

Abstract
Slit guidance ligand 2 (SLIT2) is a large, secreted protein that binds roundabout (ROBO) receptors on multiple cell types, including neurons and kidney podocytes. SLIT2-ROBO–mediated signaling regulates neuronal migration and ureteric bud (UB) outgrowth during kidney development as well as glomerular filtration in adult kidneys. Additionally, SLIT2 binds Gremlin, an antagonist of bone morphogenetic proteins (BMPs), and BMP–Gremlin signaling also regulates UB formation. However, direct cross-talk between the ROBO2–SLIT2 and BMP–Gremlin signaling pathways has not been established. Here, we report the discovery of negative feedback between the SLIT2 and BMP–Gremlin signaling pathways. We found that the SLIT2–Gremlin interaction inhibited both SLIT2–ROBO2 signaling in neurons and Gremlin antagonism of BMP activity in myoblasts and fibroblasts. Furthermore, BMP2 down-regulated SLIT2 expression and promoter activity through canonical BMP signaling. Gremlin treatment, BMP receptor inhibition, and SMAD family member 4 (SMAD4) knockdown rescued BMP-mediated repression of SLIT2. BMP2 treatment of nephron progenitor cells derived from human embryonic stem cells decreased SLIT2 expression, further suggesting an interaction between the BMP2–Gremlin and SLIT2 pathways in human kidney cells. In conclusion, our study has revealed direct negative cross-talk between two pathways, previously thought to be unassociated, that may regulate both kidney development and adult tissue maintenance.
Keywords: bone morphogenetic protein (BMP), development, fibrosis, kidney, protein–protein interaction, transcription regulation, Gremlin, ROBO2, SLIT2
Introduction
Slit guidance ligands are ∼200-kDa proteins that contain four N-terminal leucine-rich repeat domains (LRR),2 seven to nine epidermal growth factor (EGF)-like domains, a laminin G domain, and a C-terminal cysteine-rich domain (1–3). A cleavage site within the EGF domain of SLIT2 results in an N-terminal fragment (SLIT2N) that binds roundabout (ROBO) receptors, via an active site within the second LRR domain (D2), and a C-terminal fragment (SLIT2C) that can bind PlexinA1 and Glypican-1 (3–9). SLIT–ROBO signaling was originally identified as a repulsive axon guidance cue during nervous system development (6, 10, 11), and the interaction of SLIT2 with ROBO receptors has also been shown to regulate forebrain development (12) and metanephric kidney development (13, 14). Disruption of SLIT2–ROBO2 signaling during kidney development can lead to congenital anomalies of kidney and urinary (CAKUT) (14–18). In adult tissue, the SLIT–ROBO interaction can inhibit nephrin-induced actin polymerization and the attachment of glomerular podocytes that are essential for glomerular filtration (19, 20), mediate fibroblast activation and fibrotic disease progression (21–24), regulate tumor growth (25–27), and promote angiogenesis (28, 29).
In addition to binding to ROBO receptors, SLIT2 was discovered to bind to the secreted protein Gremlin in a yeast two-hybrid screen (30). Gremlin, as well as its family member Noggin, is a secreted cysteine knot protein that antagonizes bone morphogenetic protein (BMP) signaling by preventing BMP ligands from binding to their receptors (31–35). BMP–Gremlin signaling has many known roles in the embryonic development of the kidney and urinary tract (36, 37), osteoblast differentiation (38), renal function (39), and fibrotic disease progression (33, 40, 41).
Metanephric kidney development is driven by epithelial and mesenchymal cell interactions between nephric ducts (ND), also referred to as Wolffian ducts (WD), and the metanephric mesenchyme (MM), which both originate from the intermediate mesoderm (42). Signals from the MM promote the outgrowth of the ureteric bud (UB) from the ND (42). The UB then invades the MM and begins to branch repeatedly to give rise to the fully formed nephron and metanephric kidney (42–44). UB outgrowth and branching is tightly regulated by the spatial and temporal expression of a number of signaling pathways and transcription factors (42, 43). A key mechanism of regulation is the formation of a complex between glial cell-derived neurotrophic factor (GDNF), which is expressed in the MM adjacent to the ND (45, 46), its receptor RET, and co-receptor GDNF family receptor α1 (GFRα1), which are expressed by the ND (44, 46–51).
UB outgrowth is also regulated by SLIT2, which is expressed in the tips of the UB, and ROBO2, which is expressed by the MM in the area surrounding the UB (13). ROBO2 knockout mice and SLIT2 knockout mice form supernumerary ureteric buds (14). SLIT2–ROBO2 signaling restricts the nephrogenic field and limits cell–cell interactions between the MM and ND, possibly by limiting GDNF–RET signaling by regulating ND-MM separation (14, 52). Interestingly, BMP–Gremlin signaling is also an important regulator of UB outgrowth and branching. Gremlin-mediated inhibition of BMP is essential to induce UB outgrowth and establish RET–GDNF feedback signaling by mediating epithelial–mesenchymal signaling (37). In Gremlin-deficient mouse embryos, the UB fails to invade the MM because of uncontrolled BMP signaling resulting in renal agenesis (36, 37). Conversely, recombinant Gremlin protein is able to induce ectopic UB outgrowth from the ND by up-regulating GDNF expression (36).
Given the roles of both SLIT2–ROBO2 signaling and BMP–Gremlin signaling in fibrotic disease progression and UB outgrowth and branching, as well as the opposite effects of SLIT2 and Gremlin genetic loss in mouse kidney development, we hypothesized that the interaction between SLIT2 and Gremlin serves to regulate their signaling. Here, we report the identification of direct negative cross-talk between SLIT2 and BMP–Gremlin signaling. We found that SLIT2 bound to Gremlin via its D2 domain and that this interaction inhibited SLIT2–ROBO2 activity as well as Gremlin-mediated antagonism of BMP2 signaling. Furthermore, BMP2 down regulated SLIT2 expression by promoting transcriptional repression via SMAD signaling in mouse and human fibroblasts. Similar changes in SLIT2 expression were observed when nephron progenitor cells derived from human embryonic stem cells were treated with BMP2. These studies unveil a regulatory feedback loop between two previously unassociated pathways that could have implications for both kidney development and adult tissue maintenance in human disease.
Results
Gremlin binds to the D2 domain of SLIT2
It had been previously reported that rat Gremlin interacts directly with the repulsive guidance cue SLIT2 (30). To determine whether human Gremlin also interacts with the N-terminal domain of SLIT2, we performed an ELISA with recombinant, biotinylated human SLIT2N and human Gremlin on a streptavidin-coated plate. We found that Gremlin bound to SLIT2N, beginning at 62.5 nm, and binding increased with higher doses (Fig. 1A). These results were confirmed by conducting surface plasmon resonance (SPR) experiments with human SLIT2N and human Gremlin (Fig. 1B). SLIT2N was the ligand and was covalently immobilized to a C1 chip surface. It was then exposed to a dose range of Gremlin (the analyte in solution) to measure association and dissociation kinetics. A sensorgram (Fig. 1B) and residuals graph (Fig. S1A) show that the maximal residual difference of the observed data from a 1:1 Langmuir binding model is less than 10% of the global maximum capacity (Rmax). The average global ka of human Gremlin for human SLIT2N was 3.99 × 106 ± 2.87 × 105 m−1 s−1, and the average global kd was 8.25 × 10−2 ± 1.15 × 10−2 s−1 for three independent biosensor surfaces (Table 1 and Table S1). The KD value of Gremlin for SLIT2N was mathematically derived from the ka and kd data, and the average kinetic KD value across all independent sensor surfaces was 20.6 ± 2.3 nm (Table 2 and Table S2). Steady-state analysis of Gremlin binding to SLIT2N (Fig. S1B) revealed an average KD of 15.1 ± 2.7 nm (Table 2 and Table S3) and confirmed our kinetic analysis. These data suggest that human SLIT2N interacts with human Gremlin with relatively low affinity.
Figure 1.

Human Gremlin binds to human SLIT2N at its second leucine-rich repeat domain. A, 45 nm biotinylated recombinant human SLIT2N (purified from 293 cells) was immobilized onto streptavidin-coated plates. The wells were blocked and incubated with increasing amounts of human Gremlin (0, 32.5, 62.5, 125, 250, 500, and 1000 nm), followed by anti-Gremlin antibody, rabbit IgG-HRP, and 3,3′,5,5′-tetramethylbenzidine substrate. The reaction was quenched with 0.18 m sulfuric acid, and binding was measured by reading the absorbance at 450 nm. Data are presented as binding minus anti-Gremlin antibody background binding. Mean ± S.D., n = 3 ***, p < 0.001, and ****, p < 0.0001, as compared with 0 nm Gremlin binding by one-way ANOVA. Data are representative of at least three independent experiments. B, representative BIAcore sensorgram displaying binding of human Gremlin (analyte in solution) to human SLIT2N coupled to an anti-human C1 chip surface (ligand). Association was monitored for 100 s, and dissociation was monitored for 30 s. Data for six concentrations of Gremlin are shown. The order of signal intensity (top to bottom) is 100, 50, 25, 12.5, 6.3, and 3.1 nm. Association and dissociation phases were fit to a 1:1 Langmuir binding model with global rate constants, global maximum capacity (Rmax), and local RI parameters. C, representative BIAcore sensorgram displaying binding of Gremlin (analyte in solution) to human SLIT2D2 coupled to an anti-human C1 chip surface (ligand). Association was monitored for 180 s, and dissociation was monitored for 30 s. Data for five concentrations of Gremlin are shown. The order of signal intensity (top to bottom) is 100, 50, 25, 12.5, and 6.3 nm. Association and dissociation phases were fit to a 1:1 Langmuir binding model with global rate constants, global Rmax, and local RI parameters. D, representative BIAcore sensorgram displaying binding of SLIT2D2 (analyte in solution) to Gremlin coupled to an anti-human C1 chip surface (ligand). Association was monitored for 180 s, and dissociation was monitored for 30 s. Data for five concentrations of SLIT2D2 are shown. The order of signal intensity (top to bottom) is 50, 25, 12.5, 6.3, and 3.1 nm. Association and dissociation phases were fit to a 1:1 Langmuir binding model with global rate constants, global Rmax, and local RI parameters. Each binding assay was performed three independent times on three independent biosensor surfaces.
Table 1.
Average ka and kd values
Observed average association rate constant (a) molar/s (m−1 s−1) and observed average dissociation rate constant (kd)/s (s−1) for each indicated ligand (protein covalently bound to chip surface)–analyte (protein in solution that flowed over ligand-coated surface) pairing across three independent biosensor surfaces are shown.
| Ligand (bound to chip) | Analyte (in solution) | Mean ka | S.E. ka | Mean kd | S.E. kd |
|---|---|---|---|---|---|
| m−1 s−1 | s−1 | ||||
| SLIT2N | Gremlin | 3.99 × 106 | 2.87 × 105 | 8.25 × 10−2 | 1.15 × 10−2 |
| SLIT2D2 | Gremlin | 5.76 × 106 | 7.17 × 105 | 1.69 × 10−1 | 5.48 × 10−2 |
| Gremlin | SLIT2D2 | 1.85 × 106 | 2.03 × 105 | 2.37 × 10−2 | 2.26 × 10−3 |
Table 2.
Average kinetic and steady-state KD values
Average mathematically derived equilibrium dissociation constants (KD) in nanomolars (nm) were calculated from association rate constants and dissociation rate constants for each indicated ligand–analyte pairing across three independent biosensor surfaces. Average steady-state KD values were calculated from the EC50 values of the concentration of the analyte plotted on the x axis and the corresponding Rmax values plotted on the y axis for each indicated ligand–analyte pairing across three independent biosensor surfaces.
| Ligand (bound to chip) | Analyte (in solution) | Kinetic |
Steady state |
||
|---|---|---|---|---|---|
| Mean KD | S.E. KD | Mean KD | S.E. KD | ||
| nm | nm | ||||
| SLIT2N | Gremlin | 20.6 | 2.3 | 15.1 | 2.7 |
| SLIT2D2 | Gremlin | 27.9 | 5.5 | 17.5 | 2.5 |
| Gremlin | SLIT2D2 | 13.0 | 1.6 | 18.8 | 0.6 |
The LRR D2 domain of SLIT2 (SLIT2D2) binds to ROBO family members and mediates ROBO signaling (4). To determine whether the SLIT2–Gremlin interaction also occurs at the same D2 domain, we performed SPR experiments by exposing immobilized human SLIT2D2 (ligand) to a range of Gremlin doses in solution (analyte) to measure association and dissociation kinetics. Interestingly, the D2 fragment of SLIT2 bound to immobilized Gremlin on three independent biosensor surfaces. A sensorgram (Fig. 1C) and residuals graph (Fig. S1C) show that the maximal residual difference of the observed data from a 1:1 Langmuir binding model is less than 10% of the Rmax. The average global ka of Gremlin for SLIT2D2 was 5.76 × 106 ± 7.17 × 105 m−1 s−1, and the average global kd was 1.69 × 10−1 ± 5.48 × 10−2 s−1 (Table 1 and Table S1). The mathematically derived average KD of Gremlin for SLIT2D2 was 27.9 ± 5.5 nm (Table 2 and Table S2). The average steady-state KD (Fig. S1D) was 17.5 ± 2.5 nm (Table 2 and Table S3) and agreed with kinetic analysis. We also reversed the orientation and exposed immobilized Gremlin (ligand) to a range of SLIT2D2 concentrations in solution (analyte) to monitor association and dissociation kinetics. A sensorgram (Fig. 1D) and residuals graph (Fig. S1E) show that the observed data deviate minimally from a 1:1 Langmuir binding model <10% of the Rmax. The average global ka of SLIT2D2 for Gremlin was 1.85 × 106 ± 2.03 × 105 m−1 s−1, and the average global kd was 2.37 × 10−2 ± 2.26 × 10−3 s−1 (Table 1). The mathematically derived average KD value was 13.0 ± 1.6 nm (Table 2), and the steady-state average KD value (Fig. S1F) was 18.8 ± 0.6 nm (Table 2). Because similar steady-state and kinetic KD values were calculated when Gremlin was the analyte and when SLIT2D2 was the analyte, our data demonstrate that the D2 domain of SLIT2 interacts with Gremlin. Furthermore, because the KD values were similar when immobilized SLIT2N and SLIT2D2 were exposed to Gremlin, it is likely that the D2 domain of SLIT2 alone is sufficient for Gremlin binding.
