ClpP +/− mice fed a HFD increased 125 and 168%, respectively, whereas ClpP −/− mice did not show a significant increase in gWAT weight, compared to respective control mice fed a LFD (Fig 5C, left panel). H&E staining of the gWAT showed hypertrophied adipocytes in HFD‐fed WT mice, whereas in HFD‐fed ClpP −/− mice adipocytes were smaller, similar to ClpP −/− mice fed LFD (Fig 5D). sWAT weight as a percentage of body weight in HFD‐fed WT and ClpP +/− mice weighed 168 and 115% more, compared to respective control mice fed a LFD. Interestingly, HFD‐fed ClpP −/− mice showed a 140% increase in sWAT weight compared to ClpP −/− mice fed a LFD (Fig 5C, middle panel). BAT weight as a percentage of body weight was significantly higher in ClpP −/− mice, compared to ClpP +/− or WT mice (Fig 5C, right panel). BAT weight as a percentage of body weight of LFD‐ and HFD‐fed ClpP +/− and WT mice was similar, however BAT weight of HFD‐fed ClpP −/− mice was reduced by 63%, compared to ClpP −/− mice fed a LFD (Fig 5C, right panel). Gastrocnemius muscle weight as a percentage of body weight showed a significant reduction in WT (36%) and ClpP +/− mice (27%) fed a HFD, compared to mice a LFD, whereas in HFD‐fed ClpP −/− mice gastrocnemius muscle weight was similar to ClpP −/− mice fed a LFD (Fig 5E, left panel). Similarly, tissue weight of quadriceps muscle weight as a percentage of body weight showed a significant reduction in HFD‐fed WT (42%) and ClpP +/− (37%) mice; however, in HFD‐fed ClpP −/− mice quadriceps weight was reduced by only 7% and this reduction is not statistically significant (Fig 5E, right panel). Ten weeks of HFD feeding increased liver weight of WT and ClpP +/− mice by 18 and 9%, respectively, whereas liver weight of ClpP −/− mice was reduced (12%) by HFD feeding, compared to ClpP −/− mice fed LFD (Fig 5F, left panel). Consistent with this, H&E staining of liver sections revealed increased lipid accumulation only in HFD‐fed WT mice, not in ClpP −/− mice (Fig 5F, right panel). Thus, ClpP −/− mice are protected against diet‐induced obesity and hepatic steatosis.
High‐fat diet induced glucose intolerance and insulin resistance in WT and ClpP +/− mice, compared to mice fed a LFD (Fig 5G and H). In contrast, glucose tolerance and insulin sensitivity in HFD‐fed ClpP −/− mice were similar to LFD‐fed ClpP −/− mice (Fig 5G and H). HFD feeding also elevated levels of circulating glucose (53%), insulin (23%), and triglyceride (22%) and reduced adiponectin (35%) in WT mice (Fig 5I). However, HFD‐fed ClpP −/− mice had similar blood glucose, insulin, and triglyceride levels to ClpP −/− mice fed LFD (Fig 5I).
Mitochondrial respiration is increased in gWAT of HFD‐fed ClpP −/− mice
To understand the effect of HFD feeding on mitochondrial function in gWAT, mitochondrial respiration was measured using the OROBOROS Oxygraph 2K. Measurement of complex I‐linked oxidative phosphorylation (OXPHOS) using glutamate/malate as ETC complex I substrates did not show a significant difference in respiration for WT, ClpP +/−, or ClpP −/− mice fed LFD or HFD (Fig 6A). However, gWAT from LFD‐fed or HFD‐fed ClpP −/− mice showed increased oxygen consumption, compared to LFD‐fed or HFD‐fed WT or ClpP +/− mice, both with the addition of succinate/rotenone to measure complex II‐linked OXPHOS (Fig 6B) and the complex II substrate succinate to measure OXPHOS capacity through complex II (Fig 6C). Comparing levels of OXPHOS linked to complex I and II substrates, it can be concluded that complex II exhibits higher activity than complex I. Measurement of proton leak‐linked oxygen consumption in the absence of substrates did not show any significant difference between LFD‐ or HFD‐fed WT, ClpP +/−, and ClpP −/− mice (Fig 6D). However, proton leak‐linked oxygen consumption following ATP synthase inhibition was significantly increased in both LFD‐fed and HFD‐fed ClpP −/− mice compared to WT or ClpP +/− mice (Fig 6E). Oligomycin increases membrane potential and thus increases the driving force for proton leak, likely causing the discrepancy in these proton leak measurements 29. Thus, gWAT from ClpP −/− mice has significantly increased respiratory capacity when fed a LFD or HFD, compared to WT or ClpP +/− mice.
Figure 6. Mitochondrial respiration is increased in gWAT of HFD‐fed ClpP −/− mice.
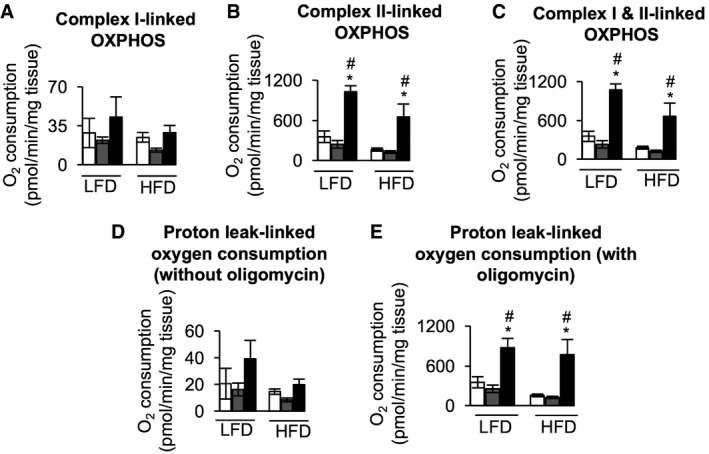
- Electron transport chain (ETC) complex I‐linked OXPHOS measured with the substrate combination glutamate, malate, and ADP (n = 6).
- ETC complex II‐linked OXPHOS measured with the substrate combination glutamate, malate, ADP, succinate, cytochrome c, and rotenone (n = 6).
- ETC complex I&II‐linked OXPHOS, or maximum OXPHOS capacity (P) of the ETC, measured with the substrate combination glutamate, malate, ADP, succinate, and cytochrome c (n = 6).
