

Abstract
Aberrant G protein–coupled receptor (GPCR) expression and activation has been linked to tumor initiation, progression, invasion, and metastasis. However, compared with other cancer drivers, the exploitation of GPCRs as potential therapeutic targets has been largely ignored, despite the fact that GPCRs are highly druggable. Therefore, to advance the potential status of GPCRs as therapeutic targets, it is important to understand how GPCRs function together with other cancer drivers during tumor progression. We now report that the α-arrestin domain–containing protein-3 (ARRDC3) acts as a tumor suppressor in part by controlling signaling and trafficking of the GPCR, protease-activated receptor-1 (PAR1). In a series of highly invasive basal-like breast carcinomas, we found that expression of ARRDC3 is suppressed whereas PAR1 is aberrantly overexpressed because of defective lysosomal sorting that results in persistent signaling. Using a lentiviral doxycycline-inducible system, we demonstrate that re-expression of ARRDC3 in invasive breast carcinoma is sufficient to restore normal PAR1 trafficking through the ALG-interacting protein X (ALIX)-dependent lysosomal degradative pathway. We also show that ARRDC3 re-expression attenuates PAR1-stimulated persistent signaling of c-Jun N-terminal kinase (JNK) in invasive breast cancer. Remarkably, restoration of ARRDC3 expression significantly reduced activated PAR1-induced breast carcinoma invasion, which was also dependent on JNK signaling. These findings are the first to identify a critical link between the tumor suppressor ARRDC3 and regulation of GPCR trafficking and signaling in breast cancer.
Keywords: beta-arrestin, breast cancer, c-Jun N-terminal kinase (JNK), protease, thrombin, Rho (Rho GTPase), trafficking, protease-activated receptor, GPCR, trafficking
Introduction
G protein–coupled receptors (GPCRs)3 are a large family of cell-surface signaling receptors that play a critical role in cancer growth and development by regulating cellular proliferation, invasion, migration, immune cell–mediated functions, angiogenesis, and survival at metastatic sites (1–3). GPCR function can be altered in cancer through aberrant overexpression, gain-of-function activating mutations, mutations in downstream G protein signaling effectors, and increased production and secretion of GPCR-activating ligands by both tumor cells and surrounding stromal cells (4–7). As cell-surface receptors with highly druggable sites, GPCRs are the largest class of drug targets, with over 30% of current Food and Drug Administration–approved drugs targeting GPCRs (8, 9). Despite the success and promise of GPCRs as therapeutic targets, there are currently no drugs in the clinic used for the treatment of cancer that specifically target GPCRs. Moreover, GPCRs are known to function in metastasis (2, 3), and there are limited targeted treatment options for patients with metastatic cancer. Thus, understanding how GPCRs function together with other drivers of cancer in tumor progression is important for the development of new effective treatment strategies for metastatic cancer.
Several GPCRs implicated in metastatic cancer, including protease-activated receptor-1 (PAR1), have also been proven to be highly druggable (10, 11). PAR1 is overexpressed in invasive ductal breast carcinoma but not in normal mammary epithelial tissue (12, 13). In addition, overexpression of PAR1 in breast cancer patient biopsies correlates with increased rates of metastasis and poor prognosis (12, 14). PAR1 is also highly expressed in invasive breast carcinoma cell lines but not in non-invasive breast cancer cells, and high PAR1 expression correlates with increased cellular invasion in vitro and tumor growth in vivo (12, 15, 16). Moreover, loss of PAR1 function by either knockdown of PAR1 or inhibition of PAR1 signaling in highly invasive breast carcinoma cells results in decreased cellular migration and invasion in vitro and tumor growth in vivo (15–17). In contrast, ectopic overexpression of PAR1 in non-invasive MCF7 breast carcinoma is sufficient to drive cellular invasion in vitro and tumor growth in vivo possibly by initiating epithelial–mesenchymal transition (18, 19), suggesting that PAR1 expression is both necessary and sufficient to promote breast cancer progression. Together, these studies strongly support a role for PAR1 as a key mediator of breast cancer progression.
PAR1 is activated by irreversible proteolytic cleavage of the N terminus, revealing a new N-terminal domain that acts as a tethered ligand that binds intramolecularly to the receptor to elicit transmembrane signaling (20, 21). Thrombin is the main effector protease for PAR1 activation in most cell types; however, other proteases can cleave and activate the receptor in different cellular contexts. In fact, PAR1 senses and responds to multiple proteases generated in the tumor microenvironment, including thrombin, plasmin, and matrix metalloproteinase-1 (MMP-1) (18, 22, 23). Once activated, PAR1 signals to distinct heterotrimeric G protein subtypes, including Gq, Gi, and G12/13, and triggers RhoGEF-mediated RhoA signaling, an increase in intracellular Ca2+, MAP kinase activation, and signaling by multiple other effectors (24, 25). However, previous studies have shown that PAR1 signaling through G12/13 is the primary driver of breast carcinoma invasion and metastasis in vitro and in vivo (26). In addition, PAR1-G12/13 signaling to RhoA and subsequent activation of c-Jun N-terminal kinase (JNK) is required for thrombin-induced breast carcinoma invasion (27). However, the defects that engender PAR1 and other GPCRs the capacity to promote breast cancer invasion and metastasis are not known.
One mechanism that contributes to PAR1 dysregulation in cancer is defective endocytic trafficking. Similar to most GPCRs, activated PAR1 signaling to heterotrimeric G proteins is rapidly desensitized at the plasma membrane (28, 29). Unlike most GPCRs, however, internalization and lysosomal sorting of proteolytically activated PAR1 is critical for termination of G protein signaling (28). In fibroblasts, a chimeric PAR1 that internalizes and recycles back to the cell surface displayed persistent signaling after activation and removal of thrombin (28, 30). PAR1 persistent signaling was caused by recycling and continued signaling by proteolytically activated PAR1 that returned to the cell surface with its tethered ligand intact. Importantly, activated PAR1 trafficking is also severely altered in metastatic breast cancer but not in non-metastatic or normal breast epithelial cells (15). Consequently, dysregulated PAR1 trafficking and signaling drives breast cancer invasion and tumor growth (15, 16). Thus, dysregulated GPCR trafficking is utilized as a gain-of-function mechanism to prolong GPCR signaling in invasive breast carcinomas.
Most classic GPCRs sort to lysosomes for degradation through the endosomal sorting complexes required for transport (ESCRT) pathway, which functions sequentially to sort ubiquitinated receptors to intraluminal vesicles of multivesicular bodies/lysosomes (31). However, we showed previously that several GPCRs, including PAR1, sort to lysosomes independently of receptor ubiquitination and components of the ubiquitin-binding ESCRT machinery (32). Rather, activated PAR1 engages ALG-interacting protein X (ALIX), an ESCRT-III–interacting protein, for lysosomal degradation (33, 34). We further identified α-arrestin domain-containing protein-3 (ARRDC3) as a key regulator of ALIX-dependent PAR1 lysosomal trafficking in HeLa cells (35). ARRDC3 has also been shown to regulate endosomal sorting of other GPCRs, including the β2-adrenergic receptor (β2AR) (36–38) through mechanisms that remain poorly defined.
Importantly, ARRDC3 is a newly identified tumor suppressor in metastatic breast cancer (39, 40). ARRDC3 expression is either lost or suppressed in basal-like invasive breast cancer that results from either gene deletion or epigenetic silencing (40, 41). In addition, low ARRDC3 expression correlates with tumor recurrence and patient poor prognosis (39, 42), further suggesting a role for ARRDC3 in tumor suppression. Given that ALIX and ARRDC3 are key regulators of PAR1 trafficking, we examined whether dysregulation of PAR1 trafficking in invasive breast carcinoma was due to defective ALIX or ARRDC3 function. Here, we report that loss of ARRDC3 expression in highly invasive breast carcinoma is responsible for defective ALIX-dependent lysosomal sorting of PAR1. In addition, loss of ARRDC3 expression not only perturbs PAR1 trafficking, but also is responsible for persistent signaling that drives thrombin-stimulated breast carcinoma cellular invasion. These findings provide the first evidence that ARRDC3 functions as a tumor suppressor by modulating GPCR trafficking and signaling in invasive breast cancer.
Results
PAR1, ARRDC3, and ALIX expression in non-invasive luminal versus invasive basal-like breast carcinoma
To determine the mechanisms responsible for dysregulated PAR1 trafficking in invasive breast carcinoma, we profiled the expression of PAR1 and two key regulators, ARRDC3 and ALIX (33, 35), in a series of human mammary luminal non-invasive and basal-like invasive breast carcinoma cell lines. The expression of ARRDC3 and ALIX in breast carcinoma was detected by immunoblotting, and PAR1 expression was determined by cell-surface ELISA. Immunoblot analysis of equivalent amounts of cell lysates revealed high ARRDC3 expression in non-invasive luminal breast carcinoma and HER2-positive SKBR3 breast cancer cells (Fig. 1A, lanes 1–4 and 9), whereas ARRDC3 expression was minimally detected in invasive basal-like breast carcinoma (Fig. 1A, lanes 5–8). These findings are consistent with loss of ARRDC3 expression previously reported in invasive breast carcinoma (39–41). Interestingly, PAR1 expression was high in basal-like invasive breast carcinoma with low ARRDC3 expression and low in non-invasive luminal breast carcinoma with high ARRDC3 expression (Fig. 1, A and B), including luminal ZR75-1 cells as reported previously (15). In contrast to ARRDC3 and PAR1, ALIX expression was variable and detected in all cell lines irrespective of luminal versus basal subtype of breast carcinoma (Fig. 1A, lanes 1–9). Whereas ALIX is expressed in BT474 cells, its size is shifted possibly due to post-translational modifications, such as ubiquitination (35, 43). Nonetheless, these findings reveal an inverse correlation between PAR1 and ARRDC3 expression and raise the intriguing idea that loss of ARRDC3 expression may be responsible for aberrant PAR1 expression in invasive breast carcinoma.
Figure 1.