Gremlin blocks SLIT2-induced inhibition of neuronal migration
SLIT2 inhibits neuronal migration by binding to its receptor ROBO2 through the D2 domain (12, 15, 19, 53, 54). Because Gremlin also interacts with SLIT2 via its D2 domain, we hypothesized that Gremlin may interfere with SLIT2 binding to ROBO and block SLIT2–ROBO2-mediated inhibition of neuronal migration. To test this hypothesis, we performed neuronal migration assays using explants from the anterior subventricular zone (SVZa) of rat brains as reported previously (19). In control media neurons migrate out from the explant (Fig. 2A). In agreement with earlier findings, human SLIT2N treatment almost completely inhibited all neuronal migration from the SVZa explants (Fig. 2B) (19). Gremlin treatment alone had no effect on neuronal migration, and neurons migrated normally in the presence of 10 to 1000 nm Gremlin (Fig. 2, C, E, and G). When explants were grown in the presence of SLIT2N and 10 nm Gremlin (Fig. 2D), there was no change in neuronal migration as compared with the neurons grown in SLIT2N alone (Fig. 2B). 100 nm Gremlin had a small inhibitory effect on SLIT2N blockade of neuronal migration (Fig. 2F). When SVZa explants were incubated in media containing SLIT2N and 1000 nm Gremlin (Fig. 2H), significantly more neurons migrated out from the SVZa center as compared with explants grown in SLIT2N alone (Fig. 2, B, H and I). Additionally, when the explants were grown in media containing SLIT2N and 1000 nm Gremlin, total neuronal migration distance was also increased as compared with neurons grown in SLIT2N alone (Fig. 2J). Taken together, these results suggest that Gremlin inhibits SLIT2–ROBO2 signaling at high concentrations in SVZa neurons.
Figure 2.

Gremlin blocks SLIT2-mediated inhibition of neuronal migration in SVZa explants. Explants from rat SVZa were grown in media alone (A), 1 nm human SLIT2N (B), 10 nm Gremlin (C), 1 nm SLIT2N + 10 nm Gremlin (D), 100 nm Gremlin (E), 1 nm SLIT2N + 100 nm Gremlin (F), 1000 nm Gremlin (G), and 1 nm SLIT2N + 1000 nm Gremlin (H) for 48 h and stained with Hoechst 33342 to visualize neuronal nuclei. Scale bar represents 500 μm and applies to all images, A–H. Images were taken on the Operetta High Content Imager with a ×10 high NA objective. Fifteen fields per well with 5% overlap were taken. A Z-stack of six planes for each field was acquired with 1 μm between each plane. Images were analyzed using Volocity software. All fields in each well were stitched together. The area of the tissue explant in the center and each nucleus outside of the tissue explant were detected by Hoechst 33342 staining. I, quantification of migrating SVZa neuron cell number from A to H. Individual nuclei were counted outside the edge of the tissue explant. The total number of nuclei from each well that was not treated with SLIT2N from five individual experiments was averaged. Then the number of total nuclei from each well was divided by the average from its corresponding control group to calculate the relative fold change of the total number of migrating cells. J, quantification of migrating distance of SVZa neurons from A to H. The distance from the center of each nucleus to the closest edge of the tissue explant was measured in micrometers. The mean migration distance of nuclei in the well was multiplied by nuclei count to obtain total migration distance for each explant. The total migration from each well that was not treated with SLIT2N from five individual experiments was averaged. Then, the total migration from each well was divided by the average from its corresponding control group to calculate the relative fold change of total migration. Bar graphs represent five pooled experiments, mean ± S.D., each with n = 3. **, p < 0.01, and ***, p < 0.001, by two-way ANOVA.
SLIT2 inhibits Gremlin-induced BMP antagonism by reducing SMAD1 phosphorylation and nuclear translocation
Because Gremlin is a known BMP antagonist and also inhibits SLIT2 activity in neuronal migration assays, we next investigated whether SLIT2 affects Gremlin–BMP signaling. C2C12 mouse myoblasts stably expressing the BMP-response element (BRE) from the Id1 promoter in a luciferase expression vector (C2C12BRE) have increased luciferase activity in response to BMP, which can be inhibited by Gremlin or Noggin (55, 56). In the presence of buffer alone, BMP2 treatment stimulated luciferase activity in C2C12BRE myoblasts, and Gremlin blocked this increase (Fig. 3A). 50 nm SLIT2N had no effect on Gremlin antagonism of BMP2 signaling. However, in the presence of 100 and 200 nm SLIT2, Gremlin failed to block BMP2 activity, and luciferase activity was significantly increased compared with cells grown in BMP2 + Gremlin in media containing buffer only, suggesting that SLIT2N inhibits Gremlin antagonism of BMP in this luciferase assay. Furthermore, 200 nm SLIT2N blocked Noggin inhibition of BMP2 activity in C2C12BRE myoblasts (Fig. 3B). Because BMP2 regulates Id1 promoter activity in C2C12BRE myoblasts, we measured ID1 gene expression in MRC5 fibroblasts to confirm the effects of SLIT2N on Gremlin antagonism of BMP2 signaling (Fig. 3C). BMP2 treatment causes a significant up-regulation of ID1 gene expression in fibroblasts, and Gremlin blocks this increase. In agreement with the luciferase data presented above, SLIT2N treatment blocked Gremlin inhibition of BMP2-induced ID1 expression.
Figure 3.

SLIT2 inhibits Gremlin-induced blockade of BMP2 signaling. A, C2C12BRE myoblasts were treated with vehicle (Cont), 0.8 nm human BMP2, 2.8 nm human Gremlin (Grem), or 0.8 nm BMP2 + 2.8 nm Gremlin in the presence of buffer, 50, 100, or 200 nm human SLIT2N for 48 h, and luminescence was measured. Bar graph represents fold change of mean luminescence relative to buffer-treated Control cells ± S.D., n = 3. ****, p < 0.0001 by two-way ANOVA. B, C2C12BRE myoblasts were treated with vehicle (Cont), 0.5 nm human BMP2, 0.25 nm human Noggin (Nog), or 0.5 nm BMP2 + 0.25 nm Noggin in the presence of buffer or 200 nm human SLIT2N for 48 h, and luminescence was measured. Bar graph represents fold change of mean luminescence relative to buffer-treated Control cells ± S.D., n = 4. ****, p < 0.0001 by two-way ANOVA. C, MRC5 fibroblasts were treated with vehicle (Cont), 1 nm human BMP2, 1 nm BMP2 + 1.25 nm Gremlin in the presence of buffer or 100 nm human SLIT2N for 24 h. RNA was isolated, and ID1 expression (normalized to ZNF592 expression) was analyzed by RT-qPCR. Bar graphs represent mean relative quantification values relative to control-treated cells ± S.D., n = 3. ****, p < 0.0001 compared with untreated cells by one-way ANOVA.
Gremlin regulates ID1 expression by inhibiting BMP2-induced SMAD1 phosphorylation and nuclear translocation (31, 57–59). To further confirm the effects of SLIT2 on Gremlin activity, we treated 3T3 mouse fibroblasts with BMP2 in the presence of buffer control or SLIT2N. In media containing buffer only, BMP2 treatment increased SMAD1/5/9 phosphorylation, and Gremlin blocked this phosphorylation (Fig. 4A, left). In the presence of SLIT2N, Gremlin was unable to inhibit BMP2-induced SMAD1/5/9 phosphorylation (Fig. 4A, right). In the presence of buffer, BMP2 stimulated an increase in mean nuclear intensity of SMAD1 staining in MRC5 human fibroblasts, and Gremlin inhibited BMP2-induced SMAD1 nuclear staining (Fig. 4, B, top, and C, left). In the presence of SLIT2N, Gremlin could not fully inhibit BMP2-induced SMAD1 nuclear translocation (Fig. 4, B, bottom, and C, right). To confirm the effects of SLIT2N on Gremlin inhibition of BMP2-induced SMAD1 signaling, we also stained MRC5 fibroblasts for phospho-SMAD1/5/9 following treatment with BMP2, Gremlin, and SLIT2N. In media containing buffer only, BMP2 stimulated an increase in mean nuclear intensity of phospho-SMAD1/5/9 nuclear staining, and this was blocked by co-treatment with Gremlin (Fig. 4, D, top, and E, left). In the presence of SLIT2N, Gremlin could not inhibit BMP2-induced phospho-SMAD1/5/9 nuclear translocation (Fig. 4, D, bottom, and E, right). Taken together, these data suggest that the interaction between SLIT2 and Gremlin inhibits Gremlin-induced antagonism of BMP2 signaling in fibroblasts and myoblasts.
Figure 4.

SLIT2 inhibits Gremlin antagonism of BMP2-stimulated SMAD signaling. A, 3T3 fibroblasts were treated with 0.5 nm BMP2 or 0.5 nm BMP2 + 3 nm Gremlin in the presence of buffer or 100 nm SLIT2N for 30 min. Protein lysates were analyzed by SDS-PAGE and Western blotting using antibodies against phospho-SMAD1/5/9 (p-SMAD1/5/9) and total SMAD1. To quantify phospho-SMAD1/5/9 levels, total SMAD1 was used as a loading control. Bar graph represents the fluorescent intensity of each phospho-SMAD1/5/9 band divided by its corresponding relative value of its total SMAD1 band. B, MRC5 fibroblasts were treated with 0.3 nm BMP2, 3 nm Gremlin, or 0.3 nm BMP2 + 3 nm Gremlin in the presence of buffer or 200 nm SLIT2N for 30 min. Cells were fixed and immunostained with anti-SMAD1 antibody followed by goat anti-rabbit IgG Alexa Fluor 647 to measure localization. Images were acquired on the Operetta High Content Imager. Scale bar, 120 μm and applies to all images. C, nuclear intensity of SMAD1 staining was measured using Harmony software. Bar graph represents mean nuclear intensity ± S.D. of at least 1000 cells in three different wells. *, p < 0.05 by two-way ANOVA. D, MRC5 fibroblasts were treated with 0.3 nm BMP2, 0.75 nm Gremlin, or 0.3 nm BMP2 + 0.75 nm Gremlin in the presence of buffer or 100 nm SLIT2N for 30 min. Cells were fixed and immunostained with anti-phospho-SMAD1/5/9 antibody followed by goat anti-rabbit IgG Alexa Fluor 647 to measure localization. Images were acquired on the Operetta High Content Imager. Scale bar, 120 μm and applies to all images. E, nuclear intensity of phospho-SMAD1/5/9 staining was measured using Harmony software. Bar graph represents mean nuclear intensity ± S.D. of at least 1000 cells in five different wells. ****, p < 0.0001 by two-way ANOVA. Data are representative of at least three independent experiments.
BMP2 down-regulates SLIT2 expression via canonical BMP signaling
The data presented here so far reveal two novel downstream effects of the Gremlin–SLIT2 interaction. Gremlin antagonizes SLIT2–ROBO2 signaling to restore neuronal migration, and SLIT2 blocks Gremlin to promote BMP2 activity in fibroblasts and myoblasts. We next asked whether there was any effect of BMP on SLIT2. Interestingly, when we treated MRC5 human fibroblasts with BMP2 for 48 h, we observed a dose-dependent decrease in SLIT2 mRNA (Fig. 5A) and protein (Fig. 5B) expression. Because Gremlin is a well-established BMP2 antagonist, we hypothesized that Gremlin would inhibit BMP2-induced SLIT2 down-regulation. When we treated MRC5 fibroblasts with both Gremlin and BMP2, we observed a significant increase in SLIT2 mRNA (Fig. 5C) and protein (Fig. 5D) expression relative to cells treated with BMP2 alone. Furthermore, Gremlin treatment alone also slightly increased SLIT2 mRNA expression, suggesting that inhibition of endogenous BMP2 could increase SLIT2 expression.