- Mitochondrial innermembrane proton leak‐linked oxygen consumption in the presence of substrates (glutamate and malate) and absence of ADP (n = 6).
- Mitochondrial innermembrane proton leak‐linked oxygen consumption in the presence of substrates (glutamate, malate, and succinate) and ADP but addition of oligomycin to inhibit ATP synthase (n = 6).
Elevated mitochondrial biogenesis markers are preserved in gWAT of HFD‐fed ClpP −/− mice
Mitochondrial dysfunction in adipose tissue is characterized by reduced mitochondrial number and is associated with the development of insulin resistance under obese conditions 30. HFD feeding reduced the expression of mitochondrial biogenesis markers [PGC‐1α (64%), Tfam (44%), and VDAC (64%)] in the gWAT of WT mice (Fig 7A). LFD‐fed ClpP −/− mice have increased expression of mitochondrial biogenesis markers PGC‐1α (2.5‐fold), Tfam (1.7‐fold), and VDAC (2.5‐fold) in gWAT and HFD feeding preserved the expression of these proteins (Fig 7A). Because skeletal muscle is the major site for glucose utilization, we tested the effect of HFD on mitochondrial biogenesis markers in this tissue. In WT mice, HFD increased the expression of PGC‐1α (47%) in skeletal muscle and expression of Tfam was unchanged; however, HFD did not affect the expression of PGC‐1α or Tfam in ClpP −/− mice (Fig 7B).
Figure 7. Elevated mitochondrial biogenesis markers are preserved in gWAT of HFD‐fed ClpP −/− mice.
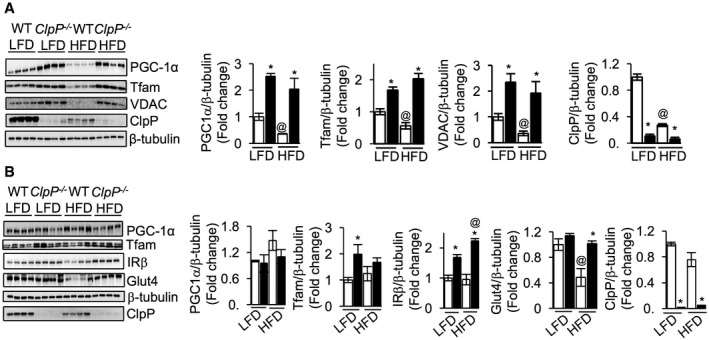
- Western blots showing protein expression of PGC‐1α, Tfam, VDAC, ClpP, and β‐tubulin in gWAT of LFD‐ or HFD‐fed WT and ClpP −/− mice (left panel) (n = 4). Quantification of proteins normalized to β‐tubulin is shown in the right panel.
- Western blots showing protein expression of PGC‐1α, Tfam, IRβ, Glut4, ClpP, and β‐tubulin in skeletal muscle of LFD‐ or HFD‐fed WT and ClpP −/− mice (left panel) (n = 4). Quantification of proteins normalized to β‐tubulin is shown in the right panel.
Protein expression of insulin receptor beta (IRβ) was elevated by 68% in LFD‐fed ClpP −/− mice skeletal muscle, compared to LFD‐fed WT mice, and could contribute to the improved insulin sensitivity in LFD‐fed ClpP −/− mice. HFD feeding further increased the expression of IRβ in ClpP −/− mice by 29% (Fig 7B). WT and ClpP −/− mice have similar levels of glucose transporter 4 (Glut4) when fed LFD; however, HFD reduced Glut4 expression in WT by 52%, whereas Glut4 expression in ClpP −/− mice skeletal muscle was unaffected by HFD (Fig 7B). Thus, increased expression of IRβ and preservation of Glut4 in skeletal muscle with HFD feeding could contribute to the improved insulin sensitivity in ClpP −/− mice. It is noteworthy that in HFD‐fed WT mice, ClpP protein expression was reduced by 73% in gWAT, but not in muscle (Fig 7A and B). It is possible that the reduction in mitochondrial content in gWAT could account for this drastic reduction in ClpP levels.
Discussion
The UPRmt is an important pathway that maintains mitochondrial matrix proteostasis through up‐regulation of mitochondrial chaperones, yet the effect of UPRmt on mammalian metabolism is not known. Because ClpP is proposed to play an important role in UPRmt, we used mice deficient in ClpP (ClpP −/− mice) to understand the role of defective UPRmt in metabolism. Surprisingly, our findings revealed an unexpected effect of ClpP on UPRmt and paradoxical beneficial effect of ClpP deficiency on metabolism.
Absence of ClpP resulted in many compensatory responses in ClpP −/− mice. One such response is increase in the expression of mitochondrial biogenesis markers, suggesting increased mitochondrial mass, in WAT of ClpP −/− mice. Gispert et al 17 measured the protein expression of VDAC/porin, Tfam, and ETC complex subunits in testis, heart, liver, and brain of ClpP −/− mice and did not find any difference in their expression compared to WT mice 17. We found a fourfold increase in mtDNA content, another mitochondrial biogenesis marker, in WAT. Interestingly, Gispert et al 17 also reported two‐ to fourfold accumulation of mtDNA in testis, ovary, heart, and brain of ClpP −/− mice compared to WT mice. However, it should be noted that in the tissues that showed an increase in mtDNA, none of the mitochondrial biogenesis markers (Tfam, porin, or ETC subunits) were increased. It is proposed that elevated levels of ClpX, the binding partner of ClpP, could contribute mtDNA accumulation 17, because ClpX has been shown to contribute to the maintenance of mitochondrial genome distribution 31. Thus, our finding that mitochondrial biogenesis markers increase only in adipose tissue, but not in other tissues, suggests that mitochondrial biogenesis is selectively occurring in WAT. The increase in mitochondrial mass could contribute to increased respiration by WAT, because respiration is reduced in the heart of ClpP −/− mice, whereas respiration in muscle and brain is similar to WT mice 14, 17. Thus, ClpP deficiency shows a differential effect on respiration in tissues. Our findings also suggest that increase in ROS level and AMPK activation could be the driver of the mitochondrial biogenesis in WAT, because ROS is known to induce PGC‐1α expression through AMPK that is a known activator of PGC‐1α 18.
