

Abstract
The cyclooxygenases COX-1 and COX-2 oxygenate arachidonic acid (AA) to prostaglandin H2 (PGH2). COX-2 also oxygenates the endocannabinoids 2-arachidonoylglycerol (2-AG) and arachidonoylethanolamide (AEA) to the corresponding PGH2 analogs. Both enzymes are targets of nonsteroidal anti-inflammatory drugs (NSAIDs), but NSAID-mediated COX inhibition is associated with gastrointestinal toxicity. One potential strategy to counter this toxicity is to also inhibit fatty acid amide hydrolase (FAAH), which hydrolyzes bioactive fatty acid ethanolamides (FAEs) into fatty acids and ethanolamine. Here, we investigated the mechanism of COX inhibition by ARN2508, an NSAID that inhibits both COXs and FAAH with high potency, target selectivity, and decreased gastrointestinal toxicity in mouse models, presumably due to its ability to increase levels of FAEs. A 2.27-Å–resolution X-ray crystal structure of the COX-2·(S)-ARN2508 complex reveals that ARN2508 adopts a binding pose similar to that of its parent NSAID flurbiprofen. However, ARN2508's alkyl tail is inserted deep into the top channel, an active site region not exploited by any previously reported NSAID. As for flurbiprofen, ARN2508's potency is highly dependent on the configuration of the α-methyl group. Thus, (S)-ARN2508 is more potent than (R)-ARN2508 for inhibition of AA oxygenation by both COXs and 2-AG oxygenation by COX-2. Also, similarly to (R)-flurbiprofen, (R)-ARN2508 exhibits substrate selectivity for inhibition of 2-AG oxygenation. Site-directed mutagenesis confirms the importance of insertion of the alkyl tail into the top channel for (S)-ARN2508's potency and suggests a role for Ser-530 as a determinant of the inhibitor's slow rate of inhibition compared with that of (S)-flurbiprofen.
Keywords: cyclooxygenase (COX), enzyme catalysis, enzyme inhibitor, enzyme kinetics, enzyme structure, fatty acid metabolism, inflammation, polyunsaturated fatty acid (PUFA)
Introduction
The enzyme cyclooxygenase (COX)3 catalyzes the committed step in prostanoid biosynthesis, the bis-dioxygenation and cyclization of AA to form the endoperoxy-hydroperoxide intermediate prostaglandin (PG) G2. Then, via its peroxidase activity, the enzyme reduces the 15-hydroperoxy group of PGG2 to produce the final product, PGH2 (1). PGH2 is a substrate for various terminal prostanoid synthases that catalyze the formation of PGs as well as thromboxane A2. PGs exert various biological effects including platelet aggregation, gastrointestinal and cardiovascular regulation, parturition, and modulation of the inflammatory response (2).
There are two COX isoforms, COX-1 and COX-2, distinguished primarily by their patterns of expression. Specifically, the expression of COX-2 is induced in response to proinflammatory cytokines and growth factors, whereas the expression of COX-1 is constitutive in most tissues (3). In addition, because of its larger active site, COX-2 can oxygenate analogs of AA that are poor substrates for COX-1. These include the endocannabinoids 2-arachidonoylglycerol (2-AG) and arachidonoylethanolamide (AEA), oxygenation of which yields PGH2-glyceryl ester and PGH2-ethanolamide, respectively (4–6).
Both COX enzymes, but particularly COX-2, are the major sites of action of the widely used non-steroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen, indomethacin, and naproxen (7). A major limitation to the use of most NSAIDs is gastrointestinal toxicity due to blockade of the biosynthesis of cytoprotective PGs (8, 9). This problem was addressed by the development of COX-2–selective inhibitors that preserved COX-1–dependent gastrointestinal PGE2 production. These drugs exhibit good anti-inflammatory activity with reduced gastrointestinal side effects; however, subsequent clinical trials uncovered adverse cardiovascular side effects associated with inhibition of COX-2 (10, 11). An alternative approach to NSAID-mediated gastrointestinal toxicity is to increase the levels of AEA and other fatty acyl ethanolamides (FAEs), such as palmitoylethanolamide, in individuals receiving NSAID therapy. AEA and palmitoylethanolamide exert anti-inflammatory and cytoprotective effects in the gastrointestinal tract that can compensate for loss of beneficial PGs (12). However, during inflammation, this FAE-dependent signaling may be compromised through increased expression of fatty acid amide hydrolase (FAAH), the primary degradative enzyme for FAEs, and COX-2, which may also contribute to AEA depletion because of its oxidation. These considerations have led to the hypothesis that simultaneous blockade of COX and FAAH might promote FAE-mediated prevention of the gastrointestinal side effects of NSAIDs while preserving most of their anti-inflammatory effects.
To test this hypothesis, dual inhibitors that simultaneously target both the COX enzymes and FAAH with high potency and oral bioavailability were synthesized. One compound from this class, ARN2508 (see Fig. 1), decreases intestinal inflammation and protects the gastrointestinal tract from NSAID-induced toxicity in vivo (13). ARN2508 combines key structural features of the compound URB597, an FAAH inhibitor, and flurbiprofen (Fig. 1), a member of the 2-arylpropionic acid class of NSAIDs. Like flurbiprofen, ARN2508 inhibits PGE2 formation in the gastric mucosa, but unlike flurbiprofen, ARN2508 was found to protect the epithelial lining in the stomach of mice, likely through its ability to increase levels of AEA and other FAEs.
Figure 1.

Structures of (R)- and (S)-flurbiprofen and (S)-ARN2508.
Inhibition of both FAAH and COX by ARN2508 was found to be functionally irreversible based on dialysis experiments (13). In the case of FAAH, ARN2508-mediated inhibition was attributed to a covalent bond formed between the carbamate moiety of the inhibitor and the enzyme's catalytic serine (14). Thus, the carbamate functional group of ARN2508 is required for FAAH inhibition; however, it is not necessary for inhibition of COX. In fact, the carboxylate moiety is necessary for COX inhibition by ARN2508 as is seen in the case of its parent compound, flurbiprofen, which inhibits COX by a noncovalent mechanism. These findings suggest that covalent modification is not required for ARN2508-mediated inhibition of COX, but they do not completely rule out this possibility.
Flurbiprofen contains one chiral center α to the carboxylic acid. Potent COX inhibitory activity is associated only with the S-enantiomer of flurbiprofen; however, the R-enantiomer is a substrate-selective inhibitor of endocannabinoid oxygenation by COX-2 (15). ARN2508 shares this chiral carbon, but the initial kinetic evaluation of its COX-inhibitory activity was carried out using the racemic mixture (13). Here, we report the characterization of the S- and R-enantiomers of ARN2508 with regard to potency, time dependence, isoform selectivity, and substrate selectivity of COX inhibition. We also explore structural determinants of ARN2508-mediated inhibition of COX and report the crystal structure of COX-2 complexed with (S)-ARN2508. Our findings reveal that the enantioselectivity of ARN2508 is similar to that of flurbiprofen. Also, like (S)-flurbiprofen, (S)-ARN2508 is a time-dependent inhibitor of COX-2, although its rate of inhibition is substantially slower than that of its parent. Furthermore, whereas the flurbiprofen moiety of ARN2508 binds in the COX-2 active site in a pose very similar to that of flurbiprofen, the inhibitor's alkyl chain occupies the same channel of COX-2 as the ω-tail of AA in a region of the active site that has not been exploited by any other NSAID for which structural data are available. Thus, in addition to its novel pharmacology as a dual inhibitor of FAAH and COX, ARN2508 exhibits binding properties not previously observed for any of the numerous selective and nonselective COX inhibitors described previously.
