

Abstract
Low-conductance, highly calcium-selective channels formed by the Orai proteins exist as store-operated CRAC channels and store-independent, arachidonic acid–activated ARC channels. Both are activated by stromal interaction molecule 1 (STIM1), but CRAC channels are activated by STIM1 located in the endoplasmic reticulum membrane, whereas ARC channels are activated by the minor plasma membrane–associated pool of STIM1. Critically, maximally activated CRAC channel and ARC channel currents are completely additive within the same cell, and their selective activation results in their ability to each induce distinct cellular responses. We have previously shown that specific ARC channel activation requires a PKA-mediated phosphorylation of a single threonine residue (Thr389) within the cytoplasmic region of STIM1. Here, examination of the molecular basis of this phosphorylation-dependent activation revealed that phosphorylation of the Thr389 residue induces a significant structural change in the STIM1-Orai–activating region (SOAR) that interacts with the Orai proteins, and it is this change that determines the selective activation of the store-independent ARC channels versus the store-operated CRAC channels. In conclusion, our findings reveal the structural changes underlying the selective activation of STIM1-induced CRAC or ARC channels that determine the specific stimulation of these two functionally distinct Ca2+ entry pathways.
Keywords: A-kinase anchoring protein (AKAP), calcium release-activated calcium channel protein 1 (ORAI1), phosphorylation, stromal interaction molecule 1 (STIM1), structural biology, Orai channels, calcium signaling
Introduction
Although originally identified as a tumor suppressor gene (1), stromal interaction molecule 1 (STIM1)2 is now known to be the essential mediator of activation of the highly calcium-selective Orai channels, both the store-operated CRAC channels and the store-independent ARC channels. Importantly, these channels represent key components in the activation and modulation of diverse signaling events in a multitude of different cell types. Although less extensively studied, the store-independent ARC channels have been shown to play key roles in a variety of different cell types including pancreatic and parotid acinar cells (2), pancreatic β cells (3), and avian nasal exocrine cells (4). The ARC channels are also present in multiple cell lines including HEK-293 cells, HeLa cells, and rat basophilic leukemia (RBL) cells (5) as well as SY5Y neuroblastoma cells (6) and K562 erythroleukemia cells (7). Correspondingly, STIM1 is known to be almost ubiquitously expressed in most tissues including the pancreas, skeletal muscle, brain, heart, liver, and lung.
Although the molecule STIM1 is essential for the activation of both CRAC channels and ARC channels, the way in which this activation is achieved is fundamentally distinct. Activation of the CRAC channels is regulated by the pool of STIM1 that is resident in the endoplasmic reticulum (ER) membrane and physiologically is activated on depletion of the ER calcium, typically as a result of agonist-induced activation of inositol trisphosphate receptors. This fall in ER calcium levels is sensed by STIM1 via an N-terminal calcium-binding EF-hand domain located within the ER lumen, and it is the consequent loss of calcium from the EF-hand of STIM1 that induces the translocation of the ER-resident STIM1 to sites close to the plasma membrane where it initiates the eventual activation of CRAC channels. In marked contrast, ARC channels are regulated by the minor pool of STIM1 that is constitutively resident in the plasma membrane (8). Here, the N-terminal calcium-binding EF-hand of STIM1 is exposed to the extracellular medium where the essentially stable calcium concentration (∼2 mm) would mean that it would never normally lose its bound calcium. Clearly then, the mode of activation of this PM-located pool of STIM1 must be entirely distinct from that of the ER STIM1. Consistent with this, complete deletion of the N-terminal portion of STIM1 that includes both the EF-hand region and the adjacent sterile α motif domain has no effect on its ability to activate the store-independent ARC channels (9). In addition, for the ARC channels, the PM location of this pool of STIM1 means that both proteins (STIM1 and the Orai proteins that form the channel) are oriented in a parallel (cis) orientation in the same membrane, whereas the Orai proteins that form the CRAC channel are in an antiparallel (trans) orientation with STIM1 in the ER membrane. Together, these features suggest that there should be fundamental differences in the molecular and/or structural basis for the activation of these two distinct channels by the STIM1 molecule. In this context, several studies have established the so-called STIM1-Orai–activating region (SOAR; residues 344–442) (10), also known as the “CRAC activation domain” (11), “Orai-activating small fragment” (12), and “coiled-coil domain–containing region b9” (13) of STIM1, as the essential region of the molecule responsible for inducing CRAC channel activation. Subsequently, we showed that this was also true for the store-independent ARC channels (9). Hence, we will use the term SOAR, if only to specify that the activity of this domain applies to both CRAC and ARC channels. Structurally, STIM1 exists as a homodimer (12), a feature that is primarily driven by interactions between the respective SOAR domains. Subsequent analysis of the crystal structure of this domain (14) revealed that it comprises three distinct helical sequences identified as CC1, CC2, and CC3 of which the CC2 and CC3 sequences of each monomer form a dimeric antiparallel “hairpin” construct under resting conditions and represent the minimal region required for effective activation of the Orai channels (11, 12, 14).
Critically, studies have demonstrated that the specific residues essential for initial interactions with Orai are located at the extreme C-terminal region of the CC2 domain. This sequence includes a so-called “basic region,” which has been shown to be critical in the ability of STIM1 to effectively induce the activation of the store-operated CRAC channels (15). This region comprises the four lysine residues (Lys382, Lys384, Lys385, and Lys386 in the human sequence) along with an additional basic residue (Arg387) immediately following the lysine-rich sequence that may also impact the overall activity. In the STIM1 homodimer that represents the endogenous resting conformation (14), these basic residues line the V-shaped cleft formed by the CC2-CC3 domains of each STIM1 monomer. This sequence is highly conserved from insects to humans with only occasional substitution of an arginine for a lysine as seen, for example, in the Drosophila STIM (16). Originally, the specific function of this region in the regulation of CRAC channel activation was thought to reflect its role in part of an intramolecular electrostatic clamp that acted to prevent spontaneous activation of STIM1 by interacting with a corresponding acidic sequence in the CC1 domain of STIM1 (Glu318–Glu322) (17). However, subsequent studies revealed that the basic region represents the key site of interactions with an acidic coiled-coil region in the C-terminal tail of Orai1 that are necessary to induce activation of the CRAC channel (18). Consistent with this, mutation of the positively charged lysine residues within the basic region (BR) sequence to either alanines or glycines results in the failure of any STIM1-Orai colocalization and the loss of effective CRAC channel activation (14, 19). In a similar manner, we have shown that introducing the same lysine mutations in this basic region also results in the loss of activation of the store-independent ARC channels (9).
In this context, we have recently demonstrated that the ability to specifically activate ARC channels versus the molecularly similar CRAC channels depends on the PKA-mediated phosphorylation status of a single threonine residue (Thr389) in the ∼450-residue cytosolic domain of STIM1 (20). Interestingly, this Thr389 residue lies at the apex of the predicted “hairpin bend” located at the transition of the CC2 to CC3 domains in the SOAR coiled-coil sequence of STIM1, a location that is immediately downstream of the above basic region in STIM1. Given this, we chose to investigate the potential functional relationships and respective contributions of the residues in this region of STIM1 that might influence the selective activation of ARC channels versus CRAC channels.