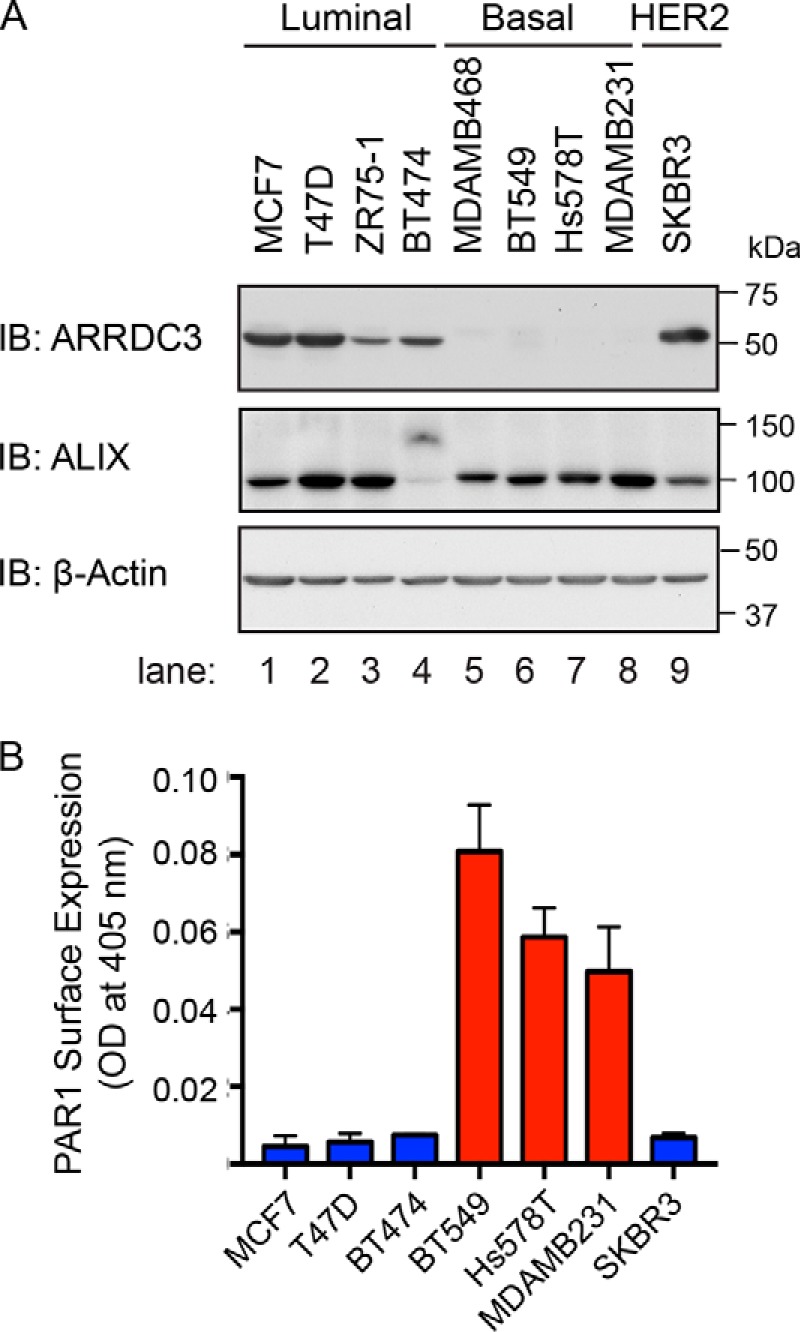
PAR1 and ARRDC3 protein expression are inversely correlated in breast carcinoma cell lines. A, equivalent amounts (20 μg) of cell lysates from various breast cancer cell lines were immunoblotted for ALIX and ARRDC3 expression. β-Actin expression was determined as a loading control. B, PAR1 cell-surface expression was determined by ELISA. Data shown (mean ± S.D., n = 3) are representative of three independent experiments.
Ectopic expression of ARRDC3 restores activated PAR1 degradation in invasive breast carcinoma
In invasive breast carcinoma, PAR1 trafficking is dysregulated and fails to sort to a lysosomal degradation pathway (15, 16). However, the mechanism responsible for defective PAR1 trafficking in breast cancer is not known. To determine whether ARRDC3 expression is sufficient to restore proper trafficking of PAR1 in invasive breast carcinoma, we used a lentiviral single lentivector for inducible knockdown (pSLIK) tetracycline-inducible expression vector system encoding ARRDC3 containing an N-terminal HA epitope tag (Fig. 2A). MDA-MB-231 pSLIK breast carcinoma cells stably expressing HA-ARRDC3 were generated and examined for doxycycline (DOX)-inducible expression of ARRDC3. An ∼12–16-fold induction of HA-ARRDC3 expression was detected in cells incubated with 1 or 10 μg/ml doxycycline for 48 h, respectively, compared with untreated control cells (Fig. 2B, lanes 4–6). The expression of ARRDC3 was determined by immunoblotting using either anti-HA or anti-ARRDC3 antibody (Fig. 2B, top and middle, respectively). As expected, parental MDA-MB-231 cells incubated with similar concentrations of doxycycline failed to induce ARRDC3 expression (Fig. 2B, lanes 1–3). In addition, incubation of MDA-MB-231 HA-ARRDC3 pSLIK cells with doxycycline using the same conditions did not alter the high level of PAR1 cell-surface expression basally as detected by ELISA (Fig. 2C). These results indicate that doxycycline specifically induces expression of HA-ARRDC3 in highly invasive MDA-MB-231 breast carcinoma.
Figure 2.
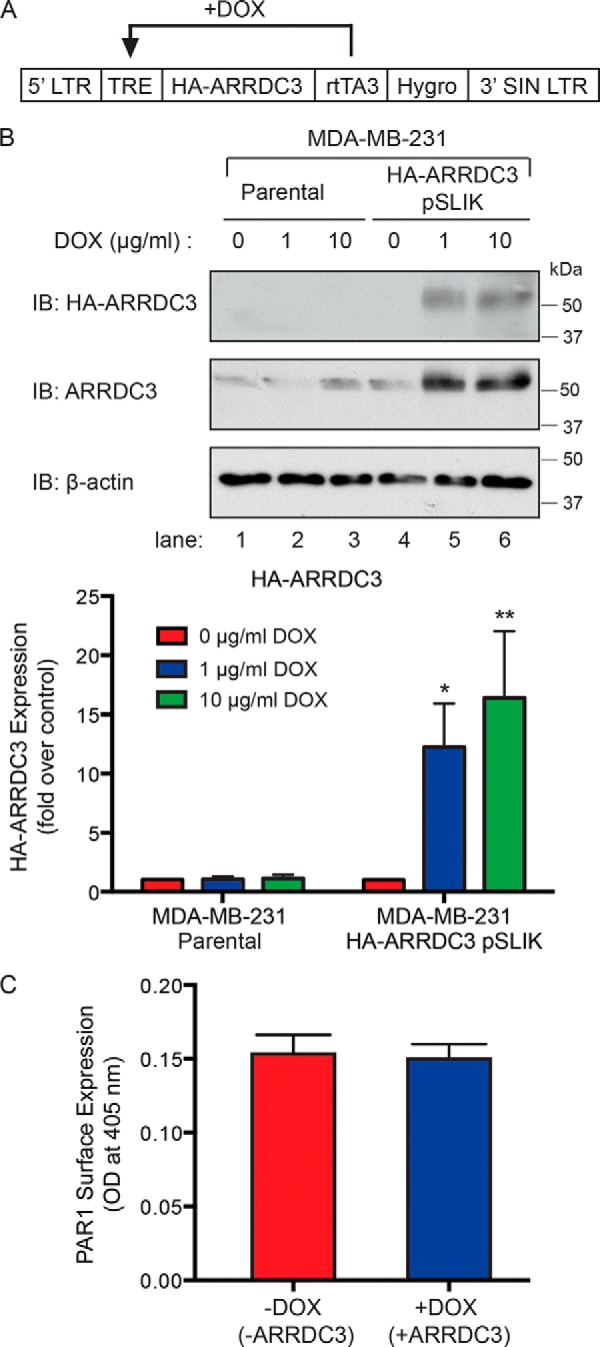
Induction of ARRDC3 expression in MDA-MB-231 breast carcinoma cell line using pSLIK vector system. A, schematic of the tetracycline-inducible pSLIK system for HA-tagged ARRDC3 expression. rtTA3, reverse tetracycline transactivator; TRE, tetracycline response element; LTR, long terminal repeat. B, cell lysates from MDA-MB-231 parental and HA-ARRDC3 pSLIK–expressing cells were collected after 48 h of incubation with DOX treatment at 0, 1, or 10 μg/ml. Cell lysates were immunoblotted to detect endogenous ARRDC3 or HA-ARRDC3 expression. β-Actin expression was detected as a control. The data shown (mean ± S.D. (error bars), n = 3) were quantified by densitometry and shown as the -fold change in ARRDC3 expression relative to 0 min control following doxycycline incubation. Statistical significance was determined by one-way ANOVA (*, p < 0.05; **, p < 0.01; n = 3). C, expression of PAR1 on the cell surface was determined before and after DOX (1 μg/ml) treatment for 48 h and determined by ELISA. The data (mean ± S.D., n = 3) shown as optical density determined at 405 nm are representative of three independent experiments.
Using the HA-ARRDC3 pSLIK doxycycline-inducible system in MDA-MB-231 cells, we next examined whether ARRDC3 expression is sufficient to restore agonist-stimulated PAR1 lysosomal degradation. MDA-MB-231 HA-ARRDC3 pSLIK cells were preincubated with or without doxycycline for 48 h and then treated with or without agonist, lysed, immunoprecipitated with anti-PAR1–specific antibodies, and then immunoblotted to detect PAR1 protein. In cells not exposed to agonist, endogenous PAR1 migrated as a high-molecular mass ∼75-kDa protein that was not detected in IgG control immunoprecipitates (Fig. 3A, lanes 2 and 7 and lanes 1 and 6). In HA-ARRDC3 pSLIK–expressing MDA-MB-231 cells not incubated with doxycycline and lacking ARRDC3 expression, prolonged treatment with the PAR1-specific agonist peptide failed to promote substantial loss of PAR1 protein as expected (Fig. 3A, lanes 2–5). These results are consistent with a defect in sorting of activated PAR1 from endosomes to lysosomes in invasive breast cancer, as reported previously (15, 16). However, in MDA-MB-231 cells expressing doxycycline-induced ARRDC3, agonist peptide induced a significant loss of PAR1 protein following 60, 90, or 120 min of stimulation (Fig. 3A, lanes 7–10). The kinetics of activated PAR1 degradation is reminiscent of a typical time course of receptor degradation observed in other cell types (33). To determine whether ARRDC3 affects PAR1 internalization, agonist-induced loss of PAR1 from the cell surface was measured by ELISA. In MDA-MB-231 HA-ARRDC3 pSLIK cells not treated with doxycycline and not expressing ARRDC3, activation of PAR1 with peptide agonist resulted in a robust ∼60% loss of receptor from the cell surface (Fig. 3B), which was indistinguishable from agonist-induced loss of cell-surface PAR1 observed in MDA-MB-231 cells re-expressing ARRDC3. These findings suggest that ARRDC3 is required for agonist-promoted PAR1 degradation but not for receptor internalization.
Figure 3.