Results
Structural analysis of COX-2 with (S)-ARN2508
To better understand the basis for inhibitor binding, we obtained the three-dimensional structure of COX-2 in complex with (S)-ARN2508 via X-ray crystallography by molecular replacement using the high-resolution monomer model of mCOX-2 in complex with naproxen (Protein Data Bank code 3NT1) (16). The COX-2·(S)-ARN2508 complex was determined to 2.27-Å resolution in the space group of I4122. The omit difference map (Fo − Fc) clearly revealed that the ligand in the crystal is indeed the S-enantiomer of ARN2508 (Fig. 2 and Table 1). We have not been able to obtain high quality crystals of the COX-2·(R)-ARN2508 complex, possibly due to the low potency of this inhibitor.
Figure 2.

Stereodiagram of the electron density map of (S)-ARN2508 in the COX-2 channel. The omit Fo − Fc map is contoured at 3 σ, the inhibitor is colored in yellow sticks, and the interacting residues of COX-2 are illustrated in light cyan. Potential H-bonds are presented with dashed lines.
Table 1.
X-ray data collection and refinement statistics
Number of crystals is 1. The values in parentheses are for the highest resolution shell. Rmerge = (ΣhklΣi|Ii(hkl) − Ii(hkl)|)/(ΣhklΣiIi(hkl)) × 100% and R = (Σhkl‖Fo| − |Fc‖)/Σhkl|Fo|) × 100% where Fo and Fc are the observed and calculated structure factors and Rfree test set is 3.0%. r.m.s., root mean square. CC½, the correlation coefficient between random half data sets.
| COX-2·(S)-ARN2508 (Protein Data Bank code 5W58) | |
|---|---|
| Data collection | |
| Wavelength (Å) | 0.9792 |
| Resolution range (Å) | 66.05–2.27 (2.35–2.27) |
| Space group | I4122 |
| Unit cell (a, b, c) | 173.8, 173.8, 203.3 |
| Total reflections | 1,048,952 (101,264) |
| Unique reflections | 71,733 (6,799) |
| Multiplicity | 14.6 (14.4) |
| Completeness (%) | 0.99 (0.99) |
| Mean I/σ(I) | 19.41 (0.82) |
| Wilson B-factor (Å2) | 48.02 |
| Rmerge | 0.1551 (2.12) |
| CC1/2 | 0.999 (0.336) |
| Refinement | |
| Rwork/Rfree | 16.9/18.9 (33.3/33.4) |
| Number of atoms (total/protein/ligands/solvents) | 4,967/4,512/181/275 |
| r.m.s. (bond/angle) | 0.012/1.26 |
| Ramachandran favored/outliers (%) | 98.0/0 |
| Average B-factor (total/protein/ligands/solvents) | 55.86/55.06/75.81/55.92 |
The cyclooxygenase active site of COX-2 comprises a long L-shaped channel beginning at the membrane-binding domain and terminating deep within the catalytic domain. The active site is separated from the membrane-binding domain and an enlarged “lobby” immediately above it by a constriction formed by Arg-120, Tyr-355, and Glu-524. Above the constriction, the channel is lined predominantly by hydrophobic residues that favor binding of fatty acid substrates to the active site. Notable exceptions are Tyr-385, the catalytic residue that, in radical form, initiates the first step in PG biosynthesis, and Ser-530. Both of these residues are located at the bend in the channel (Fig. 3). AA binds in this channel with the carboxyl group in close proximity to Arg-120 and Tyr-355 at the constriction site, carbon 13 aligned with the catalytic Tyr-385, and the ω-tail deep in the alcove above Ser-530 (17, 18). Flurbiprofen also binds in the active site with the carboxylate close to the polar constriction site residues; however, this more compact molecule does not extend into the active site above Ser-530 (19–23).
Figure 3.

Crystal structure overlay of (S)-ARN2508 and AA in the COX-2 active site. The side chains of corresponding residues of the COX-2·AA complex were omitted for clarity. (S)-ARN2508 is illustrated in yellow, and AA (Protein Data Bank code 3HS5) is illustrated in gray.
ARN2508 is an alkyl carbamic acid biphenyl conjugate. As might be predicted, the flurbiprofen moiety of ARN2508 sits in the hydrophobic channel of the enzyme's active site with the same binding mode that is adopted by flurbiprofen in COX-1 and COX-2 (19–23). Thus, the carboxylate moiety of ARN2508 lies at the constriction of the active site, ion-pairing to Arg-120 and hydrogen-bonding to Tyr-355, and the aromatic rings project upward toward Tyr-385. However, hydrogen bond formation with ARN2508's carbamate group requires an upward displacement of the side chain of Ser-530 in the COX-2·(S)-ARN2508 complex relative to its position in the COX-2·flurbiprofen complex. Additional hydrogen bonds form between Tyr-385 and the oxygen atoms of ARN2508's carbamate group. The alkyl chain of ARN2508 reaches deeply into the distal portion of the cyclooxygenase channel between helices 2 and 17, making hydrophobic interactions with various residues including Phe-205, Phe-209, Phe-210, Val-228, Ile-341, Val-344, Ile-377, Phe-381, and Leu-534 (Fig. 2).
Potency, isoform selectivity, and substrate selectivity of ARN2508 enantiomers
It was previously reported that racemic ARN2508 has an IC50 value of ∼12 nm for COX-1 and 430 nm for COX-2 (13). To further assess the potency of the inhibitor, each enantiomer was individually tested for both COX-1 and COX-2 inhibition. The results (Fig. 4) indicated that the potency of the S-enantiomer of ARN2508 (IC50 = 7.0 nm) for COX-1 inhibition is much greater than that of the R-enantiomer (IC50 = 4.6 μm) (Table 2). Similarly, using 5 μm AA as substrate, the S-enantiomer was the more potent of the two against COX-2, achieving complete inhibition of AA oxygenation with an IC50 of ∼39 nm (Fig. 5A and Table 2), whereas the R-enantiomer inhibited the enzyme by ∼50% at 10 μm and failed to reach complete inhibition at the highest concentration tested (100 μm). Thus, as previously reported for the racemic mixture (13), each enantiomer of ARN2508 exhibits a small degree of COX-1 selectivity. The potency of the S-enantiomer for 2-AG oxygenation by COX-2 (IC50 = 21 nm) was greater than that for AA oxygenation but only by ∼2-fold (Fig. 5B and Table 2). In contrast, (R)-ARN2508 exhibited an ∼30-fold higher inhibitor potency for 2-AG (IC50 = 0.34 μm) versus AA oxygenation and completely blocked 2-AG oxygenation. These results indicate that the R-enantiomer is a substrate-selective COX-2 inhibitor.
Figure 4.

Inhibition of oCOX-1 with ARN2508. Oxygenation of 5 μm AA by oCOX-1 was assessed by quantification of enzymatic product formation utilizing LC-MS/MS as described under “Experimental procedures.” Enzyme was preincubated with each enantiomer of ARN2508 for 10 min. AA (5 μm) was allowed to react for 10 s before quenching with organic solvent containing deuterated internal standards. Results are the mean ± S.D. of triplicate determinations. Some error bars are shorter than the height of the symbol.
Table 2.
(±)-ARN2508 inhibition IC50 values
| Enzyme | Substrate | Preincubation | IC50 |
|
|---|---|---|---|---|
| (S)-ARN2508 | (R)-ARN2508 | |||
| min | μm | |||
| COX-1 WT | AA | 10 | 0.007 | 4.6 |
| COX-2 WT | AA | 10 | 0.039 | 10 |
| COX-2 WT | AA | 0 | n/aa | 11 |
| COX-2 WT | 2-AG | 10 | 0.021 | 0.34 |
| COX-2 WT | 2-AG | 0 | n/a | 0.54 |
| COX-2 Y355F | AA | 10 | 0.099 | 29 |
| COX-2 S530A | AA | 10 | 0.026 | n/a |
| COX-2 S530T | AA | 10 | 0.291 | n/a |
a n/a, unable to obtain fitting due to incomplete enzyme inhibition.
Figure 5.