Results
Distinct roles of the basic region of STIM1 in ARC channel activation versus CRAC channel activation
We began by re-examining the key role of the basic region (BR) lysine residues (Lys382 and Lys384–Lys386) in CRAC channel activation. As noted, previous studies had demonstrated that mutation of these four lysine residues to either alanines or glycines (13, 17) or to glutamates (18) results in the loss of CRAC channel activation. Interestingly, our further examination of the essential role of the BR sequence in CRAC channel activation revealed that just a single lysine-to-glutamine mutation (K386Q) in this BR sequence was sufficient to essentially eliminate store-operated CRAC channel activity (mean ± S.D. =0.06 ± 0.03 pA/pF at −80 mV) (Fig. 1). Moreover, similar data were obtained for mutations of the same Lys386 residue to either alanine, valine, or glutamate (Fig. 1). Together, these findings demonstrate the acute specificity of this region for effective CRAC channel activation. As noted above, we have previously shown that mutation of the Lys382 and Lys384–Lys386 residues also results in an inhibition of the ARC channel activity (9), potentially suggesting that such inhibition again results from a loss of STIM1-Orai interactions. However, consideration of the location of this STIM1 basic region spanning residues Lys382–Lys386 shows that it lies immediately upstream of the Thr389 residue whose phosphorylation we have shown to be essential for specific activation of the ARC channels, whereas CRAC channel activity only occurs in the absence of phosphorylation of the Thr389 residue as in the T389A mutant (20). In this context, we would predict that mutation of the lysines in the Lys382–Lys386 sequence would likely disrupt the required motif for successful phosphorylation of the adjacent Thr389 region. To examine this, we compared the arachidonic acid (AA)–induced ARC channel currents in a STIM1 construct bearing the BR mutations K382A, K384G, K385A, and K386G with an identical construct bearing the same lysine mutations but incorporating the phosphomimetic T389E mutation. To ensure that the measured currents could be exclusively assigned to the ARC channels, we incorporated the above mutations in a Lck-STIM1 construct, which specifically records ARC channel activity induced by plasma membrane–located STIM1 (9). The results obtained showed that cells expressing the above BR (Lys → Ala/Gly) mutations displayed markedly reduced AA-activated ARC channel currents to 0.20 ± 0.05 pA/pF at −80 mV (mean ± S.D.), a value virtually identical to that obtained under the same conditions and reported earlier (9) (Fig. 2). However, expression of an identical BR-mutant STIM1 construct incorporating the phosphomimetic T389E mutation resulted in ARC channel currents of 0.46 ± 0.08 pA/pF at −80 mV (mean ± S.D.), a value that was not significantly different from those seen with wildtype cells bearing the T389E mutation (mean ± S.D. = 0.49 ± 0.09 pA/pF at −80 mV) (Fig. 2).
Figure 1.
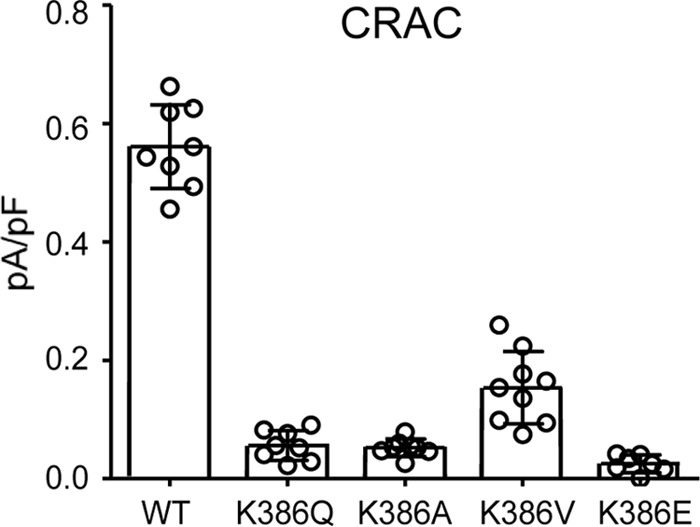
The effect of mutation of the Lys386 residue located in the BR of STIM1 on CRAC channel currents. The data shown are the whole-cell inward CRAC channel currents (mean ± S.D., n = 8) measured at −80 mV in WT cells and in the same cells bearing the indicated mutations of the Lys386 residue in STIM1 (n = 6–9). Error bars represent the mean ± S.D. channel currents induced by inclusion of adenophostin A (2 μm) in the patch pipette solution with individual measurements indicated by open circles.
Figure 2.
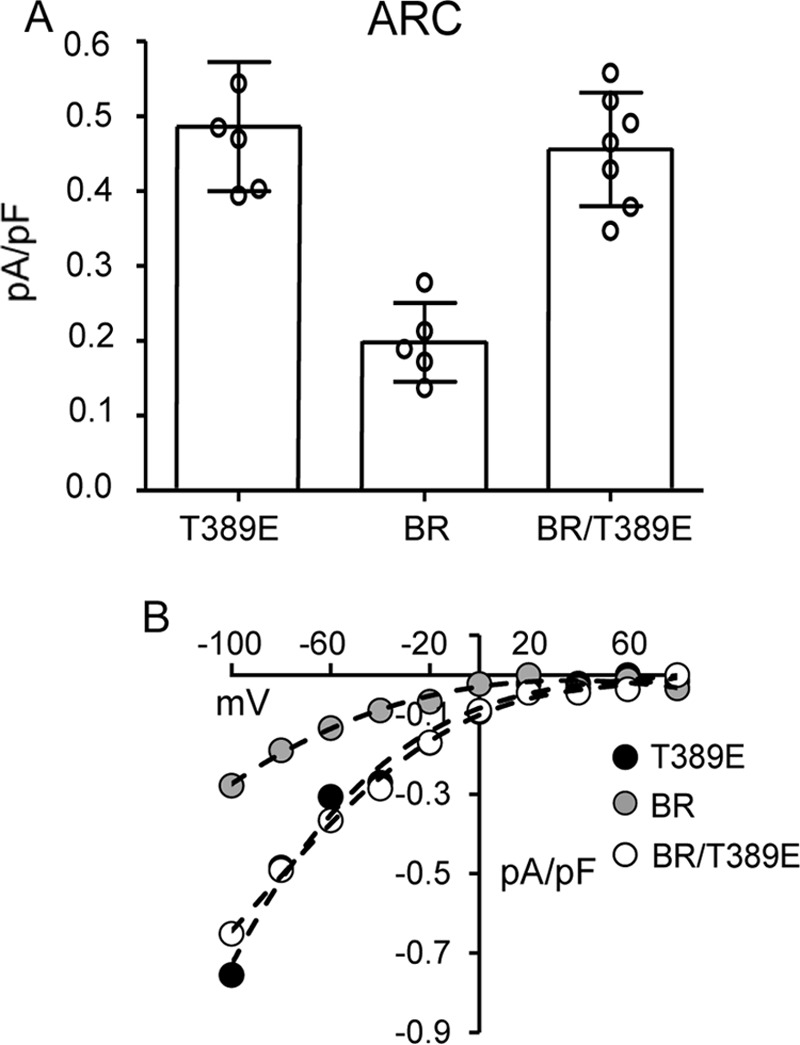
The effect of BR mutations (Lys382 and Lys384–Lys386) on ARC channel currents either alone or in cells incorporating the T389E phosphomimetic mutation. Currents in cells expressing the ARC-specific T389E-STIM1 are shown for comparison. Channel currents were activated by exogenous addition of arachidonic acid (8 μm) in cells expressing a Lck-STIM1 construct, which specifically records ARC channel activity induced by plasma membrane–located STIM1 (8). A, error bars represent the mean ± S.D. current magnitudes measured at −80 mV (n = 5–7) with individual measurements indicated by open circles. B, representative current–voltage relationship for each of the three constructs.
Based on the above data, we conclude that the residues that form the basic region in STIM1 (Lys382–Lys386) and have been shown to be essential for both CRAC and ARC channel activation have entirely distinct roles in determining the activity of these two distinct, coexisting Orai channels. Thus, the lysine residues in this BR region are, themselves, essential for effective CRAC channel activation. However, their only role in activation of the ARC channel is to provide a specific sequence necessary for the effective PKA-dependent phosphorylation of the adjacent Thr389 residue, a phosphorylation that is an absolute requirement for ARC channel activation. However, what is less clear is exactly how phosphorylation of this single threonine in the almost 690-residue sequence of STIM1 might act to play such a unique role in this activation.
Functional effects of phosphorylation of the Thr389 and adjacent residues
In this context, we considered that phosphorylation of the Thr389 residue would make it less hydrophobic due to the methyl group in its side chain, potentially inducing a shift in the orientation or proximity of the adjacent Leu390 residue. Based on this, we would predict that the hydrophobic/hydrophilic nature of the residue at position 390 would likely also impact the potential phosphorylation of Thr389 with phosphorylation of Thr389 requiring a hydrophobic residue at position 390. To evaluate this possibility, we began by introducing various mutant versions of the Leu390 residue and examining their effects on the ARC channel and CRAC channel activities. Consistent with the predicted requirement of a hydrophobic residue at position 390 in STIM1 to form a “phosphorylation-compatible” sequence necessary for ARC channel activation, expression of STIM1 bearing the hydrophobic-to-hydrophilic L390Q mutation resulted in the absence of any significant ARC channel activity (mean ± S.D. = 0.08 ± 0.04 pA/pF at −80mV versus 0.46 ± 0.05 pA/pF for the wildtype) (Fig. 3A). However, further examination revealed that this was more complex than originally thought. We would have predicted that incorporating the phosphomimetic T389E mutation in STIM1 should overcome the inhibition of ARC channel currents by the L390Q mutation; however, this proved not to be the case as the expression of the T389E/L390Q-STIM1 mutation resulted in negligible ARC channel currents (mean ± S.D. = 0.08 ± 0.02 pA/pF at −80 mV) (Fig. 3A). Based on these data, we conclude that the key role of the Leu390 residue is not exclusively to complete the necessary phosphorylation motif but that it likely serves additional roles related to a specific requirement of a hydrophobic residue at this location.