In addition to the increased expression of mitochondrial transcription factor PGC‐1α, up‐regulation of mitochondrial uncoupling proteins (and “beiging or browning” of WAT) and OPA1 could also be compensatory responses due to ClpP deficiency in WAT. Increased expression of these proteins could be metabolically beneficial in ClpP −/− mice, because studies have shown that overexpression of human PGC‐1α in human subcutaneous white adipocytes increases the expression of respiratory chain proteins, UCP1, and fatty acid oxidation 32, and WAT‐specific loss of PGC‐1α in mice has been shown to reduce expression of mitochondrial OXPHOS and fatty acid oxidation genes and these mice develop insulin resistance when challenged with HFD 31. UCP2 uncouples OXPHOS only after induction by cold or ROS and we found increased ROS level in gWAT of ClpP −/− mice 33. UCP2 gene expression is reduced in the WAT of patients with obesity and type 2 diabetes 34, and moderate overexpression of human UCP2 in mice has been shown to reduce fat mass 35, and three common polymorphisms in UCP2 gene are possibly associated with DM2 and/or obesity 36. Proteomic analysis of gWAT revealed increased expression of enzymes/proteins involved in glycolysis, TCA cycle, ETC, and beta‐oxidation, in ClpP −/− mice. These changes in gWAT are associated with increased mitochondrial number, and elevated respiration in gWAT. Recently, it was shown that absence of ClpP can increase respiration; for example, DARS2 mice, a mouse model of dysregulated mitochondrial translation, have a strong up‐regulation of the UPRmt and heart‐specific deletion of ClpP in DARS2 mice increases respiration 37. OPA1 is critical in regulating mitochondrial cristae structure that in turn determines respiratory efficiency, and deletion of OPA1 reduces supercomplex assembly and respiration, whereas its overexpression favors supercomplex formation and increases respiration 38, 39. It is possible that increased OPA1 protein is contributing to increased respiration in WAT of ClpP −/− mice. Increased expression of PGC‐1α and OPA1 is specific to WAT of ClpP −/− mice; however, the reason(s) for this tissue‐specific effect is not known. Thus, our findings signify a beneficial metabolic effect of increased adipose tissue metabolism due to ClpP deficiency. This was further supported by the finding that increased oxygen consumption and energy expenditure rates of ClpP −/− mice were reduced when normalized to lean rather than total body mass.
Up‐regulation of mitochondrial chaperones in ClpP −/− mice might be yet another compensatory response that will help to stabilize unfolded proteins generated due to ClpP deficiency 3. In WAT depots of ClpP −/− mice, it is difficult to differentiate up‐regulation of mitochondrial chaperones from mitochondrial biogenesis. It is possible that the increase in mitochondrial chaperones is a reflection of elevated mitochondrial number in WAT. However, in other tissues that showed an increase in mitochondrial chaperones (testis, heart, liver, and brain) mitochondrial biogenesis markers are not elevated suggesting that mitochondrial chaperones are induced in those tissues 17. Similarly, in ClpP +/− mice WAT, mitochondrial chaperones Hsp60 and HSp40 are elevated whereas expression of mitochondrial biogenesis markers in ClpP +/− mice was similar to WT mice. The transcription factors CHOP and C/EBPβ are the proposed transcription factors for mitochondrial chaperones in mammals, and they bind to the conserved regulatory element in promoters of the UPRmt‐related genes when UPRmt is initiated 40. How these transcription factors are activated to increase the expression of mitochondrial chaperones in the absence of ClpP is not known. Induction of mitochondrial chaperones in the absence of ClpP might suggest that ClpP is dispensable for mammalian UPRmt induction. One possible explanation for the accumulation of mitochondrial chaperones in ClpP −/− mice is that ClpP is the peptidase that is specifically responsible for mitochondrial chaperone turnover; therefore, absence of ClpP would lead to chaperone accumulation. This is not unlikely, because mitochondrial chaperones ClpX and Eral1 are degraded by ClpP 14, 17. Mitochondrial matrix protease Lon is yet another QC protease that showed a tissue‐specific adaptive response in ClpP −/− mice. Lon is also increased in both WAT and BAT of ClpP −/− mice, and a similar increase is previously reported in brain, but not in other tissues 17. Lon protease is mainly involved the degradation of oxidized proteins, in addition to its role in the turnover of specific mitochondrial enzymes and in the regulation of mtDNA replication 41. Whether Lon can compensate for the absence of ClpP is not known.
In our previous study in C2C12 muscle cells, we performed an acute knockdown of ClpP and found that decline in ClpP (70% down‐regulation) expression can cause mitochondrial dysfunction 13. However, in ClpP −/− mice, such an effect was not observed in skeletal muscle. A likely explanation is that loss of ClpP is a chronic effect in ClpP −/− mice that is compensated by molecular adaptations. Thus, the lack of adaptations in acute knockdown could explain the differential outcome. The finding that ClpP is critical for the initiation of UPRmt and UPRmt initiation will shift cell metabolism from respiration to glycolysis was made using C. elegans as a model organism 8, 42. In contrast, many aspects of mammalian UPRmt are less well understood, even though loss of mitochondrial proteostasis is shown to increase the expression of Hsp60 and ClpP 10, 11. The role of ClpP in the initiation of mammalian UPRmt and how UPRmt affects metabolism in mammals is not known. Recent study by Seiferling et al 37 suggests that ClpP is neither required for, nor it regulates the UPRmt in mammals. Their study demonstrated that a strong mitochondrial cardiomyopathy and diminished respiration due to DARS2 deficiency can be alleviated by the loss of ClpP. Thus, further studies are needed to understand the role of ClpP in mammalian UPRmt and UPRmt‐associated metabolic shift.