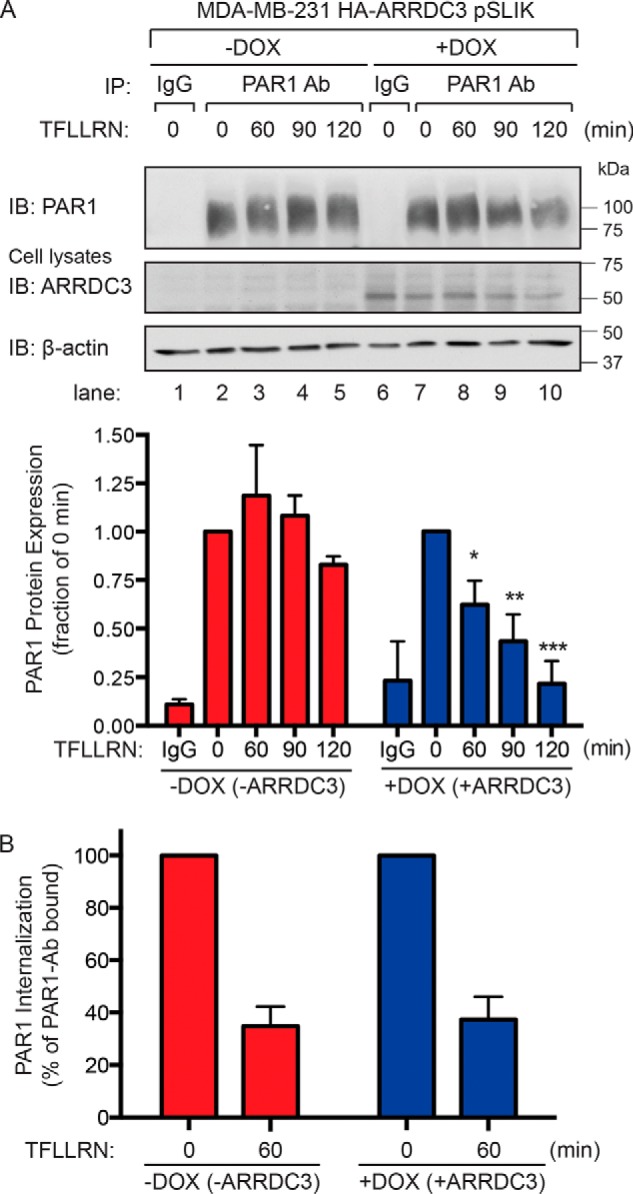
Agonist peptide–induced PAR1 lysosomal degradation is restored in cells re-expressing ARRDC3. A, MDA-MB-231 HA-ARRDC3 pSLIK cells were treated with or without 1 μg/ml DOX for 48 h. Cells were then stimulated with 100 μm TFLLRN peptide agonist for the indicated times, lysed, and immunoprecipitated using the anti-PAR1 WEDE antibody or anti-IgG antibody as a control. Immunoprecipitates were immunoblotted with anti-PAR1 rabbit polyclonal antibody. Cell lysates were immunoblotted with anti-HA antibody to detect ARRDC3. β-Actin expression was determined as a control. The data shown (mean ± S.D. (error bars), n = 3) were quantified by densitometry and represented as PAR1 expression relative to unstimulated control (0 min). Statistical significance was determined by one-way ANOVA (*, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 3). B, MDA-MB-231 HA-ARRDC3 pSLIK cells were treated with or without 1 μg/ml DOX for 48 h, labeled with anti-PAR1 WEDE antibody at 4 °C, and then stimulated with or without 100 μm TFLLRN for the indicated times. Cells were then fixed, and the amount of cell-surface PAR1 was determined by ELISA. The data (mean ± S.D., n = 3) are represented as the percentage of PAR1 remaining on the cell surface relative to untreated control (0 min) and representative of three independent experiments.
To further assess ARRDC3 function as a key mediator of PAR1 degradation, we examined whether ARRDC3 regulates PAR1 degradation when proteolytically activated by its natural ligand thrombin. In MDA-MB-231 HA-ARRDC3 pSLIK cells not treated with doxycycline and lacking ARRDC3 expression, thrombin stimulation resulted in a shift in PAR1 mobility indicative of receptor cleavage but failed to cause a substantial loss of PAR1 protein (Fig. 4A, lanes 2 and 3). PAR1 protein was not detected in IgG immunoprecipitates (Fig. 4A, lanes 1 and 4). In contrast, thrombin caused a significant ∼50% loss of PAR1 protein in MDA-MB-231 cells expressing ARRDC3 (Fig. 4A, lanes 5 and 6), suggesting that ARRDC3 is necessary for receptor lysosomal trafficking. However, thrombin-promoted rapid and robust PAR1 internalization in both MDA-MB-231 HA-ARRDC3 pSLIK cells with and without ARRDC3 expression (Fig. 4B), indicating that receptor internalization occurs independently of ARRDC3 expression. These results suggest that ARRDC3 is required for agonist-induced PAR1 trafficking from endosomes to a lysosomal degradative pathway in invasive breast carcinoma.
Figure 4.
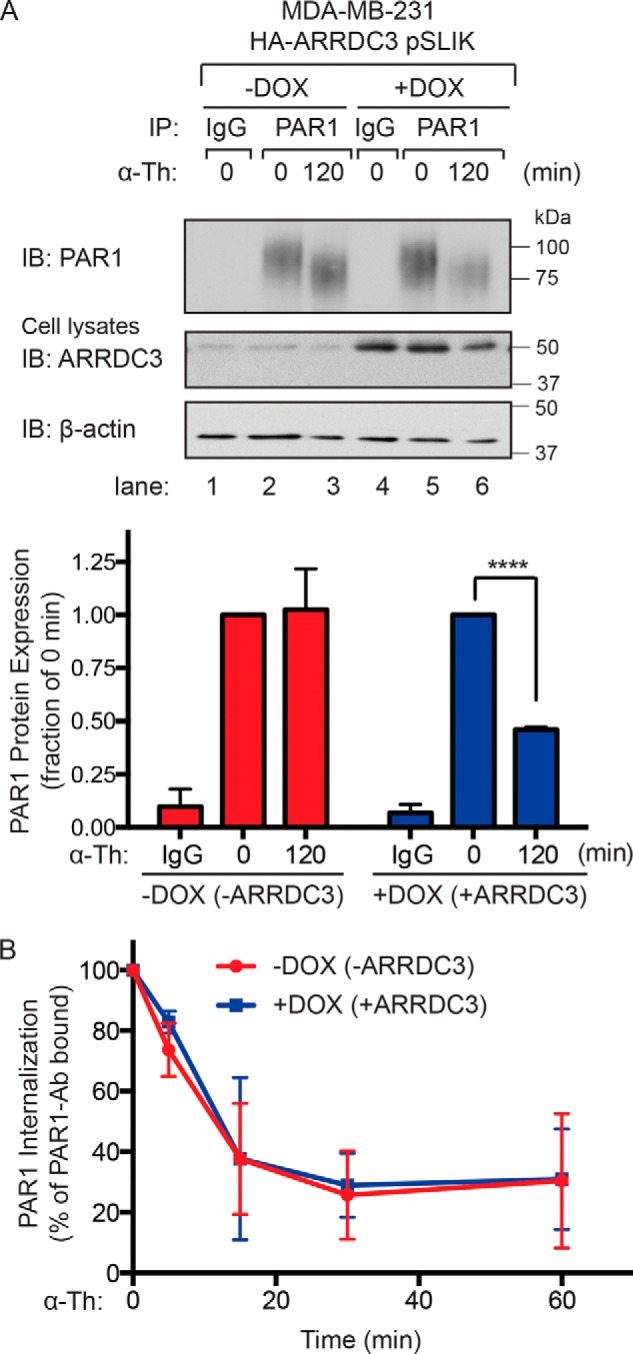
Thrombin-induced PAR1 lysosomal degradation is restored in cells re-expressing ARRDC3. A, MDA-MB-231 HA-ARRDC3 pSLIK cells incubated with or without 1 μg/ml DOX for 48 h were treated with 10 nm α-thrombin for the indicated times, lysed, and immunoprecipitated with anti-PAR1 WEDE antibody or anti-IgG control. Cell lysates were immunoblotted for HA-ARRDC3 and β-actin expression. The data (mean ± S.D. (error bars), n = 3) are represented as the fraction of PAR1 protein remaining relative to 0 min control. Statistical significance was determined using an unpaired t test (****, p < 0.0001; n = 3). B, MDA-MB-231 HA-ARRDC3 pSLIK cells were incubated with or without 1 μg/ml DOX for 48 h, prelabeled with anti-PAR1 WEDE antibody at 4 °C, and then treated with or without 10 nm α-thrombin. The amount of PAR1 remaining on the cell surface was determined by ELISA. The data (mean ± S.D., n = 3) are expressed as the percentage of PAR1 remaining relative to 0 min control and representative of three independent experiments.
ALIX is required for ARRDC3-mediated PAR1 degradation in invasive breast carcinoma
We showed previously that ARRDC3 regulates PAR1 degradation in HeLa cells by modulating the function of ALIX, an adaptor protein that binds directly to PAR1 and ESCRT-III to facilitate lysosomal sorting (33, 35). Thus, we next determined whether ARRDC3 regulates PAR1 degradation in invasive breast carcinoma through an ALIX-dependent pathway. To assess ALIX function, MDA-MB-231 HA-ARRDC3 pSLIK cells were transfected with ALIX-specific siRNAs to deplete cells of endogenous ALIX expression (Fig. 5, middle). In MDA-MB-231 HA-ARRDC3 pSLIK cells transfected with nonspecific siRNA and not treated with doxycycline, ALIX expression was easily detectable, whereas ARRDC3 expression was markedly low and thrombin failed to induce PAR1 degradation (Fig. 5, lanes 1 and 2). In contrast, nonspecific siRNA–transfected MDA-MB-231 HA-ARRDC3 pSLIK cells expressing ALIX and re-expressing ARRDC3 showed a significant ∼40% decrease in PAR1 protein following thrombin stimulation (Fig. 5, lanes 3 and 4). These findings suggest that ARRDC3 expression is sufficient to restore agonist-induced PAR1 degradation in MDA-MB-231 cells with ALIX expression. However, siRNA-mediated knockdown of ALIX blocked thrombin-stimulated PAR1 degradation in cells expressing ARRDC3 (Fig. 5, lanes 7 and 8). These findings indicate that ALIX expression is required for ARRDC3-mediated agonist-stimulated PAR1 degradation in invasive breast carcinoma.
Figure 5.