Figure 3.
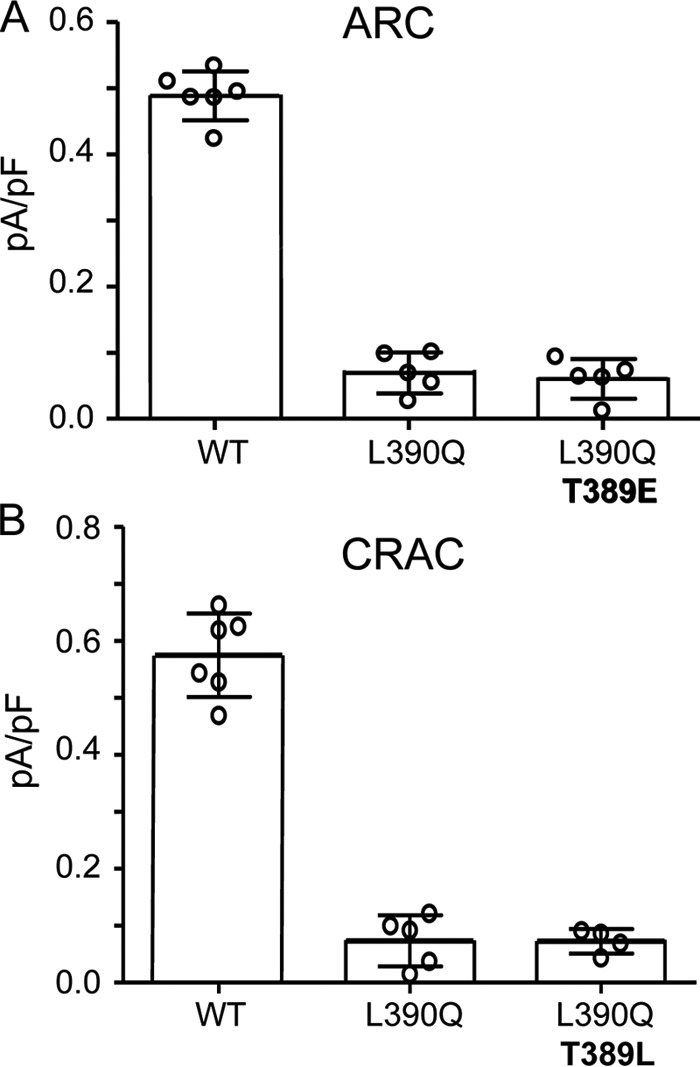
A, effects of the STIM1 L390Q mutation on AA-activated ARC channel currents either alone or when included with T389E mutation. B, effects of the STIM1 L390Q mutation on adenophostin-activated CRAC channel activity either alone or when included with T389L mutation. Whole-cell inward ARC and CRAC channel currents (error bars represent the mean ± S.D.; n = 4–6) were measured at −80 mV using the standard protocols in WT cells and in the same cells bearing the STIM1 L390Q mutation and the same mutation along with either the T389E mutation (ARC channels; A) or the T389L mutation (CRAC channels; B). Individual measurements are indicated by open circles. ARC channel and CRAC channel currents were activated by standard procedures (see “Experimental procedures”).
More surprising, however, was the finding that the same L390Q single mutation also resulted in the failure to support significant CRAC channel activity (mean ± S.D. = 0.07 ± 0.02 pA/pF compared with 0.57 ± 0.04 pA/pF at −80 mV for the wildtype) (Fig. 3B). One possible explanation for this loss of CRAC channel activity is that the leucine-to-glutamine mutation would effectively introduce a switch from a hydrophobic to hydrophilic residue at this location, and this may impact activation of the CRAC channels. To assess this possibility, we introduced a L390Q/T389L double mutation into STIM1, thereby retaining the overall hydrophilic-hydrophobic relationship between the 389 and 390 residues but simply reversing their sequence order. However, examination of the resulting CRAC channel activity induced by the standard store-depletion protocol again revealed negligible currents (mean ± S.D. = 0.07 ± 0.02 pA/pF at −80 mV) (Fig. 3B).
Given the above, we sought to further examine the possible effects of various mutations at the Leu390 site of STIM1 on both CRAC and ARC channel activities. In this, we focused on the apparent requirement of a hydrophobic residue at position 390 for successful CRAC channel activity. Mutation of the leucine at residue 390 to an isoleucine (L390I mutation) resulted in the activation of both ARC and CRAC channels to normal levels (mean ± S.D. currents at −80 mV of 0.53 ± 0.02 pA/pF for ARC and 0.55 ± 0.03 pA/pF for CRAC channels) (Fig. 4). This result was not particularly surprising given the structural and biochemical similarity between the leucine and isoleucine residues. In contrast, however, mutation of the Leu390 to either an alanine (L390A) or a valine (L390V) resulted in markedly reduced CRAC channel currents (mean ± S.D. currents at −80 mV of 0.05 ± 0.02 pA/pF for the L390A mutation and 0.16 ± 0.05 pA/pF for the L390V mutation). In contrast, the corresponding ARC channel currents were unaffected (mean ± S.D = 0.49 ± 0.04 and 0.53 ± 0.02 pA/pF, respectively, measured at −80 mV) (Fig. 4). In this context, it should be noted that both hydrophobic and conformational requirements at position 390 may be necessary for normal CRAC channel activity, requirements that are met by isoleucine and leucine but not by valine or alanine. For example, the side chains of alanine and valine are significantly less hydrophobic than those of leucine and isoleucine (relative side-chain hydrophobicity scales of 41 and 76 versus 97 and 99, respectively) (21, 22). In addition, both alanine and valine have significantly smaller side chains than leucine and isoleucine (solvent-accessible surface areas of 129 and 174 Å2 versus 197 and 201 Å2, respectively) (23). Thus, it is possible that a combination of both lower hydrophobicity and smaller side chains could underlie the inability of the alanine and valine mutants to elicit significant CRAC channel activity.
Figure 4.
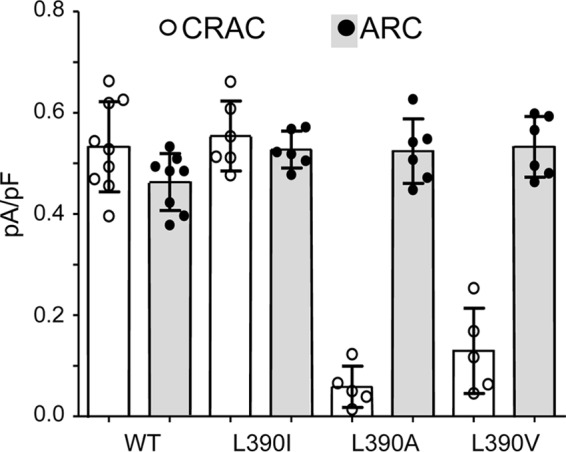
The effects of mutations of the Leu390 residue in STIM1 on adenophostin-activated CRAC channel currents and AA-activated ARC channel currents compared with the same in the wildtype channels. Data shown are individual measurements (circles) of inward currents (with mean ± SD, n = 6–9, indicated by error bars) measured at −80 mV in cells expressing the wildtype STIM1 or STIM1 bearing the indicated mutations of the Leu390 residue. ARC channel and CRAC channel currents were activated by standard procedures (see “Experimental procedures”).