Inhibition of mCOX-2 with ARN2508 with or without preincubation. Oxygenation of 5 μm AA (A) or 5 μm 2-AG (B) by mCOX-2 was assessed by quantification of enzymatic product formation utilizing LC-MS/MS as described under “Experimental procedures.” Each enantiomer of ARN2508 was preincubated for 10 min (10 m) or added simultaneously (0 m) with substrate. Each substrate was allowed to react for 10 s before quenching with organic solvent containing deuterated internal standards. Results are the mean ± S.D. of triplicate determinations. Some error bars are shorter than the height of the symbol.
Time dependence of AA and 2-AG oxygenation inhibition by ARN2508 enantiomers
As most highly potent COX inhibitors are time-dependent, initial experiments included an arbitrarily chosen 10-min preincubation period prior to substrate addition. To further explore the time dependence of ARN2508, various concentrations of each enantiomer were added simultaneously with either AA or 2-AG to COX-2, reactions were quenched after 10 s, and products were analyzed using LC-MS/MS. The data indicate that COX-2 inhibition by the R-enantiomer is essentially time-independent for both AA and 2-AG oxygenation as preincubation had minimal effect on the observed IC50 values or extent of inhibition observed. In contrast, COX-2 inhibition by the S-enantiomer is strongly time-dependent as indicated by IC50 values in the nm range for both AA and 2-AG oxygenation following preincubation but failure to reach even 50% inhibition in the absence of preincubation at the concentrations tested (Fig. 5, A and B, and Table 2).
The vast majority of COX-2 inhibitors bind to the enzyme in a non-covalent manner, a notable exception being aspirin, which acetylates Ser-530 (24). Most time-dependent, non-covalent inhibitors exhibit kinetics consistent with a two-step binding model that includes rapid, reversible formation of an initial complex followed by a slower step that leads to a much stronger enzyme-inhibitor interaction (25). To assess the kinetic mechanism of ARN2508-mediated COX-2 inhibition, various concentrations of the S-enantiomer were preincubated with wildtype COX-2 for different time periods, and the activity of the enzyme was measured. A plot of enzyme activity versus time for each inhibitor concentration exhibited pseudo-first order kinetics (Fig. 6A), and the observed first order rate constants for each curve (kobs) were plotted against inhibitor concentration. The resulting data (Fig. 6B) failed to yield the hyperbolic curve that is expected from the two-step model. A possible explanation is that the two-step model is correct, but insufficient concentrations of inhibitor were used to fully delineate the hyperbola. Indeed, fitting of the data to this model yields a first step dissociation constant of 220 μm and inhibition rate constant of 41 min−1, corresponding to an overall kinetic efficiency (kon/KI) of 0.18 min−1·μm−1. These values are, at best, only estimates as evaluation of much higher inhibitor concentrations is necessary to fully define the curve. Unfortunately, solubility limitations prevented this evaluation. An alternative explanation is that the plot is linear rather than hyperbolic, suggesting that ARN2508 does not follow the typical two-step kinetic mechanism. In this case, the binding process would more likely occur in a single step, or if multistep, the first step is rate-limiting. The slope of the line (0.11 min−1·μm−1) provides an estimate of the second order rate constant for the binding of inhibitor to enzyme in this model. Regardless of which model is correct, the formation of the inhibitory (S)-ARN2508·COX-2 complex occurs substantially more slowly than formation of the inhibitory complex of (S)-flurbiprofen with COX-2, which exhibits a reported overall kinetic efficiency of 6 min−1·μm−1 (26).
Figure 6.

Kinetics of the time-dependent inhibition of mCOX-2 by (S)-ARN2508. A, various concentrations of (S)-ARN2508 were incubated with wildtype mCOX-2 for the indicated preincubation times before the addition of 5 μm AA. Each enzymatic reaction proceeded for 10 s before being quenched with organic solvent containing deuterated internal standards. Oxygenation of AA was assessed by quantification of enzymatic product formation utilizing LC-MS/MS as described under “Experimental procedures.” B, observed first order rate constants were plotted against inhibitor concentration to obtain the kinetic parameters for the enzyme-substrate interaction. Both linear and hyperbolic fits are shown. Results are the mean ± S.D. of triplicate determinations. Some error bars are shorter than the height of the symbol.
Importance of Tyr-355 in determining the potency of ARN2508
The Tyr-355 residue of COX-2 forms part of the constriction site that separates the lobby region from the active site, placing it within hydrogen-bonding distance of the carboxylate group of many NSAIDs including ARN2508 (7). To evaluate the importance of this interaction, ARN2508 was tested for its ability to inhibit AA oxygenation by Y355F. As seen in Table S1 and Fig. S1, the Y355F mutation had only modest effects on the kinetics of the enzyme with AA as substrate. The potency of (S)-ARN2508 decreased slightly for the Y355F mutant as indicated by an ∼2.5-fold increase in IC50. The potency of the R-enantiomer decreased more substantially; failure to achieve more than 50% inhibition at the highest inhibitor concentration tested (50 μm) precluded determination of an IC50 value (Fig. 7 and Table 2). These findings differ significantly from those obtained with other previously reported carboxylate-containing NSAIDs including indomethacin, meclofenamic acid, piroxicam, ketorolac, and ibuprofen, which exhibit substantially reduced potency against the COX-2 Y355F enzyme (27–29), and they support the hypothesis that hydrogen bond formation with Tyr-355 contributes only modestly to the binding affinity of (S)-ARN2508.
Figure 7.

Inhibition of mCOX-2 mutants with ARN2508. Oxygenation of 5 μm AA by wildtype, Y355F, S530A, or S530T mCOX-2 enzyme was assessed by quantification of enzymatic product formation utilizing LC-MS/MS as described under “Experimental procedures.” Each enzyme was preincubated with the indicated enantiomer of ARN2508 for 10 min. AA was allowed to react for 10 s before quenching with organic solvent containing deuterated internal standards. Results are the mean ± S.D. of triplicate determinations. Some error bars are shorter than the height of the symbol.
Role of Ser-530 in ARN2508 binding to COX-2
The presence of a reactive carbamoyl group and the known ability of ARN2508 to covalently modify FAAH suggested the possibility that the inhibitor also covalently modifies COX-2. The proximity of the carbamoyl group of ARN2508 to Ser-530 observed in the crystal structure led us to hypothesize that a covalent modification might occur at that residue in solution. However, LC-MS/MS analysis of the tryptic peptides of COX-2 that had been incubated with the inhibitor failed to reveal the mass shift expected from the addition of ARN2508 to Ser-530. Despite obtaining ample sequence coverage (84%) that includes Ser-530 (Fig. S2), no detectable modifications were observed. These results do not support covalent bond formation between the inhibitor and Ser-530.
Although our data did not support covalent bond formation between the carbamoyl group of (S)-ARN2508 and Ser-530 of COX-2, the crystal structure reveals the presence of a hydrogen bond between these moieties. To determine the importance of this interaction to ARN2508's potency and to further rule out a covalent interaction, the ability of each enantiomer to inhibit a COX-2 S530A mutant enzyme was evaluated. As in the case of Y355F, the S530A mutation had minimal effect on enzymatic activity using AA as substrate (Table S1 and Fig. S1). Furthermore, this mutation did not have a major effect on the potency of (S)-ARN2508 and caused only a mild reduction in the potency of the R-enantiomer using AA as substrate and a 10-min preincubation (Fig. 7). These findings do not support the hypothesis that an interaction (covalent or noncovalent) with the hydroxyl group of Ser-530 plays a significant role in ARN2508's potency.
Although the potency of (S)-ARN2508 (as measured by IC50 values) was nearly identical for wildtype and S530A COX-2 following our standard 10-min preincubation time, the position of S530A at the bend of the active site suggested that this residue might interfere with the insertion of ARN2508's hydrophobic tail into the upper part of the channel. To test this hypothesis, we compared the rate of inhibition of the mutant with that of the wildtype enzyme at two concentrations of (S)-ARN2508 (Fig. 8A). The data clearly showed a much higher rate of inhibition in the case of the mutant enzyme. Unfortunately, the very rapid rates at high (S)-ARN2508 concentrations precluded a full kinetic analysis.