Figure 5.

BMP2 promotes the down-regulation of SLIT2 mRNA and protein expression via canonical BMP signaling. A, MRC5 fibroblasts were treated with 0, 0.1, 1, or 10 nm human BMP2 for 48 h. RNA was isolated, and SLIT2 expression (normalized to ZNF592 expression) was analyzed by RT-qPCR. Bar graphs represent mean relative quantification values relative to control-treated cells ± S.D., n = 3. ***, p < 0.001 compared with untreated cells by one-way ANOVA. B, MRC5 fibroblasts were treated with 0, 0.1, 0.5, 1, 5, or 10 nm BMP2 for 48 h. Protein lysates were analyzed by SDS-PAGE and Western blotting with antibodies against SLIT2 and tubulin. To quantify SLIT2 levels, tubulin was used as a loading control. Bar graph represents the fluorescent intensity of each SLIT2 band divided by its corresponding relative value of its tubulin band. C, MRC5 fibroblasts were treated with 1 nm BMP2, 3 nm Gremlin (Grem), or 1 nm BMP2 + 3 nm Gremlin for 48 h. RNA was isolated, and SLIT2 expression (normalized to ZNF592 expression) was analyzed by RT-qPCR. Bar graphs represent mean relative quantification values relative to control-treated cells ± S.D., n = 3. ****, p < 0.0001 by one-way ANOVA. D, MRC5 fibroblasts were treated with 5 nm BMP2 in the presence of 10 or 50 nm Gremlin. Protein lysates were analyzed by SDS-PAGE and Western blotting using antibodies against SLIT2 and tubulin. Bar graph represents the fluorescent intensity of each SLIT2 band divided by its corresponding relative value of its tubulin band. E, MRC5 cells were treated with 5 nm BMP2 in the presence of DMSO or 0.1, 1, or 10 nm DMH1 for 48 h. RNA was isolated, and SLIT2 expression (normalized to ZNF592 expression) was analyzed by RT-qPCR. Bar graphs represent mean relative quantification values relative to DMSO-treated wells ± S.D., n = 3. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 by two-way ANOVA. F, MRC5 fibroblasts were treated with 5 nm BMP2 in the presence of DMSO or 0.1, 1, or 10 nm DMH1 for 48 h. Protein lysates were analyzed by SDS-PAGE and Western blotting with antibodies against SLIT2 and tubulin. Bar graph represents the fluorescent intensity of each SLIT2 band divided by its corresponding relative value of its tubulin band. MRC5 fibroblasts were transfected with 18 pmol of siRNA targeting SMAD4 or negative control siRNA, then treated with 5 nm BMP2 for 48 h. RNA was isolated. G, SMAD4 expression (normalized to ZNF592 expression); H, SLIT2 expression (normalized to ZNF592 expression) was analyzed by RT-qPCR. Bar graphs represent mean relative quantification values relative to control-treated cells transfected with negative control siRNA ± S.D., n = 3. **, p < 0.01, and ****, p < 0.0001 compared with control-treated cells transfected with control siRNA by two-way ANOVA. I, protein lysates from MRC5 cells transfected with 25 pmol of siRNA targeting SMAD4 or negative control siRNA and treated with 5 nm BMP2 for 48 h were analyzed by SDS-PAGE and Western blotting with antibodies against SLIT2, SMAD4, and tubulin. Top bar graph represents the fluorescent intensity of each SLIT2 band divided by its corresponding relative value of its tubulin band. Bottom bar graph represents the fluorescent intensity of each SMAD4 band divided by its corresponding relative value of its tubulin band. Each experiment was performed at least three independent times.
To determine whether canonical BMP signaling is responsible for SLIT2 down-regulation, we pretreated MRC5 fibroblasts with increasing amounts of DMH1, a BMP receptor kinase activity inhibitor (60), followed by BMP2. Interestingly, DMH1 treatment alone significantly increased SLIT2 mRNA levels, compared with vehicle-treated cells (Fig. 5E, left). Furthermore, co-treatment of MRC5 cells with DMH1 and BMP2 restored SLIT2 mRNA (Fig. 5E, right) and protein expression (Fig. 5F) as compared with cells treated with BMP2 alone. BMP signals through its receptors to phosphorylate SMAD1, and SMAD4 then binds to phosphorylated SMAD1 to promote its translocation into the nucleus (61). Therefore, to inhibit BMP2 signaling, we transfected MRC5 cells with siRNA targeting SMAD4. Although SMAD4 also binds to SMAD2/3, we did not detect an increase in phospho-SMAD2/3 in response to BMP2 treatment in MRC5 or 3T3 fibroblasts (data not shown). Similar to BMP receptor inhibition, SMAD4 knockdown (Fig. 5G) alone increased SLIT2 mRNA (Fig. 5H, left) and protein (Fig. 5I) expression as compared with cells transfected with control siRNA. BMP2 failed to down-regulate SLIT2 mRNA (Fig. 5H, right) and protein (Fig. 5I) expression in SMAD4 knockdown cells. Taken together, these results demonstrate that SLIT2 expression is repressed by canonical BMP signaling. Exogenous BMP2 treatment reduces SLIT2 mRNA and protein expression. SLIT2 expression is rescued when cells are treated with BMP2 in the presence of Gremlin, BMP receptor inhibition, or SMAD4 knockdown. Furthermore, inhibition of basal BMP signaling by Gremlin, BMP receptor blockade, or SMAD4 knockdown increases SLIT2 expression.
BMP2 down-regulates SLIT2 promoter activity via canonical BMP-signaling pathways
To determine whether BMP2 signaling represses SLIT2 expression by regulating SLIT2 promoter activity, we cloned a portion of the human SLIT2 promoter from −1430 to +69 into a luciferase reporter gene expression vector (Fig. 6A and Fig. S2) (62). 3T3 mouse fibroblasts stably expressing the human SLIT2 promoter luciferase construct (3T3-hSLIT2P-luc) were generated, and BMP2 treatment of these cells induced a dose-dependent decrease in luciferase activity (Fig. 6B). SLIT2 transcriptional activity was rescued when the cells were treated with BMP2 + Gremlin (Fig. 6C). We also observed that DMH1-mediated inhibition of BMP receptor kinase activity significantly increased basal levels of SLIT2 promoter activity as compared with cells treated with vehicle (Fig. 6D, left). As expected, BMP2 failed to reduce SLIT2 promoter activity in the presence of DMH1 (Fig. 6D, right). To determine whether SMAD signaling mediates BMP2 regulation of SLIT2 promoter activity, we transfected 3T3-hSLIT2P-luc cells with siRNA targeting SMAD4. siRNA-mediated knockdown of Smad4 (Fig. 6E) increased luciferase activity in control-treated 3T3-hSLIT2P-luc cells, and BMP2 failed to down-regulate luciferase activity in reporter cells lacking Smad4 (Fig. 6F). These data suggest that BMP2 down-regulates the SLIT2 gene and protein expression by repressing SLIT2 transcription through canonical BMP-signaling pathways, which is consistent with the data presented in Fig. 5.
Figure 6.
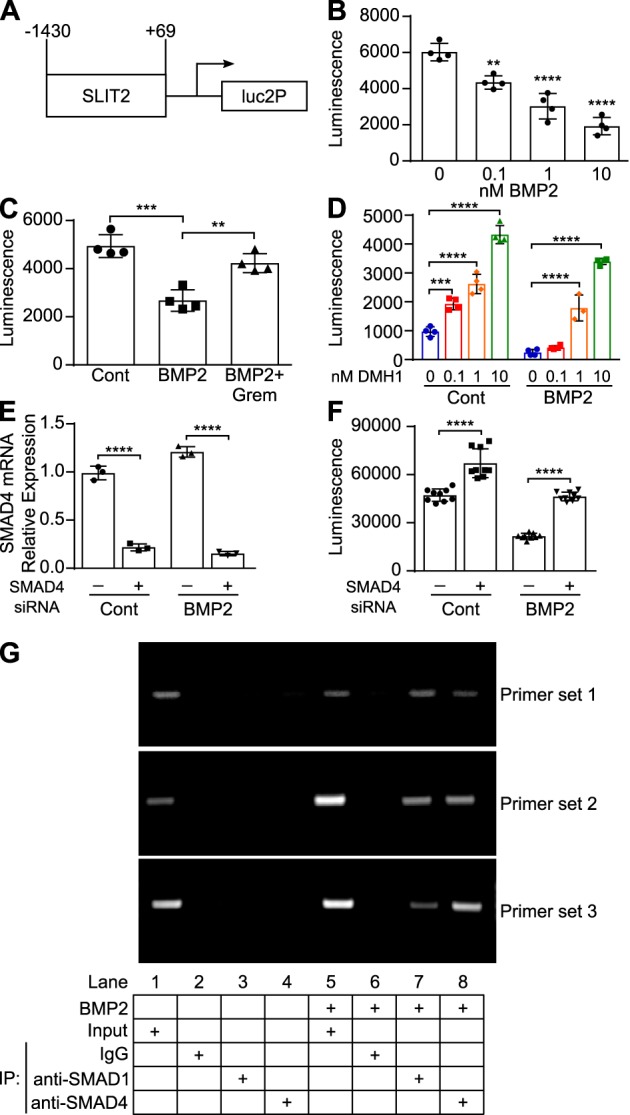
BMP2 down-regulates SLIT2 promoter activity by binding to the SLIT2 promoter. A, schematic of human SLIT2 promoter construct cloned into the pGL4.26[luc2/minP/Hygro] vector. B, 3T3 cells stably expressing a portion of the human SLIT2 promoter in a luciferase construct (3T3-hSLIT2P-luc) were treated with 0, 0.1, 1, or 10 nm BMP2 for 24 h. Bar graphs represent mean luciferase values ± S.D., n = 4. **, p < 0.01, and ****, p < 0.0001 as compared with control-treated cells by one-way ANOVA. C, 3T3-hSLIT2P-luc cells were treated with 3 nm BMP2 or 3 nm BMP2 + 12 nm Gremlin. Bar graphs represent mean luciferase values ± S.D., n = 4. **, p < 0.01, and ***, p < 0.001, as compared with control-treated cells by one-way ANOVA. D, 3T3-hSLIT2P-luc cells were treated with 3 nm BMP2 in the presence of DMSO or 0.1, 1, or 10 nm DMH1. Bar graphs represent mean luciferase values ± S.D., n = 4. ***, p < 0.01, and ****, p < 0.0001 by two-way ANOVA. E, 3T3-hSLIT2P-luc cells were transfected with 18 pmol of siRNA targeting Smad4 or negative control siRNA and then treated with 5 nm BMP2 for 24 h. RNA was isolated, and Smad4 expression (normalized to Znf592) was analyzed by RT-qPCR. Bar graphs represent mean relative quantification values relative to control-treated cells transfected with control siRNA ± S.D., n = 3. ****, p < 0.0001 by two-way ANOVA. F, luciferase activity was measured following Smad4 siRNA knockdown and BMP2 treatment. Bar graphs represent mean luciferase values ± S.D., n = 3. ****, p < 0.0001 by two-way ANOVA. G, 3T3-hSLIT2P-luc were treated with 5 nm BMP2 for 4 h. Chromatin from these cells was subjected to ChIP assays using antibodies against SMAD1, SMAD4, or normal rabbit IgG as a control. DNA was amplified in PCRs using primers designed against two predicted SMAD1-binding sites (primer sets 1 and 3) and one predicted SMAD4-binding site (primer set 2) and run on an 1.5% agarose gel to visualize bands. Each experiment was performed at least three independent times. IP, immunoprecipitation.
To confirm the role of SMAD signaling in SLIT2 repression, we performed ChIP assays in BMP2-treated 3T3-hSLIT2P-luc cells using antibodies against SMAD1 and SMAD4. PCR primers were designed to amplify three different regions of the SLIT2 promoter, each containing a predicted SMAD1- or SMAD4-binding site (Fig. S2). BMP2 treatment stimulated the binding of SMAD1 and SMAD4 to the two predicted SMAD1-binding sites and the predicted SMAD4-binding site on the SLIT2 promoter (Fig. 6G, right), whereas control-treated 3T3-hSLIT2P-luc cells exhibited no such binding (Fig. 6G, left). These data suggest that BMP2 down-regulates SLIT2 expression by stimulating the binding of SMAD1 and SMAD4 to the SLIT2 promoter.
BMP2 down-regulates SLIT2 expression in human nephron progenitor cells
The data we have generated thus far suggest that there is a negative feedback loop between the SLIT2 and BMP–Gremlin signaling pathways in mouse and human fibroblasts. Because both SLIT2–ROBO2 signaling and Gremlin–BMP signaling are each known to regulate early mouse kidney development (14, 36, 37, 52, 63), we wanted to determine whether this negative feedback may also regulate signaling in human nephron progenitor cells. Recently, a number of studies have used human embryonic stem cells and induced pluripotent stem cells to generate nephron-like structures and kidney organoids made up of different types of cells to further study human kidney development, disease, and toxicology (64–69). Lineage tracing has been used to identify markers of each stage of metanephric kidney development and has enabled advances in in vitro kidney organogenesis (68, 70). A recently published study outlined a protocol to promote the differentiation of embryonic stem cells into nephron progenitor cells that will eventually form nephron-like structures, enabling recapitulation of human metanephric kidney development in vitro (69). We used this protocol to differentiate human embryonic stem cells (H9) into nephron progenitor cells (Fig. 7A) (69). Before beginning any experiment, we confirmed the mRNA expression of markers of the late primitive streak, posterior intermediate mesoderm, metanephric mesenchyme, pre-tubular aggregate, renal vesicle, and nephron (Fig. S3), as reported previously (69).