ClpP −/− mice fed ad libitum exhibited improved insulin sensitivity compared to WT mice. In contrast, glucose clearance in response to GTT in ClpP −/− mice fed ad libitum was similar to WT mice. This would suggest that the ClpP −/− mice have a lower or slower insulin release in response to the glucose challenge, and in support of this, insulin levels in ClpP −/− mice in fed state are lower than WT mice (Fig 4D). Thus, a reduction in glucose‐induced insulin secretion could be a potential reason why we do not see improved glucose clearance in chow‐fed animals. However, when fed HFD, WT mice develop glucose intolerance, and therefore, the difference between WT and ClpP −/− mice in glucose clearance becomes more obvious. Reduced fat mass could account for the improved metabolic parameters in ClpP −/− mice when fed ad libitum or HFD. Previous studies have correlated reduced fat mass with beneficial metabolic outcomes with HFD feeding 43, 44, 45. ClpP −/− mice have minimal fat deposition when fed a HFD, and interestingly, this fat accumulation occurred only in the sWAT, but not in gWAT, and it is known that visceral adipose tissue mass, not subcutaneous adipose tissue mass, correlates with the development of insulin resistance 46, 47. Importantly, HFD‐fed ClpP −/− mice retained elevated levels of PGC‐1α and increased respiration in gWAT that could contribute to increased fat utilization. In addition to reduced fat mass in ClpP −/− mice, increased expression of IRβ and GLUT4 in skeletal muscle could also contribute to improved insulin sensitivity in HFD‐fed conditions. Reduced insulin signaling is a well‐established defect in obesity‐mediated insulin resistance 48, and studies have shown that patients with insulin receptor mutation exhibit insulin resistance 49, and muscle‐specific knockout of IR can cause systemic insulin resistance in mice 50. Thus, cellular levels of IR itself, rather than downstream signaling, are critical for insulin signaling. Muscle‐specific deletion of GLUT4 in mice causes insulin resistance and glucose intolerance 51, and transgenic overexpression of GLUT4 enhances glucose tolerance in lean and obese mice 52. Adiponectin is a well‐known insulin sensitizer and increased levels of adiponectin are associated with improved insulin sensitivity, whereas a reduction in adiponectin is associated with insulin resistance condition 53. Surprisingly, ClpP −/− mice have reduced levels of circulating adiponectin when fed a LFD or HFD, suggesting that adiponectin does not contribute to the improved insulin sensitivity in ClpP −/− mice. In support of this, studies have shown that fat‐specific Tfam knockout mice have reduced levels of circulating adiponectin, yet have improved insulin sensitivity when fed a HFD 44.
The importance of WAT mitochondria in metabolism is highlighted by the fact that a reduction in mitochondrial number, respiration, or antioxidant levels is associated with metabolic disease conditions in mice and humans 54, 55. Compounds such as TZD that can stimulate mitochondrial biogenesis in WAT have beneficial metabolic outcome 56. WAT‐specific genetic manipulations targeting mitochondrial biogenesis or mitochondrial fatty acid oxidation have improved metabolic phenotype when challenged with HFD 44, 57, 58. Thus, adipose tissue mitochondria are an ideal organelle for targeting in obesity and related metabolic disorders. Generation of fat‐specific ClpP knockout mice will help to understand whether the beneficial metabolic effects in ClpP −/− mice are due to the metabolic changes specifically in WAT. In future, identification of ClpP inhibitor(s) and targeting ClpP using these inhibitor(s) to increase mitochondrial respiration in adipose tissue will make our findings translationally important that will help to compact the obesity pandemic.
Materials and Methods
Animals
All experiments were approved by the Institutional Animal Care and Use Committee at the Oklahoma Medical Research Foundation. ClpP −/− mice were generated as described previously and obtained from Georg Auburger (Goethe University Medical School, Frankfurt am Main, Germany) 17. All experiments, except HFD feeding, were performed in 5‐month‐old male ClpP −/−, ClpP +/−, and control littermates in C57BL/6 background. The mice were group housed (five animals per cage) in ventilated cages 20 ± 2°C, 12‐h/12‐h dark/light cycle and were fed ad libitum.
Quantitative magnetic resonance imaging
Body composition (fat mass and lean mass) of non‐anesthetized mice was analyzed by quantitative magnetic resonance (QMR) imaging during the light phase using QMR imaging [EchoMRI (Echo Medical Systems, Houston, TX, USA)] as described before 59.
WAT and liver histology
WAT and liver tissue were fixed in 10% formalin and embedded in paraffin. Sections (7 μm) were stained with hematoxylin and eosin (H&E), and images were visualized and captured with Nikon Element software (Nikon Inc., Melville, NY, USA). ImageJ software (NIH image) was used to quantify adipocyte cell area.
Glucose tolerance test and insulin tolerance test
For GTT, mice were given an intraperitoneal injection of 2 g/kg body weight of glucose (Sigma, St. Louis, MO, USA) after a 6‐h fast during the light cycle. For ITT, mice were given an intraperitoneal injection of 0.5 U of insulin (Novolin R; Novo Nordisk, Princeton, NJ, USA) after fasting for 5 h during the light cycle. Before injection and at indicated time points after injection, blood glucose levels were measured using a One‐Touch Ultra glucometer (Life Scan, Inc., Milpitas, CA, USA) 59.
Primary adipocyte culture
Stromal vascular fraction (SVF) from sWAT was differentiated to mature adipocytes as described before 27, 60. In brief, adipose tissue was digested with dispase II (Roche) and 1.5 U/ml collagenase D (Roche), and the SVF obtained after centrifugation and filtration was cultured in complete stromal vascular culture medium (DMEM/F12 [1:1; Invitrogen] plus glutamax, pen/strep, and 10% FBS). For adipocyte differentiation assays, SVF was plated and grown to confluency and exposed to the adipogenic cocktail (1 μM dexamethasone, 5 μg/ml insulin, 0.5 mM isobutylmethylxanthine (DMI), and 1 μM rosiglitazone) in stromal vascular culture medium, followed by addition of 5 μg/ml insulin in stromal vascular culture medium after 48 h. At day 6 of differentiation, cells were ready for analysis.
Measurement of mitochondrial respiration in differentiated adipocytes
Mitochondrial respiration in differentiated primary adipocyte cultures or 3T3‐L1 differentiated adipocytes was measured using a Seahorse Bioscience XF24 Extracellular Flux Analyzer (North Billerica, MA, USA). SVF isolated from WT, ClpP +/−, and ClpP −/− mice sWAT was seeded at a density of 20,000 cells/well kept in a 37°C incubator with 5% CO2. Once the cells reached confluency, the pre‐adipocytes were differentiated to mature adipocytes and respiration was assessed as described before. In brief, adipocytes were metabolically perturbed by the sequential injections of oligomycin, carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (FCCP), and antimycin A (1 mM, final concentration) and oxygen consumption rate (OCR) was recorded. The following measurements were made from the OCR values and normalized to protein concentration in each well: Third basal measurement indicates basal respiration; the difference between basal respiration and oligomycin‐induced respiration represents ATP‐linked respiration; the difference between oligomycin‐induced and antimycin A‐induced respirations is proton leak; the OCR after FCCP injection represents maximal respiration; and the difference between maximal respiration and basal respiration is reserve capacity 13.