Figure 8.

Kinetics of the time-dependent inhibition of S530A and S530T mCOX-2 by (S)-ARN2508. A, two concentrations of (S)-ARN2508 were incubated with wildtype or S530A mCOX-2 for the indicated preincubation times before the addition of 5 μm AA. Each enzymatic reaction proceeded for 10 s before being quenched with organic solvent containing deuterated internal standards. Oxygenation of AA was assessed by quantification of enzymatic product formation utilizing LC-MS/MS as described under “Experimental procedures.” B, two concentrations of (S)-ARN2508 were incubated with wildtype or S530T mCOX-2 for the indicated preincubation times before the addition of 5 μm AA. Results are the mean ± S.D. of triplicate determinations. Some error bars are shorter than the height of the symbol.
For a time-dependent inhibitor, the measured IC50 value is dependent on the length of the preincubation period. Longer preincubations provide time for the enzyme·inhibitor complex to form, making the inhibitor appear more potent and reducing the IC50. Consistently, when the potency of (S)-ARN2508 was determined for S530A and wildtype COX-2 following a 30-s preincubation period, the IC50 values obtained for both enzymes were substantially higher (0.31 μm for S530A and 4.51 μm for wildtype) than those obtained following a 10-min preincubation (Table 2 and Fig. 9). Notably, at this short preincubation time, (S)-ARN2508 is significantly more potent against the mutant than the wildtype enzyme. This is due to the much more rapid development of inhibition with the mutant.
Figure 9.

Dose response of (S)-ARN2508 with 30-s preincubation. The indicated concentrations (S)-ARN2508 were preincubated with either wildtype or S530A mCOX-2 for 30 s before the addition of 5 μm AA. Each enzymatic reaction proceeded for 10 s before being quenched with organic solvent containing deuterated internal standards. Oxygenation of AA was assessed by quantification of enzymatic product formation utilizing LC-MS/MS as described under “Experimental procedures.” Results are the mean ± S.D. of triplicate determinations. Some error bars are shorter than the height of the symbol.
Because the S530A mutant decreases steric bulk from the bend in the COX-2 active site and eliminates hydrogen bonding with the inhibitor, we evaluated the effects of a COX-2 S530T mutation on inhibitor potency to observe how additional steric bulk in that region might alter the rate or potency of inhibition. This mutation had substantial effects on enzyme activity, both raising the Km (3-fold) and lowering the Vmax (3-fold) (Table S1 and Fig. S1). The mutation was also associated with a reduction in potency of (S)-ARN2508 (Table 2). Somewhat unexpectedly, however, the rate of inhibition of S530T observed with two concentrations of (S)-ARN2508 was faster than that of the wildtype enzyme (Fig. 8B). These results suggest that the larger threonine residue does not substantially impede the inhibitor's access to the top channel of the COX-2 active site.
Inhibition of COX-2 Gly-533 mutants by (S)-ARN2508
As observed from the crystal structure, AA binds in an L-shaped conformation with its ω-tail inserted into the top channel of the COX-2 active site (17, 18). Gly-533 lies near the end of the channel, and mutations of this residue to bulkier side chains inhibit the oxygenation of AA. However, Gly-533 mutants including G533A, G533V, and G533L can oxygenate the 18-carbon fatty acid α-linolenic acid (LNA) (30). One of the major products formed from this reaction is 12-hydroxyoctadecatrienoic acid (31), which contains a conjugated double bond that can be detected by UV-visible spectroscopy at 235 nm. The oxygenation of LNA by wildtype mCOX-2 and the mutant G533L in the presence of (S)-ARN2508 or (S)-flurbiprofen was evaluated to determine whether disruption of the structure of the top channel would significantly impact ARN2508's potency due to the extension of the inhibitor's hydrophobic tail to the end of the upper active site channel. Differences were observed between the wildtype enzyme and the G533L mutant with respect to inhibition by (S)-ARN2508 versus (S)-flurbiprofen. In Fig. 10A, oxygenation of LNA by wildtype mCOX-2 is shown to be completely inhibited by a 10 μm concentration of both (S)-ARN2508 and (S)-flurbiprofen following a preincubation of 3 min or greater. With a 1-min preincubation, there was slight substrate oxygenation in the presence of (S)-ARN2508 but not (S)-flurbiprofen. In contrast, although (S)-flurbiprofen completely inhibited mCOX-2 G533L following a 3-min preincubation, (S)-ARN2508 was only partially effective. Even following a 15-min preincubation, (S)-ARN2508 inhibited oxygenation by G533L COX-2 less efficiently than did (S)-flurbiprofen preincubated for just 1 min (Fig. 10B). These findings suggest that the bulky side chain of G533L interferes with the binding of (S)-ARN2508's side chain in the active site channel and that this binding interaction is important to the inhibitor's potency.
Figure 10.
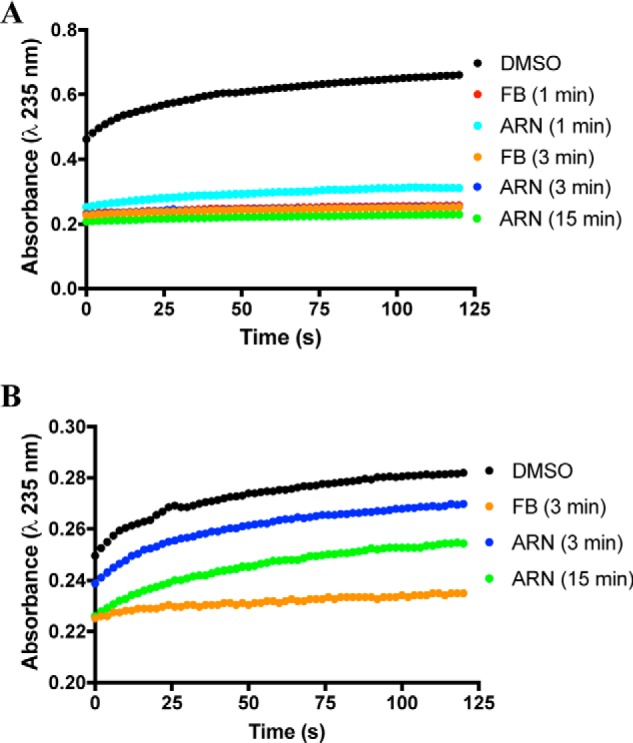
Kinetics of LNA inhibition by (S)-ARN2508 and (S)-flurbiprofen. A, wildtype mCOX-2 (200 nm monomer) with 200 nm heme was used to oxygenate 50 μm LNA. (S)-ARN2508 (ARN) or (S)-flurbiprofen (FB) from a DMSO stock was preincubated with enzyme for the indicated time before the addition of substrate. B, G533L mCOX-2 (200 nm monomer) with 200 nm heme was used to oxygenate 50 μm LNA. Reactions were monitored by increasing absorbance at 235 nm. Results are from one experiment.
Discussion
ARN2508 is a designed dual inhibitor of COX enzymes and FAAH. Here, we show that each enantiomer of the compound exhibits distinct kinetics of inhibition of substrate oxygenation by both COX isoforms. (S)-ARN2508 is a time-dependent, highly potent, tightly bound inhibitor that is slightly more potent against COX-1 than COX-2. In contrast, (R)-ARN2508 is a weak, rapidly reversible inhibitor of AA oxygenation that is slightly more potent against COX-1 than COX-2 and a substrate-selective inhibitor for COX-2–dependent 2-AG oxygenation. This kinetic behavior of the two ARN2508 enantiomers is similar to that of the enantiomers of its parent compound, flurbiprofen (15, 26, 32, 33).