To further examine the uniquely distinct requirements of this specific region of STIM1 for the selective action of these two related Orai channels, we sought to investigate the molecular basis of their interactions with the Orai protein. Studies have shown that the essential step in the STIM1-dependent activation of CRAC channels is the ability of the STIM1 SOAR domain to interact with a coiled-coil region located in the C-terminal tail of Orai1. Consistent with this, it was shown that a single L373S mutation within the CC2 domain of STIM1 was sufficient to essentially eliminate CRAC channel activation (24). In contrast, however, a double mutation, L373S/A376S, in the corresponding CC2 domain of STIM1 was required to induce a similar elimination of ARC channel activity (9). Given the apparent difference in the relative “affinity” of this critical coiled-coil interaction, we considered the possibility of any potential effect that the phosphorylation status of the Thr389 residue might have on STIM1-Orai interactions in this region. Examination of the available crystal structure of the SOAR domain (14) shows that the Thr389 side chain would be solvent-exposed, directed away from the coiled-coil backbone structure, features that would suggest that the phosphorylation of Thr389 would not significantly impact the overall SOAR structure. In contrast, however, primary sequence analysis using COILS (25) suggests that a T389E mutation could increase the propensity for coiled-coil formation.
Examination of the potential structural effects of the phosphorylation of Thr389
To experimentally test the potential structural effects of phosphorylation in more detail, we used a recombinant human STIM1 protein corresponding to residues 234–491, which encompasses all three coiled-coil domains. Comparison of the elution volumes by size exclusion chromatography (SEC) indicated similar molecular weights and/or conformations for the wildtype, T389E, T389A, and L390Q proteins with elution volumes ranging between 12.7 and 13.0 ml (i.e. stoichiometries of ∼2.3–2.5) for all proteins (Fig. 5A). This observation stands in marked contrast with similar analyses of other mutations such as L251S, L419S, and L423S, which have been shown to induce a pronounced decrease in the elution volume of STIM1(234–491) by as much as ∼0.8 ml compared with wildtype, an effect reflecting the rearrangement of the coiled-coil domains (26). Incorporation of mutations that elicit a conformational extension in this STIM1 cytosolic domain construct, as detected by FRET in live cells and in vitro by SEC in full-length STIM1, results in constitutively active CRAC channels (26). Moreover, the SEC data suggest that the T389E/A and L390Q mutations do not elicit a conformational extension and do not appreciably perturb the dimer state also adopted by full-length STIM1 and domain fragments in cells (11, 14, 17, 18, 27–31).
Figure 5.
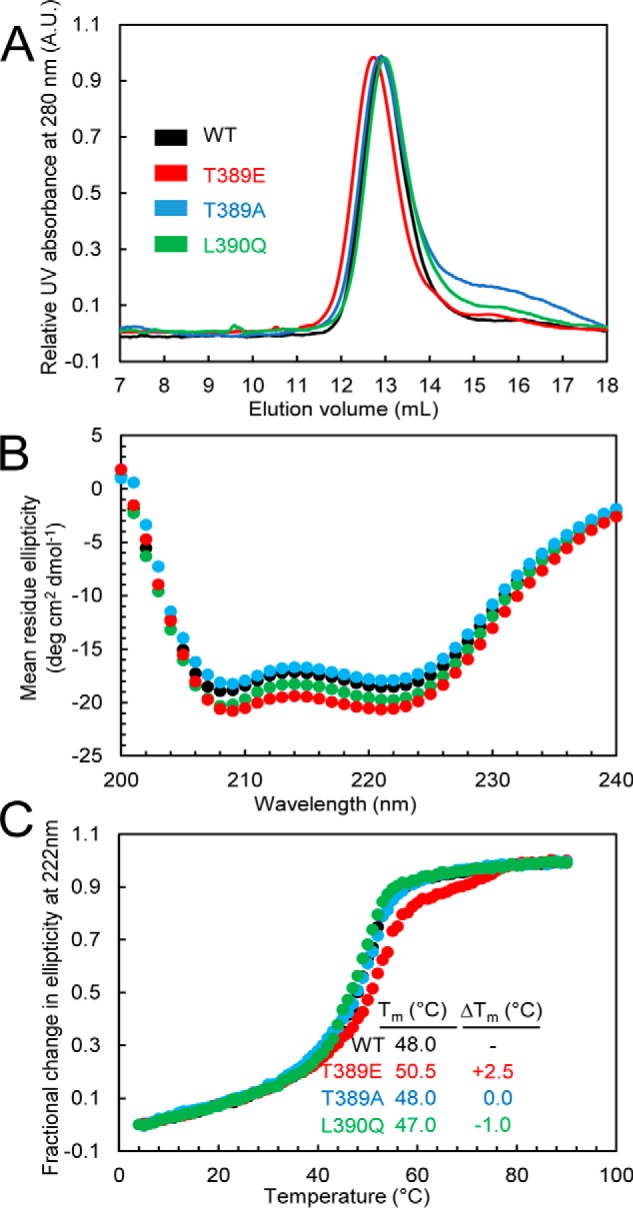
A, SEC of the STIM1 sequence 234–491 in wildtype STIM1 and the T389A-, T389E-, or L390Q-STIM1 mutant. Apparent molecular masses of WT, T389E, T389A, and L390Q were 2.4, 2.5, 2.4, and 2.3× the theoretical monomeric molecular mass of STIM1(234–491) (30.8 kDa), consistent with the well-established notion that the cytosolic domains of STIM1 recapitulate the dimeric nature of full-length STIM1 when expressed in live mammalian cells. Data are representative of at least two distinct protein preparations. B, secondary structure obtained by far-UV CD spectroscopy for the STIM1 sequence 234–491 in wildtype STIM1 and the T389A, T389E, or L390Q mutant. Far-UV CD spectra of STIM1(234–491) were acquired at 0.3–0.5 mg ml−1. Data are representative of at least two distinct protein preparations. C, thermal stability analyses by changes in far-UV CD spectroscopy at 222 nm for the STIM1 sequence 234–491 in wildtype STIM1 and the T389E-, T389A-, or L390Q-STIM1 mutants. Thermal stability was measured at protein concentrations of 0.3–0.5 mg ml−1. The apparent Tm values were 48.0, 50.5, 48.0, and 47.0 °C for the wildtype, T389E, T389A, and L390Q proteins, respectively. Data are representative of at least two distinct protein preparations. A.U., absorbance units.
We next examined the secondary structure of the wildtype and T389E, T389A, and L390Q mutant STIM1(234–491) proteins using far-UV circular dichroism (CD) spectroscopy (Fig. 5B). Here, all constructs displayed ellipticity minima at 208 and 222 nm, indicative of α-helical structures. However, the T389E mutant consistently revealed more negative ellipticity at those wavelengths, suggesting an increase in the α-helicity relative to the other proteins (Fig. 5B). As confirmation that the T389E mutation enhanced the α-helicity of STIM1(234–491), we assessed the thermal stability by monitoring the changes in the far-UV CD signal at 222 nm as a function of temperature. Consistent with the apparent increased α-helicity of the T389E mutant, the midpoint of temperature denaturation (Tm) of the T389E phosphomimetic mutant was ∼2.5–3.5 °C higher than each of the other proteins examined (Fig. 5C). Thus, the SEC and far-UV CD data together suggest that the T389E phosphomimetic mutant uniquely induces an increase in the helical structure and stability of this region of the SOAR sequence without any major alterations in the coiled-coil arrangement, which would have been detectable by SEC.
Molecular dynamics analysis of the structural effects of Thr389 phosphorylation
To explore the possible effect(s) of the phosphorylation of the Thr389 residue at atomic resolution, we utilized molecular dynamics (MD) simulations. Such an approach, when used appropriately, can provide important information enabling predictions of potential structural changes resulting from secondary modifications such as phosphorylation or mutations. Thus, in these simulations, we compared wildtype STIM1, phosphorylated Thr389 (“phos-STIM1”) constructs, and mutant phosphomimetic T389E constructs. These analyses were considered critical given reports indicating that the structural features of phosphomimetic mutant constructs may not entirely conform to the “native” phosphorylated protein (32). Critically, molecular dynamics simulations have the advantage that they can resolve dynamic structure with atomic resolution, allowing us to directly distinguish the conformational differences between proteins that underlie their distinct functional profiles. In this case, the resulting simulations indicated that both phosphorylation of the Thr389 residue and introduction of the phosphomimetic T389E mutation induce an extension of the CC2 coiled-coil region beyond the Thr389 residue. This is illustrated in the side-chain contact probability maps generated over the course of the simulation for wildtype STIM1 (WT-STIM1), STIM1 in which the Thr389 residue is phosphorylated (phos-STIM1), and STIM1 bearing the phosphomimetic T389E mutation (T389E-STIM1) (Fig. 6). For reference, the average contact map for phos-STIM1 is shown in Fig. S1. The maps are broadly similar, showing that the structures of the native and modified proteins are similar, but parsing the specific details is difficult. For this reason, in Fig. 7, we present the differences between these contact maps, specifically focusing on the region of greatest interest, namely between residues Lys386 and Ser399. These maps highlight the fact that the side chain–side chain contact probabilities in the Phe394–Ser399 region increase in both the phosphorylated and T389E mutant proteins relative to native STIM1 (Fig. 7). Specifically, these contacts indicate that the helix in this region extends by approximately one to two turns; Fig. 9 demonstrates this visually using representative snapshots from late in the trajectories.