Figure 7.
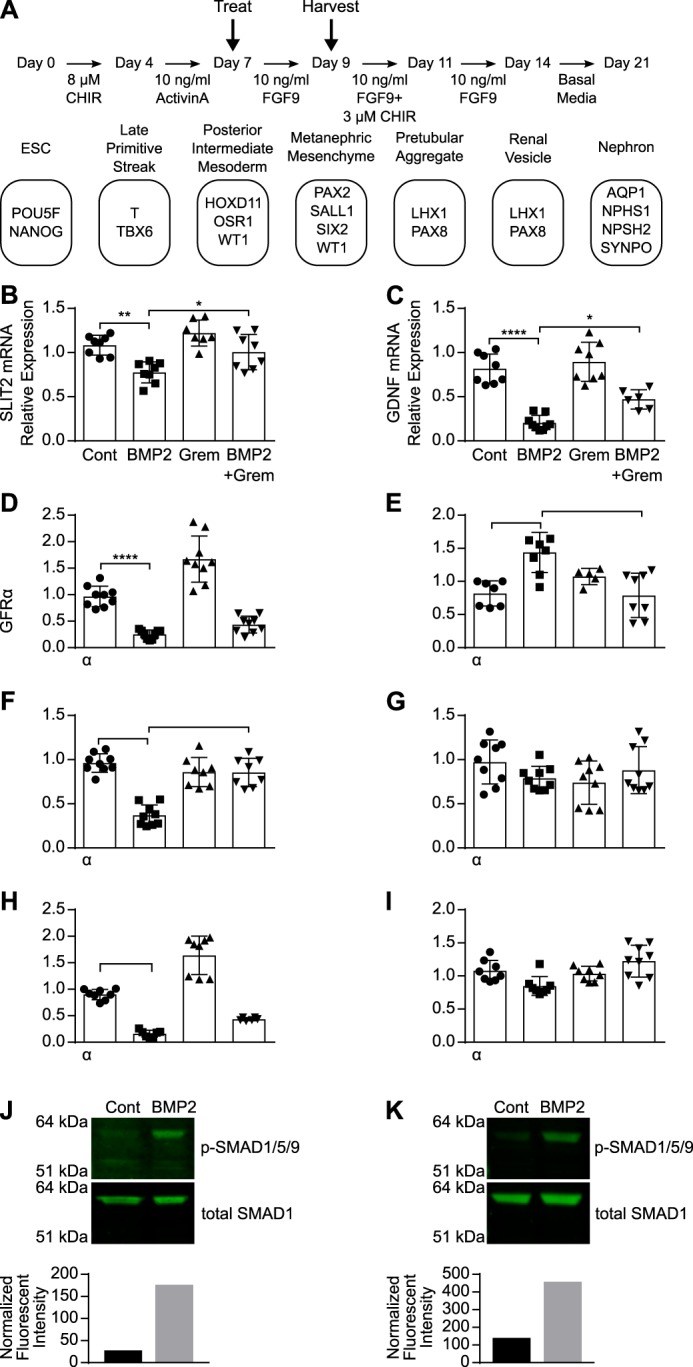
BMP2 down-regulates SLIT2 expression during kidney differentiation. A, protocol used to promote differentiation of H9 stem cells into nephron-like cells. Cells were treated on day 7 of differentiation containing 10 ng/ml FGF9 with 2.5 nm BMP2, 5 nm Gremlin, or 2.5 nm BMP2 + 5 nm Gremlin. RNA was harvested on day 9 and analyzed by RT-qPCR to measure SLIT2 (B), GDNF (C), GFRα1 (D), RET (E), PAX2 (F), SALL1 (G), SIX2 (H), and WT1 (I) mRNA expression (all normalized to ZNF592 expression). Bar graphs represent mean relative quantification values relative to control-treated wells ± S.D., n = 3. *, p < 0.05; **, p < 0.01; ***, p < 0.001, and ****, p < 0.0001 by two-way ANOVA. H9 stem cells were treated on day 7 (J) or day 9 (K) of differentiation with 5 nm BMP2 for 30 min. Protein lysates were analyzed by SDS-PAGE and Western blot using antibodies against phospho-SMAD1/5/9 (p-SMAD1/5/9) and total SMAD1. To quantify phospho-SMAD1/5/9 levels, total SMAD1 was used as a loading control. Bar graphs represent the fluorescent intensity of each phospho SMAD1/5/9 band divided by its corresponding relative value of its total SMAD1 band. Data are representative of at least three independent experiments.
SLIT2 is known to be expressed in the nephric duct, and ROBO2 is expressed by metanephric mesenchymal cells during ureteric bud outgrowth (13, 14). However, exogenous SLIT2 does not affect kidney branching morphogenesis or nephron formation during kidney development (71). Gremlin is expressed by the metanephric mesenchyme surrounding the UB (37). We confirmed that nephron progenitor cells express SLIT2 and ROBO2, as well as other regulators of UB outgrowth and branching morphogenesis, including GDNF, GFRα1, Gremlin, and RET throughout differentiation from H9 human embryonic stem cells into nephron-like cells (Fig. S4). To determine the effects of BMP2 and Gremlin treatment on SLIT2 expression in human nephron progenitor cells, we treated these cells while they were in the posterior intermediate mesoderm stage (day 7) with differentiation media (ARPMI + 10 ng/ml FGF9) (69) containing BMP2, Gremlin, or BMP2 + Gremlin for 48 h until they were in the metanephric mesenchyme stage of development (day 9) (Fig. 7A). BMP2 treatment of posterior intermediate mesoderm cells resulted in a statistically significant decrease in SLIT2 mRNA expression, and Gremlin treatment rescued SLIT2 mRNA down-regulation by BMP2 (Fig. 7B).
Because both SLIT2–ROBO2 and BMP2–Gremlin signalings regulate the expression and activity of GDNF (14, 36, 37, 52) and the formation of a complex of GDNF with RET and GFRα1 is the initiating factor in UB outgrowth during kidney development (51), we also investigated the effects of BMP2 treatment on GDNF, RET, and GFRα1 mRNA expression in posterior intermediate mesenchyme-like cells. BMP2 treatment of cells during differentiation from the posterior intermediate mesoderm stage to the metanephric mesenchyme (days 7–9) caused a significant decrease in mRNA expression of GDNF (Fig. 7C) and GFRα1 (Fig. 7D) while increasing RET mRNA expression (Fig. 7E). Gremlin treatment inhibited BMP2 activity and restored levels of GDNF (Fig. 7C) and RET gene expression (Fig. 7E) (64). In addition, BMP2 treatment reduced PAX2 (Fig. 7F) and SIX2 gene expression (Fig. 7H) while having little to no effect on SALL1 (Fig. 7G) or WT1 gene expression (Fig. 7I). BMP2 also promoted SMAD1 phosphorylation in posterior intermediate mesoderm-like cells (day 7) (Fig. 7J), as well as in metanephric mesoderm-like cells (day 9) (Fig. 7K), confirming that canonical BMP2 signaling pathways are active in these nephron progenitor cells. Taken together, these data suggest that BMP–Gremlin signaling regulates SLIT2 expression, as well as the expression of GDNF and RET, in human nephron progenitor-like cells differentiated from ES cells.
Discussion
In this study, we have confirmed that SLIT2N binds Gremlin, and we determined that this interaction occurs via the D2 domain of SLIT2. We have shown that Gremlin inhibits SLIT2N blockade of neuronal migration and that SLIT2N inhibits Gremlin antagonism of BMP signaling in fibroblasts and myoblasts. BMP2, in turn, down-regulates SLIT2 expression in fibroblasts by repressing SLIT2 transcriptional activity via its canonical SMAD signaling cascade. BMP2 treatment also decreases SLIT2 expression in nephron progenitor cells derived from human embryonic stem cells. These findings reveal three new molecular mechanisms of SLIT2 signaling: 1) Gremlin inhibits SLIT2–ROBO2 signaling in neuronal migration; 2) SLIT2N inhibits Gremlin antagonism of BMP activity; and 3) BMP2 inhibits SLIT2 expression by down-regulating SLIT2 promoter activity via SMAD-mediated signaling pathways. Therefore, we demonstrated for the first time the existence of direct negative cross-talk between the SLIT2 and BMP–Gremlin signaling pathways mediated by protein–protein interactions and transcriptional repression (Fig. 8).
Figure 8.
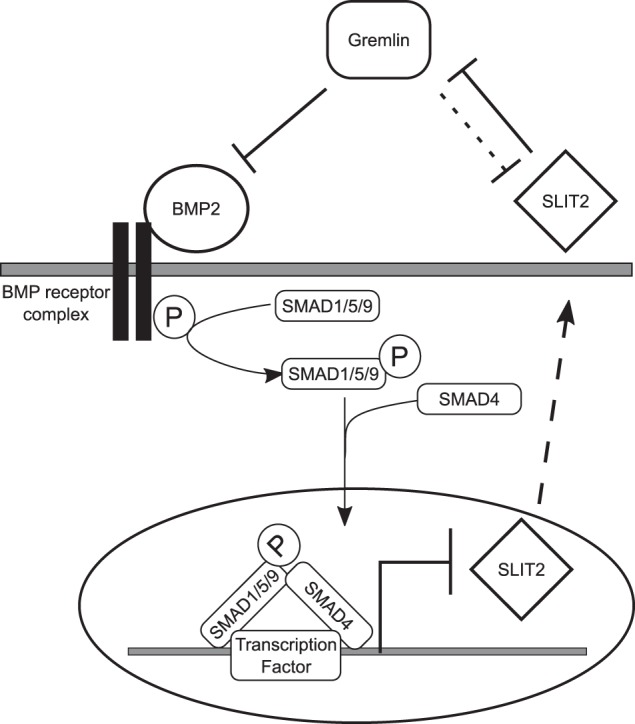
Proposed direct negative cross-talk between SLIT2 and BMP2–Gremlin signaling. Gremlin antagonizes BMP2 signaling to block BMP2 repression of SLIT2 transcriptional activity. BMP2 down-regulates SLIT2 expression by facilitating the binding of SMAD1 and SMAD4 to the SLIT2 promoter through canonical BMP-signaling pathways. SLIT2 binds to Gremlin and inhibits its antagonism of BMP2. High concentrations of Gremlin also block SLIT2 inhibition of neuronal migration (indicated by dashed line).
The majority of the research on SLIT2 has focused on its interaction with ROBO and downstream signaling events. There is still much unknown about what other proteins to which SLIT2 binds, whether SLIT2N has any ROBO2-independent activities, and how SLIT2 itself is regulated. SLIT2 was found to bind Gremlin in a yeast two-hybrid assay, and functional data suggested that the two proteins cooperate to negatively regulate monocyte chemotaxis (30). We confirmed this finding by measuring SLIT2N–Gremlin binding using both ELISA and SPR. Further analysis revealed that Gremlin binds to the D2 domain of SLIT2, the same domain that binds ROBO2 (3–5). However, the average KD of SLIT2N for Gremlin ranges from 13 to 28 nm, whereas the affinity of ROBO2 for SLIT2N is much higher with a KD of about 4 nm (6, 72). This could explain why 1000 nm Gremlin blocked SLIT2N-induced inhibition of neuronal migration. A high local concentration of Gremlin is needed to overcome the tighter interaction between SLIT2 and ROBO2. Given how UB branching and outgrowth depend on spatial and temporal expression of key protein factors and that affinity and local concentrations effect protein–protein interactions, the different affinities of ROBO2 and Gremlin for SLIT2N may play a specific role in metanephric kidney development.
SLIT2 has been shown to interact with proteins other than ROBO2 to regulate developmental processes. The C-terminal domain of SLIT2 binds PlexinA1 to regulate commissural axon guidance during nervous system development (7). Full-length SLIT2, SLIT2N, and SLIT2C all bind Glypican-1 to regulate nervous system growth and development (8, 9). Although SLIT2 is considered to be an activating ligand for Glypican-1, PlexinA1, and ROBO2, our data demonstrate that SLIT2N antagonizes Gremlin activity, thereby promoting BMP2 signaling. Interestingly, a recent study found that Robo2 signaling via Slit2 is required for BMP activity in Drosophila blood cell differentiation and lymph gland regulation (73). These findings reveal a novel mechanism of SLIT2 signaling, and future work will focus on determining whether SLIT2 inhibits Gremlin in the context of kidney development in animal models and human stem cell culture systems.
