

Abstract
Tyrosyl‐DNA phosphodiesterase 2 (TDP2) repairs abortive topoisomerase II cleavage complexes. Here, we identify a novel short isoform of TDP2 (TDP2S) expressed from an alternative transcription start site. TDP2S contains a mitochondrial targeting sequence, contributing to its enrichment in the mitochondria and cytosol, while full‐length TDP2 contains a nuclear localization signal and the ubiquitin‐associated domain in the N‐terminus. Our study reveals that both TDP2 isoforms are present and active in the mitochondria. Comparison of isogenic wild‐type (WT) and TDP2 knockout (TDP2 −/−/−) DT40 cells shows that TDP2 −/−/− cells are hypersensitive to mitochondrial‐targeted doxorubicin (mtDox), and that complementing TDP2 −/−/− cells with human TDP2 restores resistance to mtDox. Furthermore, mtDox selectively depletes mitochondrial DNA in TDP2 −/−/− cells. Using CRISPR‐engineered human cells expressing only the TDP2S isoform, we show that TDP2S also protects human cells against mtDox. Finally, lack of TDP2 in the mitochondria reduces the mitochondria transcription levels in two different human cell lines. In addition to identifying a novel TDP2S isoform, our report demonstrates the presence and importance of both TDP2 isoforms in the mitochondria.
Keywords: DNA repair, mitochondria, mitochondrial DNA, topoisomerase
Subject Categories: DNA Replication, Repair & Recombination
Introduction
Type IIA topoisomerases (TOP2α and TOP2β) are essential for decatenating DNA and removing DNA torsional stress generated by transcription, replication, and chromosomal segregation 1, 2. All these reactions are carried out by the formation of transient DNA double‐strand breaks that are covalently linked to the catalytic tyrosine residues of TOP2 homodimers 1, 2. The transient covalent DNA‐TOP2 complexes are termed TOP2 cleavage complexes (TOP2cc). Endogenous DNA alterations and anti‐cancer drugs, including doxorubicin and etoposide, trap TOP2α and TOP2β cleavage complexes 3, 4, 5 and extend the normally very short half‐lives of TOP2cc 1, 2, 4, 5, 6, 7. If the TOP2‐generated DNA breaks fail to reseal, the abortive TOP2 protein adducts require specific repair carried out by tyrosyl‐DNA phosphodiesterase 2 (TDP2) 8. TDP2 can also act on TOP2 trapped on RNA 9 and is known to process replication intermediates for a broad range of viruses (e.g., picornavirus Vpg unlinkase 10 and hepatitis B virus 11).
A recent study revealed that TDP2 deficiency in humans leads to intellectual disability, epilepsy and ataxia, and that Tdp2 knockout mice have significantly reduced interneuron density in the molecular layer of the cerebellum 12. The same study employed an inducible transcription system to show that TDP2 is required for normal TOP2‐dependent transcription, indicating that normal transcription generates abortive TOP2cc 13. Genome‐wide expression analysis demonstrated that lack of TDP2 in brain tissues leads to downregulation of many genes critical for nervous system development and function 12. These results establish that endogenous levels of TOP2cc are significant, and the removal of such TOP2cc by TDP2 is critical for normal cellular functions.
In addition to their nuclear localization, TOP2α and TOP2β localize to mitochondria 14, implying that specific repair mechanisms might be required to remove TOP2cc from mitochondrial DNA (mtDNA). Furthermore, DNA alterations generated by reactive oxidative species in mitochondria and unrepaired ribonucleotides in mtDNA are both known to enhance the trapping of TOP2cc 6, 9, 15. Here, we demonstrate that full‐length vertebrate TDP2 is present not only in the nucleus but also in mitochondria. We reveal that a short isoform of TDP2, which we term TDP2S, is expressed in human and mouse cells and is excluded from the nucleus and selectively targeted to mitochondria and cytosol. We also show that both TDP2 isoforms in mitochondria protect against detrimental effects of mitochondrial TOP2cc and are important for mitochondrial transcription.
Results
Two isoforms of TDP2: TDP2 and TDP2S with different N‐terminal sequences
Immunoblotting analysis consistently reveals two bands for TDP2 in different cell lines (a panel of human lung cancer cells is shown in Fig 1A). Indeed, in addition to the higher band, which corresponds to the reported full‐length TDP2 (362 a.a.; ~41 kDa but running at ~48 kDa) 8, we consistently observe a second band migrating at ~37 kDa (Fig 1A).
Figure 1. A short isoform of TDP2, TDP2S, arises from an alternative transcription start site.
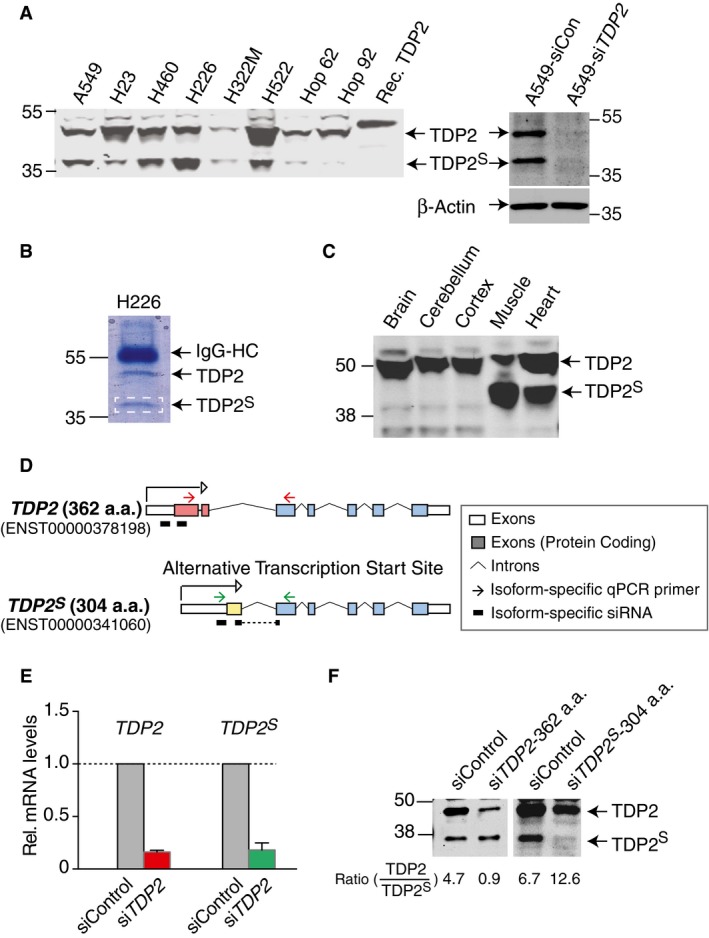
- Left: Representative TDP2 immunoblot of whole cell lysates from a panel of lung cancer cell lines (106 cells loaded per lane). Recombinant TDP2 bearing a His6‐tag was loaded as control. Right: TDP2 immunoblot of whole cell lysates from A549 cells (25 μg per lane) treated with either control siRNA (siCon) or a cocktail of siRNA targeting TDP2 (siTDP2); β‐actin immunoblot serves as loading control.
- Coomassie blue staining of proteins from TDP2 pull‐down in H226 whole cell lysate, resolved on a 10% SDS–PAGE. The band at 37 kDa was excised (marked by the white dashed box) and subjected to tandem mass spectrometry analysis (detected peptide sequences listed in red in Appendix Fig S1A).
- Representative TDP2 immunoblot of murine tissues (20 μg per lane).
- Scheme showing the two transcripts of TDP2. Protein‐coding exons unique to full‐length TDP2 are marked by red boxes, protein‐coding exon unique to TDP2 S is marked by a yellow box, and other protein‐coding exons shared by both transcripts are marked by blue boxes. Locations of isoform‐specific qPCR primers for TDP2 and TDP2 S are marked by red and green arrows, respectively. Locations of isoform‐specific siRNA are marked by black bars underneath each transcript.
- Quantitative real‐time PCR using isoform‐specific primers for TDP2 and TDP2 S shows that the levels of both transcripts are reduced in response to siTDP2 treatment for 48 h in A549 cells. Average of three independent experiments is shown (error bars represent SEM).
- Representative TDP2 immunoblot of whole cell lysates from A549 cells (25 μg per lane) either treated with control siRNA (siCon) or siRNA specifically targeting each TDP2 isoform (siTDP2‐362 a.a. or siTDP2 S‐304 a.a.). The ratio of TDP2 to TDP2S is quantified using Li‐COR and shown below each sample.
Source data are available online for this figure.
To determine whether the two bands reflect different TDP2 isoforms, we knocked down TDP2 using a cocktail of 4 siRNA targeting the portion of transcript encoding the catalytic domain of TDP2 (siTDP2). Treatment with siTDP2 eliminates both bands (Fig 1A, right panel), consistent with the possibility that the two bands correspond to two different isoforms of TDP2. We refer to the previously reported long isoform as TDP2 8 and the 37‐kDa isoform as TDP2‐short (TDP2S).
Immunoprecipitation with antibody against TDP2 in H226 lung cancer cells, in which TDP2S is particularly abundant (see Fig 1A), pulled down both isoforms (Fig 1B), and mass spectrometry identified the 37‐kDa protein as TDP2 (58% coverage, 12 unique peptides, Appendix Fig S1). We extended the immunoblotting analysis to different murine tissues, where the muscle and heart tissues show particularly high expression of both TDP2 isoforms (Fig 1C).