The identical enantioselectivity of ARN2508 and flurbiprofen is not surprising as the orientation of (S)-ARN2508's flurbiprofen moiety in the COX-2 active site (Fig. 2) is nearly identical to that observed for (S)-flurbiprofen in complex with COX-1 and COX-2 (19–23). The α-methyl group of the flurbiprofen core of ARN2508 is adjacent to Tyr-355, which forms part of the constriction at the entrance into the active site. A similar binding interaction is observed for flurbiprofen, and previous studies have indicated that Tyr-355 is involved in flurbiprofen's time-dependent inhibition (34). A clash between Tyr-355 and the α-methyl group of the R-enantiomer of profen class inhibitors has been predicted to interfere with binding, thus explaining the poor potency of these compounds (35). However, a crystal structure of (R)-flurbiprofen complexed with COX-2 revealed a shift in the inhibitor's carboxylate group, α-carbon, and fluorophenyl ring that enables accommodation of the α-methyl group with retention of polar interactions with both Arg-120 and Tyr-355. Nevertheless, the overall result is a reduction in total enzyme-inhibitor contacts that likely explains the R-enantiomer's lower potency (15). It is likely that a similar conformational change is required to accommodate the (R)-methyl group of (R)-ARN2508, providing an explanation for its reduced potency relative to that of the S-enantiomer. However, as a crystal structure of (R)-ARN2508 complexed with COX was not obtained, this hypothesis could not be confirmed.
Despite its structural and kinetic similarities to flurbiprofen, the alkyl carbamate substituent of ARN2508 imparts it with properties that are not observed in any other previously reported NSAID. These include FAAH inhibition and utilization of the upper portion of the cyclooxygenase active site channel. Indeed, our crystal structure data reveal a number of interesting aspects to the binding interactions of ARN2508 in the active site of COX-2. The catalytic Tyr-385 residue forms hydrogen bonds with both oxygen atoms of the carbamate moiety of ARN2508 reminiscent of the hydrogen bond formed between Tyr-385 and the carbonyl oxygen of aspirin. This interaction places the aspirin carbonyl in a position that is hypothesized to be favorable for nucleophilic attack by the oxygen of Ser-530 and stabilizes the negatively charged tetrahedral intermediate formed in the transition state (36). However, in our COX-2·(S)-ARN2508 complex crystal structure, an additional interaction is observed between the amide nitrogen of ARN2508 and Ser-530, thus making a nucleophilic attack by the Ser-530 hydroxyl group on the carbonyl carbon of the inhibitor less favorable. This is consistent with previous molecular modeling and simulation studies that predicted unfavorable geometry for covalent modification of COX by ARN2508 (37) and with our own site-directed mutagenesis data and tryptic peptide analysis by LC-MS/MS, which do not support covalent modification of Ser-530 by the inhibitor. In fact, there is no major loss of (S)-ARN2508 potency for S530A COX-2, suggesting that the hydrogen bond between the inhibitor and Ser-530 also plays a minimal role in inhibitor potency. Nevertheless, the S530A mutation clearly exerts a subtle effect on the enzyme-inhibitor interaction. Specifically, (S)-ARN2508 inhibits the S530A mutant more rapidly than the wildtype enzyme.
Although slow-binding COX inhibitors have consistently been seen to exhibit multistep kinetic mechanisms in which one or more rapidly reversible steps are followed by the rate-limiting formation of a much more tightly bound enzyme·inhibitor complex (7), the structural basis for the initial enzyme-inhibitor interaction in most cases is poorly understood. For some NSAIDs, such as indomethacin and the diarylheterocycle class of COX-2–selective inhibitors, the slow step has been attributed to insertion of a portion of the molecule into a specific binding pocket in the enzyme active site (7). Such mechanistic data are not available for flurbiprofen or ARN2508. Clearly, however, the alkyl side chain of ARN2508 impacts its binding kinetics in that a longer preincubation period is required for (S)-ARN2508 than for (S)-flurbiprofen to reach the same level of inhibition. Although kinetic analysis failed to clearly delineate the mechanism of (S)-ARN2508–mediated inhibition of COX-2, the available data suggest that, if a classic two-step mechanism is applicable, much higher inhibitor concentrations are needed to form the initial complex for (S)-ARN2508 (KI ≈ 220 μm) than (S)-flurbiprofen (KI = 0.17 μm) (26). Alternatively, the (S)-ARN2508·COX-2 complex may form in a single slow step or in multiple steps of which the first is rate-limiting. The only structural difference between flurbiprofen and ARN2508 is the presence of the alkyl carbamate side chain on the latter. Thus, it is reasonable to hypothesize that insertion of ARN2508's alkyl chain into its position in the active site is responsible for their kinetic differences. Our data also support the hypothesis that Ser-530, which is present at the bend of the channel, plays a role in the insertion process. Mutation of Ser-530 to Ala shortens the time required for (S)-ARN2508 to inhibit COX-2, whereas it does not affect the rate of binding of flurbiprofen (data not shown). Hydrogen bond interactions between Ser-530 and the carbamoyl group of the inhibitor may also contribute to its COX-2 binding interaction, although removal of this hydrogen bond does not substantially alter potency as measured in our standard IC50 assay.
To further investigate the effects of Ser-530, we mutated it to a bulkier threonine residue. This mutation resulted in a 90% reduction in catalytic efficiency (kcat/Km) (Table S1), suggesting that the added bulk significantly impairs active site structure and/or dynamics. It also resulted in a mild reduction in potency of (S)-ARN2508, although the rate of inhibition was, surprisingly, slightly higher than that of the wildtype enzyme (Fig. 8B). Structural studies have shown that the threonine side chain of the mutant overlays one of several alternative conformations adopted by serine in the wildtype enzyme (38). This orientation must provide sufficient room for the ω-end of the substrate to access the hydrophobic top channel. The hydroxyl group of threonine can also hydrogen-bond to the inhibitor as does that of the serine residue based on an assessment of the previously deposited S530T crystal structure.
Our data suggest substantial importance of the nonpolar interactions established between the alkyl chain of ARN2508 and the top channel of COX-2, which is lined with hydrophobic residues. This region of the active site, where the ω-tail of AA normally binds, has not previously been reported as the site of a COX protein-inhibitor interaction. The interactions observed in our crystal structure are consistent with previous structure–activity relationship studies (39) and in accordance with those predicted by molecular simulations that showed a nearly identical statistical distribution of hydrophobic contacts between COX-1 and AA when compared with COX-1 and ARN2508 (37). In the structure–activity relationship studies, increasing the length of the alkyl chain up to eight carbons correlated with an increase in potency toward FAAH. However, the trend differed with respect to inhibition of COX-1 and COX-2. Short alkyl chains with two or three carbons exhibited low inhibitor potency. Increasing the chain length resulted in an increase followed by a decrease in potency for both isoforms. Inhibition of COX-1 was maximized at a chain length of six carbons, whereas that for COX-2 was optimal and similar for chain lengths of five to seven carbons. The current crystal structure reveals that the additional steric bulk of longer alkyl chains cannot be accommodated in the hydrophobic channel (39). Kinetic experiments with the G533L mutant of COX-2 further support the hypothesis that the alkyl chain of ARN2508 binds in the top channel and that this interaction is important to inhibitory kinetics in a way that is specific to ARN2508. In particular, a G533L mutation markedly reduces the potency of ARN2508 but has little effect on the potency of flurbiprofen. Mutation of this residue to valine is known to abolish oxygenation of AA due to the inability of the substrate to access the top channel and bind close enough to Tyr-385 to initiate the enzymatic reaction (40).