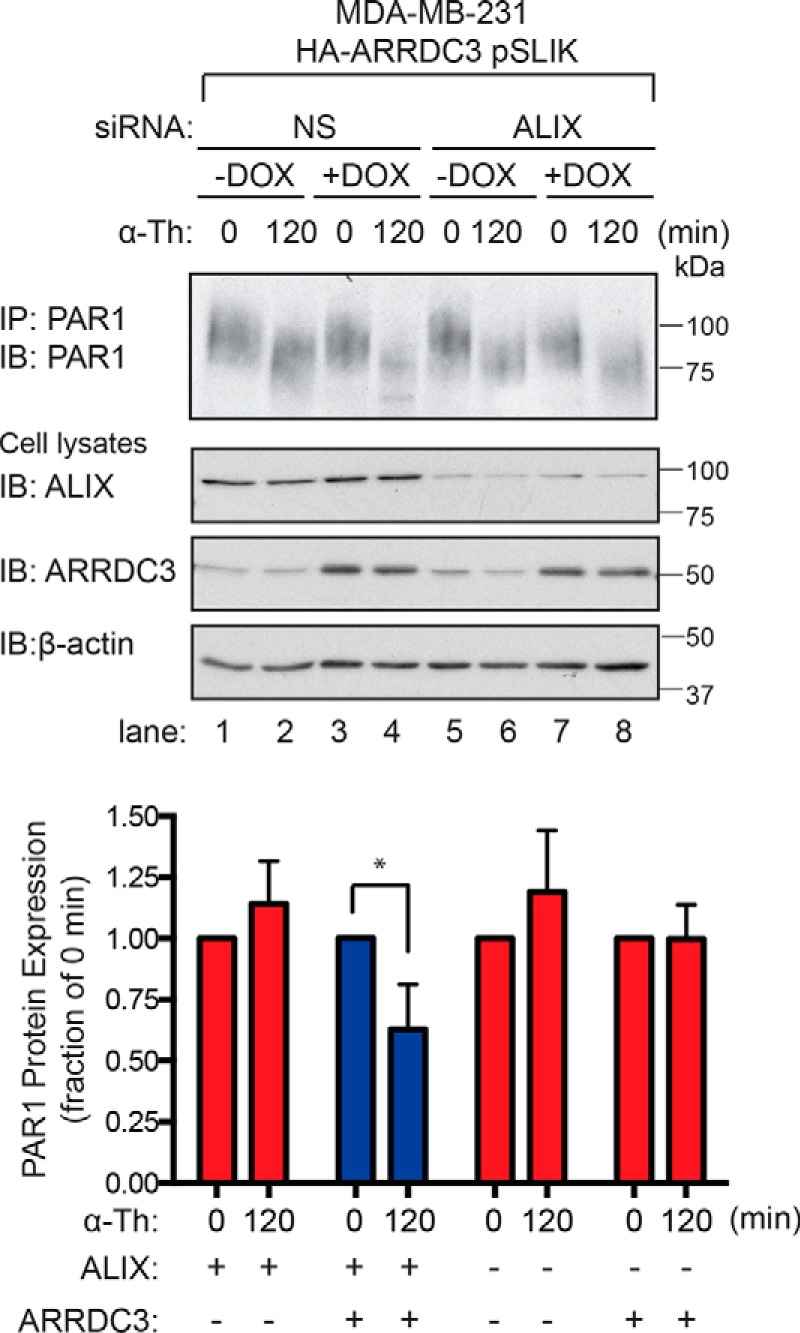
ALIX is required for ARRDC3-mediated degradation of activated PAR1. MDA-MB-231 HA-ARRDC3 pSLIK cells were transfected with 25 nm nonspecific (NS) or 25 nm of ALIX siRNA using a combination of 12.5 nm ALIX 1 and 12.5 nm ALIX 3 siRNAs and then incubated with or without 1 μg/ml DOX for 48 h. Cells were then stimulated with 10 nm α-thrombin for the indicated times, lysed, and immunoprecipitated using the anti-PAR1 WEDE antibody. Immunoprecipitates were immunoblotted with anti-PAR1 rabbit antibody to detect PAR1 expression. ALIX, HA-ARRDC3, and β-actin expression were determined by immunoblotting cell lysates. The data (mean ± S.D. (error bars), n = 3) are represented as the fraction of PAR1 remaining relative to 0 min control and are representative of three independent experiments. Statistical significance was determined by unpaired t test (*, p < 0.05; n = 3).
Sorting of activated PAR1 to lysosomes requires ARRDC3
To determine whether ARRDC3 directly mediates activated PAR1 sorting to multivesicular bodies/lysosomes, we used immunofluorescence confocal microscopy and LysoTracker to label lysosomes. To ensure that the cell-permeable LysoTracker probe labeled acidic lysosomal organelles, we immunostained MDA-MB-231 HA-ARRDC3 pSLIK cells with antibodies targeted against the lysosome-associated membrane protein-1 (LAMP1), a resident lysosomal membrane protein. LysoTracker accumulated in the lumen of organelles with distinct LAMP1 expression at the limiting membrane, as detected by confocal microscopy and line-scan analysis (Fig. 6A), suggesting that LysoTracker is a valid marker of lysosomes in MDA-MB-231 cells. MDA-MB-231 HA-ARRDC3 pSLIK cells incubated with or without doxycycline and LysoTracker were then labeled with anti-PAR1 antibodies at 4 °C. Under these conditions, only the cell-surface cohort of PAR1 binds antibody. Cells were washed and then stimulated with the PAR1-specific agonist peptide for 90 min. In untreated 0 min control cells, PAR1 localized mainly to the cell surface and not in LysoTracker-labeled organelles, irrespective of doxycycline induction of ARRDC3 (Fig. 6 (B and C), top panels). After 90 min of agonist stimulation, PAR1 redistributed from the cell surface to endocytic puncta and failed to co-localize with LysoTracker in cells that displayed minimal ARRDC3 expression (Fig. 6, B and D). In contrast, however, in cells treated with doxycycline and expressing HA-ARRDC3 (Fig. 6C), activated and internalized PAR1 accumulated within LysoTracker-labeled organelles, as shown by confocal microscopy and line-scan analysis (Fig. 6, C and E). Collectively, these data indicate that ARRDC3 expression is both necessary and sufficient to target activated PAR1 to lysosomes for degradation.
Figure 6.

ARRDC3 expression is required for activated PAR1 lysosomal trafficking. A, MDA-MB-231 HA-ARRDC3 pSLIK cells incubated with 100 nm LysoTracker were fixed, processed, immunostained for LAMP1, and imaged by confocal microscopy. Images are representative of many cells examined in three independent experiments. Scale bars, 10 μm. Line-scan analysis of the white dotted line region is plotted as the fraction of maximum pixel intensity versus pixel number (distance) and demonstrates that LysoTracker (red) accumulates in the lumen of LAMP1-positive lysosomes (green). MDA-MB-231 HA-ARRDC3 pSLIK cells treated without (B) or with (C) 1 μg/ml DOX for 48 h were incubated with 2 mm leupeptin and 100 nm LysoTracker at 37 °C. Cells were then incubated with anti-PAR1 antibody to label the surface cohort, stimulated with 100 μm TFLLRN, fixed, processed, and imaged by confocal microscopy. Images are representative of many cells examined in three independent experiments. Scale bars, 10 μm. Line-scan analysis of the white dotted line region is plotted as described above and indicates that activated PAR1 (green) accumulates in the lumen of LAMP1-positive lysosomes (red) in the presence of ARRDC3 (blue) (E) but not in lysosomes in the absence of ARRDC3 expression (D).
ARRDC3 attenuates PAR1-mediated persistent signaling and cellular invasion
Dysregulated PAR1 trafficking in invasive breast carcinoma results in persistent signaling and contributes to cellular invasion (15, 16). Because ARRDC3 expression is sufficient to restore activated PAR1 lysosomal sorting, we examined whether ARRDC3 expression was sufficient to attenuate activated PAR1 signaling. In these studies, we focused on activation of JNK, an effector of PAR1-stimulated G12/13 signaling and known mediator of PAR1-stimulated breast carcinoma invasion (26, 27). MDA-MB-231 cells express JNK1 and JNK2 (27), which are phosphorylated on threonine 183 and tyrosine 185 residues following activation of upstream MAP kinases induced by RhoA signaling (44). To assess the function of ARRDC3 on PAR1 signaling, MDA-MB-231 HA-ARRDC3 pSLIK cells were treated with or without doxycycline and stimulated with thrombin, and phosphorylation of JNK1/2 was determined by immunoblotting. In MDA-MB-231 HA-ARRDC3 pSLIK cells without doxycycline treatment and minimal ARRDC3 expression, thrombin induced a prolonged and robust ∼7-fold increase in JNK1/2 phosphorylation that peaked at 20 min (Fig. 7), consistent with enhanced and persistent PAR1 signaling. However, in MDA-MB-231 HA-ARRDC3 pSLIK cells treated with doxycycline and re-expressing ARRDC3, thrombin induced a transient and ∼3-fold increase in JNK1/2 phosphorylation (Fig. 7), a response substantially diminished compared with cells lacking ARRDC3 expression. These results suggest that the impact of ARRDC3 on PAR1 trafficking is important for regulating appropriate cellular signaling.
Figure 7.
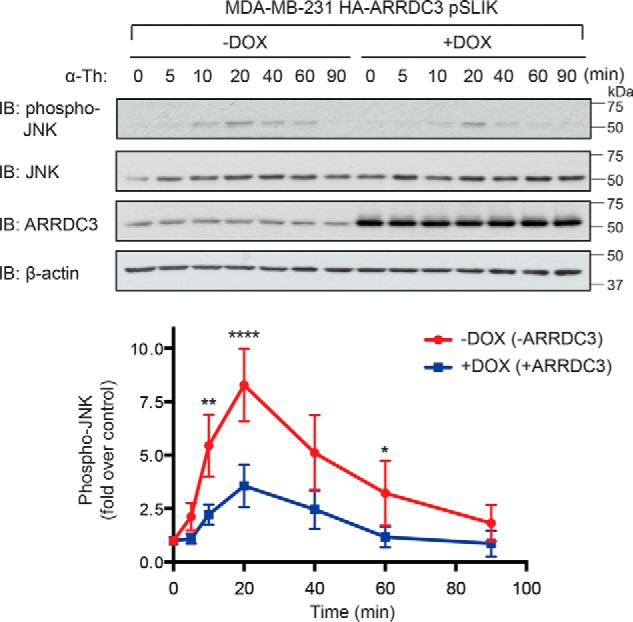
ARRDC3 re-expression attenuates PAR1-stimulated JNK signaling. MDA-MB-231 HA-ARRDC3 pSLIK cells treated with or without 1 μg/ml DOX for 48 h were stimulated with 10 nm α-thrombin for the indicated times. Cells were lysed and immunoblotted for phospho-JNK1/2, total JNK, HA-ARRDC3, and β-actin expression. The data shown (mean ± S.D. (error bars), n = 3) were quantified by densitometry and are represented as the -fold increase in JNK1/2 phosphorylation relative to 0 min control. Statistical significance was determined by one-way ANOVA (**, p < 0.01; ****, p < 0.0001; n = 3).