Figure 6.

Side-chain contact probability maps determined from MD simulations. The probability that a particular pair of side chains is in contact over the course of the simulation is indicated by the grayscale (lighter indicates the contact is more probable). The results are the average of results from the four replicas for each protein type. Note the increased contact probability in both the phos-STIM1 and the T389E-STIM1 data in the region spanning residues Phe394–Ser400 compared with the same region in wildtype STIM1.
Figure 7.

Contact probability “difference maps” obtained from MD simulations. These plots are the differences between the maps shown in Fig. 6, focusing on residues 386–399. Red indicates that a particular contact is more likely in the first system listed, whereas blue indicates that the contact becomes less populated. A represents the difference (diff) between the phosphorylated STIM1 sequence and the same sequence in wildtype STIM1, whereas B represents the difference between the same sequence in the phosphomimetic T389E mutant STIM1 and wildtype STIM1. The image in C shows the difference between the data for the phosphorylated STIM1 and the corresponding data for the T389E-STIM1.
Figure 9.
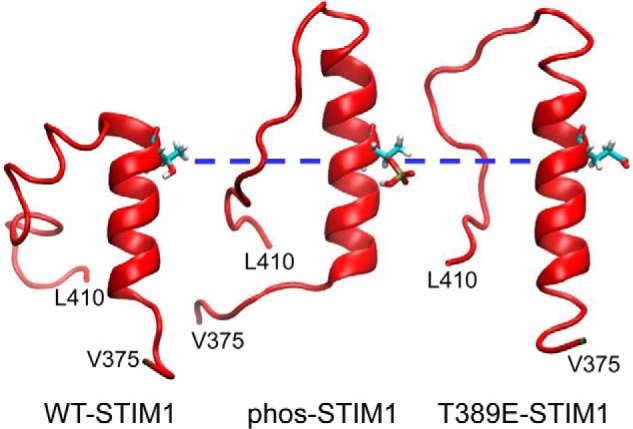
Representative molecular dynamic snapshots of the STIM1 CC2 terminal helical region. The individual images demonstrate the STIM1 CC2 helical region in wildtype STIM1 and the elongation of this region upon phosphorylation of residue Thr389 (phos-STIM1) or introduction of the T389E phosphomimetic mutation (T389E-STIM1). For comparison, the three images have been aligned at the position of the Thr389 residue (blue dashed line).
Similarly, the resulting difference map for T389E-STIM1 also demonstrated enhanced contacts localized in this Phe394–Ser399 region (Fig. 7B). Critically, the similarity of the results for the phosphorylated STIM1 and T389E-STIM1 contacts in these difference maps is illustrated in an additional difference map comparing these two constructs (Fig. 7C). This shows only a minimal increase in the extent of intramolecular interactions for phosphorylated STIM1 compared with the T389E-STIM1 and that this is limited to just the Phe394–His395 region.
When calculating quantities of this type, it is crucial to ask whether the simulations were run long enough so that the differences are statistically significant. To test this, we computed difference probability maps as shown in Fig. S2, A–C. Treatment of each individual trajectory as a single measurement of the probability of a particular side chain–side chain contact allows an estimation of the likelihood that the differences between two different systems (e.g. wildtype STIM1 and phos-STIM1) would occur randomly using a standard Welch's t test. Plotting the computed p values such that only those side-chain contacts with p < 0.05 are shown reveals that the contact changes in this region are unlikely to be coincidental.
Collectively, the results from our in vitro and in silico approaches indicate that both the phosphorylation of Thr389 and expression of the T389E phosphomimetic induce an extension of the C-terminal helical region of the CC2 region of STIM1, resulting in an increase in the stability of the helix beyond the Thr389 residue. As such, this finding raises the question of whether it is the phosphorylation status of the Thr389 residue per se that is critical for the exclusive activity of the ARC channels or, alternatively, whether it is the resulting C-terminal extension of the CC2 helix region that is the critical component. In other words, is the observed ARC channel–specific response a “biochemically mediated” effect reflecting the phosphorylation status of the Thr389 residue, or does it result from an essentially “physical” effect induced by the N-terminal extension of the CC2 helix?
Selective activation of the ARC channels results from an extension of the STIM1 CC2 helix
To attempt to distinguish between these two possibilities, we examined the effect of mutating the adjacent Gly392 residue, located just three residues upstream of the critical Thr389 residue, to an alanine with the prediction that this mutation would induce a significant increase in the helix stability (33, 34). To examine this, we introduced the G392A mutation in full-length STIM1 and recorded the resulting CRAC and ARC channel currents activated by depletion of the ER calcium store or addition of exogenous arachidonic acid, respectively. As shown in Fig. 8, when compared with the wildtype STIM1 cells (Fig. 8A), the G392A mutation resulted in AA-activated ARC channel currents that were indistinguishable from those recorded in wildtype cells (mean ± S.E. = 0.59 ± 0.06 pA/pF at −80 mV). Critically, however, the same G392A mutation in STIM1 completely eliminated store-operated CRAC channel currents (0.04 ± 0.01 pA/pF at −80 mV) (Fig. 8B). Together, these data indicate that it is not the phosphorylation status of Thr389 per se that is critical for ARC channel activity, but rather it is the extension of the helical region that is induced by the phosphorylation of residue Thr389. If this is the case, we would predict that the G392A mutation should be capable of inducing the exclusive activation of the ARC channels even in the absence of any phosphorylation of the Thr389 residue. To examine this, we expressed STIM1 incorporating the G392A mutation along with the phosphomutant T389A mutation that exclusively allows only CRAC channel activity (i.e. a T389A/G392A double mutation). The results obtained showed that arachidonic acid–activated ARC channel currents were indistinguishable from normal wildtype currents (mean ± S.E. = 0.54 ± 0.05 pA/pF at −80 mV), whereas CRAC channel currents were, once again, essentially negligible (0.02 ± 0.01 pA/pF at −80 mV) (Fig. 8C). Together, these data indicate that the predicted CC2 extension of the coiled-coil region induced by the G392A mutation fully supports normal ARC channel currents, even in the absence of any phosphorylation of the Thr389 residue, while simultaneously eliminating CRAC channel currents. Thus, we conclude that it is specifically the extension of the helical region at the apex of the CC2-CC3 junction of STIM1 that enables the selective activation of the ARC channels versus the CRAC channels, and physiologically this extension is induced by phosphorylation of the Thr389 residue.
Figure 8.
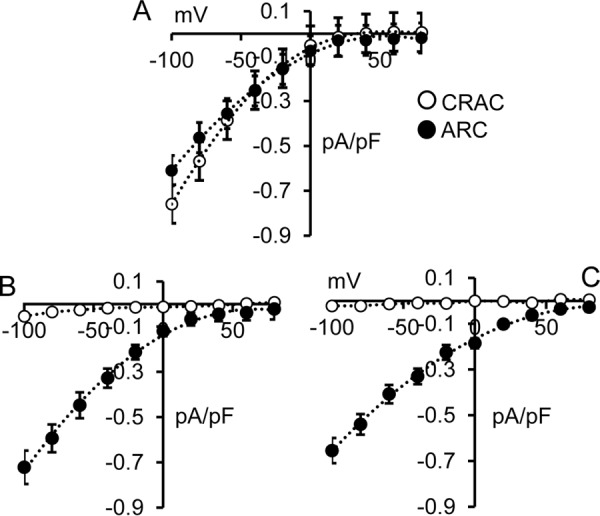
A, whole-cell current–voltage relationships for both CRAC channels and ARC channels in WT cells. B, the same currents in cells bearing the G392A-STIM1 mutation. C, the same currents obtained with G392A along with the “CRAC-specific” T389A mutation. The data shown are the whole-cell adenophostin-activated CRAC channel currents (open circles) and AA-activated ARC channel currents (filled circles). Error bars represent the mean ± S.E. (n = 6–7) in each case.