COX-2 and FAAH are critical enzymes in the inflammatory response; they regulate the presence of proinflammatory eicosanoids and degrade endocannabinoids that have analgesic and anti-inflammatory effects. ARN2508 is a pharmacologically distinctive compound in that it can simultaneously block the activity of both enzymes. Here, we show that the in vitro efficacy of ARN2508 for inhibition of PG synthesis is primarily attributable to the S-enantiomer, whereas both enantiomers may effectively block COX-2–dependent oxygenation of endocannabinoids. In contrast, ARN2508-mediated inhibition of FAAH is not affected by the orientation of the compound's α-methyl group. Rather, ARN2508 irreversibly blocks FAAH through covalent modification of the enzyme via a reaction between the carbamate moiety of the inhibitor and the catalytic Ser-241 of FAAH (14). Here, we further show that a covalent modification of COX-2 by ARN2508 does not occur based on crystal structure, site-directed mutagenesis, and analysis of digested peptides by mass spectrometry. Although their mechanisms of inhibition differ, FAAH and COX enzymes both contain long, hydrophobic channels, allowing favorable binding of ARN2508 (41). In conclusion, ARN2508 inhibits COX via structural determinants contributed primarily by its flurbiprofen moiety, whereas the alkyl carbamate substituent, which is required for FAAH inhibition, imparts distinct attributes to its COX inhibitor kinetics and binding interactions.
Experimental procedures
Materials
AA was purchased from Nu-Chek Prep, Inc. 2-AG, PGE2-d4, (S)-flurbiprofen, and α-linolenic acid were purchased from Cayman Chemical. PGE2-G-d5 was synthesized as described previously using chemicals from Sigma-Aldrich (4). Linearized baculovirus DNA and related reagents were purchased from Expressions Systems, LLC. Detergents were purchased from Affymetrix.
COX-1 and -2 expression and purification
The expression and purification of oCOX-1 from sheep seminal vesicles were performed as described previously (42). The expression and purification of recombinant mCOX-2 in Sf9 cells were performed as described previously (30) with modification. The pVL-1393 transfer vector containing the gene sequence of wildtype mCOX-2 and a C-terminal His6 tag was cotransfected with linearized baculovirus DNA and used for overexpression in Sf9 cells. Following an infection period of 72 h, cells were harvested by centrifugation and lysed with sonication and solubilization in 1.5% n-decyl β-d-maltoside. The cell lysate was incubated with nickel-nitrilotriacetic acid beads and washed with buffer containing 50 mm imidazole. The enzyme was eluted with buffer containing 150 mm imidazole. The final purification step was performed using a size exclusion column and buffer exchange with buffer containing 0.3% n-octyl β-d-glucoside. Protein purity was evaluated by SDS-PAGE and Western blotting, and the enzyme was found to be >95% pure.
AA and 2-AG kinetics with mCOX-2
For mCOX-2 kinetics and inhibition with ARN2508, the method utilized was similar to that reported previously (43). Briefly, 15 nm purified mCOX-2 (monomer) was reconstituted with 30 nm heme in 190 μl of assay buffer (100 mm Tris-HCl, pH 8.0, and 500 μm phenol). For the assessment of activity with no preincubation of inhibitor, the desired quantities of substrate and inhibitor were added together in 10 μl of DMSO to the enzyme solution and quickly vortexed. For the assessment of activity with various preincubation times, the desired quantity of inhibitor in 5 μl of DMSO was added to the enzyme solution and incubated for various time points at 37 °C. After the incubation, the desired quantity of substrate in 5 μl of DMSO was added to the mixture, which was quickly vortexed. After 10 s, reactions were quenched with 200 μl of ice-cold ethyl acetate containing 0.5% acetic acid and 0.3 μm PGE2-d4 or PGE2-G-d5 as internal standard. Products were analyzed using LC-MS/MS operating in positive ion mode using selected reaction to monitor levels of PGE2 and PGD2 relative to those of the internal standards. Kinetic data and fitting were performed using Prism 7. The kinetic constants for (S)-ARN2508 were analyzed as described previously by Rome and Lands (25).
Inhibition of COX-2 Gly-533 mutants by (S)-ARN2508
Inhibition kinetics with Gly-533 mutants were analyzed using UV-visible spectroscopy on a Beckman-Coulter DU800 spectrophotometer. Gly-533 mutants were expressed and purified as described previously (30). Enzyme (200 nm monomer) was resuspended in 590 μl of assay buffer with 1 eq of heme. LNA (50 μm) was then added and mixed, and the absorbance at 235 nm was recorded for 2 min after ∼5 s of dead time. Inhibitors in a DMSO stock were added for various preincubation times prior to adding substrate.
Crystallization, X-ray data collection, and structural determination
The crystal structures of murine COX-2 in complex with ARN2508 compound were obtained following published methods (44–46). The crystals were grown in a solution created by combining 3.5 μl of protein·inhibitor complex and 3.5 μl of crystallization solution (50 mm EPPS, pH 8.0, 80–140 mm MgCl2, and 21–26% PEG monomethylether 550) via the hanging drop method at 18 °C. Full-size crystals (obtained following 5 ∼ 6 weeks of incubation) were transferred to 50 mm EPPS, pH 8.0, 100 mm MgCl2, and 28% PEG monomethylether 550 for 30 s and then flash frozen in liquid N2 for transportation and data collection. Diffraction data were collected using the synchrotron radiation X-ray source with 100 K liquid N2 streaming at beamline 24-ID-E in the Advance Photon Source at Argonne National Laboratory. Diffraction data were collected and processed with XDS (47). The crystal complex was determined as the I4122 space group. Initial phases were determined by molecular replacement using a search model (chain A of Protein Data Bank code 3NT1) with Phaser (48). The models were improved with several rounds of model building in Coot (49) and PHENIX with F ≥ 1.34 (50). Random selected data (3%) were set aside for test and quality control. Ligand constraints were computed using eBLOW with PHENIX (50); the ligand molecule was built in Coot (49) and refined with PHENIX (50). Water molecules were added during the last cycles of refinement, and TLS (Translation-Libration-Screw) refinement was applied in the last cycle of refinement (50). The potential of phase bias was examined by simulated annealing using PHENIX (51). The values of the Ramachandran plot for the final refinement of the structure were obtained by use of the PHENIX suite. Data collection and refinement statistics are reported in Table 2. All illustrations were prepared with PyMOL (Schrödinger, LLC).
COX-2 tryptic digest and peptide adduct analysis
Purified COX-2 was incubated with or without 100 μm (S)-ARN2508 for 2 h at 37 °C. Samples were precipitated with ice-cold acetone overnight at −20 °C. Specifically, 300 μl of acetone was added to 50 μl of each protein sample. Following precipitation, samples were centrifuged at 18,000 × g at 4 °C, and precipitates were washed with cold acetone and allowed to dry for 15 min. Protein pellets were reconstituted in 20 μl of 100 mm ammonium bicarbonate, reduced with 2 μl of 45 mm DTT, and treated with 2 μl of 100 mm iodoacetamide to alkylate cysteine residues. COX-2 samples were digested with 1 μg of sequencing-grade trypsin overnight at 37 °C. Peptides were analyzed by LC-MS/MS. An analytical column was packed with 20 cm of C18 reverse-phase material (Jupiter, 3-μm beads, 300 Å, Phenomenex) directly into a laser-pulled emitter tip. Peptides were loaded on the capillary reverse-phase analytical column (360-μm outer diameter × 100-μm inner diameter) using a Dionex Ultimate 3000 NanoLC and autosampler. The mobile-phase solvents consisted of 0.1% formic acid and 99.9% water (solvent A) and 0.1% formic acid and 99.9% acetonitrile (solvent B). Peptides were gradient-eluted at a flow rate of 350 nl/min using a 90-min gradient. The gradient consisted of the following: 1–3 min, 2% B (sample loading from autosampler); 3–65 min, 2–40% B; 65–75 min, 40–99% B; 75–79 min, 99% B; 79–80 min, 99–2% B; and 80–90 min (column re-equilibration), 2% B. A Q Exactive Plus mass spectrometer (Thermo Scientific) equipped with a nanoelectrospray ionization source was used to mass-analyze the eluting peptides using a data-dependent method. The instrument method consisted of MS1 using an MS automatic gain control target value of 3e6 followed by up to 20 MS/MS scans of the most abundant ions detected in the preceding MS scan. A maximum MS/MS ion time of 80 ms was used with an MS2 automatic gain control target of 1e5. Dynamic exclusion was set to 15 s, high energy collision dissociation was set to 28% of the normalized collision energy, and peptide match and isotope exclusion were enabled. For identification of peptides, tandem mass spectra were searched with Sequest (Thermo Fisher Scientific) against a Mus musculus database created from the UniProtKB protein database (www.uniprot.org).4 Variable modification of +15.9949 on Met (oxidation), +57.0214 on Cys (carbamidomethylation), and +386.1722 on Ser were included for database searching. Search results were assembled using Scaffold 4.3.2 (Proteome Software).