We next queried the human genome databank for reported alternative TDP2 transcripts. One previously uncharacterized transcript resulting from an alternative transcription start site is predicted to contain 304 a.a. residues with a MW of 35 kDa, consistent with the observed MW of TDP2S. The reported alternative transcript contains a unique protein‐coding first exon distinctive from the full‐length TDP2 transcript (Fig 1D). We designed isoform‐specific PCR primers for the two TDP2 isoform transcripts (primers pairs are marked by arrows above each transcript in Fig 1D) and confirmed their specificity for each TDP2 isoform (Appendix Fig S2A). Using these isoform‐specific PCR primers, we find that siTDP2 treatments attenuate the levels of both transcripts in quantitative real‐time PCR analysis (Fig 1E). To confirm that the alternative transcript encodes TDP2S, we then designed isoform‐specific siRNA targeting sequences within the unique regions of each transcript (marked by black bars below each transcript in Fig 1D) and analyzed the TDP2 isoform expression levels in cells transfected with each isoform‐specific siRNA. The siRNA specific for the canonical full‐length TDP2 selectively reduces the upper TDP2 band, and the siRNA specific for the alternative transcript nearly abolishes the lower TDP2S band (Fig 1F), demonstrating that TDP2S is encoded by the variant transcript (ENST00000341060) with an alternative transcription start site. The identity of the transcript for TDP2 S is further confirmed by Sanger sequencing of the resulting PCR product, where the exon–exon junction unique to the novel transcript is unambiguously resolved (Appendix Fig S2B).
We next examined RNA sequencing data from the panel of lung cancer cells tested in Fig 1A. The reads mapped to the TDP2 locus were fitted to the transcripts for two TDP2 isoforms to compare their relative expression levels in each cell line (Appendix Fig S2B). Transcripts for both TDP2 isoforms display a large dynamic range, and the transcript level of TDP2 S tends to be lower than full‐length TDP2. The relative abundance of TDP2 and TDP2S protein obtained from immunoblotting analysis (see Fig 1A) does not correlate with relative transcript levels in different cells, suggesting that additional downstream mechanisms are involved in governing the steady‐state levels of the two TDP2 isoforms.
Both TDP2 isoforms localize to mitochondria in human cells
Prediction software for peptide localization revealed that the full‐length TDP2 contains a nuclear localization sequence (NLS) 16, whereas the alternative isoform TDP2S includes a potential mitochondrial targeting sequence (MTS) (Fig 2A and full sequence in Appendix Fig S1).
Figure 2. Both TDP2 and TDP2S localize to human mitochondria.
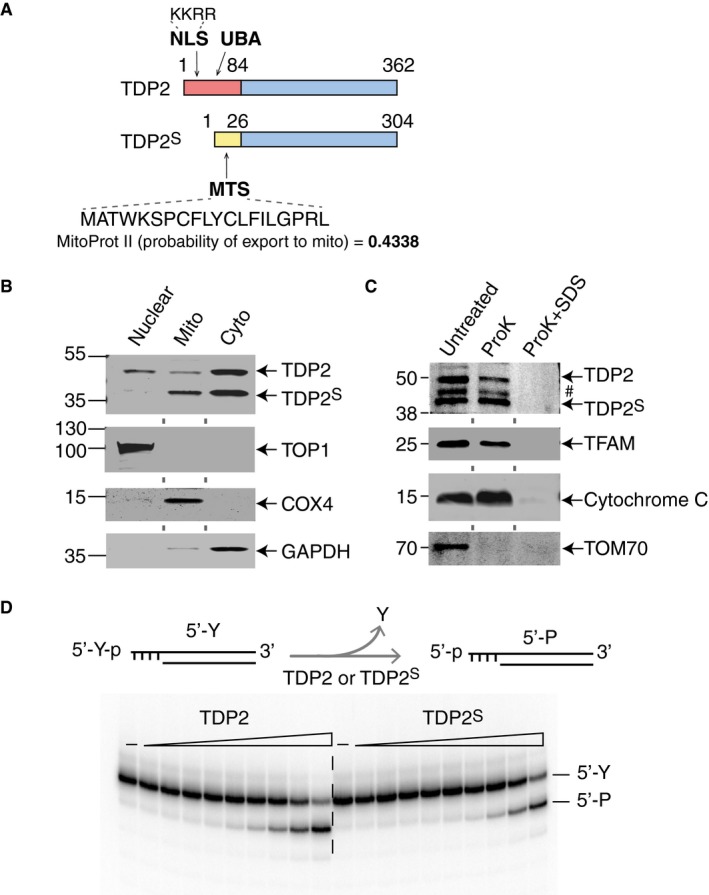
- Schematic representations of TDP2 and TDP2S protein isoforms (full amino acid sequences are shown in Appendix Fig S1). The color scheme is the same as in Fig 1D. NLS and MTS are the nuclear localization and mitochondrial targeting sequences, respectively. UBA is the ubiquitin‐associated domain.
- Representative immunoblots of H226 cell fractions (50 μg nuclear extract and 30 μg mitochondrial and cytosolic extracts). Upper panel probes for TDP2, and the lower panels probe for fractionation markers, TOP1, COX4, and GAPDH.
- Proteinase K protection assay of purified mitochondria from H226 cells. Upper panel probes for TDP2 (#: non‐specific band). Lower panels probe for TFAM, cytochrome C, and TOM70, which are fractionation controls for mitochondria.
- Representative biochemical assay for 5′‐phosphotyrosine cleavage activities of both recombinant TDP2 and TDP2S. Double‐strand DNA substrate bearing a 5′‐tyrosine (5′‐Y) on a four base overhang is radiolabeled on the 3′‐end and incubated with increasing concentrations of recombinant TDP2 or TDP2S (1:3 serial dilutions ranging from 3 pM to 18 μM) for 2 h. 80 μM of cold single‐strand DNA is included as competitors to reduce non‐specific degradation. The rates of DNA product (5′‐P, with a 5′‐phosphate end) generation by both isoforms are comparable.
Source data are available online for this figure.
To examine the cellular localization of TDP2 and TDP2S, we carried out cellular fractionation followed by immunoblotting analysis for TDP2 in H226 lung cancer cells. Our results show that full‐length TDP2 is present in the mitochondrial and cytosolic fractions in addition to the nuclear fraction (Fig 2B). They also show that TDP2S is present in both the mitochondrial and cytosolic fractions, with particular enrichment in mitochondria (Fig 2B), consistent with the software prediction that TDP2S bears a MTS. Immunoblotting for TOP1, COX4, and GAPDH were used as markers for nuclear, mitochondrial and cytosolic fractions, respectively (Fig 2B). All samples were treated with 0.1 mg/ml digitonin prior to fractionation to minimize contamination of mitochondrial fraction from non‐mitochondrial proteins.
Next, we subjected purified intact mitochondria to proteinase K (ProK) treatment in the presence or absence of SDS to determine the presence of TDP2 and TDP2S inside mitochondria (Fig 2C). In this assay, proteins within the mitochondrial membranes are resistant to ProK digestion, and disruption of the mitochondrial membrane by SDS renders them sensitive to ProK. The representative immunoblot shown in Fig 2C shows that both TDP2 isoforms in mitochondria are resistant to ProK digestion in the absence of SDS. We probed the same fraction with a compartmental marker, TOM70, which is degraded by ProK in the absence of SDS, consistent with its location on the outer membrane of mitochondria. Cytochrome C and TFAM, which served as protein markers for intermembrane space and matrix of mitochondria, respectively, are resistant to ProK digestion in the absence of SDS. These results show that both TDP2S and full‐length TDP2 are present inside human mitochondria.
We also examined the cellular localization of TDP2 and TDP2S in two other human cell lines, kidney embryo HEK293T and colon carcinoma HCT116 cells. We included control cells treated with siTDP2 to unambiguously identify both TDP2 isoforms in our immunoblotting analysis. HEK293T cells show both isoforms in whole cell extract (WCE) and only full‐length TDP2 with no detectable TDP2S in the nuclear fractions (Appendix Fig S3A). By contrast, both TDP2 isoforms are present in the mitochondrial and cytosolic fractions and are downregulated by siTDP2 (Appendix Fig S3A), consistent with results from H226 cells. HCT116 cells express the lowest level of TDP2S in the cell lines examined and show strong signal for full‐length TDP2 in both the mitochondrial and nuclear fractions, while a weak band for TDP2S is visible only in the mitochondrial fraction (Appendix Fig S3B), consistent with the enrichment of TDP2S in the mitochondria observed for the other human cell lines.
Next, we overexpressed and purified recombinant TDP2 and TDP2S and compared their biochemical activities. TDP2 specifically cleaves off the tyrosine residue covalently linked to the 5′‐end of the DNA and leaves a phosphate group (scheme in Fig 2D) 8, 9, 17. Judging from the apparent rate of 5′‐phosphotyrosyl cleavage, recombinant TDP2 and TDP2S show comparable activity (lower panel in Fig 2D), demonstrating that the N‐terminal leading sequences do not impact the biochemical activity of the TDP2 isoforms, consistent with prior biochemical analyses of N‐terminus‐truncated TDP2 (a.a. 110–362) 18, 19, 20. Based on these results, we conclude that TDP2S contains a different N‐terminal leading sequence that leads to its localization to the mitochondrial and cytosolic fractions while maintaining similar biochemical activity as full‐length TDP2.