Our findings that Gremlin blocks SLIT2 inhibition of neuronal migration, SLIT2 inhibits Gremlin-antagonism of BMP signaling, and BMP2 down-regulates SLIT2 promoter activity and expression unveil cross-talk between two signaling pathways, previously thought to be distinct, that have been individually shown to regulate metanephric kidney development. Both SLIT2–ROBO2 and BMP–Gremlin signaling have been identified as regulators of GDNF–RET signaling that induce UB outgrowth (14, 36, 37, 51). Furthermore, like mutations in SLIT2 and ROBO2, mutations in BMP and Gremlin have been identified in CAKUT patients (17, 18, 74, 75). Gremlin knockdown in mice disrupts RET–GDNF epithelial–mesenchymal feedback signaling (36, 37), and the role of SLIT2–ROBO2 signaling appears to limit GDNF–RET signaling by regulating WD-nephrogenic cord separation (14, 52). In human nephron progenitor-like cells differentiated from ES cells, BMP2-induced down-regulation of SLIT2 expression is concurrent with a decrease in GDNF and GFRα1 expression and an increase in RET expression. The decrease in GDNF expression is consistent with the effects of BMP4 treatment of kidney rudiments from a mouse around embryonic day 11 (76). Although there are fewer ureteric buds in rudiments grown in the presence of BMP4, RET mRNA is detected in ureteric tips (76). BMP2 treatment, in combination with retinoic acid, induced GDNF mRNA in Schwann cells (77). BMP2 can affect human pluripotent stem cell differentiation into ureteric bud kidney progenitor-like cells (64); therefore, future work will be needed to examine how the down-regulation of SLIT2 and GDNF and up-regulation of RET are interrelated following BMP2 treatment (76). Our data suggest that BMP2 stimulates canonical SMAD signaling in these nephron progenitor cells. More studies are needed to determine whether SMAD signaling drives changes in SLIT2 in these cells or whether other pathways are active. Because the temporal and spatial expression and localization of these proteins drive their roles in metanephric kidney development, it will be important to also consider the types of cells expressing these proteins to determine whether and how these pathways interact to regulate UB outgrowth and branching.
Although SLIT2 and BMP–Gremlin signalings regulate kidney branching morphogenesis, they also have established roles in adult tissue. Our findings reveal a negative feedback loop in fibroblasts where SLIT2 inhibits Gremlin activity and BMP down-regulates SLIT2 expression by suppressing SLIT2 promoter activity through canonical BMP-signaling pathways (Fig. 8). SLIT2, BMP, and Gremlin have all been shown to play a role in the differentiation of cells into myofibroblasts whose accumulation contributes to fibrotic disease. Gremlin is overexpressed in the lungs of patients with idiopathic pulmonary fibrosis (41), whereas BMPs oppose pro-fibrotic transforming growth factor β (TGFβ) signaling and are considered to be protective against myofibroblast accumulation (78–81). Gremlin has also been shown to aggravate podocyte injury caused by high levels of glucose by activating canonical TGFβ signaling, suggesting that Gremlin may play a role in renal disease initiation or progression (39). Recently, there have been a number of opposing reports documenting SLIT2 expression in the context of fibrotic diseases. SLIT2 expression levels are low in fibrotic lesions in patients with advanced pulmonary fibrosis (22). It has also been shown that SLIT2 can inhibit TGFβ-induced stress fiber formation, SMAD2/3 signaling, and collagen synthesis in renal fibroblasts (21) as well as LPS-induced renal inflammation and fibrosis (24). In contrast, SLIT2 secretion and expression were up-regulated in patients with liver fibrosis, and SLIT2 overexpression exacerbated liver fibrosis and increased α-smooth muscle actin and collagen expression in mouse models (23). Therefore, more work is needed to understand the possible effects of the SLIT2–Gremlin interaction on myofibroblast activation and accumulation and its role in fibrotic disease.
In conclusion, our data suggest the existence of a negative feedback loop where SLIT2 blocks Gremlin inhibition of BMP2, and canonical BMP2 signaling down-regulates SLIT2 expression. The data presented here are the first demonstration of SLIT2 transcriptional regulation by a signaling pathway. We have shown that BMP2 represses SLIT2 promoter activity through canonical BMP signaling, leading to down-regulation of mRNA and protein expression (Fig. 8). Although it has been shown that an Sp1 site on the SLIT2 promoter is necessary for maximal promoter activity (62) and we have identified possible SMAD-binding sites on the SLIT2 promoter, more work is needed to determine precisely how BMP2 regulates SLIT2 promoter activity (82–86). Our data also suggest that Gremlin weakly antagonizes SLIT2 inhibition of neuronal migration. Taken together, these results further our understanding of how SLIT2 expression and signaling are mediated; however, other aspects of SLIT2 regulation are still unknown (5, 6, 72). Because SLIT2 is a large ligand with many conserved domains, it will be necessary to identify other binding partners to further elucidate SLIT2 signaling mechanisms. These findings reveal cross-talk between two pathways that further our understanding of their regulation and mechanisms of downstream signaling.
Experimental procedures
SLIT2N purification
To generate recombinant SLIT2N, 293 cells were transfected with a SLIT2 construct corresponding to amino acids 1–1118 of the N-terminal domain with a C-terminal His6 tag for detection and purification. These cells were cultured in serum-free conditions. Conditioned media were harvested and filtered, and protein was bound to nickel-Sepharose HP resin (GE Healthcare, 175268-02). The resin was washed extensively in 0.5 m NaCl, 20 mm Tris, pH 7.1, followed by 30 mm imidazole, 0.5 m NaCl, pH 8.0. SLIT2N was eluted off the resin with 225 mm imidazole, 0.5 m arginine, 375 mm NaCl, pH 8.0. Preparative size-exclusion chromatography in 0.5 m arginine, 0.5 m NaCl, 50 mm sodium phosphate, pH 8.0, was used to further purify the protein. Protein purity was confirmed by SDS-PAGE followed by Coomassie staining and analytical size-exclusion high-performance-liquid chromatography.
ELISA
Recombinant SLIT2N was biotinylated using EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific, 21327) according to the manufacturer's instructions. Briefly, 10 nm biotin was prepared, added to SLIT2N in 20-fold molar excess, and incubated at room temperature for 1 h. Excess biotin was removed by dialyzing the protein into PBS. 45 nm biotinylated SLIT2 was incubated onto a streptavidin-coated 96-well plate (Thermo Fisher Scientific, 15500) in wash buffer (TBS containing 0.1% BSA + 0.05% Tween 20). The next day, the plate was washed with wash buffer and then blocked in TBS containing 1% BSA + 0.05% Tween 20. The wells containing immobilized SLIT2 and control wells were then incubated with indicated amounts of Gremlin (Sigma, SRP3285) overnight, followed by incubation with anti-Gremlin antibody (Cell Signaling, 4383S, lot 1, 1:100), anti-rabbit-IgG horseradish peroxidase-linked whole antibody (GE Healthcare, NA934V, lot 9653124, 1:1000), and 3,3′, 5,5′-tetramethylbenzidine one-component HRP microwell substrate (Surmodics, TMBW-1000-01). The reaction was quenched using 0.18 m sulfuric acid, and absorbance was measured at 450 nm. Data were normalized to background binding of anti-Gremlin antibody to plates.
SPR
SPR was used to measure binding affinity and kinetics of Gremlin to SLIT2N and SLIT2D2. The SLIT2–Gremlin interaction was analyzed in both possible analyte/ligand orientations where the ligand was the interacting partner that was covalently immobilized on the chip surface, and the analyte was the protein that was titrated in solution and flowed over the ligand-coated surface. A BIAcore T200 C1 chip (GE Healthcare, BR100535) was docked into a BIAcore T200 system. The chip was equilibrated with HEPES-buffered saline (HBS) containing 10 mm HEPES and 150 mm NaCl (HBS-N) (GE Healthcare, BR100670). After equilibration, the chip was pre-conditioned with 100 mm glycine, pH 12, containing 0.3% Triton X-100 to prepare the surface for ligand coupling. Then, about 20 resonance units (RU) equivalent of SLIT2D2 was covalently attached to flow cell 2; about 100 RU equivalent of SLIT2N (Sigma, SRP3153) was covalently attached to flow cell 3, and about 20 RU equivalent of Gremlin (Sigma, SRP3285) was covalently attached to flow cell 4 using amine coupling chemistry and the target RU method. Briefly, SLIT2N and SLIT2D2 were diluted in HBS-N to a concentration of 1 μg/ml, and Gremlin was diluted to a concentration of 0.5 μg/ml. 1-Ethyl-3-(3-dimethylamino-propyl)carbodiimide hydrochloride (EDC) and NHS from the amine coupling kit (GE Healthcare, BR10005) were mixed in equal parts and used to activate the chip surface. The diluted ligands were then exposed to the activated chip surface to allow for amine coupling to take place. Finally, the surface was inactivated using ethanolamine as supplied in the amine coupling kit.
To determine kinetic and affinity binding, the amine-coupled C1 chip was primed in HBS containing 10 mm HEPES, 3 mm EDTA, and 0.05% surfactant p20 (HBS-EP+) (GE Healthcare, BR1006-69) and 1% BSA (Sigma, A7030) and allowed to equilibrate for ∼2 h. Once the chip was fully equilibrated, an automatic method was used to run the kinetic analysis. Briefly, a 2-fold dilution series was performed on the SLIT2D2 and Gremlin analytes ranging from 200 to 0.4 nm in HBS-EP+ with 1% BSA. The immobilized ligands on the chip were exposed to analytes in solution at a rate of 100 μl/min with an association time of 100–180 s and dissociation time of 30–180 s. After each binding cycle, the chip was regenerated with three consecutive injections of 3 m MgCl2 for 30 s each at a flow rate of 20 μl/min. After the third injection, the flow cells and injection apparatus were rinsed with running buffer. Multiple iterative cycles through the above method were performed to permute through all analyte concentrations from 200 to 0.4 nm. Blank injections were also performed where only running buffer flowed over the flow cells. This was to control for baseline drift. An additional control was flowing each analyte over flow cell 1, which had no ligand covalently bound. Therefore, each analyte concentration was exposed to a reference flow cell to account for non-specific binding and system noise.
The BIAcore T200 BIA evaluation software (GE Healthcare) version 1.0 was used to analyze the data. After automated pre- and post-analyte injection trimming and both x and y axis data set alignment, the kinetic data were double referenced by subtracting flow cell 1 data as well as the blank buffer only injection data. Association and dissociation phases were fit to a 1:1 Langmuir binding model with global rate constants, global maximum capacity (Rmax), and local refractive index (RI) parameters. Three sets of data were produced for each ligand, originating from flow cells 2–4. The equilibrium dissociation constant (KD) was derived mathematically from the observed association rate constant (ka) and the observed dissociation rate constant (kd). In addition to the mathematically derived KD, we also calculated the steady-state KD by plotting the concentration of the analyte on the x axis versus the corresponding RU at equilibrium on the y axis and determining the EC50 of that curve.
SVZa neuronal migration assay
An in vitro neuronal migration assay with postnatal SVZa neurons has been described previously (15, 19, 87). Briefly, P1–5 Sprague-Dawley rat (obtained from Charles River Laboratories) olfactory bulbs devoid of meninges were coronally sectioned by a vibratome. Migrating SVZa cells were dissected, and an explant of 200–300 μm in diameter was embedded in a collagen (Corning, CB-40236)–Matrigel (Corning, CB-40234) gel (3:2:1 collagen/Matrigel/medium) in the center of each well of 6-well glass-bottom tissue culture plates (MatTek, P06G-1.5-14-F) and incubated at 37 °C, 5% CO2 for 30 min to allow the gels to polymerize. Explants were incubated in DMEM (Corning, MT10013CM) supplemented with 10% FBS (Corning, MT35011CV) containing 1 nm SLIT2, 10 nm Gremlin, 10 nm Gremlin + 1 nm SLIT2, 100 nm Gremlin, 100 nm Gremlin + 1 nm SLIT2, 1000 nm Gremlin, or 1000 nm Gremlin + 1 nm SLIT2 at 37 °C in 5% CO2. SLIT2 and Gremlin were diluted and incubated together at 37 °C for 20 min before transferring the mixtures to the neurons embedded in polymerized collagen–Matrigels. The SVZa assays were incubated for 48 h at 37 °C in 5% CO2. Cells were then washed in PBS and fixed overnight at 4 °C in 4% paraformaldehyde (Boston Bioproducts, BM-155). Fixed neurons were stained overnight in PBS containing Hoechst 33342 (Thermo Fisher Scientific, H3570, lot 1639150, 1:200). Wide-field fluorescence images were taken on the Operetta High Content Imager (PerkinElmer Life Sciences) with a 10× 0.4 NA objective. Fifteen fields per well with 5% overlap were taken to capture the entire center area of the well. A Z-stack for each field was acquired consisting of six planes with 1 μm distance between each plane to capture the full depth of the tissue explant. Images were analyzed using Volocity software (PerkinElmer Life Sciences). All fields in each well were stitched together. The area of the tissue explant in the center and each nucleus outside of the tissue explant were detected by Hoechst 33342 staining. Individual nuclei were counted, and the distance of the center of each nucleus to the closest edge of the tissue explant was measured in micrometers. The mean migration distance of nuclei in the well was multiplied by the number of nuclei to obtain the total migration distance for each well. SVZa experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Boston University Medical Center (protocol 14388).