We next examined the function of ARRDC3 and JNK signaling on thrombin-induced breast carcinoma cellular invasion. In MDA-MB-231 HA-ARRDC3 pSLIK cells with no doxycycline treatment and low ARRDC3 expression, incubation with thrombin induced an ∼3-fold increase in cellular invasion compared with untreated control cells (Fig. 8A, top panels). This is consistent with previously reported effects of thrombin on breast carcinoma cellular invasion (15, 18). However, in MDA-MB-231 cells exhibiting doxycycline-induced ARRDC3 expression, thrombin-induced invasion was markedly reduced and equivalent to unstimulated control cells (Fig. 8A, bottom panels), suggesting that ARRDC3 suppresses thrombin-activated PAR1-driven breast carcinoma invasion. To determine whether JNK1/2 is important for thrombin-induced cellular invasion, MDA-MB-231 HA-ARRDC3 pSLIK cells were pretreated with the selective JNK inhibitor SP600125 or DMSO vehicle control. Thrombin stimulated a marked increase in JNK1/2 phosphorylation in MDA-MB-231 cells that was virtually abolished in cells treated with the JNK inhibitor SP600125, whereas thrombin-induced p38 phosphorylation was unaffected (Fig. 8B, inset). Importantly, thrombin-induced breast carcinoma invasion was markedly inhibited in cells preincubated with the JNK inhibitor SP600125 compared with DMSO-treated control cells (Fig. 8B). Together, these findings indicate that ARRDC3 functions as a tumor suppressor by controlling GPCR trafficking and consequently signaling that promotes breast cancer invasion (Fig. 9).
Figure 8.

PAR1-stimulated breast carcinoma invasion is suppressed by ARRDC3 and JNK inhibition. A, MDA-MB-231 HA-ARRDC3 pSLIK cells treated with or without 1 μg/ml DOX for 48 h were serum-starved cells, seeded onto transwells coated with Matrigel, and incubated with or without 1 pm α-thrombin (α-Th) for 5 h at 37 °C. Cells were fixed, stained, and imaged. Images shown are representative of three independent experiments. The data (mean ± S.D. (error bars), n = 3) were quantified from nine different fields of view at ×10 magnification for each condition and are represented as the -fold change over untreated control cells. Statistical significance was determined by unpaired t test (***, p < 0.001; n = 3). B, MDA-MB-231 HA-ARRDC3 pSLIK cells were preincubated with DMSO or 20 μm SP600125 JNK inhibitor for 2 h at 37 °C, seeded onto transwells coated with Matrigel, and then treated with 1 pm α-Th. Cells were processed, and statistical significance was determined by unpaired t test (****, p < 0.0001; n = 3). The inset shows effects of DMSO and SP600125 on α-Th–stimulated phosphorylation of JNK and p38; cell lysates were immunoblotted for total JNK and p38 as a control (Ctrl).
Figure 9.
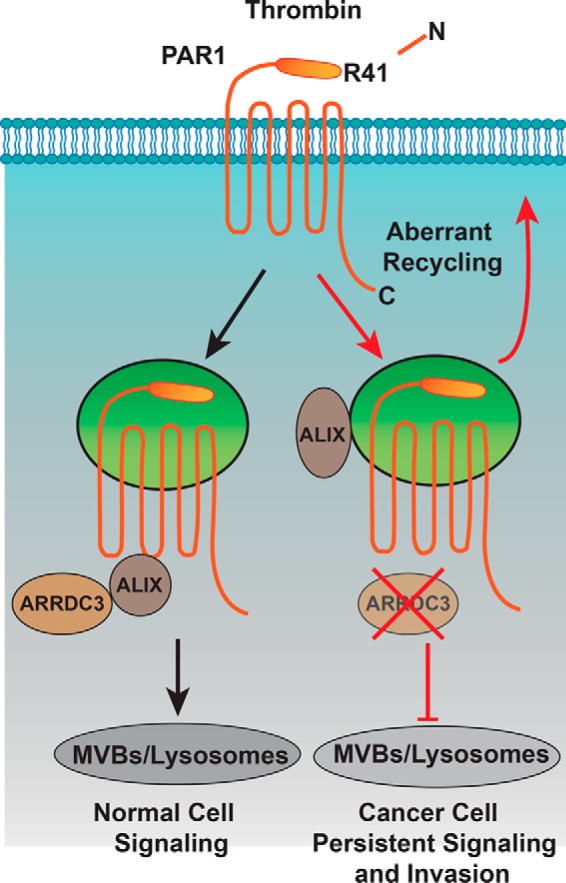
Model of ARRDC3 and PAR1 trafficking. Thrombin binds to and cleaves the PAR1 N terminus at arginine 41, exposing a new N-terminal domain that acts like a tethered ligand. Due to the irreversible proteolytic mechanism of PAR1 activation, internalization and lysosomal sorting is critical for termination of G protein signaling. Unlike most classic GPCRs, activated PAR1 is rapidly sorted from endosomes to lysosomes through a non-canonical pathway mediated by ARRDC3 and ALIX. In invasive breast cancer, activated PAR1 is internalized and recycled and fails to sort to lysosomes for degradation and consequently signals persistently, which promotes tumor cell invasion and growth. We discovered that loss of ARRDC3 of expression in invasive breast cancer is responsible for defective PAR1 trafficking that results in persistent signaling and cellular invasion.
Discussion
Whereas the survival rate for patients diagnosed at early stage breast cancer has improved, this is not the case for patients diagnosed with advanced metastatic breast cancer, which lacks druggable targets. We propose that highly druggable GPCRs are a promising class of targets for metastatic cancer. In fact, several lines of evidence indicate that the GPCR PAR1 is an attractive therapeutic target for advanced breast cancer (11, 45). PAR1 confers tumor cell motility, invasiveness, survival, and self-renewal in breast cancer and other cancer types (46). In addition, expression of PAR1 correlates with high breast tumor grade and poor patient prognosis (14). Despite compelling evidence supporting the oncogenic function of PAR1 in driving breast cancer progression, the defects that contribute to dysfunction of PAR1 in cancer remain largely unknown. We previously showed that dysregulation of PAR1 trafficking results in persistent signaling and consequently promotes tumor invasion and growth (15, 16), but the underlying defects resulting in aberrant PAR1 trafficking are not known. In the present study, we sought to determine the mechanism responsible for aberrant PAR1 trafficking and signaling in invasive breast carcinoma. We found that expression of the α-arrestin ARRDC3, a key regulator of PAR1 trafficking (35), is suppressed in basal-like invasive breast carcinoma, which exhibit high PAR1 expression. We further show that re-expression of ARRDC3 in basal-like invasive breast carcinoma is sufficient to restore normal PAR1 lysosomal trafficking, which occurs through an ALIX-dependent endosomal-lysosomal sorting pathway (Fig. 9). Importantly, ARRDC3 re-expression also attenuated thrombin-stimulated JNK signaling and breast carcinoma invasion. These studies are the first to demonstrate a role for the tumor suppressor ARRDC3 in regulation of GPCR trafficking and signaling in invasive basal-like breast carcinoma.
The α-arrestin ARRDC3 shares structural homology to the β-arrestin family of adaptor proteins (47), which have important regulatory roles in mammalian GPCR signaling and trafficking (37). ARRDC3 has been shown to regulate endosomal sorting of the β2AR, a classic GPCR (36–38). However, the precise role of ARRDC3 in this pathway remains controversial, given the dominant role of β-arrestins in regulation of β2AR function (36, 38). ARRDC3 possesses arrestin-like N- and C-domains and C-terminal PPXY motifs that bind to WW domains of HECT domain–containing E3 ubiquitin ligases (48, 49). Despite the presence of arrestin-like domains, the α-arrestin ARRDC3 appears to lack a polar core, which is essential for β-arrestin binding to activated and phosphorylated GPCRs (50). In addition, β-arrestins lack C-terminal PPXY motifs, indicating that α-arrestin and β-arrestin proteins probably have divergent functions. We previously showed that ARRDC3 plays a critical role in regulating PAR1 lysosomal trafficking in HeLa cells. Unlike the β2AR, however, ARRDC3 appears to regulate PAR1 lysosomal sorting by modulating ALIX ubiquitination via recruitment of the E3 ubiquitin ligase WWP2 (35). Ubiquitination of ALIX enhances its dimerization and binding to activated PAR1 and ESCRT-III to facilitate receptor lysosomal degradation (33, 35). However, there is limited knowledge as to how loss of ARRDC3 disrupts cellular homeostasis to promote breast cancer progression. In the present study, we found that ARRDC3 expression is suppressed in basal-like invasive breast carcinoma, which exhibit high PAR1 expression and dysregulated receptor trafficking (15, 16). We further report that re-expression of ARRDC3 is sufficient to restore normal agonist-induced PAR1 lysosomal degradation in invasive breast carcinoma. In addition, ARRDC3 controls PAR1 lysosomal sorting through an ALIX-dependent pathway in invasive breast carcinoma, consistent with results from our previous HeLa cell studies (35). These findings indicate that ARRDC3 tumor suppressor function is linked to regulation of GPCR trafficking.
ARRDC3 has been identified as a tumor suppressor in breast and prostate cancer (39–41, 51). Using combined high-resolution analysis of genome copy number and gene expression in primary basal and luminal breast cancers, ARRDC3 expression was reported to be low or absent in basal-like breast cancer, the most aggressive and metastatic subtype of breast carcinoma, which lack targeted therapies (40). ARRDC3 expression was also shown to decrease with tumor grade, metastasis, and recurrence and was found in a gene cluster on chromosome 5 deleted in 17% of basal-like breast cancers, compared with 0% deletion in luminal breast cancers (39, 40). In addition to gene deletion, ARRDC3 expression is suppressed by epigenetic silencing in basal-like breast carcinoma cells through ARRDC3 promoter deacetylation or hypermethylation that results in inactivation of the tumor-suppressor gene (41, 52). Endogenous small non-coding microRNAs that control ARRDC3 gene expression have also been reported to function in regulation of ARRDC3 gene expression in prostate cancer (53). The strong correlation between ARRDC3 expression and tumor progression indicates that loss of ARRDC3 expression is linked to dysregulation of important cancer drivers. One target of ARRDC3 described previously is the membrane protein integrin β4 (39), which is enriched in triple-negative breast cancer and a marker of poor prognosis (54, 55). Here, we now report that in addition to functioning in regulation of integrin β4, loss of ARRDC3 in invasive breast carcinoma results in aberrant PAR1 lysosomal trafficking, persistent signaling, and consequent breast carcinoma cellular invasion. These findings indicate that the tumor suppressor function of ARRDC3 is linked to the regulation of both integrin receptors and GPCRs. Although there is currently no known direct link between integrin β4 and PAR1, integrin β1 has been implicated in PAR1-promoted chemotaxis (56), invasion (57), and proliferation and ERK1/2 activation (58) in skin, bone, and brain tumor cells, respectively. In addition, ανβ5 cooperates with PAR1 in thrombin-mediated lung cancer cell invasion (59). Thus, future studies are important to determine whether integrin β4 is integrated in PAR1-driven breast cancer progression.