TDP2S contains a mitochondrial targeting sequence
To establish the role of the N‐terminal leading sequences in the cellular localization of TDP2 and TDP2S, we made a series of GFP‐tagged protein constructs. Fluorescence confocal microscopy shows that full‐length TDP2 predominately localizes to the nucleus with limited signals in the cytosol (Fig 3, top row). Mutating the predicted NLS in TDP2 (a.a. 23–26: KKRR) results in loss of nuclear localization with TDP2 becoming diffusely distributed throughout the cell (Fig 3, 2nd row) 16. By contrast, TDP2S displays a cytosolic distribution pattern, where it partially co‐localizes with MitoTracker (Fig 3, 3rd and 4th rows). Furthermore, a peptide bearing only the predicted MTS of TDP2S (MitoProt II, a.a. 1–20) localizes exclusively to the mitochondria (Fig 3, 5th row), while removing the MTS from TDP2S (a.a. 21–304) gives rise to a diffused pattern throughout the cell (Fig 3, 6th row). The difference between the distribution patterns of TDP2S and its MTS peptide suggests that additional competing localization signals may be present elsewhere in TDP2S, leading to its dual‐localization to the mitochondrial and cytosolic compartments. Fluorescence microscopy images of HCT116 cells, which express low TDP2S isoform (Appendix Fig S3), show mainly nuclear distribution with limited foci in the cytoplasm that partially colocalized with mitochondrial oxidative phosphorylation (OXPHOS) complexes (Appendix Fig S4).
Figure 3. TDP2S bears a mitochondrial targeting sequence.
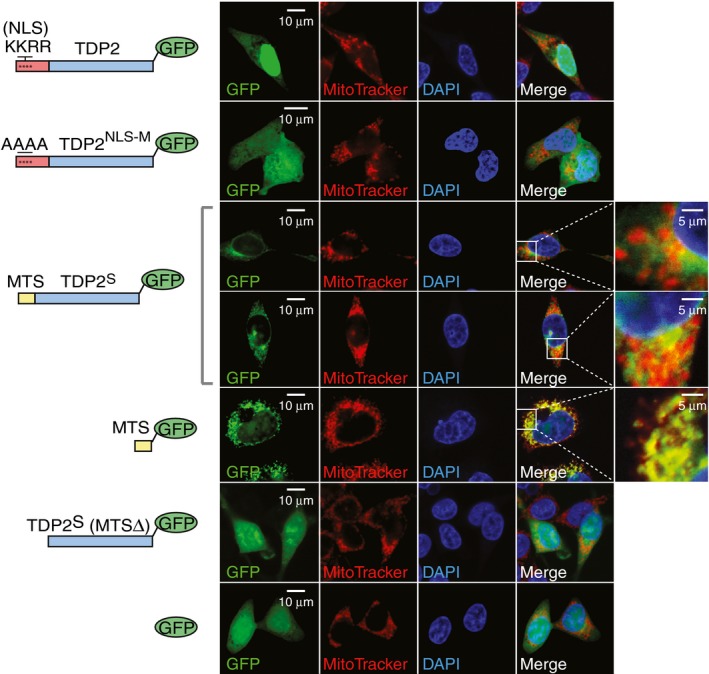
Fluorescence microscopy images of HCT116 cells transfected with the indicated TDP2 constructs tagged with C‐terminal GFP (schemes at left of each panel). Columns from left to right show GFP (green), MitoTracker® Deep Red (red), DAPI (blue) and merged images with selective zoomed‐in fields. NLS is the nuclear localization sequence, NLS‐M is the nuclear localization sequence‐mutated, MTS is the mitochondrial targeting sequence, and TDP2S (MTSΔ) is TDP2S lacking the N‐terminal MTS.
Combined, these results show that the different N‐terminal leading sequences in the TDP2 isoforms direct their distinctly different cellular localization patterns. The highly expressed full‐length TDP2 contains a NLS, making it the primary nuclear isoform. In addition, limited yet detectable amount of full‐length TDP2 localizes to the mitochondria, joining a long list of mitochondrial proteins without well‐defined MTS‐dependent translocation mechanism. Expressed at lower levels, TDP2S lacks the NLS and contains a MTS, which excludes it from the nucleus and drives its localization to the mitochondria and cytosol.
TDP2 is present and active in mitochondria of chicken DT40 cells
To examine TDP2 activity in mitochondria, we performed genetic experiments by complementing the previously generated TDP2 knockout (TDP2 −/−/−) DT40 cells 16, 21 with full‐length human TDP2 (hTDP2 cells). We first verified that TDP2 is present in the mitochondria of DT40 cells. Immunoblotting shows the presence of TDP2 in both the mitochondrial and nuclear fractions of hTDP2 cells (Fig 4A, Lanes 2 and 5), consistent with human cells. Note that chicken TDP2 protein in WT DT40 cells is not detected with the available anti‐TDP2 antibody (Fig 4A, Lanes 4 and 7). As a marker, recombinant TDP2 was loaded in the same gel (Fig 4A, Lane 1); the different tags (His6‐tag on recombinant TDP2 vs. FLAG‐tag on the complementation TDP2 construct in hTDP2 cells) likely accounts for the slight difference in migration (Fig 4A, Lanes 1 and 2). TOP1 and COX4 serve as controls to demonstrate the lack of contamination between the mitochondrial and nuclear fractions. Consistent with our results in human cells (Fig 2C), TDP2 in intact hTDP2 DT40 mitochondria is resistant to ProK digestion (Fig 4B), confirming that human TDP2 localizes within mitochondria in DT40 cells.
Figure 4. Recomplemented human TDP2 localizes to the mitochondria of DT40 cells and protects against the mitochondria‐specific TOP2 poison, mtDox.
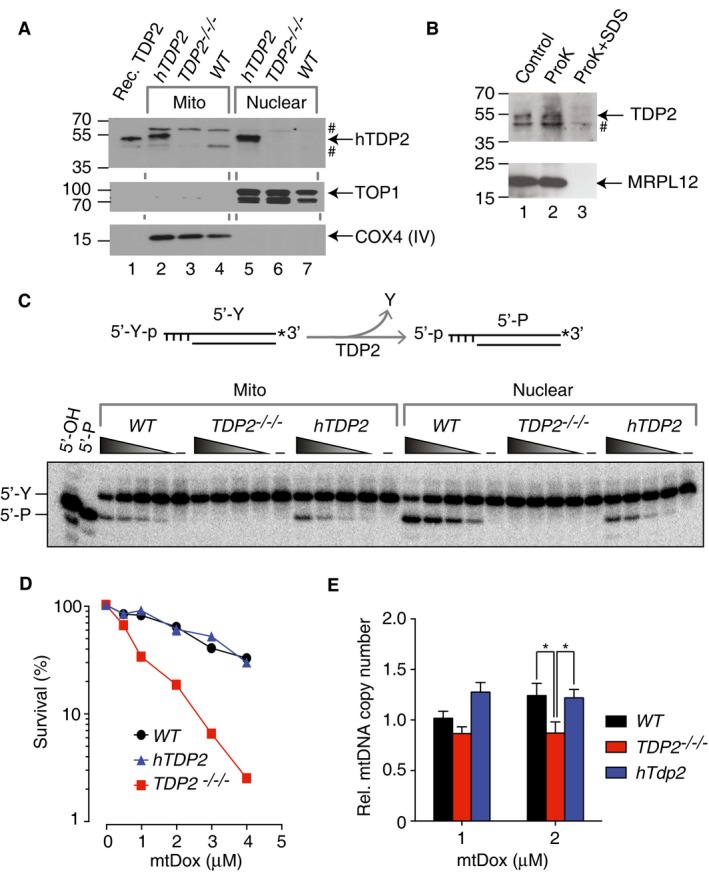
- Representative immunoblots of mitochondrial (Lanes 2–4) and nuclear extracts (Lanes 5–7) from wild‐type (WT), TDP2 knockout (TDP2 −/−/−), and human TDP2‐complemented (hTDP2) DT40 cells (15 μg per lane). Recombinant human TDP2 bearing a His6‐tag was loaded as a marker (Lane 1). Upper panel probes for TDP2 protein (#: non‐specific bands), middle panel for nuclear TOP1 (nuclear maker), and bottom panel for COX4 (mitochondrial marker). The 70‐kDa TOP1 band represents a commonly observed proteolytic truncation product.
- Proteinase K protection assay of purified mitochondria from hTDP2 DT40 cells. Upper panel probes for TDP2 (#: non‐specific band). Lower panel probes for MRPL12, an internal mitochondrial marker.
- TDP2 activity in mitochondrial (Mito) and nuclear fractions. Upper panel: schematic representation of the assay: TDP2 cleaves the 5′‐phosphotyrosine bond (5′‐Y) and generates a product bearing a 5′‐phosphate group (5′‐P). Lower panel: the same samples as in (A) were tested for TDP2 activity. Indicated extracts were serially diluted (1:3 serial dilution starting at 0.5 μg/μl) and incubated with 32P‐3′‐end‐labeled duplex DNA constructs for 2 hours. Left lanes: markers bearing a 5′‐phosphate or a 5′‐hydroxyl group.