Cell culture
C2C12 myoblasts stably expressing the BMP-response element from the Id1 gene cloned into the pGL3 luciferase vector (C2C12BRE) (55) and NIH3T3 fibroblasts (ATCC CRL-1658) were grown in DMEM (Gibco, 11995-065) containing 10% FBS (Gibco, 16140-071) and 1% penicillin/streptomycin (Gibco, 15140-122) and incubated in a 5% CO2 atmosphere at 37 °C. MRC5 fibroblasts (ATCC, CCL-171) were grown in Eagle's minimal essential medium (Corning, 10-009-CV) containing 10% FBS and 1% penicillin/streptomycin. Where indicated, cells were serum-starved overnight and treated in low-serum medium containing 0.5% FBS and 1% penicillin/streptomycin.
Luciferase activity assays
To measure BMP2 activity, C2C12BRE cells were plated in 96-well plates (Corning, 3903), serum-starved overnight, and treated as indicated for 48 h with BMP2 or BMP2 in combination with Gremlin (Sigma, SRP3285) or Noggin (R&D Systems, 6057-NG-025) in low-serum media containing SLIT2 (Sigma, SRP3155) or equivalent amounts of reconstitution buffer. Luciferase activity was determined by incubating cells in BriteLite luciferase substrate (PerkinElmer Life Sciences, 6066766) and measuring luminescence on a PHERAstar FS microplate reader (BMG Labtech). To measure SLIT2 promoter activity, a 1499-bp fragment of the human SLIT2 promoter (hSLIT2P) (−1430 to +69) was cloned into the pGL4.26[luc2/minP/Hygro] vector (Promega, E844A) (62). 3T3 fibroblasts were transfected with 2.5 μg of hSLIT2P-pGL4.26 DNA using Lipofectamine 2000 (Thermo Fisher Scientific, 11668). Cells expressing the vector (3T3-hSLIT2P-luc) were selected using 0.1 μg/ml hygromycin (Thermo Fisher Scientific, 10687010). Cells were treated as described in 96-well plates, and luciferase activity was determined by incubating cells with ONE-Glo luciferase assay substrate (Promega, E6110), and luminescence was measured on an EnVision microplate reader (PerkinElmer Life Sciences).
SDS-PAGE and Western blotting
Total protein lysates were isolated from cells using 1× Cell Lysis Buffer (Cell Signaling Technology, catalog no. 9803) containing protease inhibitors (Roche Applied Science, 4693116001) and phosphatase inhibitors (Roche Applied Science, 4906845001) according to the manufacturer's directions. Protein concentration was determined by BCA assay (Thermo Fisher Scientific, 23227) against a BSA standard curve. Equal amounts of protein were loaded onto 4–12% BisTris gels (Thermo Fisher Scientific, NP0335) or 3–8% Tris acetate gels (Thermo Fisher Scientific, EA0378) and subjected to electrophoresis. The proteins were transferred from the gels onto nitrocellulose membranes (Thermo Fisher Scientific, IB23001 or IB23002) using an iBlot2 (Thermo Fisher Scientific). Membranes were incubated in blocking buffer (LICOR, 927-50000) and probed with antibodies against phospho-Smad1 (Ser-463/465)/Smad5 (Ser-463/465)/Smad9 (Ser-465/467) (D5B10) (Cell Signaling Technology, 13820S, lot 1, 1:1000), total Smad1 (D59D7) XP (Cell Signaling Technology, 6944S, lot 5, 1:1000), SLIT2 (Novus, NBP2-20398, lot 40302, 1:1000), α/β tubulin (Cell Signaling Technology, 2148S, lot 6, 1:1000), and SMAD4 (Novus, NBP2-24951, lot AB102003A-1, 1:1000). Signal was detected using IRDye 800CW goat ant-rabbit IgG secondary antibody (LICOR, 926–32211, lot C60113-05, 1:15,000). Western blottings were imaged on a LICOR Odyssey CLx using infrared fluorescent detection. After probing with antibodies against SMAD4, SLIT2, and phospho-SMAD1/5/9, the blots were stripped with New Blot Nitro Stripping Buffer (LICOR, 928-40030) and reprobed with loading control antibodies. To quantify SMAD4 and SLIT2 expression, α/β tubulin was used as a loading control. To quantify phospho-SMAD1/5/9 levels, total-SMAD1 was used as a loading control. Fluorescent intensity of the loading control was measured for each sample, and a relative value was calculated by dividing each value by the highest intensity. Then the fluorescence intensity of each band of the target protein was divided by its corresponding relative value of its loading control.
Immunofluorescence
MRC5 fibroblasts were grown in black 96-well plates (PerkinElmer Life Sciences, 6005182), serum-starved overnight, and treated as indicated with BMP2, Gremlin, and SLIT2. Cells were fixed in 4% formaldehyde (Thermo Fisher Scientific, 28908) and blocked in PBS containing 5% goat serum (Sigma, G9023) and 0.3% Triton X-100. Cells were stained overnight in PBS containing 1% BSA and 0.3% Triton X-100 with total-Smad1 (D59D7) XP (Cell Signaling Technology, 6944S, lot 5, 1:200) or phospho-Smad1 (Ser-463/465)/Smad5 (Ser-463/465)/Smad9 (Ser-465/467) (D5B10) (Cell Signaling Technology, 13820S, lot 1, 1:200), followed by goat anti-rabbit IgG Alexa Fluor 647 (Thermo Fisher Scientific, A21244, lot 1726547, 1:1000) and Hoechst 33342 (Thermo Fisher Scientific, H3570, lot 1639150, 1:5000) to visualize nuclei and HCS Cell Mask Orange stain (Thermo Fisher Scientific, H32713, lot 1503627, 1:10,000) to visualize the entire cell. Cells were imaged on an Operetta High Content Imager (PerkinElmer Life Sciences) with a 20 × 0.45 NA objective at room temperature in PBS. Nuclear intensity was measured in Harmony software by calculating the intensity of SMAD1 staining in the nucleus as defined by Hoechst 33342 staining. Whole cells were defined by HCS Cell Mask Orange stain.
Quantitative real time PCR (RT-qPCR)
RNA was harvested from cells using the RNeasy Plus mini kit (Qiagen, 74136) according to the manufacturer's instructions, and cDNA was generated using a high-capacity RNA-to-cDNA kit (Thermo Fisher Scientific, 4387406). Between 5 and 20 μg of cDNA were amplified using TaqMan Assays (Table S4) and TaqMan Master Mix (Thermo Fisher Scientific, 4369016) on a Viia7 real-time PCR system (Thermo Fisher Scientific). Amplification results were quantified using the comparative Ct method. ZNF592 expression served as an internal endogenous control.
BMP receptor inhibition
To inhibit BMP receptor activity, MRC5 fibroblasts and 3T3-hSLIT2P-luc fibroblasts were pre-treated for 1 h with the indicated amounts of DMH1 (Sigma, D8946), an ALK2 inhibitor (60). Equivalent amounts of DMSO were used as controls. Cells were then treated with the indicated amounts of BMP2 for 48 h.
siRNA
MRC5 fibroblasts were transfected with siRNA targeting SMAD4 (Sigma, EHU01867) or negative control siRNA (Sigma, SIC001). 3T3 fibroblasts stably expressing the human SLIT2 promoter luciferase construct were transfected with siRNA targeting Smad4 (Thermo Fisher Scientific, 155234) or negative control siRNA (Thermo Fisher Scientific, AM4611). Cells were transfected using Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13778150) according to the manufacturer's instructions. Knockdown was confirmed using both Western blotting and RT-qPCR.
Chromatin immunoprecipitation assays
3T3-hSLIT2P-luc were serum-starved overnight and treated with 5 nm BMP2 for about 4 h. The cells were cross-linked with 1% formaldehyde, and chromatin was harvested, digested, and analyzed using a SimpleChip plus kit (Cell Signaling Technology, 9005) according to the manufacturer's directions. Immunoprecipitations were performed with antibodies against normal rabbit IgG (Cell Signaling Technology, 2729, lot 7), SMAD1, and SMAD4 (Cell Signaling Technology, 38454, lot 2) according to manufacturer's directions. Primer sets were designed against various regions of the human SLIT2 promoter (Fig. S2), and DNA was amplified by PCR using a hot start DNA polymerase (Thermo Fisher Scientific, EP1712). Amplified DNA was loaded onto 1.5% agarose gels containing 1× SYBR Safe DNA gel stain (Thermo Scientific, S33102) and run at 150 V for about 45 min. Bands were imaged on a E-Gel Imager (Life Technologies, Inc.).
Stem cell differentiation
Undifferentiated H9 ES cells were licensed from Wisconsin Alumni Research Foundation, which has been verified as compliant with Pfizer policies. Stem cells were cultured in a feeder-free system on ES cell-qualified Matrigel-coated tissue culture plates (Corning, 354277). Cells were plated in mTESR1 media (Stem Cell Technologies, 05850) containing 2 μm Thiazovivin (Tocris, 3845) for 24 h. Cells were refed every day with fresh mTESR1 media without added Thiazovivin. To differentiate stem cells into nephron-like cells, 8 × 104 cells were plated onto Matrigel-coated 6-well plates in mTESR1 media containing 2 μm Thiazovivin. Cells were cultured in mTESR1 for 72 h, with media changes every day (Thiazovivin was only added at the time of plating). After 72 h in mTESR1, cells were cultured in differentiation media made up of Advanced RPMI 1640 medium (ARPMI) (Thermo Fisher Scientific, 12633-020) and 2 mm GlutaMax (Thermo Fisher Scientific, 35050-061) and supplemented as described previously (Fig. 6A) (69). Briefly, cells were cultured for 96 h in 8 μm CHIR99021 (Tocris, 4423) in ARPMI to induce differentiation toward the late primitive streak. Then the cells were cultured in 10 ng/ml activin A (R&D Systems, 338-AC-10) in ARPMI to promote the late intermediate mesoderm stage. After 72 h, cells were switched to 10 ng/ml FGF9 (R&D Systems, 273-F9–025) in ARPMI for 48 h to promote metanephric mesenchyme induction. To promote pre-tubular aggregate formation, cells were cultured in 10 ng/ml FGF9 and 3 μm CHIR99021 in ARPMI for 48 h. Then the cells were switched back to 10 ng/ml FGF9 to induce renal vesicle formation for 72 h. Finally, the cells were maintained in basal ARPMI media for 7 more days to promote nephron-like cell differentiation. Expression of markers of each stage of differentiation was measured by RT-qPCR.
Statistical analyses
Each experiment was performed a minimum of three independent times. Data are represented as mean ± S.D. Statistical analyses were performed by using one-way ANOVA or two-way ANOVA, and significance was determined at p < 0.05.
Author contributions
K. E. T. and S. P. B. conceived and designed the experiments. K. E. T., N. H. S., X. F., K. M. K., P. B., and M. S. performed the experiments and acquired the data. A. J. C. and W. L. provided critical analysis. K. E. T. and S. P. B. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Cindy Ross for technical support with transient transfections and Leigh Zawel for critical review of the manuscript.
This work was supported by Pfizer Worldwide Research and Development and in part by National Institutes of Health Grant R01DK078226 (to W. L.) and a research grant from the Pfizer Centers for Therapeutic Innovation (to W. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Tables S1–S4.
- LRR
- leucine-rich repeat domain
- EGF
- epidermal growth factor
- ROBO
- roundabout
- UB
- ureteric bud
- BMP
- bone morphogenetic protein
- MM
- metanephric mesenchymel
- WD
- Wolffian duct
- ND
- nephric duct
- GDNF
- glial cell-derived neurotrophic factor
- CAKUT
- congenital anomaly of kidney and urinary tract
- ANOVA
- analysis of variance
- SPR
- surface plasmon resonance
- RU
- resonance unit
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- RT-qPCR
- quantitative real-time PCR
- BRE
- BMP-response element
- HBS
- HEPES-buffered saline
- NHS
- N-hydroxysuccinimide
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- SVZa
- anterior subventricular zone
- ARPMI
- advanced RPMI 1640 medium
- RI
- refractive index
- ES
- embryonic stem.