Targeted quantitative proteomics
Quantitative proteomics was used to determine changes in mitochondrial enzymes in gWAT as previously described 61, 62. Briefly, 20 μg total adipose tissue homogenate from gWAT was run 1.5 cm into a 12.5% SDS–PAGE gel (Criterion, Bio‐Rad) followed by fixation and staining with GelCode Blue (Pierce). The entire lane was cut into ~1 mm3 pieces, washed, reduced with DTT, alkylated with iodoacetamide, and digested with trypsin. The peptides generated were extracted with 50% methanol/10% formic acid in water, dried, reconstituted in 1% acetic acid, and analyzed using selected reaction monitoring (SRM) with a triple quadrupole mass spectrometer (ThermoScientific TSQ Vantage) configured with a splitless capillary column HPLC system (Eksigent). Data processing was done using the program Pinpoint (ThermoScientific), which aligned the various collision‐induced dissociation reactions monitored for each peptide and determines the chromatographic peak areas. The response for each protein was taken as the total response for all peptides monitored. Changes in the relative abundance of the proteins were determined by normalization to the BSA internal standard, with confirmation by normalization to the housekeeping proteins.
Transmission electron microscopy
The electron microscopy experiment was carried out at the Oklahoma Medical Research Foundation Imaging Facility using established procedures, as described before 13.
In vivo insulin action
Mice at 5 months of age were fasted for 16 h and given an intraperitoneal injection of insulin (1 U/kg animal body weight (Novolin; Novo Nordisk) or an equal volume of saline. Ten minutes after the injection, mice were euthanized via cervical dislocation, and WAT, liver, and gastrocnemius muscle were collected and snap‐frozen in liquid nitrogen, and kept at −80°C until further analysis 59.
Oil red O staining
Differentiated adipocytes in 6‐well culture dishes were fixed with 10% formalin, washed with 60% isopropanol, and stained with oil red O. After washing with water, images were taken and the content of oil red O in each well was quantified by extracting with 100% isopropanol and measuring absorbance at 550 nm 63.
High‐fat diet feeding
Three‐ to four‐month‐old WT (n = 8/group), ClpP +/− (n = 10/group), and ClpP −/− (n = 8/group) female mice were fed a defined diet containing 10% fat diet [low fat diet (LFD), Research Diets, Cat#D12450J] or 60% kcal from fat (HFD, Research Diets, Cat#D12492). Mice were fed the diets for 10 weeks, and food consumption and body weight were monitored weekly as previously described 45. GTT and ITT were performed at 8 and 9 weeks after feeding the diets. At the end of experimental period, mice were sacrificed and tissues were snap‐frozen in liquid nitrogen and stored at −80°C until used.
Antibodies and ELISA kits
The following primary antibodies were used for Western blotting: Anti‐PGC‐1 alpha (1:1,000, ab54481, Abcam, rabbit polyclonal); Anti‐VDAC1/Porin (1:1,000, ab15895, Abcam, rabbit polyclonal); Anti‐Tfam (1:1,000, ab131607, Abcam, rabbit polyclonal); Anti‐Hsp60 (1:1,000, ab46798, Abcam, rabbit polyclonal); Anti‐Hsp40 (1:1,000, ab69402, Abcam, rabbit polyclonal); Anti‐Hsp10 (Cpn10) (1:1,000, ab53106, Abcam, rabbit polyclonal); Anti‐OPA1 (1:1,000, ab42364, Abcam, rabbit polyclonal); Anti‐UCP1 (1:1,000, ab23842, Abcam, rabbit polyclonal); Anti‐Parkin (1:500, ab77924, Abcam, mouse monoclonal); Anti‐PINK1 (1:500, ab75487, Abcam, mouse monoclonal); Phospho‐AMPKα (Thr172) (1:1,000, 2531, Cell Signaling Technology, rabbit polyclonal); AMPKα (1:1,000, 2532, Cell Signaling Technology, rabbit polyclonal); Insulin Receptor β (1:1,000, 3020, Cell Signaling Technology, mouse monoclonal); Glut4 (1:500, 2213, Cell Signaling Technology, mouse monoclonal); Phospho‐Akt (T308) (1:1,000, 9275, Cell Signaling Technology, rabbit polyclonal); Akt2 (1:1,000, 2964, Cell Signaling Technology, rabbit monoclonal); β‐Actin (1:1,000, 4970, Cell Signaling Technology, rabbit monoclonal); Anti‐CLPP (1:1,000, WH0008192M1, Sigma, mouse monoclonal); Anti‐β‐Tubulin (1:2,000, T5201, Sigma, mouse monoclonal); Anti‐CLPX (1:1,000, AP10767b, Abgent, rabbit polyclonal). ELISA kits for insulin and adiponectin were from Crystal Chem (Downers Grove, IL, USA); kits for triglyceride were from Cayman Chemical (Ann Arbor, MI, USA); and the nonesterified fatty acid (NEFA) kit was from Wako USA (Richmond, VA, USA).
Western blotting
The tissues collected during sacrifice were immediately frozen in liquid nitrogen and stored at −80°C until use. Homogenization of tissues and Western blotting was performed as previously described 59. Images were taken using a G:BOX imaging system (Syngene) and quantified using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
Detection of 4‐HNE adducts
For the detection of 4‐HNE modified proteins, equal amounts of protein (30 μg/lane) were subjected to SDS–PAGE, and separated proteins were transferred to PVDF membrane and treated with 250 mM sodium borohydride in 100 mM MOPS, pH 8.0 for 15 min to chemically reduce the adduct for antibody recognition. This was followed by washing the membrane with water, TBS‐T, blocking with 5% non‐fat milk and overnight incubation with 1:2,000 dilution of polyclonal antibody made against 4‐HNE 64. The antibody recognizes cysteine, lysine, and histidine 4‐HNE protein adducts and is highly specific to 4‐HNE derived protein adducts (gift from Luke Szweda, OMRF) 65. This was followed by incubation with anti‐rabbit IgG HRP conjugated antibody and development of the blot using enhanced chemiluminescence reagent.