- Clonogenic survival assays of wild‐type (WT), TDP2 knockout (TDP2 −/−/−), and human TDP2‐recomplemented (hTDP2) DT40 cells treated with indicated concentrations of mtDox for 14 days (n = 2).
- Protective effect of TDP2 on mtDNA. The indicated DT40 cell lines were treated with mtDox for 8 h followed by genomic DNA extraction. mtDNA copy numbers were measured by quantitative real‐time PCR normalized to a nuclear housekeeping gene (18S‐rRNA). Relative mtDNA copy numbers were normalized to mock‐treated control cells (n = 7, error bars represent SEM). *P ≤ 0.05 (two‐tailed Wilcoxon test).
Source data are available online for this figure.
To demonstrate that TDP2 is active in DT40 mitochondria, we measured TDP2 biochemical activity 8, 9, 17. Using the same mitochondrial and nuclear extracts probed by immunoblotting in Fig 4A, we detect robust 5′‐tyrosine cleavage activity in both the mitochondrial and nuclear extracts of the WT and hTDP2 cells (representative gel in Fig 4C). The products generated by both extracts bear phosphate ends, as they co‐migrate with the 5′‐P marker, distinct from the 5′‐OH maker (Fig 4C, left lanes), consistent with the cleavage product generated by TDP2 8, 9, 17. Conversely, TDP2 activity is undetectable in the nuclear and mitochondrial extracts of TDP2 −/−/− cells, indicating that TDP2 is the primary source for 5′‐tyrosine removal activity in both the nucleus and mitochondria of DT40 cells.
TDP2 protects against mitochondrial TOP2 poisoning
To examine the role of TDP2 for the repair of TOP2cc in mitochondria 14, 22, we employ a mitochondrial‐targeted doxorubicin conjugate (mtDox, structure shown in Appendix Fig S5A), where a mitochondrial‐targeted peptidyl motif is chemically attached to the TOP2 poison, doxorubicin 1, 5, 6, 23. Live‐cell fluorescence microscopy shows the accumulation of mtDox in mitochondria within minutes and its retention for up to 24 h (Appendix Fig S5B, upper panel). By contrast, unmodified doxorubicin accumulates mostly in the nucleus (Appendix Fig S5B, lower panel). These results confirm that mtDox specifically localizes to mitochondria in our cellular system 23. We also verified that mtDox does not induce nuclear DNA damage, as opposed to unmodified doxorubicin under our experimental conditions (Appendix Fig S5C). Next, clonogenic assays showed that TDP2 −/−/− cells are hypersensitive to mtDox, and that TDP2 recomplementation in the hTDP2 cells restored resistance close to levels of WT cells (Fig 4D and Appendix Fig S6A), indicating that TDP2 is important in protecting against mtDox in the mitochondria. As expected, we observed similar hypersensitivities to doxorubicin, a known nuclear TOP2 poison (Appendix Fig S6B) 4. Because TDP1, the counterpart of TDP2, has been reported to repair trapped TOP2 24 as well as to translocate to mitochondria 25, we investigated the role of TDP1 in repairing mtDox‐induced damage. As shown in Appendix Fig S6C, TDP1 knockout cells (TDP1 −/−) showed similar hypersensitivity to mtDox as the WT cells, and the double‐knockout cells (TDP1 −/− , TDP2 −/−/−) showed similar hypersensitivity as the TDP2 −/−/− cells. These results are consistent with the role of mitochondrial TDP2 as the primary enzyme in repairing mitochondrial TOP2cc.
To determine whether TDP2 protects mtDNA against mtDox, we also measured mtDNA copy number in the panel of DT40 cells in response to mtDox. Quantitative real‐time PCR measurement for each sample was carried out and normalized to a nuclear housekeeping gene, 18S‐rRNA. The mtDNA copy number was then normalized to the mock‐treated cells to determine the effect of mtDox on the relative mtDNA copy number. Compared to the WT cells, the relative mtDNA copy number of TDP2 −/−/− cells is reduced after 8 h of mtDox treatment and complementation with hTDP2 reversed this effect (Fig 4E). Together, the results from clonogenic assay and mtDNA copy number measurement are consistent with a role of mitochondrial TDP2 in repairing mitochondrial TOP2cc.
The mitochondrial TDP2S isoform protects against mitochondrial TOP2 poisoning
To specifically investigate the role of the mitochondrial TDP2S isoform, we applied CRISPR gene editing technology to selectively disrupt the TDP2 gene in an isoform‐specific manner in human lymphoblastoid TK6 cells. We targeted a site in the first exon unique to the full‐length TDP2 isoform, which would not disrupt TDP2 S (red asterisk in Fig 5A). We screened the resulting clones for those that lost expression of TDP2 but not TDP2S, which we termed TDP2 S cells (Fig 5B). These cells express TDP2S in mitochondria but not full‐length TDP2 in the nucleus (Fig 5C, Lanes 5–6), while the WT clones express full‐length TDP2 both in the mitochondrial and nuclear fractions as well as TDP2S in mitochondrial fraction (Fig 5C, Lanes 1–2). TK6 TDP2 −/− cells previously generated by disrupting the last exon of TDP2 18 lack both TDP2 isoforms in the mitochondrial or nuclear fractions (Fig 5C, Lanes 3–4).
Figure 5. TDP2S is selectively expressed in mitochondria and protects against mtDox.
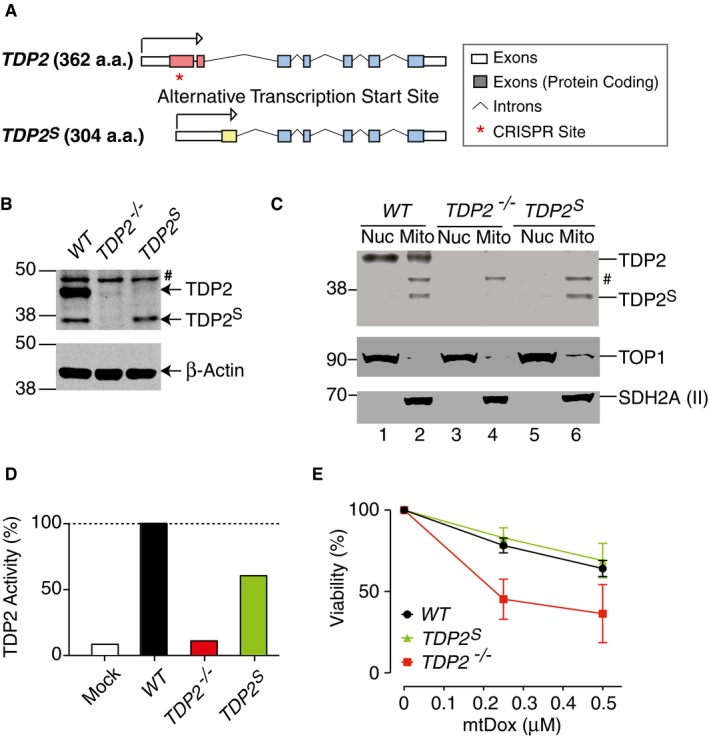
- Scheme showing the design of the CRISPR site (marked by red asterisk) used to generate isoform‐specific TDP2 knockout cell lines in human TK6 cells.
- Representative TDP2 immunoblot of whole cell lysates from wild‐type (WT), TDP2 knockout (TDP2 −/−), and TDP2 S isoform‐specific (TDP2 S) TK6 cells (#: non‐specific band) (30 μg per lane). β‐Actin served as loading control.
- Representative TDP2 immunoblot after cellular fractionations of the indicated TK6 cells (#: non‐specific band) (30 μg protein per lane). TOP1 and SDH2A (II) served as fractionation and loading controls.
- Comparison of TDP2 biochemical activity of mitochondrial extracts of TK6 WT, TDP2 −/−, and TDP2 S cells. Processing of radiolabeled substrates (shown in the upper panel of Fig 2D) by 0.3 μg/μl of mitochondrial extract for 2 h is compared between the three isogenic cell lines. Control mock samples contained only buffer. 80 μM of unlabeled single‐strand DNA is present in all samples to minimize degradation of specific TDP2 substrates. The amounts of product generated were quantified and normalized to the WT cells (n = 2).
- Viability assays of wild‐type (WT), TDP2 knockout (TDP2 −/−), and TDP2S isoform‐specific (TDP2 S) TK6 cells treated with mtDox (n = 3, error bars represent SEM).
Source data are available online for this figure.
Next, we tested the TDP2 biochemical activities from mitochondrial extracts purified from these cells. TDP2 activity is nearly completely attenuated in the mitochondrial extracts of TDP2 −/− cells, while mitochondrial extracts of TDP2 S cells retained ~70% of TDP2 activity compared to WT cells (Fig 5D). These results are consistent with the selective localization of biochemically active TDP2S in mitochondria. They also show the dual‐localization of full‐length TDP2 in both the nucleus and mitochondria. Viability assays with these three isogenic cell lines show that TDP2 −/− cells are hypersensitivity to mtDox, while TDP2 S cells are similarly resistant to mtDox as the WT cells (Fig 5E). By contrast, TDP2 S and TDP2 −/− cells showed similar hypersensitivity to unmodified doxorubicin compared to WT cells, as expected, since both of these cell lines lack nuclear TDP2 (Appendix Fig S7). These results demonstrate that both the TDP2S and TDP2 isoforms protect against mitochondrial TOP2cc.