References
- 1. Rothberg J. M., Jacobs J. R., Goodman C. S., and Artavanis-Tsakonas S. (1990) Slit: an extracellular protein necessary for development of midline glia and commissural axon pathways contains both EGF and LRR domains. Genes Dev. 4, 2169–2187 10.1101/gad.4.12a.2169 [DOI] [PubMed] [Google Scholar]
- 2. Rothberg J. M., and Artavanis-Tsakonas S. (1992) Modularity of the slit protein. Characterization of a conserved carboxy-terminal sequence in secreted proteins and a motif implicated in extracellular protein interactions. J. Mol. Biol. 227, 367–370 10.1016/0022-2836(92)90891-M [DOI] [PubMed] [Google Scholar]
- 3. Morlot C., Thielens N. M., Ravelli R. B., Hemrika W., Romijn R. A., Gros P., Cusack S., and McCarthy A. A. (2007) Structural insights into the Slit-Robo complex. Proc. Natl. Acad. Sci. U.S.A. 104, 14923–14928 10.1073/pnas.0705310104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Howitt J. A., Clout N. J., and Hohenester E. (2004) Binding site for Robo receptors revealed by dissection of the leucine-rich repeat region of Slit. EMBO J. 23, 4406–4412 10.1038/sj.emboj.7600446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang K. H., Brose K., Arnott D., Kidd T., Goodman C. S., Henzel W., and Tessier-Lavigne M. (1999) Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell 96, 771–784 10.1016/S0092-8674(00)80588-7 [DOI] [PubMed] [Google Scholar]
- 6. Brose K., Bland K. S., Wang K. H., Arnott D., Henzel W., Goodman C. S., Tessier-Lavigne M., and Kidd T. (1999) Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 96, 795–806 10.1016/S0092-8674(00)80590-5 [DOI] [PubMed] [Google Scholar]
- 7. Delloye-Bourgeois C., Jacquier A., Charoy C., Reynaud F., Nawabi H., Thoinet K., Kindbeiter K., Yoshida Y., Zagar Y., Kong Y., Jones Y. E., Falk J., Chédotal A., and Castellani V. (2015) PlexinA1 is a new Slit receptor and mediates axon guidance function of Slit C-terminal fragments. Nat. Neurosci. 18, 36–45 10.1038/nn.3893 [DOI] [PubMed] [Google Scholar]
- 8. Ronca F., Andersen J. S., Paech V., and Margolis R. U. (2001) Characterization of Slit protein interactions with glypican-1. J. Biol. Chem. 276, 29141–29147 10.1074/jbc.M100240200 [DOI] [PubMed] [Google Scholar]
- 9. Liang Y., Annan R. S., Carr S. A., Popp S., Mevissen M., Margolis R. K., and Margolis R. U. (1999) Mammalian homologues of the Drosophila slit protein are ligands of the heparan sulfate proteoglycan glypican-1 in brain. J. Biol. Chem. 274, 17885–17892 10.1074/jbc.274.25.17885 [DOI] [PubMed] [Google Scholar]
- 10. Li H. S., Chen J. H., Wu W., Fagaly T., Zhou L., Yuan W., Dupuis S., Jiang Z. H., Nash W., Gick C., Ornitz D. M., Wu J. Y., and Rao Y. (1999) Vertebrate slit, a secreted ligand for the transmembrane protein roundabout, is a repellent for olfactory bulb axons. Cell 96, 807–818 10.1016/S0092-8674(00)80591-7 [DOI] [PubMed] [Google Scholar]
- 11. Kidd T., Bland K. S., and Goodman C. S. (1999) Slit is the midline repellent for the robo receptor in Drosophila. Cell 96, 785–794 10.1016/S0092-8674(00)80589-9 [DOI] [PubMed] [Google Scholar]
- 12. Hu H. (1999) Chemorepulsion of neuronal migration by Slit2 in the developing mammalian forebrain. Neuron 23, 703–711 10.1016/S0896-6273(01)80029-5 [DOI] [PubMed] [Google Scholar]
- 13. Piper M., Georgas K., Yamada T., and Little M. (2000) Expression of the vertebrate Slit gene family and their putative receptors, the Robo genes, in the developing murine kidney. Mech. Dev. 94, 213–217 10.1016/S0925-4773(00)00313-0 [DOI] [PubMed] [Google Scholar]
- 14. Grieshammer U., Le Ma, Plump A. S., Wang F., Tessier-Lavigne M., and Martin G. R. (2004) SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev. Cell 6, 709–717 10.1016/S1534-5807(04)00108-X [DOI] [PubMed] [Google Scholar]
- 15. Lu W., van Eerde A. M., Fan X., Quintero-Rivera F., Kulkarni S., Ferguson H., Kim H. G., Fan Y., Xi Q., Li Q. G., Sanlaville D., Andrews W., Sundaresan V., Bi W., Yan J., et al. (2007) Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am. J. Hum. Genet. 80, 616–632 10.1086/512735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bertoli-Avella A. M., Conte M. L., Punzo F., de Graaf B. M., Lama G., La Manna A., Polito C., Grassia C., Nobili B., Rambaldi P. F., Oostra B. A., and Perrotta S. (2008) ROBO2 gene variants are associated with familial vesicoureteral reflux. J. Am. Soc. Nephrol. 19, 825–831 10.1681/ASN.2007060692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hwang D. Y., Dworschak G. C., Kohl S., Saisawat P., Vivante A., Hilger A. C., Reutter H. M., Soliman N. A., Bogdanovic R., Kehinde E. O., Tasic V., and Hildebrandt F. (2014) Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 85, 1429–1433 10.1038/ki.2013.508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hwang D. Y., Kohl S., Fan X., Vivante A., Chan S., Dworschak G. C., Schulz J., van Eerde A. M., Hilger A. C., Gee H. Y., Pennimpede T., Herrmann B. G., van de Hoek G., Renkema K. Y., Schell C., et al. (2015) Mutations of the SLIT2–ROBO2 pathway genes SLIT2 and SRGAP1 confer risk for congenital anomalies of the kidney and urinary tract. Hum. Genet. 134, 905–916 10.1007/s00439-015-1570-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fan X., Yang H., Kumar S., Tumelty K. E., Pisarek-Horowitz A., Rasouly H. M., Sharma R., Chan S., Tyminski E., Shamashkin M., Belghasem M., Henderson J. M., Coyle A. J., Salant D. J., Berasi S. P., and Lu W. (2016) SLIT2/ROBO2 signaling pathway inhibits nonmuscle myosin IIA activity and destabilizes kidney podocyte adhesion. JCI Insight 1, e86934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan X., Li Q., Pisarek-Horowitz A., Rasouly H. M., Wang X., Bonegio R. G., Wang H., McLaughlin M., Mangos S., Kalluri R., Holzman L. B., Drummond I. A., Brown D., Salant D. J., and Lu W. (2012) Inhibitory effects of Robo2 on nephrin: a cross-talk between positive and negative signals regulating podocyte structure. Cell Rep. 2, 52–61 10.1016/j.celrep.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuen D. A., Huang Y. W., Liu G. Y., Patel S., Fang F., Zhou J., Thai K., Sidiqi A., Szeto S. G., Chan L., Lu M., He X., John R., Gilbert R. E., Scholey J. W., and Robinson L. A. (2016) Recombinant N-terminal Slit2 inhibits TGF-β-induced fibroblast activation and renal fibrosis. J. Am. Soc. Nephrol. 27, 2609–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pilling D., Zheng Z., Vakil V., and Gomer R. H. (2014) Fibroblasts secrete Slit2 to inhibit fibrocyte differentiation and fibrosis. Proc. Natl. Acad. Sci. U.S.A. 111, 18291–18296 10.1073/pnas.1417426112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang J., Lan T., Li C., Ji X., Zheng L., Gou H., Ou Y., Wu T., Qi C., Zhang Q., Li J., Gu Q., Wen D., Cao L., Qiao L., Ding Y., and Wang L. (2015) Activation of Slit2-Robo1 signaling promotes liver fibrosis. J. Hepatol. 63, 1413–1420 10.1016/j.jhep.2015.07.033 [DOI] [PubMed] [Google Scholar]
- 24. Zhou X., Yao Q., Sun X., Gong X., Yang Y., Chen C., and Shan G. (2017) Slit2 ameliorates renal inflammation and fibrosis after hypoxia-and lipopolysaccharide-induced epithelial cells injury in vitro. Exp. Cell Res. 352, 123–129 10.1016/j.yexcr.2017.02.001 [DOI] [PubMed] [Google Scholar]
- 25. Gu F., Ma Y., Zhang J., Qin F., and Fu L. (2015) Function of Slit/Robo signaling in breast cancer. Front. Med. 9, 431–436 10.1007/s11684-015-0416-9 [DOI] [PubMed] [Google Scholar]
- 26. Kong R., Yi F., Wen P., Liu J., Chen X., Ren J., Li X., Shang Y., Nie Y., Wu K., Fan D., Zhu L., Feng W., and Wu J. Y. (2015) Myo9b is a key player in SLIT/ROBO-mediated lung tumor suppression. J. Clin. Invest. 125, 4407–4420 10.1172/JCI81673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng Y., Feng L., Yu D., Zou J., and Huang Z. (2016) srGAP1 mediates the migration inhibition effect of Slit2-Robo1 in colorectal cancer. J. Exp. Clin. Cancer Res. 35, 191 10.1186/s13046-016-0469-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li S., Huang L., Sun Y., Bai Y., Yang F., Yu W., Li F., Zhang Q., Wang B., Geng J. G., and Li X. (2015) Slit2 promotes angiogenic activity via the Robo1-VEGFR2-ERK1/2 pathway in both in vivo and in vitro studies. Invest. Ophthalmol. Vis. Sci. 56, 5210–5217 10.1167/iovs-14-16184 [DOI] [PubMed] [Google Scholar]
- 29. Rama N., Dubrac A., Mathivet T., Ní Chárthaigh R. A., Genet G., Cristofaro B., Pibouin-Fragner L., Ma L., Eichmann A., and Chédotal A. (2015) Slit2 signaling through Robo1 and Robo2 is required for retinal neovascularization. Nat. Med. 21, 483–491 10.1038/nm.3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen B., Blair D. G., Plisov S., Vasiliev G., Perantoni A. O., Chen Q., Athanasiou M., Wu J. Y., Oppenheim J. J., and Yang D. (2004) Cutting edge: bone morphogenetic protein antagonists Drm/Gremlin and Dan interact with Slits and act as negative regulators of monocyte chemotaxis. J. Immunol. 173, 5914–5917 10.4049/jimmunol.173.10.5914 [DOI] [PubMed] [Google Scholar]
- 31. Hsu D. R., Economides A. N., Wang X., Eimon P. M., and Harland R. M. (1998) The Xenopus dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol. Cell 1, 673–683 10.1016/S1097-2765(00)80067-2 [DOI] [PubMed] [Google Scholar]
- 32. Avsian-Kretchmer O., and Hsueh A. J. (2004) Comparative genomic analysis of the eight-membered ring cystine knot-containing bone morphogenetic protein antagonists. Mol. Endocrinol. 18, 1–12 10.1210/me.2003-0227 [DOI] [PubMed] [Google Scholar]
- 33. Katagiri T., and Watabe T. (2016) Bone morphogenetic proteins. Cold Spring Harb. Perspect. Biol. 8(6). pii 10.1101/cshperspect.a021899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brazil D. P., Church R. H., Surae S., Godson C., and Martin F. (2015) BMP signalling: agony and antagony in the family. Trends Cell Biol. 25, 249–264 10.1016/j.tcb.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 35. Zimmerman L. B., De Jesús-Escobar J. M., and Harland R. M. (1996) The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell 86, 599–606 10.1016/S0092-8674(00)80133-6 [DOI] [PubMed] [Google Scholar]
- 36. Michos O., Gonçalves A., Lopez-Rios J., Tiecke E., Naillat F., Beier K., Galli A., Vainio S., and Zeller R. (2007) Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signalling during kidney branching morphogenesis. Development 134, 2397–2405 10.1242/dev.02861 [DOI] [PubMed] [Google Scholar]
- 37. Michos O., Panman L., Vintersten K., Beier K., Zeller R., and Zuniga A. (2004) Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development 131, 3401–3410 10.1242/dev.01251 [DOI] [PubMed] [Google Scholar]
- 38. Wan M., and Cao X. (2005) BMP signaling in skeletal development. Biochem. Biophys. Res. Commun. 328, 651–657 10.1016/j.bbrc.2004.11.067 [DOI] [PubMed] [Google Scholar]
- 39. Li G., Li Y., Liu S., Shi Y., Chi Y., Liu G., and Shan T. (2013) Gremlin aggravates hyperglycemia-induced podocyte injury by a TGFβ/Smad dependent signaling pathway. J. Cell. Biochem. 114, 2101–2113 10.1002/jcb.24559 [DOI] [PubMed] [Google Scholar]
- 40. Myllärniemi M., Lindholm P., Ryynänen M. J., Kliment C. R., Salmenkivi K., Keski-Oja J., Kinnula V. L., Oury T. D., and Koli K. (2008) Gremlin-mediated decrease in bone morphogenetic protein signaling promotes pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 177, 321–329 10.1164/rccm.200706-945OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koli K., Myllärniemi M., Vuorinen K., Salmenkivi K., Ryynänen M. J., Kinnula V. L., and Keski-Oja J. (2006) Bone morphogenetic protein-4 inhibitor gremlin is overexpressed in idiopathic pulmonary fibrosis. Am. J. Pathol. 169, 61–71 10.2353/ajpath.2006.051263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Costantini F. (2012) Genetic controls and cellular behaviors in branching morphogenesis of the renal collecting system. Wiley Interdiscip. Rev. Dev. Biol. 1, 693–713 10.1002/wdev.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Costantini F., and Shakya R. (2006) GDNF/Ret signaling and the development of the kidney. BioEssays 28, 117–127 10.1002/bies.20357 [DOI] [PubMed] [Google Scholar]