RhoA signaling (16) has been implicated in tumorigenesis and cancer progression (60, 61). Several previous studies have shown that constitutively active Gα12 and Gα13 proteins as well as induction of Gα12/13 signaling through activation of PAR1 increase invasion of breast carcinoma in vitro, whereas inhibition of G12 signaling reduces metastasis and improves metastasis-free survival in vivo (26). In the present studies, we report that ARRDC3 tumor suppressor function is linked to the regulation of PAR1-stimulated G12-effector JNK signaling, which mediates breast carcinoma invasion. We discovered that ARRDC3 functions as a negative regulator of PAR1-stimulated breast carcinoma invasion. In addition, ARRDC3 appears to control breast cancer invasion though JNK signaling. In the absence of ARRDC3, PAR1 stimulated a marked and prolonged increase in JNK signaling; however, re-expression of ARRDC3 in breast cancer cells resulted in a significant reduction in both the magnitude and duration of JNK signaling induced by PAR1. JNK has been shown to contribute to malignant transformation, drug resistance, and tumor growth in various cancer types (62). JNK phosphorylates a large number of targets, mainly transcription factors, and regulates cellular proliferation, survival, and apoptosis, but precisely how JNK contributes to breast carcinoma invasion stimulated by a GPCR is not known, and this is an area of active investigation.
In summary, this study reveals a novel and important role for the tumor suppressor ARRDC3 in the regulation of GPCR trafficking and signaling in invasive basal-like breast cancer. JNK signaling makes important contributions to tumor progression and is an effector of GPCR-G12/13 RhoA signaling. The work reported here now suggests that the tumor suppressor ARRDC3 functions not only by regulating PAR1 trafficking but also through modulating appropriate JNK signaling, a critical driver of breast cancer invasion. Thus, in future studies, it will be important to determine how JNK regulates certain substrates to control GPCR-induced breast cancer invasion in vitro and the consequences for tumor invasion and growth in vivo.
Experimental procedures
Reagents and antibodies
The PAR1 agonist peptide TFLLRNPNDK (“TFLLRN”) was synthesized as the carboxyl amide and purified by reverse-phase high-pressure liquid chromatography by the Tufts University Core Facility (Boston, MA). Human α-thrombin was obtained from Enzyme Research Technologies (South Bend, IN). Protein A-Sepharose CL-4B beads were from GE Healthcare. Mouse IgG (catalog no. 010-0102) and polyclonal anti-HA antibodies (catalog no. 600-401-384) were purchased from Rockland Immunochemicals (Gilbertsville, PA). Mouse monoclonal anti-HA antibody (HA.11) (catalog no. MMS-101R) was purchased from Covance (Princeton, NJ). Mouse monoclonal anti-PAR1 WEDE antibody (catalog no. IM2584) was purchased from Beckman Coulter (Fullerton, CA). Rabbit anti-PAR1 polyclonal antibody was described previously (63). Rabbit polyclonal anti-ARRDC3 antibody (catalog no. ab64187) was purchased from Abcam (Cambridge, UK). Monoclonal anti-ALIX antibody (catalog no. sc-53538) was from Santa Cruz Biotechnology (Dallas, TX). Mouse anti-LAMP1 (catalog no. 15665), rabbit anti-phospho-JNK1/2 (catalog no. 9251), and rabbit anti-JNK (catalog no. 9252) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Monoclonal anti-β-actin antibody (catalog no. A5316) and SP600125 (JNK inhibitor) were purchased from Sigma-Aldrich, and anti-GAPDH antibody (catalog no. GTX627408) was from GeneTex (Irvine, CA). Goat anti-mouse secondary antibody conjugated to Alexa Fluor 488 (catalog no. A-11001) and goat anti-rabbit secondary antibody conjugated to Alexa Fluor 647 (A-21244) were purchased from Thermo Fisher Scientific (Waltham, MA). Goat anti-mouse (catalog no. 170-6516) and goat anti-rabbit (catalog no. 170-6515) secondary antibodies conjugated to horseradish peroxidase (HRP) were purchased from Bio-Rad. Doxycycline was purchased from Clontech Takara Bio USA (Mountain View, CA). LysoTracker, ProLong Gold, and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) were purchased from Thermo Fisher Scientific.
Cell culture and transfections
MDA-MB-231 cells were purchased from ATCC and maintained without CO2 in Leibowitz-15 medium supplemented with 10% fetal bovine serum (v/v). MCF7, BT549, T47D, BT474, Hs578T, and SKBR3 cells were all purchased from ATCC and grown according to ATCC instructions. siRNA transfections were performed using Oligofectamine (Life Technologies, Inc.) per the manufacturer's instructions. Degradation and invasion assays described were performed 48 h after transfection. All single siRNAs were purchased from Qiagen: nonspecific siRNA sequence, 5′-CUACGUCCAGGAGCGCACC-3′; ALIX 1 target sequence, 5′-AAGTACCTCAGTCTATATTGA-3′; ALIX 3 target sequence, 5′-AATCGAGACGCTCCTGAGATA-3′.
ARRDC3 lentiviral construct and stable cell lines
The pSLIK lentiviral vectors were constructed as described previously (64, 65). pSLIK-Hygro (Addgene plasmid 25737) and pEN_TmiRc3 (Addgene plasmid 25748) were gifts from Dr. Iain Fraser (National Institutes of Health, Bethesda, MD) (65). Briefly, N-terminal HA-tagged human ARRDC3 cDNA was cloned into the entry vector pEN_TmiRc3 using restriction sites SpeI and EcoRI downstream of tetracycline response element promoter. The tetracycline response element promoter and ARRDC3 cDNA were then transferred to the pSLIK-Hygro destination vector expressing reverse tetracycline transactivator and hygromycin-selection marker by Gateway recombination (Invitrogen) to generate pSLIK encoding tetracycline-inducible ARRDC3 (pSLIK-ARRDC3). Lentivirus was produced by transfecting 293T cells with pSLIK-ARRDC3 vector along with packaging plasmids pMDL, pRSV, and pVSV using polyethyleneimine. Lentivirus-containing supernatant was harvested on day 2 post-transfection, filtered, and used to transduce MDA-MB-231 cells overnight with 8 μg/ml Polybrene (EMD Millipore). MDA-MB-231 cells stably expressing pSLIK-ARRDC3 vector were selected using 200 μg/ml hygromycin (Omega Scientific).
Immunoblotting
Cell lysates were collected in 2× Laemmli sample buffer containing 200 mm DTT. Samples were resolved by SDS-PAGE, transferred to PVDF membranes, immunoblotted with appropriate antibodies, and then developed by chemiluminescence. Immunoblots were quantified by densitometry using ImageJ software (National Institutes of Health). For comparison of ARRDC3 and ALIX expression, subconfluent cells in the exponential growth stage were lysed in radioimmune precipitation assay lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 5 mm EDTA, and 1% Triton X-100) with freshly added protease inhibitors (2 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 mm phenylmethylsulfonyl fluoride) and phosphatase inhibitors (50 mm NaF, 10 mm sodium pyrophosphate) and quantified by BCA analysis (Pierce BCA Protein Assay Kit; Thermo Scientific), and equal amounts of lysates were used for immunoblotting.
Cell-surface ELISA
PAR1 surface expression and internalization assays were performed by ELISA, as described previously (66). Briefly, 2 × 105 cells were plated in triplicate on a fibronectin-coated 12-well plate. After 3 days, confluent cells were incubated with starvation medium (Dulbecco's modified Eagle's medium with 1 mm HEPES and 0.1% BSA) for 1 h at 37 °C, washed with chilled PBS, and fixed with 4% paraformaldehyde on ice for 15 min. Cells were then incubated with mouse anti-PAR1 WEDE antibody or mouse IgG for 1 h at 4 °C and followed by secondary HRP-conjugated goat anti-mouse antibody for 1 h at room temperature. Antibody bound to the cell surface was detected by incubation with one-step 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) substrate for 10–20 min at room temperature and measured at the absorbance of 405 nm using a microplate reader (SpectraMax Plus, Molecular Devices). The amount of cell-surface PAR1 was determined by subtracting the background mouse IgG from the samples labeled with anti-PAR1 antibody. For PAR1 internalization assays, 24-well plates were coated with fibronectin, and 7.5 × 104 MDA-MB-231 pSLIK cells/well were plated. Cells treated with doxycycline for 48 h were serum-starved for 1 h and then incubated on ice with anti-PAR1 antibody to label surface PAR1. Cells were stimulated with PAR1 agonist for various times, fixed, and incubated with HRP-conjugated secondary antibody. The amount of antibody remaining at the cell surface was detected and quantified as described above.
Immunoprecipitation
To assess PAR1 degradation, immunoprecipitation of endogenous PAR1 was performed as described previously (67). Briefly, cells were plated in 6-cm dishes at a density of 1 × 106 cells/dish and treated with 1 μg/ml doxycyline the following day. For siRNA knockdown, cells were seeded in 6-well plates at 5 × 105 cells/well, transfected as described, and treated with doxycycline the following day. After a 48-h doxycycline treatment, cells were serum-starved for 1 h; treated with α-thrombin; and then placed on ice, washed with PBS, and lysed with Triton X-100 lysis buffer supplemented with a mixture of protease inhibitors. Cell lysates were sonicated for 10 s at 10% amplitude (Branson model 450 sonifier) and cleared by centrifugation. A BCA assay (Thermo Fisher Scientific) was performed to determine protein concentrations. Equal amounts of normalized lysates were immunoprecipitated with appropriate antibodies and preblocked Protein A-Sepharose beads overnight at 4 °C. Immunoprecipitates were washed three times with lysis buffer, and proteins were eluted in 50 μl of 2× Laemmli sample buffer containing 200 mm DTT. Cell lysates and PAR1 immunoprecipitates were analyzed by immunoblotting.
Immunofluorescence confocal microscopy
Cells were plated at a density of 4 × 105 cells/well on fibronectin-coated glass coverslips placed in a 12-well dish and grown overnight. Cells were treated with doxycycline for 48 h and then serum-starved in Dulbecco's modified Eagle's medium containing 1 mg/ml BSA, 2 mm leupeptin, and 100 nm LysoTracker for 1 h at 37 °C. Cells were then incubated at 4 °C with anti-PAR1 WEDE antibody to label the surface population of receptors and then stimulated with 100 μm TFLLRN, fixed with 4% paraformaldehyde in PBS, permeabilized with methanol, immunostained with polyclonal anti-HA antibody, and processed as described previously (66). Coverslips were mounted with ProLong Gold reagent. Confocal images of 0.28-μm x-y sections were collected sequentially using an Olympus IX81 DSU spinning confocal microscope fitted with a Plan Apo ×60 oil objective and a Hamamatsu ORCAER digital camera using Metamorph version 7.7.4.0 software (Molecular Devices). Fluorescence intensity line-scan analysis was performed using ImageJ software.