TDP2 in the mitochondria is important for mitochondrial transcription
As a previous report showed that loss of TDP2 inhibits TOP2‐dependent nuclear gene transcription 12, we examined whether lack of TDP2 impacted mitochondrial transcription. We carried out quantitative real‐time PCR analysis to measure five mitochondrial transcripts for WT, TDP2 −/− , and TDP2 S TK6 cells. Our results show that the levels of all five mitochondrial transcripts in TDP2 −/− cells are reduced compared to WT cells, while three out of five transcripts also show reduced levels in TDP2 S cells (Fig 6A). It is possible that TDP2S has gene‐specific role in mitochondrial transcription or that the intermediate level of TDP2 enzyme present in the mitochondria of TDP2 S cells only results in partial rescue. We verified that the reduction in mitochondrial transcripts in the TDP2 −/− and TDP2 S cells is not due to the loss of mitochondrial copy numbers (Appendix Fig S8). To confirm the impact of TDP2 on mitochondrial transcription, we generated TDP2 knockout human colon carcinoma HCT116 cells using CRISPR (Appendix Fig S9). Quantitative real‐time PCR analysis revealed that the levels of all five mitochondrial transcripts in HCT116 TDP2 −/− are also reduced compared to the parental HCT116 cells (Fig 6B). Together, these results extend the observed role of TDP2 for nuclear transcription 12 to mitochondrial transcription.
Figure 6. Lack of TDP2 in mitochondria leads to reduced mitochondrial transcripts.
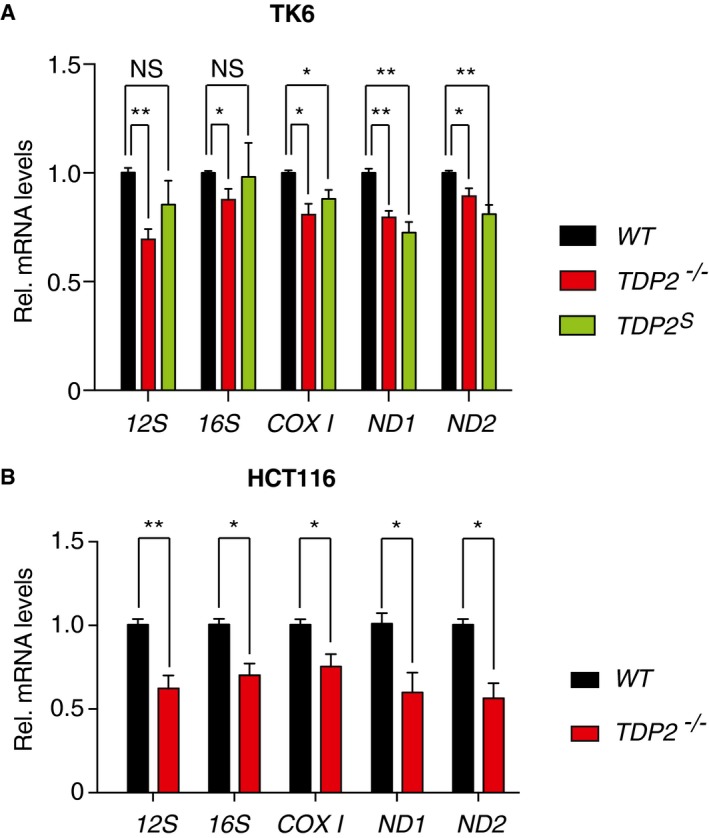
- Relative mRNA levels for indicated mitochondrial genes in TK6 WT (black bars), TDP2 −/− (red bars), and TDP2 S (green bars) cells were measured by quantitative real‐time PCR. All samples are referenced to the transcript levels for the housekeeping gene β2 microglobulin (β2M), and transcript levels of each mitochondrial gene in the WT cells are set as 1 (n = 8, error bars represent SEM). **P ≤ 0.01; *P ≤ 0.05 (two‐tailed Wilcoxon test).
- Relative mRNA levels for indicated mitochondrial genes in HCT116 WT (black bars) and TDP2 −/− (red bars) cells were measured by quantitative real‐time PCR. All samples are referenced to the transcript levels for the housekeeping gene β2 microglobulin (β2M), and transcript levels of each mitochondrial gene in the WT cells are set as 1 (n = 6, error bars represent SEM). **P ≤ 0.01; *P ≤ 0.05 (two‐tailed Wilcoxon test).
Discussion
The present study provides genetic, biochemical, cellular, and functional evidence that both full‐length TDP2 and a previously undescribed short isoform TDP2S localize to mitochondria and function to maintain mtDNA integrity and biogenesis in response to trapped TOP2cc. We show that TDP2S is excluded from the nucleus as it lacks the nuclear localization sequence of full‐length TDP2, but is present in the cytosol and mitochondria as it contains a MTS encoded by the variant transcript. Thus, TDP2 joins a growing list of DNA repair proteins that function in both the nucleus and mitochondria.
Maintenance and repair of the mitochondrial genome rely on enzymes encoded by the nuclear genome. In particular, the topological changes generated by transcription and replication of circular mtDNA are resolved by nucleus‐encoded topoisomerases. At least four different topoisomerases have been reported in mitochondria: TOP1mt, TOP2α, TOP2β, and TOP3α 14, 26, 27, 28, 29, 30. The associated danger of topoisomerases is that they can become trapped on DNA 2, 31, 32. In mitochondria, trapped topoisomerase cleavage complexes on mtDNA would require repair by the TDPs, which are nucleus‐encoded DNA repair proteins 31, 33. Although vertebrate TDP1 has been reported to localize and function in mitochondria 25, it preferentially repairs cleavage complexes of type IB topoisomerases, due to its limited activity on cleavage complexes of type II topoisomerases 34. The presence of TDP2 in mitochondria demonstrated in this study fulfills the potential need to repair TOP2cc in mitochondria.
We also report a previously uncharacterized human TDP2 isoform with full DNA phosphodiesterase activity, TDP2S, localizing to mitochondria and the cytosol. TDP2S results from an alternative transcription start site of the TDP2 gene (located on chromosome 6p22.3‐p22.1), leading to the replacement of the NLS in the canonical full‐length isoform with a MTS (see Fig 2A). An analogous transcript encoding a putative mitochondrial isoform of TDP2 with a MTS has also been reported for macaque. This type of dual‐localization strategy is common among enzymes that are required in multiple cellular compartments 35. Other strategies include gene duplications, alternative mRNA splicing and alternative translation start sites 35. Interestingly, both isoforms of TDP2 are present in mitochondria, although TDP2S is enriched in mitochondria and excluded from the nucleus. Similar phenomenon has been reported for FEN1 and FENMIT, two isoforms of the same nucleus‐encoded DNA repair enzyme 36. Only FENMIT contains a MTS, yet both isoforms localize to mitochondria 36. In the case of full‐length TDP2, which lacks a MTS, further investigation is required to understand its mitochondrial translocation mechanism. The presence of a NLS and an ubiquitin‐associated (UBA) domain 16 in the N‐terminal of the canonical TDP2 isoform is consistent with the role of this isoform to selectively remove abortive nuclear TOP2cc. By contrast, in the short isoform TDP2S, a functional MTS replaces the NLS and UBA, leading to its mitochondrial and cytosolic distribution. In addition to DNA repair, TDP2 has been reported to play a role in other cellular pathways, including multiple signal transduction and immune response pathways 33, 37. It is probable that the divergent N‐terminal domains of the two TDP2 isoforms facilitate distinct interactions and regulating mechanisms. The different expression levels of two TDP2 isoforms in different cell lines and the tight regulation of TDP2S expression support the idea that the two isoforms have specialized functions. Further studies are warranted to elucidate the potential roles of TDP2S in the cytosol, such as potentially serving as Vpg unlinkase 10.
The presence of both TDP1 and TDP2 in mitochondria highlights the importance of repairing mtDNA damage produced by abortive topoisomerase cleavage complexes, which likely form under normal conditions (reviewed in 2, 33). The physiological accumulation of TOP2cc has recently been demonstrated in neurons 12, 13 and in the promoter of actively transcribing genes 13, 38 (reviewed in 2, 39), in agreement with our finding that lack of TDP2 in the mitochondria leads to reduced mitochondrial transcript levels. In addition, oxidative radicals in mitochondria can damage mtDNA 40 and directly oxidize TOP2 41, 42, 43, both of which can lead to TOP2cc stabilization. Also, ribonucleotides, which are present in mtDNA 44, are known to trap TOP2cc that would require repair by TDP2 9, 15. The roles of both TOP2 and TDP2 in mitochondrial transcription open new perspectives for further studies.