- 44. Bridgewater D., and Rosenblum N. D. (2009) Stimulatory and inhibitory signaling molecules that regulate renal branching morphogenesis. Pediatr. Nephrol. 24, 1611–1619 10.1007/s00467-008-1048-y [DOI] [PubMed] [Google Scholar]
- 45. Hellmich H. L., Kos L., Cho E. S., Mahon K. A., and Zimmer A. (1996) Embryonic expression of glial cell-line derived neurotrophic factor (GDNF) suggests multiple developmental roles in neural differentiation and epithelial-mesenchymal interactions. Mech. Dev. 54, 95–105 10.1016/0925-4773(95)00464-5 [DOI] [PubMed] [Google Scholar]
- 46. Towers P. R., Woolf A. S., and Hardman P. (1998) Glial cell line-derived neurotrophic factor stimulates ureteric bud outgrowth and enhances survival of ureteric bud cells in vitro. Exp. Nephrol. 6, 337–351 10.1159/000020541 [DOI] [PubMed] [Google Scholar]
- 47. Pachnis V., Mankoo B., and Costantini F. (1993) Expression of the c-ret proto-oncogene during mouse embryogenesis. Development 119, 1005–1017 [DOI] [PubMed] [Google Scholar]
- 48. Cacalano G., Fariñas I., Wang L. C., Hagler K., Forgie A., Moore M., Armanini M., Phillips H., Ryan A. M., Reichardt L. F., Hynes M., Davies A., and Rosenthal A. (1998) GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron 21, 53–62 10.1016/S0896-6273(00)80514-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Enomoto H., Araki T., Jackman A., Heuckeroth R. O., Snider W. D., Johnson E. M. Jr, and Milbrandt J. (1998) GFR α1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron 21, 317–324 10.1016/S0896-6273(00)80541-3 [DOI] [PubMed] [Google Scholar]
- 50. Golden J. P., DeMaro J. A., Osborne P. A., Milbrandt J., and Johnson E. M. Jr. (1999) Expression of neurturin, GDNF, and GDNF family-receptor mRNA in the developing and mature mouse. Exp. Neurol. 158, 504–528 10.1006/exnr.1999.7127 [DOI] [PubMed] [Google Scholar]
- 51. Sainio K., Suvanto P., Davies J., Wartiovaara J., Wartiovaara K., Saarma M., Arumäe U., Meng X., Lindahl M., Pachnis V., and Sariola H. (1997) Glial-cell-line-derived neurotrophic factor is required for bud initiation from ureteric epithelium. Development 124, 4077–4087 [DOI] [PubMed] [Google Scholar]
- 52. Wainwright E. N., Wilhelm D., Combes A. N., Little M. H., and Koopman P. (2015) ROBO2 restricts the nephrogenic field and regulates Wolffian duct-nephrogenic cord separation. Dev. Biol. 404, 88–102 10.1016/j.ydbio.2015.05.023 [DOI] [PubMed] [Google Scholar]
- 53. Wu W., Wong K., Chen J., Jiang Z., Dupuis S., Wu J. Y., and Rao Y. (1999) Directional guidance of neuronal migration in the olfactory system by the protein Slit. Nature 400, 331–336 10.1038/22477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nguyen-Ba-Charvet K. T., Picard-Riera N., Tessier-Lavigne M., Baron-Van Evercooren A., Sotelo C., and Chédotal A. (2004) Multiple roles for slits in the control of cell migration in the rostral migratory stream. J. Neurosci. 24, 1497–1506 10.1523/JNEUROSCI.4729-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Herrera B., and Inman G. J. (2009) A rapid and sensitive bioassay for the simultaneous measurement of multiple bone morphogenetic proteins. Identification and quantification of BMP4, BMP6 and BMP9 in bovine and human serum. BMC Cell Biol. 10, 20 10.1186/1471-2121-10-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zilberberg L., ten Dijke P., Sakai L. Y., and Rifkin D. B. (2007) A rapid and sensitive bioassay to measure bone morphogenetic protein activity. BMC Cell Biol. 8, 41 10.1186/1471-2121-8-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kretzschmar M., Liu F., Hata A., Doody J., and Massagué J. (1997) The TGF-β family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes Dev. 11, 984–995 10.1101/gad.11.8.984 [DOI] [PubMed] [Google Scholar]
- 58. Liu F., Hata A., Baker J. C., Doody J., Cárcamo J., Harland R. M., and Massagué J. (1996) A human Mad protein acting as a BMP-regulated transcriptional activator. Nature 381, 620–623 10.1038/381620a0 [DOI] [PubMed] [Google Scholar]
- 59. Glister C., Richards S. L., and Knight P. G. (2005) Bone morphogenetic proteins (BMP)- 4, -6, and -7 potently suppress basal and luteinizing hormone-induced androgen production by bovine theca interna cells in primary culture: could ovarian hyperandrogenic dysfunction be caused by a defect in thecal BMP signaling? Endocrinology 146, 1883–1892 10.1210/en.2004-1303 [DOI] [PubMed] [Google Scholar]
- 60. Hao J., Ho J. N., Lewis J. A., Karim K. A., Daniels R. N., Gentry P. R., Hopkins C. R., Lindsley C. W., and Hong C. C. (2010) In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem. Biol. 5, 245–253 10.1021/cb9002865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lagna G., Hata A., Hemmati-Brivanlou A., and Massagué J. (1996) Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature 383, 832–836 10.1038/383832a0 [DOI] [PubMed] [Google Scholar]
- 62. Saunders J., Wisidagama D. R., Morford T., and Malone C. S. (2016) Maximal expression of the evolutionarily conserved Slit2 gene promoter requires Sp1. Cell. Mol. Neurobiol. 36, 955–964 10.1007/s10571-015-0281-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nie X., Xu J., El-Hashash A., and Xu P. X. (2011) Six1 regulates Grem1 expression in the metanephric mesenchyme to initiate branching morphogenesis. Dev. Biol. 352, 141–151 10.1016/j.ydbio.2011.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xia Y., Nivet E., Sancho-Martinez I., Gallegos T., Suzuki K., Okamura D., Wu M. Z., Dubova I., Esteban C. R., Montserrat N., Campistol J. M., and Izpisua Belmonte J. C. (2013) Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat. Cell Biol. 15, 1507–1515 10.1038/ncb2872 [DOI] [PubMed] [Google Scholar]
- 65. Takasato M., Er P. X., Becroft M., Vanslambrouck J. M., Stanley E. G., Elefanty A. G., and Little M. H. (2014) Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 16, 118–126 10.1038/ncb2894 [DOI] [PubMed] [Google Scholar]
- 66. Ciampi O., Iacone R., Longaretti L., Benedetti V., Graf M., Magnone M. C., Patsch C., Xinaris C., Remuzzi G., Benigni A., and Tomasoni S. (2016) Generation of functional podocytes from human induced pluripotent stem cells. Stem Cell Res. 17, 130–139 10.1016/j.scr.2016.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lam A. Q., Freedman B. S., Morizane R., Lerou P. H., Valerius M. T., and Bonventre J. V. (2014) Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J. Am. Soc. Nephrol. 25, 1211–1225 10.1681/ASN.2013080831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Taguchi A., Kaku Y., Ohmori T., Sharmin S., Ogawa M., Sasaki H., and Nishinakamura R. (2014) Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14, 53–67 10.1016/j.stem.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 69. Morizane R., Lam A. Q., Freedman B. S., Kishi S., Valerius M. T., and Bonventre J. V. (2015) Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 33, 1193–1200 10.1038/nbt.3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Morizane R., and Bonventre J. V. (2017) Kidney organoids: a translational journey. Trends Mol. Med. 23, 246–263 10.1016/j.molmed.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Piper M., Nurcombe V., Wilkinson L., and Little M. (2002) Exogenous Slit2 does not affect ureteric branching or nephron formation during kidney development. Int. J. Dev. Biol. 46, 545–550 [PubMed] [Google Scholar]
- 72. Nguyen Ba-Charvet K. T., Brose K., Ma L., Wang K. H., Marillat V., Sotelo C., Tessier-Lavigne M., and Chédotal A. (2001) Diversity and specificity of actions of Slit2 proteolytic fragments in axon guidance. J. Neurosci. 21, 4281–4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Morin-Poulard I., Sharma A., Louradour I., Vanzo N., Vincent A., and Crozatier M. (2016) Vascular control of the Drosophila haematopoietic microenvironment by Slit/Robo signalling. Nat. Commun. 7, 11634 10.1038/ncomms11634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kohl S., Hwang D. Y., Dworschak G. C., Hilger A. C., Saisawat P., Vivante A., Stajic N., Bogdanovic R., Reutter H. M., Kehinde E. O., Tasic V., and Hildebrandt F. (2014) Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J. Am. Soc. Nephrol. 25, 1917–1922 10.1681/ASN.2013101103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Weber S., Taylor J. C., Winyard P., Baker K. F., Sullivan-Brown J., Schild R., Knüppel T., Zurowska A. M., Caldas-Alfonso A., Litwin M., Emre S., Ghiggeri G. M., Bakkaloglu A., Mehls O., Antignac C., et al. (2008) SIX2 and BMP4 mutations associate with anomalous kidney development. J. Am. Soc. Nephrol. 19, 891–903 10.1681/ASN.2006111282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Raatikainen-Ahokas A., Hytönen M., Tenhunen A., Sainio K., and Sariola H. (2000) BMP-4 affects the differentiation of metanephric mesenchyme and reveals an early anterior-posterior axis of the embryonic kidney. Dev. Dyn. 217, 146–158 10.1002/(SICI)1097-0177(200002)217:2%3C146::AID-DVDY2%3E3.0.CO%3B2-I [DOI] [PubMed] [Google Scholar]
- 77. Kinameri E., and Matsuoka I. (2003) Autocrine action of BMP2 regulates expression of GDNF-mRNA in sciatic Schwann cells. Brain Res. Mol. Brain Res. 117, 221–227 10.1016/S0169-328X(03)00326-7 [DOI] [PubMed] [Google Scholar]
- 78. Wang Z., Zhao J., Zhang J., Wei J., Zhang J., and Huang Y. (2010) Protective effect of BMP-7 against aristolochic acid-induced renal tubular epithelial cell injury. Toxicol. Lett. 198, 348–357 10.1016/j.toxlet.2010.07.018 [DOI] [PubMed] [Google Scholar]
- 79. Zeisberg M., Hanai J., Sugimoto H., Mammoto T., Charytan D., Strutz F., and Kalluri R. (2003) BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968 10.1038/nm888 [DOI] [PubMed] [Google Scholar]
- 80. Xu Y., Wan J., Jiang D., and Wu X. (2009) BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition in human renal proximal tubular epithelial cells. J. Nephrol. 22, 403–410 [PubMed] [Google Scholar]
- 81. Izumi N., Mizuguchi S., Inagaki Y., Saika S., Kawada N., Nakajima Y., Inoue K., Suehiro S., Friedman S. L., and Ikeda K. (2006) BMP-7 opposes TGF-β1-mediated collagen induction in mouse pulmonary myofibroblasts through Id2. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L120–L126 10.1152/ajplung.00171.2005 [DOI] [PubMed] [Google Scholar]
- 82. Johnson K., Kirkpatrick H., Comer A., Hoffmann F. M., and Laughon A. (1999) Interaction of Smad complexes with tripartite DNA-binding sites. J. Biol. Chem. 274, 20709–20716 10.1074/jbc.274.29.20709 [DOI] [PubMed] [Google Scholar]
- 83. Ishida W., Hamamoto T., Kusanagi K., Yagi K., Kawabata M., Takehara K., Sampath T. K., Kato M., and Miyazono K. (2000) Smad6 is a Smad1/5-induced smad inhibitor. Characterization of bone morphogenetic protein-responsive element in the mouse Smad6 promoter. J. Biol. Chem. 275, 6075–6079 10.1074/jbc.275.9.6075 [DOI] [PubMed] [Google Scholar]
- 84. Brugger S. M., Merrill A. E., Torres-Vazquez J., Wu N., Ting M. C., Cho J. Y., Dobias S. L., Yi S. E., Lyons K., Bell J. R., Arora K., Warrior R., and Maxson R. (2004) A phylogenetically conserved cis-regulatory module in the Msx2 promoter is sufficient for BMP-dependent transcription in murine and Drosophila embryos. Development 131, 5153–5165 10.1242/dev.01390 [DOI] [PubMed] [Google Scholar]
- 85. López-Rovira T., Chalaux E., Massagué J., Rosa J. L., and Ventura F. (2002) Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J. Biol. Chem. 277, 3176–3185 10.1074/jbc.M106826200 [DOI] [PubMed] [Google Scholar]
- 86. Lewis T. C., and Prywes R. (2013) Serum regulation of Id1 expression by a BMP pathway and BMP responsive element. Biochim. Biophys. Acta 1829, 1147–1159 10.1016/j.bbagrm.2013.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ward M. E., and Rao Y. (2005) Investigations of neuronal migration in the central nervous system. Methods Mol. Biol. 294, 137–156 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.