Quantitative real‐time PCR
Total RNA was extracted using the RNeasy kit (Qiagen, Valencia, CA, USA) from 50 mg of frozen WAT as described before 45. First‐strand cDNA was synthesized using SuperScript II reverse transcriptase (Life Technologies, Grand Island, NY, USA), and quantitative real‐time PCR was performed with ABI Prism using Power SYBR Green PCR Master Mix with the primers (Applied Biosystems, Foster City, CA, USA). The following primers were used: UCP1 (forward‐ACTGCCACACCTCCAGTCATT, reverse‐CTTTGCCTCACTCAGGATTGG), UCP2 (forward‐GTGGTCGGAGATACCAGAGC, reverse‐GAGGTTGGCTTTCAGGAGAG) PGC‐1α (forward‐CCCTGCCATTGTTAAGACC, reverse‐TGCTGCTGTTCCTGTTTTC); Prdm16 (forward‐CAGCACGGTGAAGCCATTC, reverse‐GCGTGCATCCGCTTGTG); CIDEA (forward‐TGCTCTTCTGTATCGCCCAGT, reverse‐GCCGTGTTAAGGAATCTGCTG); Cox8b (forward‐GAACCATGAAGCCAACGACT, reverse‐GCGAAGTTCACAGTGGTTCC); β2‐microglobulin (forward‐CACTGACCGGCCTGTATGC, reverse‐GGGTGGCGTGAGTATACTTGAAT); β‐actin (forward‐AATCGTGCGTGACATCAAAGAG, reverse‐GCCATCTCCTGCTCGAAGTC); 18S (forward‐CTGAGAAACGGCTACCACATC, reverse‐CGCTCCCAAGATCCAACTAC). Calculations were performed by a comparative method () using β‐microglobulin, actin, and 18S.
mtDNA copy number
Total DNA was isolated from gWAT (50 mg) by proteinase K digestion for 16 h as described before 59. Relative mtDNA copy number was measured as described by Gispert et al 17 by determining the ratio of mtDNA target sequence (mitochondrial Cox3 gene: forward, TTTGCAGGATTCTTCTGAGC; reverse, TGAG CTCATGTAATTGAAACACC) to the expression of nuclear target sequence (Ndufv1gene: forward, CTTCCCCACTGGCCTCAAG; reverse, CCAAAACCCAGTGATCCAGC) by real‐time PCR using Power SYBR Green PCR Master Mix (Applied Biosystems). The reaction was initiated by 50°C for 2 min and then 94°C for 10 min, followed by 40 cycles of 95°C for 10 s, 60°C for 60 s, and melting curve ranging from 85 to 65°C was done. The C t for Ndufv1 was subtracted from C t for Cox3 to generate ΔC t. Relative differences in mtDNA copy number were determined using .
Mitochondrial respirometry
Mitochondrial respiration in gWAT was measured using high‐resolution respirometry in the OROBOROS Oxygraph 2K (Innsbruck, Austria) as previously described 66. In brief, gWAT was removed, weighed, and immediately placed in mitochondrial isolation buffer (0.21 M mannitol, 70 mM sucrose, 0.1 mM potassium EDTA, 1 mM EGTA, 10 mM Tris–HCl, 0.5% BSA, pH 7.4) on ice. 40–60 mg adipose tissue was cut into ~1 mm3 and loaded into the Oxygraph 2K in calibrated MiR05 assay buffer (100 mM sucrose, 60 mM potassium lactobionate, 20 mM HEPES, 10 mM KH2PO4, 3 mM MgCl2, 0.5 mM EGTA, 0.1% BSA, pH 7.1). Mitochondrial function was assessed using sequential additions of 10 mM glutamate, 2 mM malate, 2.5 mM ADP, 10 mM succinate, 5 μM cytochrome c, 0.5 μM rotenone, 2.5 μM oligomycin, and 5 μM Antimycin A. Data were normalized using the Complex III inhibitor Antimycin A to account for non‐mitochondrial respiration and tissue mass.
Cell culture and generation of a stable ClpP‐deficient cell line
The mouse cell line 3T3‐L1 (ATCC) was grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum and 1% penicillin–streptavidin and maintained at 37°C in 5% humidified incubator. For generation of the stable cell line, 3T3‐L1 cells were infected with mission shRNA lentiviral transduction particles for ClpP (Sigma) or shRNA control transduction particles and transduced cells were obtained by puromycin selection. 3T3‐L1 cells were differentiated to adipocytes by growing them in DMEM containing 2% horse serum and 1% penicillin–streptavidin for 4 days.
Immunofluorescence
Primary cultures of differentiated adipocytes were stained with Tom20 antibody followed by Alexa Fluor® 488 F(ab′)2 Fragment of Goat Anti‐Rabbit IgG (H + L) antibody as per manufacturer's instructions. Cell fluorescence was measured using the NIH ImageJ software. Corrected total cell fluorescence (CTCF) was calculated as CTCF = Integrated Density − (Area of selected cell × Mean fluorescence of background readings).
Indirect calorimetry
Energy expenditure was measured by indirect calorimetry in WT, ClpP +/−, and ClpP −/− mice. Animals were acclimated to the testing cages and metabolic cabinet for 48 h before data collection with ad libitum access to food and water. After acclimation, oxygen consumption and carbon dioxide production were measured using a multiple animal respirometry system (MARS) (Sable Systems, Las Vegas, NV, USA). 10‐min/animal averages were collected hourly over a continuous 20‐h period. The respiratory exchange ratio was calculated as the ratio of the average carbon dioxide produced to oxygen consumed over this time period. Energy expenditure was calculated as described by 67 using the caloric equivalent of oxygen. Data were initially subdivided into the light or dark phase time periods to evaluate potential differences in metabolic responses associated with different activity levels. Light and dark phase values were then averaged to report results. Rates of energy expenditure were normalized to either total body mass or lean body mass as determined by dual‐energy X‐ray absorptiometry analysis.
Statistics
Ordinary one‐way ANOVA or two‐way ANOVA with Tukey's post hoc test was used to analyze data.
Author contributions
SB and SSD designed and conducted the experiments, analyzed data, and prepared figures. SSD wrote the manuscript and SB helped with the manuscript preparation and statistical analysis. GP performed OROBOROS Oxygraph 2K experiments. RR, AM, and BF performed animal husbandry maintenance and assistance to diet feeding and QMR. MK performed targeted quantitative proteomic analysis, TMG performed indirect calorimetry using a multiple animal respirometry system, and SM and KMH helped with mitochondrial measurements. BCN provided suggestions for in vitro knockdown studies and gave critical suggestions for the manuscript. SG and GA provided the ClpP −/− mice and gave comments for the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Review Process File
Acknowledgements
This work was supported by American Heart Association Beginning Grant‐in‐Aid Grant [13BGIA14670024], American Federation for Aging Research Grant [A13415], and NIH COBRE Expanding Excellence in Developmental Biology Pilot Projects [5 P20 GM103636‐02 and 5 P20 GM103636‐03] to S.S.D. Imaging Core Facility, Oklahoma Medical Research Foundation is acknowledged for histology and electron microscopy experiments.