Defects in DNA repair genes are increasingly linked to neurological diseases, which might be related to critical functions in mitochondria. For example, mutation of TDP1 leads to spinocerebellar ataxia with axonal neuropathy, and aprataxin defects lead to ataxia with oculomotor apraxia 1 45, 46, 47, 48, 49. Both nucleus‐encoded enzymes are important for mtDNA repair as well as nuclear DNA repair 25, 50, 51, 52. In addition to its canonical role for removing TOP1 and TOP1mt adduct, TDP1 is functionally important for repairing DNA damage generated by oxidative species or DNA chain‐terminating anti‐viral drugs in mitochondria 25, 53. Similarly, aprataxin is found to repair abortive ligase adduct at ribonucleotide sites 54. As mtDNA contains relatively high levels of ribonucleotides 44, this type of abortive ligase adduct presumably occurs frequently in the mitochondrial genome 44, 55. It will be important to delineate the precise roles of TDP1 and aprataxin in the nucleus vs mitochondria with respect to their related neurological diseases. With the new appreciation of the role of TDP2 in mitochondrial genome maintenance, further studies on how its various functions might contribute to neurological defects in TDP2‐deficient individuals 12, 56 are warranted.
Materials and Methods
Constructs
The first exon of cDNA for TDP2 S was synthesized as a gblock (IDT) to swap out the first two exons of TDP2 in TDP2 human cDNA ORF clone (RC202015, OriGene, Rockville, MD). The ORFs of two TDP2 isoforms were cloned into pEGFP‐N2, where further truncation and mutations of each construct were made using In‐Fusion Cloning Kit (Clontech, Mountain View, CA). The human TDP2 open‐reading frame was cloned into pCMV6‐AN‐DDK using RapidShuttling Kit (OriGene, Rockville, MD) after PCR amplification from TDP2 human cDNA ORF clone (OriGene, Rockville, MD) for establishing hTDP2‐recomplemented (with a FLAG‐tag) DT40 cells. Cloning and expression of His6‐tagged recombinant human TDP2 and TDP2S is as described previously 17.
Cell culture
All human cancer cell lines were obtained from the Developmental Therapeutics Program at the National Cancer Institute. HCT116 and HEK293T (ATCC, Manassas, VA) cells were cultured at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gemini, West Sacramento, CA). All lung cancer cell lines were cultured in the same condition but with Roswell Park Memorial Institute Medium (Life Technologies, Carlsbad, CA). Silencing of TDP2 is done with ON‐TARGETplus SMARTpool targeting TDP2 (Dharmacon Lafayette, CO) and Lipofectamine® RNAiMAX transfection reagent (Life Technologies, Carlsbad, CA) following the manufacturer's instructions for 48–72 h. To silence specific‐isoform of TDP2, the following RNA sequences were transfected using Lipofectamine® RNAiMAX transfection reagent (Life Technologies, Carlsbad, CA): si‐TDP2‐362 a.a. (aggaagagggcgagccugaggugaa/uucaccucaggcucgcccucuuccu; gcguuggccuccuguuccgcuuaaa/uuuaagcggaacaggaggccaacgc) or si‐TDP2‐304 a.a. (aagcgagagguuugaaacaaaugaa/uucauuuguuucaaaccucucgcuu; ugaccggccagaauaacagagucau/augacucuguuauucuggccgguca). DT40 chicken cells were cultured at 37°C with 5% CO2 in Roswell Park Memorial Institute medium (Life Technologies, Carlsbad, CA) supplemented with 1% chicken serum (Life Technologies, Carlsbad, CA), 50 μM β‐mercaptoethanol, penicillin, streptomycin, and 10% fetal bovine serum (Gemini, West Sacramento, CA), as described previously 53. To establish stable hTDP2 DT40 clone, ScaI‐linearized (New England BioLabs, Ipswich, MA) pCMV6‐AN‐DDK‐TDP2 vector was stably transfected into TDP2 −/−/− DT40 cells 21 by electroporation (550 V, 25 μF, Bio‐Rad, Hercules, CA). The clones were selected by resistance to neomycin, and the expression levels were verified by immunoblotting (anti‐TDP2, Bethyl, Montgomery, TX). TDP1 −/− 34 and TDP1 −/− ,TDP2 −/−/− 57 DT40 cells were generated as previously described. TK6 cells were cultured in the Roswell Park Memorial Institute Medium (Life Technologies, Carlsbad, CA), supplemented with 5% horse serum (Gibco, ThermoFisher Scientific) and 200 mg/ml sodium pyruvate (Invitrogen), and TK6 TDP2 −/− cells were generated as previously described 18. To generate TK6 TDP2S cells, sequence ggcatcgcagcttgcgaccg targeting the first exon of the full‐length TDP2 was cloned into px330 58 and transfected into TK6 WT cells. Single clones were screened using Custom TaqMan SNP Genotyping Assays (ThermoFisher Scientific), and verified by sequencing and immunoblotting analysis. To generate HCT116 TDP2 −/− cells, sequence gttgggtaaaccaccacatc targeting the last exon of TDP2 was cloned into px330 58 and co‐transfected with a Puro‐resistance gene flanked by homology arms (of ~1 kb) upstream and downstream of the target site. Transfected cells were selected with 1 μg/ml of puromycin 72 h post‐initial transfection for cells with puro‐resistance gene recombined into at least one copy of the target site. Established clones from single cell were subsequently screened by immunoblotting analysis.
Immunoblotting and antibodies
Freshly prepared nuclei pellets or whole cell pellets were resuspended in high salt RIPA buffer (20 mM Tris–HCl, pH = 7.5, 550 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP‐40, and Protease inhibitor cocktail [Roche, Indianapolis, IN]), agitated at 4°C for 30 min, and clarified by centrifugation at 4°C at 15,000 × g for 15 min. The mitochondria pellets were resuspended in mitochondrial extract buffer (320 mM sucrose, 10 mM Tris–HCl, pH = 7.5, 1 mM EDTA, 0.5% Triton and 500 mM NaCl and Protease inhibitor cocktail [Roche, Indianapolis, IN]), and chilled on ice for 10 min before clarifying by centrifugation at 4°C at 15,000 × g for 15 min. Immunoblotting was done following standard procedures, and signal was detected by ECL chemiluminescence reaction (Thermo Scientific, Waltham, MA) or by Odyssey Imaging Systems by Li‐COR (Lincoln, NE). Rabbit polyclonal anti‐TDP2 was from Bethyl (Montgomery, TX), mouse monoclonal anti‐TOP1 was from BD PharmingenTM (San Jose, CA), rabbit monoclonal anti‐GAPDH was from Cell Signaling (Danvers, MA), rabbit polyclonal anti‐TFAM, mouse monoclonal anti‐cytochrome C, and mouse polyclonal anti‐TOMM70A were from Abcam (Cambridge, MA), mouse monoclonal anti‐β‐actin was from Sigma‐Aldrich (St. Louis, MO), monoclonal anti‐COX4 and mouse monoclonal anti‐SDH2A were from Life Technologies (Carlsbad, CA), and rabbit polyclonal anti‐MRPL12 was from ProteinTech (Chicago, IL).
Pull‐down from whole cell lysate from H226 cells was carried out following standard procedures with anti‐TDP2 antibody (Bethyl, Montgomery, TX). The sample was separated on a 10% SDS–PAGE followed by Coomassie blue staining. The band at 37 kDa was excised and subjected to tandem mass spectrometry analysis, performed using an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific).
Fluorescence confocal imaging
All images were taken using a confocal microscope (LSM 710 or LSM 780, Zeiss, Oberkochen, Germany). HCT116 cells were transfected with indicated GFP‐tagged constructs using Lipofectamine® 2000 transfection reagent (Life Technologies, Carlsbad, CA) for 48 h and stained with MitoTracker® Deep Red (Life Technologies, Carlsbad, CA) prior to fixing and imaging.
Nucleus and mitochondria purification and proteinase K protection assay
Nucleus and mitochondria fractionation and purification were performed as described previously 25. Briefly, cell pellets were resuspended in stabilizing buffer containing 0.1 mg/ml digitonin (225 mM mannitol, 75 mM sucrose, 10 mM HEPES, pH = 7.3, 10 mM EDTA, and Protease Inhibitor Cocktail [Roche, Indianapolis, IN]) and chilled on ice for 10 min. After homogenizing with a glass Dounce homogenizer, cell nucleus was pelleted by centrifugation at 1,000 × g for 10 min, followed by washing three times with stabilizing buffer with 0.5% Triton. The supernatant from the initial centrifugation step was further separated by spinning at 15,000 × g for 10 min. The resulting supernatant is the cytosolic fraction, and the pelleted mitochondria were washed three more times with stabilizing buffer.
Proteinase K protection assay was performed as described previously 25. Equal amounts of freshly prepared mitochondria in stabilizing buffer were treated with 0.8 mg/ml proteinase K in the presence or absence of 1% sodium dodecyl sulfate (SDS) at 25°C for 10 min. The remaining peptides were precipitated by addition of 2 volumes of 20% trichloroacetic acid at 0°C for 20 min, followed by centrifugation at 4°C at 15,000 × g for 15 min. The pellets were washed three times with ice‐cold acetone before drying and resuspension in SDS gel loading buffer for immunoblotting analysis.