Discussion
In this study, we have examined the basis for the selective activation by STIM1 of either the store-operated CRAC channels or the store-independent ARC channels. Specifically, we focused on the critical CC2-CC3 region of STIM1, which is known to represent the site for the key interactions between STIM1 and the C termini of the Orai proteins that are necessary for the activation of both CRAC (14, 19) and ARC channels (9). Using a combination of different approaches, we have revealed that activation of both the CRAC channels and the ARC channels depends on the so-called basic region of STIM1, a series of lysines spanning residues Lys382–Lys386 that lie just upstream of the hairpin bend in the SOAR sequence at the junction of the CC2 and CC3 regions. Critically, however, our data demonstrate that this essential dependence on these basic region lysines is a result of entirely distinct effects of these residues on the two Orai channel types. Thus, previous studies have shown that the STIM1-dependent activation of the CRAC channels requires not only an intact basic region (14, 18, 19) but also the absence of any phosphorylation of the Thr389 residue (20). However, we have revealed that such CRAC channel activation also requires the specific presence of either a leucine or isoleucine at position 390. Importantly, the data indicate that this requirement may not be a sole reflection of the hydrophobicity of the 390 residue; rather, the lower hydrophobicity and smaller side chains may contribute to the inability of the alanine and valine mutants to elicit CRAC channel activity, presumably by perturbing the required STIM1-STIM1 and/or STIM1-Orai binding interface(s). Homology modeling of the L390A and L390V variants showed that the SOAR crystal structure can accommodate these variations with minimal backbone perturbations, suggesting that high-resolution structural data of STIM1 in complex with Orai1 are needed to reveal the precise structural role of Leu390 in CRAC channel activation.
In marked contrast, however, activation of the ARC channels results from the necessity of the Lys382–Lys386 basic region sequence to permit the PKA-dependent phosphorylation of the adjacent Thr389 residue. As to the possible physical structural effects of the phosphorylation of the Thr389 residue, our initial analyses indicated that such phosphorylation induced a modest, but detectible, increase in α-helicity and in the stability associated with this enhanced secondary structure assessed by far-UV CD spectroscopy. Moreover, these structural changes were supported by MD simulations, which revealed a potential extension of the STIM1 CC2 helix in the T389E phosphomimetic construct as well as in the phosphorylated Thr389 residue compared with the unmodified wildtype STIM1 construct. As such, our observations are consistent with previous work showing that phosphorylation of threonine residues increases the potential for backbone conformations that promote α-helix formation (34). Moreover, contact difference maps of STIM1 versus either phosphorylated STIM1 or the phosphomimetic T389E-STIM1 revealed increased intramolecular interactions specifically in the region spanned by residues Phe394–Ser399, just downstream of the critical Thr389 residue (Fig. 7).
Interestingly, Wang et al. (35) have reported that the Phe394 residue has a strategic significance for CRAC channel activation in that mutation of this residue to a leucine substantially reduced CRAC channel activity. Moreover, alternative mutations of the Phe394 residue to either an alanine, histidine, or glycine systematically enhanced the inhibition of CRAC channel activity. It is therefore intriguing that this Phe394 residue is located precisely at the site where we observe a distinct increase in intramolecular interactions in our “contact difference” maps for both the phosphorylated Thr389 residue in STIM1 and the phosphomimetic mutant T389E-STIM1 (Fig. 7). Thus, our data are in line with the conclusions of Wang et al. (35) that this region represents a key site for the STIM1-Orai interactions necessary for channel activation. Critically, however, we also show how the key interactions required for gating are fundamentally different between the CRAC and ARC channels. In conclusion, our data collectively demonstrate that it is the extension of the helical region of the apex of the CC2 domain of STIM1 (Fig. 9) specifically that is the key feature determining the unique activation of the ARC channels versus the CRAC channels. Moreover, this extension of the apical CC2 helix is functionally induced as a result of the specific phosphorylation of the Thr389 residue in STIM1. Precisely how this extension of the CC2 helix in STIM1 induces such a profound switch in ARC channel versus CRAC channel activities remains unclear. In this context, however, it is important to emphasize that the critical interactions between STIM1 and the CRAC and ARC channels are structurally quite distinct. Thus, although activation of the CRAC channels involves Orai and STIM1 proteins in two structurally separate membranes (the ER and PM), activation of the ARC channels involves interactions of the same proteins located within the same membrane. In addition, a combination of both protein kinase A–anchoring protein and PKA activities are essential for activation of ARC channels but not CRAC channels (2, 20). Thus, we would hypothesize that the critical interactions between STIM and Orai in CRAC and ARC channels occur in entirely distinct manners. In this context, the dynamic nature of the STIM1 cytosolic domains is consistent with available high-resolution structures showing the coiled-coil domains adopting markedly different conformations in the free state versus the Orai1 C-terminal domain–bound state (13, 18). Moreover, the data presented here show that ARC channel activation is dependent upon the extended CC2 helix and likely occurs in a more complex multimolecular assembly. Consistent with this are the data presented showing that phosphorylation, phosphomimetic mutations, and other molecular variations, all of which result in the extension of the CC2 helix, uniquely allow the selective activation of the ARC channels. Ultimately, high-resolution structural elucidation of the extended CC2 conformation in complex with Orai1/Orai3 will be required for precise mechanistic insight into the unique features of ARC channel activation at the atomic level. Nevertheless, whatever their molecular nature, the divergent structural features revealed in this study, which enable the selective activation of either the CRAC channels or the ARC channels by the same STIM1 molecule, represent an important mechanism that prevents the simultaneous activation of these two distinct agonist-activated Ca2+ entry pathways, thereby enabling an entirely discrete and unique signal in each case, a feature that is clearly critical in all cell signaling pathways.
Experimental procedures
Electrophysiology
Flp-InTM-293 cells (Invitrogen) were used throughout with culture conditions and procedures for the depletion of endogenous STIM1 using siRNA duplexes as described previously (8). In the current studies, such siRNA knockdown of endogenous STIM1 resulted in an ∼70% reduction of endogenous STIM1. Cells bearing a variety of different siRNA-resistant STIM1 mutations were generated as follows. For the L390Q, L390A, L390I, L390V, and G392A mutations, a PCR approach was used where a sense primer including the EcoRI site prior to the STIM1 initiation site and a mutated antisense primer including the downstream PmlI site was used along with an siRNA-resistant STIM1 DNA template. The resulting PCR product was then cut using PmlI and EcoRI and inserted into a PmlI/EcoRI–cut, siRNA-resistant STIM1 Venus backbone. Essentially the same procedure was used to generate STIM1-resistant constructs T389A/G392A, T389E/L390Q, and T389L/L390Q using the appropriate T389A, T389E, and L390Q siRNA-resistant DNA templates. Similarly, STIM1-resistant mutant versions of the Lys384 STIM1 residue (K384Q, K384A, K384V, and K384E) were generated using an siRNA-resistant STIM1-Δ448 template. Finally, a Lck-STIM1 basic region construct incorporating the T389E mutation was generated using the previously described construct (9), terminated at residue His448 and bearing a C-terminal cerulean tag. All constructs were sequenced prior to use. Transfection of constructs into cells (0.75 μg of the relevant DNA plus 225 pmol of STIM1 siRNA) was by an Amaxa Nucleofector II device following the manufacturer's guidelines.
Procedures for patch clamp experiments were performed at room temperature (20–22 °C) using protocols described previously (9, 20). Briefly, currents were recorded at room temperature (20–22 °C) using 250-ms pulses to −80 mV from a holding potential of 0 mV, sampled at 20 kHz during the voltage steps, and low-pass Bessel filtered online at 5 kHz. Current–voltage relationships were obtained from data obtained from 10-ms pulses from −100 to +80 mV at 20-mV intervals, sampled at 10 kHz, and low-pass Bessel filtered online at 5 kHz. The external (bath) solution comprised 140 mm NaCl, 5 mm CsCl, 1.2 mm MgCl2, 10 mm CaCl2, 10 mm glucose, and 10 mm Hepes (pH 7.4). The standard internal (pipette) solution contained 140 mm cesium acetate, 3.5 mm CaCl2, 3.72 mm MgCl2, 10 mm EGTA, and 10 mm Hepes (pH 7.2), resulting in a calculated free Ca2+ concentration of 100 nm.