EMBO Reports (2018) 19: e45009
References
- 1. Wallace DC, Fan W, Procaccio V (2010) Mitochondrial energetics and therapeutics. Annu Rev Pathol 5: 297–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baker BM, Haynes CM (2011) Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci 36: 254–261 [DOI] [PubMed] [Google Scholar]
- 3. Voos W, Jaworek W, Wilkening A, Bruderek M (2016) Protein quality control at the mitochondrion. Essays Biochem 60: 213–225 [DOI] [PubMed] [Google Scholar]
- 4. Luce K, Weil AC, Osiewacz HD (2010) Mitochondrial protein quality control systems in aging and disease. Adv Exp Med Biol 694: 108–125 [DOI] [PubMed] [Google Scholar]
- 5. Jensen MB, Jasper H (2014) Mitochondrial proteostasis in the control of aging and longevity. Cell Metab 20: 214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Civitarese AE, MacLean PS, Carling S, Kerr‐Bayles L, McMillan RP, Pierce A, Becker TC, Moro C, Finlayson J, Lefort N et al (2010) Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell Metab 11: 412–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quiros PM, Ramsay AJ, Sala D, Fernandez‐Vizarra E, Rodriguez F, Peinado JR, Fernandez‐Garcia MS, Vega JA, Enriquez JA, Zorzano A et al (2012) Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J 31: 2117–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans . Dev Cell 13: 467–480 [DOI] [PubMed] [Google Scholar]
- 9. Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D (2010) The matrix peptide exporter HAF‐1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans . Mol Cell 37: 529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ (2002) A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497: 451–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, Hoogenraad N, Sans M, Sartor RB, Haller D (2012) Induction of dsRNA‐activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 61: 1269–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deepa SS, Bhaskaran S, Ranjit R, Qaisar R, Nair BC, Liu Y, Walsh ME, Fok WC, Van Remmen H (2016) Down‐regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic Biol Med 91: 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Szczepanowska K, Maiti P, Kukat A, Hofsetz E, Nolte H, Senft K, Becker C, Ruzzenente B, Hornig‐Do HT, Wibom R et al (2016) CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J 35: 2566–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jenkinson EM, Rehman AU, Walsh T, Clayton‐Smith J, Lee K, Morell RJ, Drummond MC, Khan SN, Naeem MA, Rauf B et al (2013) Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP‐dependent chambered protease. Am J Hum Genet 92: 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jukarainen S, Heinonen S, Ramo JT, Rinnankoski‐Tuikka R, Rappou E, Tummers M, Muniandy M, Hakkarainen A, Lundbom J, Lundbom N et al (2016) Obesity is associated with low NAD(+)/SIRT pathway expression in adipose tissue of BMI‐discordant monozygotic twins. J Clin Endocrinol Metab 101: 275–283 [DOI] [PubMed] [Google Scholar]
- 17. Gispert S, Parganlija D, Klinkenberg M, Drose S, Wittig I, Mittelbronn M, Grzmil P, Koob S, Hamann A, Walter M et al (2013) Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum Mol Genet 22: 4871–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Irrcher I, Ljubicic V, Hood DA (2009) Interactions between ROS and AMP kinase activity in the regulation of PGC‐1alpha transcription in skeletal muscle cells. Am J Physiol Cell Physiol 296: C116–C123 [DOI] [PubMed] [Google Scholar]
- 19. Silveira LR, Pilegaard H, Kusuhara K, Curi R, Hellsten Y (2006) The contraction induced increase in gene expression of peroxisome proliferator‐activated receptor (PPAR)‐gamma coactivator 1alpha (PGC‐1alpha), mitochondrial uncoupling protein 3 (UCP3) and hexokinase II (HKII) in primary rat skeletal muscle cells is dependent on reactive oxygen species. Biochim Biophys Acta 1763: 969–976 [DOI] [PubMed] [Google Scholar]
- 20. St‐Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W et al (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC‐1 transcriptional coactivators. Cell 127: 397–408 [DOI] [PubMed] [Google Scholar]
- 21. Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M (2009) Antioxidants prevent health‐promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 106: 8665–8670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andringa KK, Udoh US, Landar A, Bailey SM (2014) Proteomic analysis of 4‐hydroxynonenal (4‐HNE) modified proteins in liver mitochondria from chronic ethanol‐fed rats. Redox Biol 2: 1038–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i‐AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204: 919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duvezin‐Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG et al (2006) Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem 281: 37972–37979 [DOI] [PubMed] [Google Scholar]
- 25. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu J, Cohen P, Spiegelman BM (2013) Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev 27: 234–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos‐Flier E et al (2012) FGF21 regulates PGC‐1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 26: 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Montgomery MK, Turner N (2015) Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect 4: R1–R15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80: 315–360 [DOI] [PubMed] [Google Scholar]
- 30. Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C et al (2012) MitoNEET‐driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med 18: 1539–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kasashima K, Sumitani M, Endo H (2012) Maintenance of mitochondrial genome distribution by mitochondrial AAA+ protein ClpX. Exp Cell Res 318: 2335–2343 [DOI] [PubMed] [Google Scholar]
- 32. Tiraby C, Tavernier G, Lefort C, Larrouy D, Bouillaud F, Ricquier D, Langin D (2003) Acquirement of brown fat cell features by human white adipocytes. J Biol Chem 278: 33370–33376 [DOI] [PubMed] [Google Scholar]
- 33. Fisler JS, Warden CH (2006) Uncoupling proteins, dietary fat and the metabolic syndrome. Nutr Metab (Lond) 3: 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahadik SR, Lele RD, Saranath D, Seth A, Parikh V (2012) Uncoupling protein‐2 (UCP2) gene expression in subcutaneous and omental adipose tissue of Asian Indians: relationship to adiponectin and parameters of metabolic syndrome. Adipocyte 1: 101–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Horvath TL, Diano S, Miyamoto S, Barry S, Gatti S, Alberati D, Livak F, Lombardi A, Moreno M, Goglia F et al (2003) Uncoupling proteins‐2 and 3 influence obesity and inflammation in transgenic mice. Int J Obes Relat Metab Disord 27: 433–442 [DOI] [PubMed] [Google Scholar]