TDP2 biochemical activity assay
TDP2 biochemical activity assay was performed as described previously 17. Briefly, the indicated amount of recombinant proteins or extracts was incubated with 1 nM of 32P‐3′‐end‐labeled DNA constructs bearing a 5′‐tyrosine moiety on a protruding 4‐nt overhang (5′‐Y‐tccgttgaagcctgcttt[ 32P*A], underlined portion forms duplex with the complementary strand) in 10 μl reaction at 25°C for 2 h. The reaction buffer contains 80 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 40 μg/ml bovine serum albumin 50 mM Tris–HCl, pH = 7.5, and 0.01% Tween‐20, supplemented with 80 μM cold single‐strand DNA competitor. The reactions were terminated by addition of formamide gel loading buffer and analyzed by denaturing PAGE (16%). Gels were dried and exposed on PhosphorImager screens. Imaging was done using a Typhoon 8600 imager (GE Healthcare, Little Chalfont, United Kingdom).
Clonogenic assays
Clonogenic assays were performed as described previously 53. Two hundred WT or TDP2 −/−/− or 1,000 hTDP2 DT40 cells were seeded in duplicates in 4 ml of DMEM/F12 clonogenic media (Life Technologies, Carlsbad, CA) containing indicated amount of mtDox and incubated at 37°C with 5% CO2. The DMEM/F12 media was supplemented with 15 g/l methylcellulose, 2 g/l NaHCO3, 15% FBS, 50 μM β‐mercaptoethanol, 1.5% chicken serum, and 2 mM l‐glutamine (Life Technologies, Carlsbad, CA). Modification of doxorubicin to mtDox was as described previously 23. The number of colonies was counted after 14 days using FociCounter (http://focicounter.sourceforge.net/) and averaged from duplicates.
Viability assays
Ten thousand TK6 cells were seeded in 384‐well plates and treated with indicated concentration of mtDox for 72 h in triplicate before cells were assayed with the ATPlite 1‐step kit (PerkinElmer, Waltham, MA, USA).
Quantitative real‐time PCR
For mtDNA copy number measurement, DT40 cells lines were treated with indicated concentrations of mtDox at 37°C for 8 h. The genomic DNA was extracted using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) and used as templates. For TDP2 and TDP2 S transcript levels measurements, A549 cells were treated with either siControl or siTDP2 for 48 h before total RNA extraction using PureLink RNA Mini Kit (ThermoFisher Scientific). For mitochondrial transcript levels measurement, total RNA was extracted from TK6 or HCT116 cells using PureLink RNA Mini Kit (ThermoFisher Scientific). All RNA samples were treated with RNase‐Free DNase (Qiagen, Valencia, CA) during the extraction before serving as templates in reverse transcription with SuperScript™ II Reverse Transcriptase (Life Technologies, Carlsbad, CA) following the manufacturer's instructions. All quantitative real‐time PCR reactions were done with FastStart Universal SYBR Green Master Mix (Roche, Indianapolis, IN) in quintuplets on an ABI 7900 thermocycler (Life Technologies, Carlsbad, CA) following the manufacturer's instructions. For DT 40 cells, two sets of primers specific for DT40 mitochondrial DNA fragment (5′‐ccctcctcctttcatcctcatttc [sense] and 5′‐cctttttgttcaggcacgcttc [antisense]) or for nuclear 18S‐rRNA gene (5′‐tgactcaacacgggaaacctcac [sense] and 5′‐ccagacaaatcgctccaccaac [antisense]) were used. For human cells, the primer sequences are as follows: TDP2‐specific (5′‐tggagtttgcctcggtcgcaag [sense] and 5′‐gagagagaacatgctgccattttcttg [antisense]), TDP2 S‐specific (5′‐agaggtttgaaacaaatgaacgaaggc [sense] and 5′‐gagagagaacatgctgccattttcttg [antisense]), β2 microglobulin (β2M) (5′‐tgctgtctccatgtttgatgtatct [sense] and 5′‐tctctgctccccacctctaagt [antisense]), 12S‐rRNA (5′‐ggttggtcaatttcgtgc [sense] and 5′‐gagttttttacaactcaggtg [antisense]), 16S‐rRNA (5′‐agccaccaattaagaaagcg [sense] and 5′‐gcttatgcggaggagaatg [antisense]), ND1 (5′‐aagtcaccctagccatcattctac [sense] and 5′‐gcaggagtaatcagaggtgttctt [antisense]), ND2 (5′‐ctcacatgacaaaaactagcccccatctc [sense] and 5′‐gctaagattttgcgtagctgggtttgg [antisense]), and COXI (5′‐tacctattattcggcgcatgagctgga [sense] and 5′‐ tgcatgggctgt gacgataacgttgta [antisense]). The C t value for mitochondrial genome or genes is referenced to the C t value of the 18S‐rRNA or β2M for each sample (ΔC t). For mtDNA copy number measurement, the ΔC t for each sample is in turn referenced to that of the untreated control WT, TDP2 −/−/− or hTDP2 DT40 cells (ΔΔC t), with the untreated control of all cell types set at 1. For TDP2 and TDP2 S transcript levels, the ΔC t for each sample is in turn referenced to that of cells treated with siControl (ΔΔC t), which were set at 1. For mitochondrial transcription levels, the ΔC t for each sample is in turn referenced to that of WT cells, which is also set at 1. The relative mtDNA copy number, the relative mRNA levels for each TDP2 isoform, and the relative mitochondrial transcription levels for each sample are then calculated based on its ΔΔC t values.
Author contributions
SNH conducted the cellular fractionation, immunoblotting, fluorescence and immunofluorescence microscopy, cloning, quantitative real‐time PCR, in vitro biochemical activity assay, clonogenic assay, generated CRISPR‐mediated TK6 TDP2 S cells and HCT116 TDP2 −/− cells as well as data analysis; IDR and performed cloning, immunofluorescence and live‐cell imaging; SAM conducted the cellular fractionation and immunoblotting; DVT, SRJ, and SOK synthesized and characterized mtDox; KA conducted immunoblotting; SK performed immunofluorescence; SAB and VMF processed murine tissue samples; SV performed RNA sequencing data analysis; JM established hTDP2 DT40 clones; LMMJ performed mass spectrometry analyses; YP supervised the study, and SNH and YP wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work is supported by National Institutes of Health Grant Z01 BC 006150 from the NCI Intramural Program, Center for Cancer Research. The Kelley group acknowledges the National Institutes of Health for the support for the work on mitochondria‐targeted probes (R01GM116886).
EMBO Reports (2018) 19: e42139
Contributor Information
Shar‐yin N Huang, Email: shar-yin.huang@nih.gov.
Yves Pommier, Email: pommier@nih.gov.