For CRAC channel currents, this internal (pipette) solution was changed by removing all CaCl2 and increasing MgCl2 to 6.77 mm, and activation was induced by addition of the inositol trisphosphate receptor agonist adenophostin A (2 μm) via the patch pipette (36). Leak subtraction for CRAC channel currents was obtained from measurements at the end of each experiment in an external solution containing La3+ (100 μm). Activation of the ARC channel currents was induced by exogenous addition of AA (8 μm) to the bath solution (36), and leak subtraction was obtained from the initial currents before activation. Patch clamp data (mean ± S.E. or S.D. as indicated) were obtained from a minimum of four to 10 measurements made on different cells obtained from separate transfections (usually four) performed on different days.
Recombinant protein expression and purification
A construct encompassing human STIM1 (accession number AFZ76986.1) residues 234–491 was expressed in BL21 (DE3) Escherichia coli cells using a pET-28a vector as described previously (26). Briefly, the His6-tagged STIM1(234–491) protein was pulled out of inclusions using guanidine HCl solubilization buffer and HisPur nickel-nitrilotriacetic acid resin (Thermo Scientific). The protein was refolded into 20 mm Tris, 300 mm NaCl, and 1 mm DTT (pH 8.8) at 4 °C. After overnight cleavage of the His6 tag with thrombin (∼1 unit/mg of protein; EMD Millipore) the protein was purified by Q Sepharose Fast Flow (GE Healthcare) anion exchange chromatography and finally dialyzed into buffer (20 mm Tris, 250 mm NaCl, and 1 mm DTT (pH 8)). T389E, T389A, and L390Q mutations were introduced into the pET-28a STIM1(234–491) construct using the QuikChange (Agilent) PCR approach, and mutant proteins were prepared as described for wildtype. Protein concentrations were estimated using ϵ280 nm = 0.95 (mg ml−1)−1 cm−1.
Size exclusion chromatography
SEC was performed using a Superdex S200 10/300 GL (GE Healthcare) column connected to an ÄKTA FPLC (GE Healthcare) housed at 4 °C. Injection volumes were ∼800 μl for each sample, and flow rates were 0.5 ml min−1. Proteins were injected at 0.8–1.0 mg ml−1. Apparent stoichiometries of the STIM1 proteins were calculated using the regression line constructed from the elution volumes of a set of molecular mass standards: tetrameric urease (545 kDa), dimeric bovine serum albumin (132 kDa), monomeric bovine serum albumin (66 kDa), chicken albumin (45 kDa), carbonic anhydrase (29 kDa), and α-lactalbumin (14.2 kDa).
Far-UV CD spectroscopy
Far-UV CD experiments were performed using a Jasco J-810 spectrometer equipped with a PTC-423S temperature controller (Jasco, Inc.). Spectra are averages of three scans collected using a 0.1-cm quartz cuvette at 4 °C using an 8-s response time, 1-nm bandwidth, and 20 nm min−1 scanning speed. Thermal melts were acquired in the same cuvette between 4 and 95 °C using an 8-s response time and a 1 °C min−1 scanning rate. The apparent midpoints of temperature denaturation (Tm) were taken as the temperature at which the normalized fractional change in ellipticity was 0.5.
Statistical analyses
Data obtained are presented as mean ± S.D. or S.E. as appropriate based on the number of independent experiments, indicated by “n” value. Statistical significance was tested by performing unpaired, two-tailed Student's t test in GraphPad Prism 5 software.
Molecular dynamics simulations
Molecular dynamics simulations were started by extracting residues 375–410 from the crystal structure (Protein Data Bank code 3TEQ) (14). This region was chosen as the minimum stable region containing the key protein motif. Hydrogen atoms were added, and the histidine was protonated on the ϵ-carbon. For the phosphorylated simulations, a phosphate was added to Thr389 using the tool psfgen from the NAMD package (37). The T389E mutant was constructed using the “Mutate” tool from PyMOL version 1.8 (38).
Each structure was embedded in a 50 × 50 × 50-Å periodic box containing 3600 water molecules using the OptimalMembraneGenerator module from LOOS (39). Sodium and chloride ions were added to make the system electrically neutral with a free salt concentration of 100 mm. Each system was built four independent times, guaranteeing variation in the water and ion coordinates surrounding the protein. The systems were run using constant pressure and temperature (NPT) dynamics using a Langevin thermostat at 310 K with the damping set to 2 ps−1 and Langevin piston barostat (40) with the pressure set to 1 atm with a piston period of 200 fs and decay time of 100 fs. Dynamics was run using a 2-fs time step with the velocity Verlet integrator, and bonds to hydrogen were constrained using RATTLE (41). van der Waals interactions were smoothly switched to 0 over distances ranging from 10 to 12 Å, and long-range electrostatic interactions were computed using particle mesh Ewald on a 50 × 50 × 50 grid (42). Structures were recorded every 100 ps, although a 1-ns resolution was used for analysis purposes.
Each trajectory was run for ∼500 ns, following roughly 10–15 ns excluded from the beginning of each run as equilibration, to an aggregate total of ∼2 μs per system. All minimization and dynamics were performed using NAMD version 2.10 (37) with the CHARMM36 force field (43, 44). Contact map analysis was performed using custom tools developed using LOOS version 2.3 (code available upon request). Two residues were considered to be in contact if there was at least one pair of side-chain heavy atoms within 4 Å. We then averaged the contact maps for the four replicates to produce the average map for each system. Difference maps were computed by subtracting the average map for one system from another. p value maps were computed using a Student's t test for each residue pair; for these purposes, each individual trajectory was treated as a single measurement of the probability of residue–residue contact, allowing us to estimate the probability that the difference between results for two different systems (e.g. native and T389E) are due to chance. Graphs are plotted such that values larger than 0.05 are not visible.
Author contributions
T. J. S. conceived the study, wrote and edited the manuscript, and assisted with experimental design and data analyses. J. L. T. generated the various STIM1 mutants used in the electrophysiological experiments and performed the patch clamp measurements and their analyses. Y. Z. and P. B. S. designed, performed, and analyzed the biochemical/biophysical experiments involving size exclusion chromatography, far-UV CD spectroscopy, and thermal stability analyses on the STIM1 constructs and wrote the associated methods and interpretations. A. G. designed and performed all the molecular dynamics studies, analyzed the resulting data, wrote the associated methods, and developed the relevant figures (e.g. contact maps). All authors have read and approved the final manuscript.
Supplementary Material
This work was supported by NIGMS, National Institutes of Health Grant GM040457 (to T. J. S.) and Natural Sciences and Engineering Research Council Grant 250225 (to P. B. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- STIM
- stromal interaction molecule
- SOAR
- STIM1-Orai–activating region
- ARC
- arachidonate-regulated Ca2+
- CRAC
- Ca2+ release–activated Ca2+
- ER
- endoplasmic reticulum
- PM
- plasma membrane
- BR
- basic region
- AA
- arachidonic acid
- pF
- picofarad
- SEC
- size exclusion chromatography
- MD
- molecular dynamics
- phos-STIM1
- phosphorylated STIM1.