Author contributions
M. C. G. conducted kinetic experiments, analyzed data, and contributed to drafting the manuscript. S. X. solved X-ray crystal structures, assisted with design of experiments, and participated in drafting the manuscript. C. A. R. assisted with design of experiments and participated in drafting the manuscript. S. B. assisted with X-ray crystal data collection at Argonne National Laboratory. K. G. assisted with protein expression and insect cell maintenance. M. M. and D. P. synthesized ARN2508 compounds and conceived ideas for the project. L. J. M. conceived ideas for the project and participated in drafting the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Kristie Rose at the Vanderbilt Mass Spectrometry Proteomics Core for peptide adduct analysis. We thank Nathan Kett and the Vanderbilt Chemical Synthesis Core for preparation of the enantiomers of ARN2508. The X-ray crystallographic work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by National Institute of General Medical Sciences, National Institutes of Health Grant P41 GM103403; the Eiger 16M detector on 24-ID-E beam line is funded by National Institutes of Health Office of Research Infrastructure Program High-end Instrumentation Grant S10OD021527). This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
This work was supported by National Institutes of Health Grants R01 CA089450 (to L. J. M.) and R01 GM030910, issued to the late Dr. Richard Armstrong of Vanderbilt University and managed by Dr. Charles Sanders of Vanderbilt University. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and Table S1.
The atomic coordinates and structure factors (code 5W58) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- COX
- cyclooxygenase
- AA
- arachidonic acid
- PG
- prostaglandin
- 2-AG
- 2-arachidonoylglycerol
- AEA
- arachidonoylethanolamide
- FAE
- fatty acyl ethanolamide
- FAAH
- fatty acid amide hydrolase
- NSAID
- non-steroidal anti-inflammatory drug
- mCOX-2
- murine cyclooxygenase-2
- oCOX-1
- ovine cyclooxygenase-1
- LNA
- α-linolenic acid
- PGE2-G
- PGE2-glycerol ester
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid.
References
- 1. Rouzer C. A., and Marnett L. J. (2003) Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 103, 2239–2304 10.1021/cr000068x [DOI] [PubMed] [Google Scholar]
- 2. Dubois R. N., Abramson S. B., Crofford L., Gupta R. A., Simon L. S., Van De Putte L. B., and Lipsky P. E. (1998) Cyclooxygenase in biology and disease. FASEB J. 12, 1063–1073 [PubMed] [Google Scholar]
- 3. Smith W. L., DeWitt D. L., and Garavito R. M. (2000) Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182 10.1146/annurev.biochem.69.1.145 [DOI] [PubMed] [Google Scholar]
- 4. Kozak K. R., Rowlinson S. W., and Marnett L. J. (2000) Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J. Biol. Chem. 275, 33744–33749 10.1074/jbc.M007088200 [DOI] [PubMed] [Google Scholar]
- 5. Yu M., Ives D., and Ramesha C. S. (1997) Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J. Biol. Chem. 272, 21181–21186 10.1074/jbc.272.34.21181 [DOI] [PubMed] [Google Scholar]
- 6. Kozak K. R., Crews B. C., Morrow J. D., Wang L. H., Ma Y. H., Weinander R., Jakobsson P. J., and Marnett L. J. (2002) Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J. Biol. Chem. 277, 44877–44885 10.1074/jbc.M206788200 [DOI] [PubMed] [Google Scholar]
- 7. Blobaum A. L., and Marnett L. J. (2007) Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 50, 1425–1441 10.1021/jm0613166 [DOI] [PubMed] [Google Scholar]
- 8. Scarpignato C., and Hunt R. H. (2010) Nonsteroidal antiinflammatory drug-related injury to the gastrointestinal tract: clinical picture, pathogenesis, and prevention. Gastroenterol. Clin. North Am. 39, 433–464 10.1016/j.gtc.2010.08.010 [DOI] [PubMed] [Google Scholar]
- 9. Sostres C., Gargallo C. J., and Lanas A. (2013) Nonsteroidal anti-inflammatory drugs and upper and lower gastrointestinal mucosal damage. Arthritis Res. Ther. 15, Suppl. 3, S3 10.1186/ar4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Capone M. L., Tacconelli S., Rodriguez L. G., and Patrignani P. (2010) NSAIDs and cardiovascular disease: transducing human pharmacology results into clinical read-outs in the general population. Pharmacol. Rep. 62, 530–535 10.1016/S1734-1140(10)70310-8 [DOI] [PubMed] [Google Scholar]
- 11. Marnett L. J. (2009) The COXIB experience: a look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 49, 265–290 10.1146/annurev.pharmtox.011008.145638 [DOI] [PubMed] [Google Scholar]
- 12. Sanger G. J. (2007) Endocannabinoids and the gastrointestinal tract: what are the key questions? Br. J. Pharmacol. 152, 663–670 10.1038/sj.bjp.0707422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sasso O., Migliore M., Habrant D., Armirotti A., Albani C., Summa M., Moreno-Sanz G., Scarpelli R., and Piomelli D. (2015) Multitarget fatty acid amide hydrolase/cyclooxygenase blockade suppresses intestinal inflammation and protects against nonsteroidal anti-inflammatory drug-dependent gastrointestinal damage. FASEB J. 29, 2616–2627 10.1096/fj.15-270637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mileni M., Kamtekar S., Wood D. C., Benson T. E., Cravatt B. F., and Stevens R. C. (2010) Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J. Mol. Biol. 400, 743–754 10.1016/j.jmb.2010.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duggan K. C., Hermanson D. J., Musee J., Prusakiewicz J. J., Scheib J. L., Carter B. D., Banerjee S., Oates J. A., and Marnett L. J. (2011) (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat. Chem. Biol. 7, 803–809 10.1038/nchembio.663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duggan K. C., Walters M. J., Musee J., Harp J. M., Kiefer J. R., Oates J. A., and Marnett L. J. (2010) Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 285, 34950–34959 10.1074/jbc.M110.162982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malkowski M. G., Ginell S. L., Smith W. L., and Garavito R. M. (2000) The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 289, 1933–1937 10.1126/science.289.5486.1933 [DOI] [PubMed] [Google Scholar]
- 18. Vecchio A. J., Simmons D. M., and Malkowski M. G. (2010) Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 285, 22152–22163 10.1074/jbc.M110.119867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupta K., Selinsky B. S., and Loll P. J. (2006) 2.0 Å structure of prostaglandin H2 synthase-1 reconstituted with a manganese porphyrin cofactor. Acta Crystallogr. D Biol. Crystallogr. 62, 151–156 10.1107/S0907444905036309 [DOI] [PubMed] [Google Scholar]
- 20. Kurumbail R. G., Stevens A. M., Gierse J. K., McDonald J. J., Stegeman R. A., Pak J. Y., Gildehaus D., Miyashiro J. M., Penning T. D., Seibert K., Isakson P. C., and Stallings W. C. (1996) Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 384, 644–648 10.1038/384644a0 [DOI] [PubMed] [Google Scholar]
- 21. Picot D., Loll P. J., and Garavito R. M. (1994) The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 367, 243–249 10.1038/367243a0 [DOI] [PubMed] [Google Scholar]
- 22. Selinsky B. S., Gupta K., Sharkey C. T., and Loll P. J. (2001) Structural analysis of NSAID binding by prostaglandin H2 synthase: time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry 40, 5172–5180 10.1021/bi010045s [DOI] [PubMed] [Google Scholar]