Signaling assays
Signaling assays were performed essentially as described previously (3). Briefly, cells were treated with or without 1 μg/ml doxycycline for 48 h and starved for 1 h at 37 °C. Cells were stimulated with 10 nm α-thrombin for the indicated times at 37 °C, and cell lysates were collected by direct lysis in 2× Laemmli sample buffer containing 200 mm DTT. Samples were resolved by SDS-PAGE and immunoblotted with anti-phospho-JNK antibody. PVDF membranes were stripped and reprobed with anti-JNK antibody.
Invasion assays
For ARRDC3 re-expression experiments, cells were treated with doxycycline for 48 h and serum-starved overnight. For JNK inhibitor experiments, cells were serum-starved overnight and then treated with DMSO or JNK inhibitor SP600125 (20 μm) for 2 h. Cells were dissociated using Cellstripper solution (Corning, NY) and seeded onto BioCoatTM Matrigel® invasion chambers (Corning, NY) with or without 1 pm α-thrombin added. For JNK inhibitor experiments, DMSO or SP600125 (20 μm) were also added during seeding into invasion chambers. Cells were allowed to invade for 5 h at 37 °C, fixed, and stained with 0.5% crystal violet in ethanol. Membranes were dried overnight, and cells that had invaded through the Matrigel and membrane were imaged using a Leica DMi1 inverted microscope (Leica Microsystems). Cell invasion was quantified by cell count in nine fields of view at ×10 magnification for each condition, from three biological independent replicates.
Data analysis
Statistical significance was determined by unpaired t test or one-way ANOVA using Prism version 4.0 software (GraphPad).
Author contributions
A. K. S. A., W. -A. P., and J. T. conceptualization; A. K. S. A. software; A. K. S. A. formal analysis; A. K. S. A., W. -A. P., and H. L. validation; A. K. S. A., W. -A. P., and H. L. investigation; A. K. S. A. visualization; A. K. S. A., W. -A. P., and H. L. methodology; A. K. S. A., W. -A. P., and J. T. writing-original draft; A. K. S. A., W. -A. P., and J. T. writing-review and editing; J. T. supervision; J. T. funding acquisition; J. T. project administration.
Acknowledgments
We thank all members of the Trejo laboratory for comments and advice. We are especially thankful to Dr. Michael R. Dores (Hofstra University) for advice, technical expertise, and graphic design.
This work was supported, in whole or in part, by NIGMS, National Institutes of Health, Grant R01 GM090689 (to J. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GPCR
- G protein–coupled receptor
- ALIX
- ALG-interacting protein X
- ARRDC3
- α-arrestin domain-containing protein-3
- ANOVA
- analysis of variance
- β2AR
- β2-adrenergic receptor
- DOX
- doxycycline
- ESCRT
- endosomal sorting complexes required for transport
- JNK
- c-Jun N-terminal kinase
- LAMP1
- lysosome-associated membrane protein-1
- PAR1
- protease-activated receptor-1
- pSLIK
- single lentiviral vector for inducible knockdown
- IB
- immunoblotting
- IP
- immunoprecipitation
- MAP
- mitogen-activated protein
- HRP
- horseradish peroxidase
- HA
- hemagglutinin
- α-Th
- α-thrombin.
References
- 1. Singh A., Nunes J. J., and Ateeq B. (2015) Role and therapeutic potential of G-protein coupled receptors in breast cancer progression and metastases. Eur. J. Pharmacol. 763, 178–183 10.1016/j.ejphar.2015.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bar-Shavit R., Maoz M., Kancharla A., Nag J. K., Agranovich D., Grisaru-Granovsky S., and Uziely B. (2016) G protein-coupled receptors in cancer. Int. J. Mol. Sci. 17, E1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Hayre M., Vázquez-Prado J., Kufareva I., Stawiski E. W., Handel T. M., Seshagiri S., and Gutkind J. S. (2013) The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 13, 412–424 10.1038/nrc3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prickett T. D., Wei X., Cardenas-Navia I., Teer J. K., Lin J. C., Walia V., Gartner J., Jiang J., Cherukuri P. F., Molinolo A., Davies M. A., Gershenwald J. E., Stemke-Hale K., Rosenberg S. A., Margulies E. H., and Samuels Y. (2011) Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nat. Genet. 43, 1119–1126 10.1038/ng.950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kan Z., Jaiswal B. S., Stinson J., Janakiraman V., Bhatt D., Stern H. M., Yue P., Haverty P. M., Bourgon R., Zheng J., Moorhead M., Chaudhuri S., Tomsho L. P., Peters B. A., Pujara K., et al. (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873 10.1038/nature09208 [DOI] [PubMed] [Google Scholar]
- 6. Kuzumaki N., Suzuki A., Narita M., Hosoya T., Nagasawa A., Imai S., Yamamizu K., Morita H., Suzuki T., Okada Y., Okano H. J., Yamashita J. K., Okano H., and Narita M. (2012) Multiple analyses of G-protein coupled receptor (GPCR) expression in the development of gefitinib-resistance in transforming non-small-cell lung cancer. PLoS One 7, e44368 10.1371/journal.pone.0044368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Feng X., Degese M. S., Iglesias-Bartolome R., Vaque J. P., Molinolo A. A., Rodrigues M., Zaidi M. R., Ksander B. R., Merlino G., Sodhi A., Chen Q., and Gutkind J. S. (2014) Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated ρ GTPase signaling circuitry. Cancer Cell 25, 831–845 10.1016/j.ccr.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santos R., Ursu O., Gaulton A., Bento A. P., Donadi R. S., Bologa C. G., Karlsson A., Al-Lazikani B., Hersey A., Oprea T. I., and Overington J. P. (2017) A comprehensive map of molecular drug targets. Nat. Rev. Drug. Discov. 16, 19–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sriram K., and Insel P. A. (2018) GPCRs as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 10.1124/mol.117.111062 10.1124/mol.117.111062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hawes B. E., Zhai Y., Hesk D., Wirth M., Wei H., Chintala M., and Seiffert D. (2015) In vitro pharmacological characterization of vorapaxar, a novel platelet thrombin receptor antagonist. Eur. J. Pharmacol. 762, 221–228 10.1016/j.ejphar.2015.05.046 [DOI] [PubMed] [Google Scholar]
- 11. Hamilton J. R., and Trejo J. (2017) Challenges and opportunities in protease-activated receptor drug development. Annu. Rev. Pharmacol. Toxicol. 57, 349–373 10.1146/annurev-pharmtox-011613-140016 [DOI] [PubMed] [Google Scholar]
- 12. Even-Ram S., Uziely B., Cohen P., Grisaru-Granovsky S., Maoz M., Ginzburg Y., Reich R., Vlodavsky I., and Bar-Shavit R. (1998) Thrombin receptor overexpression in malignant and physiological invasion processes. Nat. Med. 4, 909–914 10.1038/nm0898-909 [DOI] [PubMed] [Google Scholar]
- 13. Korkola J. E., DeVries S., Fridlyand J., Hwang E. S., Estep A. L., Chen Y. Y., Chew K. L., Dairkee S. H., Jensen R. M., and Waldman F. M. (2003) Differentiation of lobular versus ductal breast carcinomas by expression microarray analysis. Cancer Res. 63, 7167–7175 [PubMed] [Google Scholar]
- 14. Hernández N. A., Correa E., Avila E. P., Vela T. A., and Pérez V. M. (2009) PAR1 is selectively over expressed in high grade breast cancer patients: a cohort study. J. Transl. Med. 7, 47 10.1186/1479-5876-7-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Booden M. A., Eckert L. B., Der C. J., and Trejo J. (2004) Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol. Cell Biol. 24, 1990–1999 10.1128/MCB.24.5.1990-1999.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arora P., Cuevas B. D., Russo A., Johnson G. L., and Trejo J. (2008) Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 27, 4434–4445 10.1038/onc.2008.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang E., Boire A., Agarwal A., Nguyen N., O'Callaghan K., Tu P., Kuliopulos A., and Covic L. (2009) Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res. 69, 6223–6231 10.1158/0008-5472.CAN-09-0187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boire A., Covic L., Agarwal A., Jacques S., Sherifi S., and Kuliopulos A. (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120, 303–313 10.1016/j.cell.2004.12.018 [DOI] [PubMed] [Google Scholar]
- 19. Yang E., Cisowski J., Nguyen N., O'Callaghan K., Xu J., Agarwal A., Kuliopulos A., and Covic L. (2016) Dysregulated protease activated receptor 1 (PAR1) promotes metastatic phenotype in breast cancer through HMGA2. Oncogene 35, 1529–1540 10.1038/onc.2015.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vu T. K., Hung D. T., Wheaton V. I., and Coughlin S. R. (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 10.1016/0092-8674(91)90261-V [DOI] [PubMed] [Google Scholar]
- 21. Vu T. K., Wheaton V. I., Hung D. T., Charo I., and Coughlin S. R. (1991) Domains specifying thrombin-receptor interaction. Nature 353, 674–677 10.1038/353674a0 [DOI] [PubMed] [Google Scholar]
- 22. Koizume S., Jin M. S., Miyagi E., Hirahara F., Nakamura Y., Piao J. H., Asai A., Yoshida A., Tsuchiya E., Ruf W., and Miyagi Y. (2006) Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Res. 66, 9453–9460 10.1158/0008-5472.CAN-06-1803 [DOI] [PubMed] [Google Scholar]
- 23. Kuliopulos A., Covic L., Seeley S. K., Sheridan P. J., Helin J., and Costello C. E. (1999) Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry 38, 4572–4585 10.1021/bi9824792 [DOI] [PubMed] [Google Scholar]
- 24. Coughlin S. R. (2005) Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 3, 1800–1814 10.1111/j.1538-7836.2005.01377.x [DOI] [PubMed] [Google Scholar]
- 25. Arora P., Ricks T. K., and Trejo J. (2007) Protease-activated receptor signalling, endocytic sorting and dysregulation in cancer. J. Cell Sci. 120, 921–928 10.1242/jcs.03409 [DOI] [PubMed] [Google Scholar]
- 26. Kelly P., Moeller B. J., Juneja J., Booden M. A., Der C. J., Daaka Y., Dewhirst M. W., Fields T. A., and Casey P. J. (2006) The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. U.S.A. 103, 8173–8178 10.1073/pnas.0510254103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Juneja J., Cushman I., and Casey P. J. (2011) G12 signaling through c-Jun NH2-terminal kinase promotes breast cancer cell invasion. PLoS One 6, e26085 10.1371/journal.pone.0026085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trejo J., Hammes S. R., and Coughlin S. R. (1998) Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc. Natl. Acad. Sci. U.S.A. 95, 13698–13702 10.1073/pnas.95.23.13698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paing M. M., Stutts A. B., Kohout T. A., Lefkowitz R. J., and Trejo J. (2002) β-Arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J. Biol. Chem. 277, 1292–1300 10.1074/jbc.M109160200 [DOI] [PubMed] [Google Scholar]