References
- 1. Williams R. T., Manji S. S., Parker N. J., Hancock M. S., Van Stekelenburg L., Eid J. P., Senior P. V., Kazenwadel J. S., Shandala T., Saint R., Smith P. J., and Dziadek M. A. (2001) Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem. J. 357, 673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mignen O., Thompson J. L., Yule D. I., and Shuttleworth T. J. (2005) Agonist activation of arachidonate-regulated Ca2+-selective (ARC) channels in murine parotid and pancreatic acinar cells. J. Physiol. 564, 791–801 10.1113/jphysiol.2005.085704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yeung-Yam-Wah V., Lee A. K., Tse F. W., and Tse A. (2010) Arachidonic acid stimulates extracellular Ca2+ entry in rat pancreatic β cells via activation of the noncapacitative arachidonate-regulated Ca2+ (ARC) channels. Cell Calcium 47, 77–83 10.1016/j.ceca.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 4. Shuttleworth T. J. (1996) Arachidonic acid activates the noncapacitative entry of Ca2+ during [Ca2+]i oscillations. J. Biol. Chem. 271, 21720–21725 10.1074/jbc.271.36.21720 [DOI] [PubMed] [Google Scholar]
- 5. Mignen O., Thompson J. L., and Shuttleworth T. J. (2003) Ca2+ selectivity and fatty acid specificity of the noncapacitative, arachidonate-regulated (ARC) channels. J. Biol. Chem. 278, 10174–10181 10.1074/jbc.M212536200 [DOI] [PubMed] [Google Scholar]
- 6. Yoo A. S., Cheng I., Chung S., Grenfell T. Z., Lee H., Pack-Chung E., Handler M., Shen J., Xia W., Tesco G., Saunders A. J., Ding K., Frosch M. P., Tanzi R. E., and Kim T. W. (2000) presenilin-mediated modulation of capacitative calcium entry. Neuron 27, 561–572 10.1016/S0896-6273(00)00066-0 [DOI] [PubMed] [Google Scholar]
- 7. Li Y. S., Wu P., Zhou X. Y., Chen J. G., Cai L., Wang F., Xu L. M., Zhang X. L., Chen Y., Liu S. J., Huang Y. P., and Ye D. Y. (2008) Formyl-peptide receptor like 1: a potent mediator of the Ca2+ release-activated Ca2+ current ICRAC. Arch. Biochem. Biophys. 478, 110–118 10.1016/j.abb.2008.07.002 [DOI] [PubMed] [Google Scholar]
- 8. Thompson J. L., and Shuttleworth T. J. (2012) A plasma membrane-targeted cytosolic domain of STIM1 selectively activates ARC channels, an arachidonate-regulated store-independent Orai channel. Channels 6, 370–378 10.4161/chan.21947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thompson J. L., and Shuttleworth T. J. (2013) Molecular basis of activation of the arachidonate-regulated Ca2+ (ARC) channel, a store-independent Orai channel, by plasma membrane STIM1. J. Physiol. 591, 3507–3523 10.1113/jphysiol.2013.256784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan J. P., Zeng W., Dorwart M. R., Choi Y. J., Worley P. F., and Muallem S. (2009) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343 10.1038/ncb1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., and Lewis R. S. (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890 10.1016/j.cell.2009.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muik M., Fahrner M., Derler I., Schindl R., Bergsmann J., Frischauf I., Groschner K., and Romanin C. (2009) A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to Orai1 channels. J. Biol. Chem. 284, 8421–8426 10.1074/jbc.C800229200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawasaki T., Lange I., and Feske S. (2009) A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 385, 49–54 10.1016/j.bbrc.2009.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang X., Jin H., Cai X., Li S., and Shen Y. (2012) Structural and mechanistic insights into the activation of stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. 109, 5657–5662 10.1073/pnas.1118947109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Derler I., Jardin I., and Romanin C. (2016) Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 310, C643–C662 10.1152/ajpcell.00007.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., and Cahalan M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 10.1038/nature04147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Korzeniowski M. K., Manjarrés I. M., Varnai P., and Balla T. (2010) Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci. Signal. 3, ra82 10.1126/scisignal.2001122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stathopulos P. B., Schindl R., Fahrner M., Zheng L., Gasmi-Seabrook G. M., Muik M., Romanin C., and Ikura M. (2013) STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 4, 2963 10.1038/ncomms3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Calloway N., Holowka D., and Baird B. (2010) A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry 49, 1067–1071 10.1021/bi901936q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thompson J. L., and Shuttleworth T. J. (2015) Anchoring protein AKAP79-mediated PKA phosphorylation of STIM1 determines selective activation of the ARC channel, a store-independent Orai channel. J. Physiol. 593, 559–572 10.1113/jphysiol.2014.284182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Radzicka A., and Wolfenden R. (1988) Comparing the polarities of the amino acids: side-chain distribution coefficients between the vapor phase, cyclohexane, 1-octanol, and neutral aqueous solution. Biochemistry 27, 1664–1670 10.1021/bi00405a042 [DOI] [Google Scholar]
- 22. Monera O. D., Sereda T. J., Zhou N. E., Kay C. M., and Hodges R. S. (1995) Relationship of sidechain hydrophobicity and α-helical propensity on the stability of the single-stranded amphipathic α-helix. J. Pept. Sci. 1, 319–329 10.1002/psc.310010507 [DOI] [PubMed] [Google Scholar]
- 23. Tien M. Z., Meyer A. G., Sydykova D. K., Spielman S. J., and Wilke C. O. (2013) Maximum allowed solvent accessibilities of residues in proteins. PLoS One 8, e80635 10.1371/journal.pone.0080635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frischauf I., Muik M., Derler I., Bergsmann J., Fahrner M., Schindl R., Groschner K., and Romanin C. (2009) Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1–3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem. 284, 21696–21706 10.1074/jbc.M109.018408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lupas A., Van Dyke M., and Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 10.1126/science.252.5009.1162 [DOI] [PubMed] [Google Scholar]
- 26. Muik M., Fahrner M., Schindl R., Stathopulos P., Frischauf I., Derler I., Plenk P., Lackner B., Groschner K., Ikura M., and Romanin C. (2011) STIM1 couples to Orai1 via an intramolecular transition into an extended conformation. EMBO J. 30, 1678–1689 10.1038/emboj.2011.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Covington E. D., Wu M. M., and Lewis R. S. (2010) Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol. Biol. Cell 21, 1897–1907 10.1091/mbc.E10-02-0145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Korzeniowski M. K., Wisniewski E., Baird B., Holowka D. A., and Balla T. (2017) Molecular anatomy of the early events in STIM1 activation: oligomerization or conformational change? J. Cell Sci. 130, 2821–2832 10.1242/jcs.205583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y., Ramachandran S., Oh-Hora M., Rao A., and Hogan P. G. (2010) Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. U.S.A. 107, 4896–4901 10.1073/pnas.1001169107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou Y., Srinivasan P., Razavi S., Seymour S., Meraner P., Gudlur A., Stathopulos P. B., Ikura M., Rao A., and Hogan P. G. (2013) Initial activation of STIM1, the regulator of store-operated calcium entry. Nat. Struct. Mol. Biol. 20, 973–981 10.1038/nsmb.2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou Y., Wang X., Wang X., Loktionova N. A., Cai X., Nwokonko R. M., Vrana E., Wang Y., Rothberg B. S., and Gill D. L. (2015) STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 6, 8395 10.1038/ncomms9395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Espinoza-Fonseca L. M., Colson B. A., and Thomas D. D. (2014) Effects of pseudophosphorylation mutants on the structural dynamics of smooth muscle myosin regulatory light chain. Mol. Biosyst. 10, 2693–2698 10.1039/C4MB00364K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Serrano L., Neira J. L., Sancho J., and Fersht A. R. (1992) Effect of alanine versus glycine in α-helices on protein stability. Nature 356, 453–455 10.1038/356453a0 [DOI] [PubMed] [Google Scholar]
- 34. López-Llano J., Campos L. A., and Sancho J. (2006) α-Helix stabilization by alanine relative to glycine: roles of polar and apolar solvent exposures and of backbone entropy. Proteins 64, 769–778 10.1002/prot.21041 [DOI] [PubMed] [Google Scholar]
- 35. Wang X., Wang Y., Zhou Y., Hendron E., Mancarella S., Andrake M. D., Rothberg B. S., Soboloff J., and Gill D. L. (2014) Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 5, 3183 10.1038/ncomms4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thompson J. L., and Shuttleworth T. J. (2011) Orai channel-dependent activation of phospholipase C-δ; a novel mechanism for the effects of calcium entry on calcium oscillations. J. Physiol. 589, 5057–5069 10.1113/jphysiol.2011.214437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DeLano W. L. (2015) The PyMOL Molecular Graphics System, version 1.8, Schrödinger, LLC, New York [Google Scholar]
- 39. Romo T. D., Leioatts N., and Grossfield A. (2014) Lightweight object oriented structure analysis: tools for building tools to analyze molecular dynamics simulations. J. Comput. Chem. 35, 2305–2318 10.1002/jcc.23753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feller S. E., Zhang Y. H., Pastor R. W., and Brooks B. R. (1995) Constant-pressure molecular-dynamics simulation: the Langevin piston method. J. Chem. Phys. 103, 4613–4621 10.1063/1.470648 [DOI] [Google Scholar]
- 41. Andersen H. C. (1983) A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 52, 24–34 10.1016/0021-9991(83)90014-1 [DOI] [Google Scholar]
- 42. Essmann U., Perera L., Berkowitz M., Darden T., Lee H., and Pedersen L. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 43. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 10.1021/ct300400x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang J., and MacKerell A. D. Jr. (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 10.1002/jcc.23354 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.