- 23. Sidhu R. S., Lee J. Y., Yuan C., and Smith W. L. (2010) Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry 49, 7069–7079 10.1021/bi1003298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roth G. J., Stanford N., and Majerus P. W. (1975) Acetylation of prostaglandin synthase by aspirin. Proc. Natl. Acad. Sci. U.S.A. 72, 3073–3076 10.1073/pnas.72.8.3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rome L. H., and Lands W. E. (1975) Structural requirements for time-dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc. Natl. Acad. Sci. U.S.A. 72, 4863–4865 10.1073/pnas.72.12.4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ouellet M., and Percival M. D. (1995) Effect of inhibitor time-dependency on selectivity towards cyclooxygenase isoforms. Biochem. J. 306, 247–251 10.1042/bj3060247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalgutkar A. S., Crews B. C., Rowlinson S. W., Marnett A. B., Kozak K. R., Remmel R. P., and Marnett L. J. (2000) Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc. Natl. Acad. Sci. U.S.A. 97, 925–930 10.1073/pnas.97.2.925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orlando B. J., Lucido M. J., and Malkowski M. G. (2015) The structure of ibuprofen bound to cyclooxygenase-2. J. Struct. Biol. 189, 62–66 10.1016/j.jsb.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rowlinson S. W., Kiefer J. R., Prusakiewicz J. J., Pawlitz J. L., Kozak K. R., Kalgutkar A. S., Stallings W. C., Kurumbail R. G., and Marnett L. J. (2003) A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 278, 45763–45769 10.1074/jbc.M305481200 [DOI] [PubMed] [Google Scholar]
- 30. Rowlinson S. W., Crews B. C., Lanzo C. A., and Marnett L. J. (1999) The binding of arachidonic acid in the cyclooxygenase active site of mouse prostaglandin endoperoxide synthase-2 (COX-2). A putative L-shaped binding conformation utilizing the top channel region. J. Biol. Chem. 274, 23305–23310 10.1074/jbc.274.33.23305 [DOI] [PubMed] [Google Scholar]
- 31. Laneuville O., Breuer D. K., Xu N., Huang Z. H., Gage D. A., Watson J. T., Lagarde M., DeWitt D. L., and Smith W. L. (1995) Fatty acid substrate specificities of human prostaglandin-endoperoxide H synthase-1 and -2. Formation of 12-hydroxy-(9Z,13E/Z,15Z)-octadecatrienoic acids from α-linolenic acid. J. Biol. Chem. 270, 19330–19336 10.1074/jbc.270.33.19330 [DOI] [PubMed] [Google Scholar]
- 32. Callan O. H., So O. Y., and Swinney D. C. (1996) The kinetic factors that determine the affinity and selectivity for slow binding inhibition of human prostaglandin H synthase 1 and 2 by indomethacin and flurbiprofen. J. Biol. Chem. 271, 3548–3554 10.1074/jbc.271.7.3548 [DOI] [PubMed] [Google Scholar]
- 33. Laneuville O., Breuer D. K., Dewitt D. L., Hla T., Funk C. D., and Smith W. L. (1994) Differential inhibition of human prostaglandin endoperoxide H synthases-1 and -2 by nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 271, 927–934 [PubMed] [Google Scholar]
- 34. So O.-Y., Scarafia L. E., Mak A. Y., Callan O. H., and Swinney D. C. (1998) The dynamics of prostaglandin H synthases: studies with prostaglandin H synthase 2 Y355F unmask mechanisms of time-dependent inhibition and allosteric activation. J. Biol. Chem. 273, 5801–5807 10.1074/jbc.273.10.5801 [DOI] [PubMed] [Google Scholar]
- 35. Bhattacharyya D. K., Lecomte M., Rieke C. J., Garavito M., and Smith W. L. (1996) Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2-phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 271, 2179–2184 10.1074/jbc.271.4.2179 [DOI] [PubMed] [Google Scholar]
- 36. Hochgesang G. P., Rowlinson S. W., and Marnett L. J. (2000) Tyrosine-385 is critical for acetylation of cyclooxygenase-2 by aspirin. J. Am. Chem. Soc. 122, 6514–6515 10.1021/ja0003932 [DOI] [Google Scholar]
- 37. Palermo G., Favia A. D., Convertino M., and De Vivo M. (2016) The molecular basis for dual fatty acid amide hydrolase (FAAH)/cyclooxygenase (COX) inhibition. ChemMedChem 11, 1252–1258 10.1002/cmdc.201500507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lucido M. J., Orlando B. J., Vecchio A. J., and Malkowski M. G. (2016) Crystal structure of aspirin-acetylated human cyclooxygenase-2: insight into the formation of products with reversed stereochemistry. Biochemistry 55, 1226–1238 10.1021/acs.biochem.5b01378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Migliore M., Habrant D., Sasso O., Albani C., Bertozzi S. M., Armirotti A., Piomelli D., and Scarpelli R. (2016) Potent multitarget FAAH-COX inhibitors: design and structure-activity relationship studies. Eur. J. Med. Chem. 109, 216–237 10.1016/j.ejmech.2015.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vecchio A. J., Orlando B. J., Nandagiri R., and Malkowski M. G. (2012) Investigating substrate promiscuity in cyclooxygenase-2: the role of Arg-120 and residues lining the hydrophobic groove. J. Biol. Chem. 287, 24619–24630 10.1074/jbc.M112.372243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Favia A. D., Habrant D., Scarpelli R., Migliore M., Albani C., Bertozzi S. M., Dionisi M., Tarozzo G., Piomelli D., Cavalli A., and De Vivo M. (2012) Identification and characterization of carprofen as a multitarget fatty acid amide hydrolase/cyclooxygenase inhibitor. J. Med. Chem. 55, 8807–8826 10.1021/jm3011146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Odenwaller R., Chen Y. N., and Marnett L. J. (1990) Preparation and proteolytic cleavage of apoprostaglandin endoperoxide synthase. Methods Enzymol. 187, 479–485 10.1016/0076-6879(90)87054-7 [DOI] [PubMed] [Google Scholar]
- 43. Mitchener M. M., Hermanson D. J., Shockley E. M., Brown H. A., Lindsley C. W., Reese J., Rouzer C. A., Lopez C. F., and Marnett L. J. (2015) Competition and allostery govern substrate selectivity of cyclooxygenase-2. Proc. Natl. Acad. Sci. U.S.A. 112, 12366–12371 10.1073/pnas.1507307112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blobaum A. L., Xu S., Rowlinson S. W., Duggan K. C., Banerjee S., Kudalkar S. N., Birmingham W. R., Ghebreselasie K., and Marnett L. J. (2015) Action at a distance: mutations of peripheral residues transform rapid reversible inhibitors to slow, tight binders of cyclooxygenase-2. J. Biol. Chem. 290, 12793–12803 10.1074/jbc.M114.635987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kudalkar S. N., Nikas S. P., Kingsley P. J., Xu S., Galligan J. J., Rouzer C. A., Banerjee S., Ji L., Eno M. R., Makriyannis A., and Marnett L. J. (2015) 13-Methylarachidonic acid is a positive allosteric modulator of endocannabinoid oxygenation by cyclooxygenase. J. Biol. Chem. 290, 7897–7909 10.1074/jbc.M114.634014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu S., Hermanson D. J., Banerjee S., Ghebreselasie K., Clayton G. M., Garavito R. M., and Marnett L. J. (2014) Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated H-bonding network. J. Biol. Chem. 289, 6799–6808 10.1074/jbc.M113.517987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 12 10.1107/S09074449090473375–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 10.1107/S0907444906045975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 10.1107/S090744490705024X [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.