- 36. Souza BM, Assmann TS, Kliemann LM, Gross JL, Canani LH, Crispim D (2011) The role of uncoupling protein 2 (UCP2) on the development of type 2 diabetes mellitus and its chronic complications. Arq Bras Endocrinol Metabol 55: 239–248 [DOI] [PubMed] [Google Scholar]
- 37. Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, Konig T, Kukat A, Trifunovic A (2016) Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep 17: 953–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana‐Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC et al (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155: 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee H, Smith SB, Yoon Y (2017) The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem 292: 7115–7130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aldridge JE, Horibe T, Hoogenraad NJ (2007) Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2: e874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bota DA, Davies KJ (2016) Mitochondrial Lon protease in human disease and aging: including an etiologic classification of Lon‐related diseases and disorders. Free Radic Biol Med 100: 188–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM (2012) Mitochondrial import efficiency of ATFS‐1 regulates mitochondrial UPR activation. Science 337: 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bluher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB, Kahn CR (2002) Adipose tissue selective insulin receptor knockout protects against obesity and obesity‐related glucose intolerance. Dev Cell 3: 25–38 [DOI] [PubMed] [Google Scholar]
- 44. Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Rardin MJ, An D, Lee KY, Ilkayeva OR, Zingaretti CM et al (2012) Adipose‐specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab 16: 765–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bhaskaran S, Unnikrishnan A, Ranjit R, Qaisar R, Pharaoh G, Matyi S, Kinter M, Deepa SS (2017) A fish oil diet induces mitochondrial uncoupling and mitochondrial unfolded protein response in epididymal white adipose tissue of mice. Free Radic Biol Med 108: 704–714 [DOI] [PubMed] [Google Scholar]
- 46. Wajchenberg BL (2000) Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev 21: 697–738 [DOI] [PubMed] [Google Scholar]
- 47. Yang YK, Chen M, Clements RH, Abrams GA, Aprahamian CJ, Harmon CM (2008) Human mesenteric adipose tissue plays unique role versus subcutaneous and omental fat in obesity related diabetes. Cell Physiol Biochem 22: 531–538 [DOI] [PubMed] [Google Scholar]
- 48. Boucher J, Kleinridders A, Kahn CR (2014) Insulin receptor signaling in normal and insulin‐resistant states. Cold Spring Harb Perspect Biol 6: a009191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krook A, O'Rahilly S (1996) Mutant insulin receptors in syndromes of insulin resistance. Baillieres Clin Endocrinol Metab 10: 97–122 [DOI] [PubMed] [Google Scholar]
- 50. Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR (1998) A muscle‐specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2: 559–569 [DOI] [PubMed] [Google Scholar]
- 51. Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais‐Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ et al (2000) Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med 6: 924–928 [DOI] [PubMed] [Google Scholar]
- 52. Atkinson BJ, Griesel BA, King CD, Josey MA, Olson AL (2013) Moderate GLUT4 overexpression improves insulin sensitivity and fasting triglyceridemia in high‐fat diet‐fed transgenic mice. Diabetes 62: 2249–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116: 1784–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heinonen S, Buzkova J, Muniandy M, Kaksonen R, Ollikainen M, Ismail K, Hakkarainen A, Lundbom J, Lundbom N, Vuolteenaho K et al (2015) Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes 64: 3135–3145 [DOI] [PubMed] [Google Scholar]
- 55. Chattopadhyay M, Khemka VK, Chatterjee G, Ganguly A, Mukhopadhyay S, Chakrabarti S (2015) Enhanced ROS production and oxidative damage in subcutaneous white adipose tissue mitochondria in obese and type 2 diabetes subjects. Mol Cell Biochem 399: 95–103 [DOI] [PubMed] [Google Scholar]
- 56. Wilson‐Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S (2004) Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 114: 1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kusminski CM, Park J, Scherer PE (2014) MitoNEET‐mediated effects on browning of white adipose tissue. Nat Commun 5: 3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kusminski CM, Scherer PE (2012) Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol Metab 23: 435–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Deepa SS, Pulliam D, Hill S, Shi Y, Walsh ME, Salmon A, Sloane L, Zhang N, Zeviani M, Viscomi C et al (2013) Improved insulin sensitivity associated with reduced mitochondrial complex IV assembly and activity. FASEB J 27: 1371–1380 [DOI] [PubMed] [Google Scholar]
- 60. Aune UL, Ruiz L, Kajimura S (2013) Isolation and differentiation of stromal vascular cells to beige/brite cells. J Vis Exp 73: e50191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fernandes J, Weddle A, Kinter CS, Humphries KM, Mather T, Szweda LI, Kinter M (2015) Lysine acetylation activates mitochondrial aconitase in the heart. Biochemistry 54: 4008–4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kinter CS, Lundie JM, Patel H, Rindler PM, Szweda LI, Kinter M (2012) A quantitative proteomic profile of the Nrf2‐mediated antioxidant response of macrophages to oxidized LDL determined by multiplexed selected reaction monitoring. PLoS One 7: e50016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sato T, Kotake D, Hiratsuka M, Hirasawa N (2013) Enhancement of inflammatory protein expression and nuclear factor Kappab (NF‐Kappab) activity by trichostatin A (TSA) in OP9 preadipocytes. PLoS One 8: e59702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Uchida K, Szweda LI, Chae HZ, Stadtman ER (1993) Immunochemical detection of 4‐hydroxynonenal protein adducts in oxidized hepatocytes. Proc Natl Acad Sci USA 90: 8742–8746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Uchida K, Itakura K, Kawakishi S, Hiai H, Toyokuni S, Stadtman ER (1995) Characterization of epitopes recognized by 4‐hydroxy‐2‐nonenal specific antibodies. Arch Biochem Biophys 324: 241–248 [DOI] [PubMed] [Google Scholar]
- 66. Mancuso DJ, Sims HF, Yang K, Kiebish MA, Su X, Jenkins CM, Guan S, Moon SH, Pietka T, Nassir F et al (2010) Genetic ablation of calcium‐independent phospholipase A2gamma prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J Biol Chem 285: 36495–36510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tschop MH, Speakman JR, Arch JR, Auwerx J, Bruning JC, Chan L, Eckel RH, Farese RV Jr, Galgani JE, Hambly C et al (2011) A guide to analysis of mouse energy metabolism. Nat Methods 9: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Review Process File