References
- 1. Nitiss JL (2009) DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 9: 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pommier Y, Sun Y, Huang SN, Nitiss JL (2016) Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17: 703–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pommier Y, Marchand C (2012) Interfacial inhibitors: targeting macromolecular complexes. Nat Rev Drug Discov 11: 25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9: 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pommier Y (2013) Drugging topoisomerases: lessons and challenges. ACS Chem Biol 8: 82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McClendon AK, Osheroff N (2007) DNA topoisomerase II, genotoxicity, and cancer. Mutat Res 623: 83–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang JC (2009) A journey in the world of DNA rings and beyond. Annu Rev Biochem 78: 31–54 [DOI] [PubMed] [Google Scholar]
- 8. Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW (2009) A human 5′‐tyrosyl DNA phosphodiesterase that repairs topoisomerase‐mediated DNA damage. Nature 461: 674–678 [DOI] [PubMed] [Google Scholar]
- 9. Gao R, Schellenberg MJ, Huang SN, Abdelmalak M, Marchand C, Nitiss KC, Nitiss JL, Williams RS, Pommier Y (2014) Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2‐DNA and ‐RNA covalent complexes by tyrosyl‐DNA‐phosphodiesterase 2 (TDP2). J Biol Chem 289: 17960–17969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Virgen‐Slane R, Rozovics JM, Fitzgerald KD, Ngo T, Chou W, van der Heden van Noort GJ, Filippov DV, Gershon PD, Semler BL (2012) An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc Natl Acad Sci USA 109: 14634–14639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koniger C, Wingert I, Marsmann M, Rosler C, Beck J, Nassal M (2014) Involvement of the host DNA‐repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc Natl Acad Sci USA 111: E4244–E4253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gómez‐Herreros F, Schuurs‐Hoeijmakers JHM, McCormack M, Greally MT, Rulten S, Romero‐Granados R, Counihan TJ, Chaila E, Conroy J, Ennis S et al (2014) TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat Genet 46: 516–521 [DOI] [PubMed] [Google Scholar]
- 13. Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC et al (2015) Activity‐induced DNA breaks govern the expression of neuronal early‐response genes. Cell 161: 1592–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang H, Zhang YW, Yasukawa T, Dalla Rosa I, Khiati S, Pommier Y (2014) Increased negative supercoiling of mtDNA in TOP1mt knockout mice and presence of topoisomerases IIalpha and IIbeta in vertebrate mitochondria. Nucleic Acids Res 42: 7259–7267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y, Thyssen A, Westergaard O, Andersen AH (2000) Position‐specific effect of ribonucleotides on the cleavage activity of human topoisomerase II. Nucleic Acids Res 28: 4815–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rao T, Gao R, Takada S, Al Abo M, Chen X, Walters KJ, Pommier Y, Aihara H (2016) Novel TDP2‐ubiquitin interactions and their importance for the repair of topoisomerase II‐mediated DNA damage. Nucleic Acids Res 44: 10201–10215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao R, Huang SN, Marchand C, Pommier Y (2012) Biochemical characterization of human tyrosyl‐DNA phosphodiesterase 2 (TDP2/TTRAP): a Mg2+/Mn2+‐dependent phosphodiesterase specific for the repair of topoisomerase cleavage complexes. J Biol Chem 287: 30842–30852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marchand C, Abdelmalak M, Kankanala J, Huang SY, Kiselev E, Fesen K, Kurahashi K, Sasanuma H, Takeda S, Aihara H et al (2016) Deazaflavin inhibitors of tyrosyl‐DNA phosphodiesterase 2 (TDP2) specific for the human enzyme and active against cellular TDP2. ACS Chem Biol 11: 1925–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi K, Kurahashi K, Gao R, Tsutakawa SE, Tainer JA, Pommier Y, Aihara H (2012) Structural basis for recognition of 5′‐phosphotyrosine adducts by Tdp2. Nat Struct Mol Biol 19: 1372–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schellenberg MJ, Appel CD, Adhikari S, Robertson PD, Ramsden DA, Williams RS (2012) Mechanism of repair of 5′‐topoisomerase II‐DNA adducts by mammalian tyrosyl‐DNA phosphodiesterase 2. Nat Struct Mol Biol 19: 1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zeng Z, Cortes‐Ledesma F, El Khamisy SF, Caldecott KW (2011) TDP2/TTRAP is the major 5′‐tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II‐induced DNA damage. J Biol Chem 286: 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khiati S, Dalla Rosa I, Sourbier C, Ma X, Rao VA, Neckers LM, Zhang H, Pommier Y (2014) Mitochondrial topoisomerase I (top1mt) is a novel limiting factor of Doxorubicin cardiotoxicity. Clin Cancer Res 20: 4873–4881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chamberlain GR, Tulumello DV, Kelley SO (2013) Targeted delivery of doxorubicin to mitochondria. ACS Chem Biol 8: 1389–1395 [DOI] [PubMed] [Google Scholar]
- 24. Nitiss KC, Malik M, He X, White SW, Nitiss JL (2006) Tyrosyl‐DNA phosphodiesterase (Tdp1) participates in the repair of Top2‐mediated DNA damage. Proc Natl Acad Sci USA 103: 8953–8958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Das BB, Dexheimer TS, Maddali K, Pommier Y (2010) Role of tyrosyl‐DNA phosphodiesterase (TDP1) in mitochondria. Proc Natl Acad Sci USA 107: 19790–19795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang H, Barcelo JM, Lee B, Kohlhagen G, Zimonjic DB, Popescu NC, Pommier Y (2001) Human mitochondrial topoisomerase I. Proc Natl Acad Sci USA 98: 10608–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Low RL, Orton S, Friedman DB (2003) A truncated form of DNA topoisomerase IIbeta associates with the mtDNA genome in mammalian mitochondria. Eur J Biochem 270: 4173–4186 [DOI] [PubMed] [Google Scholar]
- 28. Zhang H, Meng LH, Zimonjic DB, Popescu NC, Pommier Y (2004) Thirteen‐exon‐motif signature for vertebrate nuclear and mitochondrial type IB topoisomerases. Nucleic Acids Res 32: 2087–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sobek S, Dalla Rosa I, Pommier Y, Bornholz B, Kalfalah F, Zhang H, Wiesner RJ, von Kleist‐Retzow JC, Hillebrand F, Schaal H et al (2013) Negative regulation of mitochondrial transcription by mitochondrial topoisomerase I. Nucleic Acids Res 41: 9848–9857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y, Lyu YL, Wang JC (2002) Dual localization of human DNA topoisomerase IIIalpha to mitochondria and nucleus. Proc Natl Acad Sci USA 99: 12114–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Menon V, Povirk LF (2016) End‐processing nucleases and phosphodiesterases: an elite supporting cast for the non‐homologous end joining pathway of DNA double‐strand break repair. DNA Repair 43: 57–68 [DOI] [PubMed] [Google Scholar]
- 32. Ashour ME, Atteya R, El‐Khamisy SF (2015) Topoisomerase‐mediated chromosomal break repair: an emerging player in many games. Nat Rev Cancer 15: 137–151 [DOI] [PubMed] [Google Scholar]
- 33. Pommier Y, Huang SN, Gao R, Das BB, Murai J, Marchand C (2014) Tyrosyl‐DNA‐phosphodiesterases (TDP1 and TDP2). DNA Repair 19: 114–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y (2012) Tyrosyl‐DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J Biol Chem 287: 12848–12857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yogev O, Pines O (2011) Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochem Biophys Acta 1808: 1012–1020 [DOI] [PubMed] [Google Scholar]
- 36. Kazak L, Reyes A, He J, Wood SR, Brea‐Calvo G, Holen TT, Holt IJ (2013) A cryptic targeting signal creates a mitochondrial FEN1 isoform with tailed R‐Loop binding properties. PLoS One 8: e62340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li C, Sun SY, Khuri FR, Li R (2011) Pleiotropic functions of EAPII/TTRAP/TDP2: cancer development, chemoresistance and beyond. Cell Cycle 10: 3274–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ray S, Panova T, Miller G, Volkov A, Porter AC, Russell J, Panov KI, Zomerdijk JC (2013) Topoisomerase IIalpha promotes activation of RNA polymerase I transcription by facilitating pre‐initiation complex formation. Nat Commun 4: 1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haffner MC, De Marzo AM, Meeker AK, Nelson WG, Yegnasubramanian S (2011) Transcription‐induced DNA double strand breaks: both oncogenic force and potential therapeutic target? Clin Cancer Res 17: 3858–3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sabourin M, Osheroff N (2000) Sensitivity of human type II topoisomerases to DNA damage: stimulation of enzyme‐mediated DNA cleavage by abasic, oxidized and alkylated lesions. Nucleic Acids Res 28: 1947–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ashley RE, Osheroff N (2014) Natural products as topoisomerase II poisons: effects of thymoquinone on DNA cleavage mediated by human topoisomerase II alpha. Chem Res Toxicol 27: 787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin RK, Zhou N, Lyu YL, Tsai YC, Lu CH, Kerrigan J, Chen YT, Guan Z, Hsieh TS, Liu LF (2011) Dietary isothiocyanate‐induced apoptosis via thiol modification of DNA topoisomerase II{alpha}. J Biol Chem 286: 33591–33600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bandele OJ, Osheroff N (2007) Bioflavonoids as poisons of human topoisomerase II alpha and II beta. Biochemistry 46: 6097–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grossman LI, Watson R, Vinograd J (1973) The presence of ribonucleotides in mature closed‐circular mitochondrial DNA. Proc Natl Acad Sci USA 70: 3339–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takashima H, Boerkoel CF, John J, Saifi GM, Salih MAM, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M et al (2002) Mutation of TDP1, encoding a topoisomerase I‐dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32: 267–272 [DOI] [PubMed] [Google Scholar]
- 46. El‐Khamisy SF, Caldecott KW (2006) TDP1‐dependent DNA single‐strand break repair and neurodegeneration. Mutagenesis 21: 219–224 [DOI] [PubMed] [Google Scholar]
- 47. Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y et al (2001) Early‐onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet 29: 184–188 [DOI] [PubMed] [Google Scholar]
- 48. Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T et al (2001) The gene mutated in ataxia‐ocular apraxia 1 encodes the new HIT/Zn‐finger protein aprataxin. Nat Genet 29: 189–193 [DOI] [PubMed] [Google Scholar]
- 49. Martin LJ (2012) Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci 107: 355–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sykora P, Wilson DM, Bohr VA (2011) Repair of persistent strand breaks in the mitochondrial genome. Mech Ageing Dev 133: 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kazak L, Reyes A, Holt IJ (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 13: 659–671 [DOI] [PubMed] [Google Scholar]
- 52. Meagher M, Lightowlers RN (2013) The role of TDP1 and APTX in mitochondrial DNA repair. Biochimie 100: 121–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang SY, Murai J, Dalla Rosa I, Dexheimer TS, Naumova A, Gmeiner WH, Pommier Y (2013) TDP1 repairs nuclear and mitochondrial DNA damage induced by chain‐terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res 41: 7793–7803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tumbale P, Williams JS, Schellenberg MJ, Kunkel TA, Williams RS (2014) Aprataxin resolves adenylated RNA‐DNA junctions to maintain genome integrity. Nature 506: 111–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kasiviswanathan R, Copeland WC (2011) Ribonucleotide discrimination and reverse transcription by the human mitochondrial DNA polymerase. J Biol Chem 286: 31490–31500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McKinnon PJ (2014) TDP2 keeps the brain healthy. Nat Genet 46: 419–421 [DOI] [PubMed] [Google Scholar]
- 57. Zeng Z, Sharma A, Ju L, Murai J, Umans L, Vermeire L, Pommier Y, Takeda S, Huylebroeck D, Caldecott KW et al (2012) TDP2 promotes repair of topoisomerase I‐mediated DNA damage in the absence of TDP1. Nucleic Acids Res 40: 8371–8380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5