- 30. Trejo J., and Coughlin S. R. (1999) The cytoplasmic tails of protease-activated receptor-1 and substance P receptor specify sorting to lysosomes versus recycling. J. Biol. Chem. 274, 2216–2224 10.1074/jbc.274.4.2216 [DOI] [PubMed] [Google Scholar]
- 31. Hurley J. H., and Hanson P. I. (2010) Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat. Rev. Mol. Cell Biol. 11, 556–566 10.1038/nrm2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dores M. R., and Trejo J. (2014) Atypical regulation of G protein-coupled receptor intracellular trafficking by ubiquitination. Curr. Opin. Cell Biol. 27, 44–50 10.1016/j.ceb.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dores M. R., Chen B., Lin H., Soh U. J., Paing M. M., Montagne W. A., Meerloo T., and Trejo J. (2012) ALIX binds a YPX(3)L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J. Cell Biol. 197, 407–419 10.1083/jcb.201110031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dores M. R., Grimsey N. J., Mendez F., and Trejo J. (2016) ALIX regulates the ubiquitin-independent lysosomal sorting of the P2Y1 purinergic receptor via a YPX3L motif. PLoS One 11, e0157587 10.1371/journal.pone.0157587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dores M. R., Lin H., J. Grimsey N., Mendez F., and Trejo J. (2015) The α-arrestin ARRDC3 mediates ALIX ubiquitination and G protein-coupled receptor lysosomal sorting. Mol. Biol. Cell 26, 4660–4673 10.1091/mbc.E15-05-0284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nabhan J. F., Pan H., and Lu Q. (2010) Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the β2-adrenergic receptor. EMBO Rep. 11, 605–611 10.1038/embor.2010.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tian X., Irannejad R., Bowman S. L., Du Y., Puthenveedu M. A., von Zastrow M., and Benovic J. L. (2016) The α-arrestin ARRDC3 regulates the endosomal residence time and intracellular signaling of the β2-adrenergic receptor. J. Biol. Chem. 291, 14510–14525 10.1074/jbc.M116.716589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Han S. O., Kommaddi R. P., and Shenoy S. K. (2013) Distinct roles for β-arrestin2 and arrestin-domain-containing proteins in β2 adrenergic receptor trafficking. EMBO Rep. 14, 164–171 10.1038/embor.2012.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Draheim K. M., Chen H. B., Tao Q., Moore N., Roche M., and Lyle S. (2010) ARRDC3 suppresses breast cancer progression by negatively regulating integrin β4. Oncogene 29, 5032–5047 10.1038/onc.2010.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adélaïde J., Finetti P., Bekhouche I., Repellini L., Geneix J., Sircoulomb F., Charafe-Jauffret E., Cervera N., Desplans J., Parzy D., Schoenmakers E., Viens P., Jacquemier J., Birnbaum D., Bertucci F., and Chaffanet M. (2007) Integrated profiling of basal and luminal breast cancers. Cancer Res. 67, 11565–11575 10.1158/0008-5472.CAN-07-2536 [DOI] [PubMed] [Google Scholar]
- 41. Soung Y. H., Pruitt K., and Chung J. (2014) Epigenetic silencing of ARRDC3 expression in basal-like breast cancer cells. Sci. Rep. 4, 3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huan D. W., Hui L. P., Wang X., Gao H. J., Sun J., Sun X., Wang C. F., and Zeng X. D. (2016) Low expression of ARRDC3 is associated with tumor invasion in colorectal cancer patients. Int. J. Clin. Exp. Pathol. 9, 5588–5593 [Google Scholar]
- 43. Sette P., Jadwin J. A., Dussupt V., Bello N. F., and Bouamr F. (2010) The ESCRT-associated protein Alix recruits the ubiquitin ligase Nedd4-1 to facilitate HIV-1 release through the LYPXnL L domain motif. J. Virol. 84, 8181–8192 10.1128/JVI.00634-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kyriakis J. M., and Avruch J. (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869 10.1152/physrev.2001.81.2.807 [DOI] [PubMed] [Google Scholar]
- 45. Flaumenhaft R., and De Ceunynck K. (2017) Targeting PAR1: now what? Trends Pharmacol. Sci. 38, 701–716 10.1016/j.tips.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wojtukiewicz M. Z., Hempel D., Sierko E., Tucker S. C., and Honn K. V. (2015) Protease-activated receptors (PARs): biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 34, 775–796 10.1007/s10555-015-9599-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alvarez C. E. (2008) On the origins of arrestin and rhodopsin. BMC Evol. Biol. 8, 222 10.1186/1471-2148-8-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qi S., O'Hayre M., Gutkind J. S., and Hurley J. H. (2014) Structural and biochemical basis for ubiquitin ligase recruitment by arrestin-related domain-containing protein-3 (ARRDC3). J. Biol. Chem. 289, 4743–4752 10.1074/jbc.M113.527473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rauch S., and Martin-Serrano J. (2011) Multiple interactions between the ESCRT machinery and arrestin-related proteins: implications for PPXY-dependent budding. J. Virol. 85, 3546–3556 10.1128/JVI.02045-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Puca L., and Brou C. (2014) α-Arrestins: new players in Notch and GPCR signaling pathways in mammals. J. Cell Sci. 127, 1359–1367 10.1242/jcs.142539 [DOI] [PubMed] [Google Scholar]
- 51. Zheng Y., Lin Z. Y., Xie J. J., Jiang F. N., Chen C. J., Li J. X., Zhou X., and Zhong W. D. (2017) ARRDC3 inhibits the progression of human prostate cancer through ARRDC3-ITGβ4 pathway. Curr. Mol. Med. 17, 221–229 [DOI] [PubMed] [Google Scholar]
- 52. Wang D., Yang P. N., Chen J., Zhou X. Y., Liu Q. J., Li H. J., and Li C. L. (2014) Promoter hypermethylation may be an important mechanism of the transcriptional inactivation of ARRDC3, GATA5, and ELP3 in invasive ductal breast carcinoma. Mol. Cell Biochem. 396, 67–77 10.1007/s11010-014-2143-y [DOI] [PubMed] [Google Scholar]
- 53. Yao J., Xu C., Fang Z., Li Y., Liu H., Wang Y., Xu C., and Sun Y. (2016) Androgen receptor regulated microRNA miR-182-5p promotes prostate cancer progression by targeting the ARRDC3/ITGB4 pathway. Biochem. Biophys. Res. Commun. 474, 213–219 10.1016/j.bbrc.2016.04.107 [DOI] [PubMed] [Google Scholar]
- 54. Lu S., Simin K., Khan A., and Mercurio A. M. (2008) Analysis of integrin β4 expression in human breast cancer: association with basal-like tumors and prognostic significance. Clin. Cancer Res. 14, 1050–1058 10.1158/1078-0432.CCR-07-4116 [DOI] [PubMed] [Google Scholar]
- 55. Bierie B., Pierce S. E., Kroeger C., Stover D. G., Pattabiraman D. R., Thiru P., Liu Donaher J., Reinhardt F., Chaffer C. L., Keckesova Z., and Weinberg R. A. (2017) Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 114, E2337–E2346 10.1073/pnas.1618298114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shi X., Gangadharan B., Brass L. F., Ruf W., and Mueller B. M. (2004) Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol. Cancer Res. 2, 395–402 [PubMed] [Google Scholar]
- 57. Radjabi A. R., Sawada K., Jagadeeswaran S., Eichbichler A., Kenny H. A., Montag A., Bruno K., and Lengyel E. (2008) Thrombin induces tumor invasion through the induction and association of matrix metalloproteinase-9 and β1-integrin on the cell surface. J. Biol. Chem. 283, 2822–2834 10.1074/jbc.M704855200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sayyah J., Bartakova A., Nogal N., Quilliam L. A., Stupack D. G., and Brown J. H. (2014) The Ras-related protein, Rap1A, mediates thrombin-stimulated, integrin-dependent glioblastoma cell proliferation and tumor growth. J. Biol. Chem. 289, 17689–17698 10.1074/jbc.M113.536227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu L., Wang X., Wu J., Mao D., Xu Z., He Z., and Yu A. (2012) Cooperation of protease-activated receptor 1 and integrin ανβ5 in thrombin-mediated lung cancer cell invasion. Oncol. Rep. 28, 553–560 10.3892/or.2012.1851 [DOI] [PubMed] [Google Scholar]
- 60. Sahai E., and Marshall C. J. (2002) ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat. Cell Biol. 4, 408–415 10.1038/ncb796 [DOI] [PubMed] [Google Scholar]
- 61. Dorsam R. T., and Gutkind J. S. (2007) G-protein-coupled receptors and cancer. Nat. Rev. Cancer 7, 79–94 10.1038/nrc2069 [DOI] [PubMed] [Google Scholar]
- 62. Chen F. (2012) JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res. 72, 379–386 10.1158/0008-5472.CAN-11-1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Paing M. M., Johnston C. A., Siderovski D. P., and Trejo J. (2006) Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol. Cell Biol. 26, 3231–3242 10.1128/MCB.26.8.3231-3242.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen B., Dores M. R., Grimsey N., Canto I., Barker B. L., and Trejo J. (2011) Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J. Biol. Chem. 286, 40760–40770 10.1074/jbc.M111.299776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shin K. J., Wall E. A., Zavzavadjian J. R., Santat L. A., Liu J., Hwang J. I., Rebres R., Roach T., Seaman W., Simon M. I., and Fraser I. D. (2006) A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 13759–13764 10.1073/pnas.0606179103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smith T. H., Li J. G., Dores M. R., and Trejo J. (2017) Protease-activated receptor-4 and purinergic receptor P2Y12 dimerize, co-internalize, and activate Akt signaling via endosomal recruitment of β-arrestin. J. Biol. Chem. 292, 13867–13878 10.1074/jbc.M117.782359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Grimsey N. J., Aguilar B., Smith T. H., Le P., Soohoo A. L., Puthenveedu M. A., Nizet V., and Trejo J. (2015) Ubiquitin plays an atypical role in GPCR-induced p38 MAP kinase activation on endosomes. J. Cell Biol. 210, 1117–1131 10.1083/jcb.201504007 [DOI] [PMC free article] [PubMed] [Google Scholar]