

Abstract
Stimuli such as inflammation or hypoxia induce cytochrome P450 epoxygenase-mediated production of arachidonic acid–derived epoxyeicosatrienoic acids (EETs). EETs have cardioprotective, vasodilatory, angiogenic, anti-inflammatory, and analgesic effects, which are diminished by EET hydrolysis yielding biologically less active dihydroxyeicosatrienoic acids (DHETs). Previous in vitro assays have suggested that epoxide hydrolase 2 (EPHX2) is responsible for nearly all EET hydrolysis. EPHX1, which exhibits slow EET hydrolysis in vitro, is thought to contribute only marginally to EET hydrolysis. Using Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice, we show here that EPHX1 significantly contributes to EET hydrolysis in vivo. Disruption of Ephx1 and/or Ephx2 genes did not induce compensatory changes in expression of other Ephx genes or CYP2 family epoxygenases. Plasma levels of 8,9-, 11,12-, and 14,15-DHET were reduced by 38, 44, and 67% in Ephx2−/− mice compared with wildtype (WT) mice, respectively; however, plasma from Ephx1−/−Ephx2−/− mice exhibited significantly greater reduction (100, 99, and 96%) of those respective DHETs. Kinetic assays and FRET experiments indicated that EPHX1 is a slow EET scavenger, but hydrolyzes EETs in a coupled reaction with cytochrome P450 to limit basal EET levels. Moreover, we also found that EPHX1 activities are biologically relevant, as Ephx1−/−Ephx2−/− hearts had significantly better postischemic functional recovery (71%) than both WT (31%) and Ephx2−/− (51%) hearts. These findings indicate that Ephx1−/−Ephx2−/− mice are a valuable model for assessing EET-mediated effects, uncover a new paradigm for EET metabolism, and suggest that dual EPHX1 and EPHX2 inhibition may represent a therapeutic approach to manage human pathologies such as myocardial infarction.
Keywords: cytochrome P450, eicosanoid biosynthesis, cardiac muscle, cardiovascular disease, ischemia, epoxyeicosatrienoic acid (EETs), microsomal epoxide hydrolase, soluble epoxide hydrolase
Introduction
Cytochrome P450 epoxygenases can oxidize arachidonic acid (AA)2 to form epoxyeicosatrienoic acids (EETs) which have potent cardiovascular effects. The biological effects of EETs are short-lived in that they are rapidly hydrolyzed to less active dihydroxyeicosatrienoic acids (DHETs) by EPHX2, also known as soluble epoxide hydrolase (sEH) (1). Indeed, Ephx2−/− mice have increased EETs, decreased DHETs, and improved outcomes in vascular disease models, which provide the basis for development of pharmacological EPHX2 inhibitors (1, 2). EPHX2 inhibition increases EET levels in vivo, and leads to cardioprotective, vasodilatory, angiogenic, anti-inflammatory, and analgesic effects (1–3).
EPHX2 inhibitors, which have completed phase I clinical trials, are under investigation for treatment of neuropathic pain, and may hold promise for treatment of other ailments. However, genetic disruption of Ephx2 or EPHX2 pharmacological inhibition does not completely abolish EET hydrolysis in vivo. Among the EETs, EPHX2 has the biggest effect on levels of its preferred substrate, 14,15-EET, but has diminishing effects on 11,12- and 8,9-EET which are equal or more potent in cardiovascular physiology (4–6). More complete or more broad inhibition of fatty acid epoxide hydrolysis could further potentiate the beneficial cardiovascular effects of endogenously produced EETs.
Others have searched for additional enzymes capable of hydrolyzing EETs (7). EPHX1, also known as microsomal epoxide hydrolase (mEH), is known to be capable of EET hydrolysis; however, it is thought to play a minimal role. Compared with EPHX2, EPHX1 is tens- to thousands-fold slower in in vitro EET hydrolysis assays, and is also less abundant in vivo (1, 7, 8). In some tissues, such as the brain, which has low EPHX2 expression, EPHX1 is believed to be responsible for hydrolysis of only a small fraction of its preferred substrate, 11,12-EET (1, 8, 9). Three other enzymes, EPHX3, EPHX4, and PEG1/MEST, were identified based on homology to the catalytic sites of EPHX1 and EPHX2 (7). EPHX3 was reported to have high catalytic efficiency toward fatty acid epoxides in vitro; however, Ephx3−/− mice exhibit no alterations of fatty acid epoxide hydrolysis (10). EPHX4 and PEG1/MEST have not yet been examined for fatty acid epoxide hydrolase activity.
In this study, we demonstrate that EPHX1 is responsible for a substantial proportion of EET hydrolysis in vivo. Ephx1−/−Ephx2−/− have nearly complete loss of DHET in plasma or tissue lysates. Compared with Ephx1−/− and Ephx2−/− mice, Ephx1−/−Ephx2−/− mice have significantly increased plasma levels of nearly all fatty acid epoxide examined. Kinetic and FRET analyses suggest that EPHX1 interacts directly with P450 epoxygenases and likely participates in a coupled reaction during basal EET formation. These surprising findings explain the large residual EET hydrolysis observed in Ephx2−/− mice, reveal a mouse model with improved potentiation of EET effects, present a new paradigm for the role of EPHXs in EET metabolism, and suggest that combined inhibition of EPHX1 and EPHX2 represents a novel approach to treatment of disorders such as ischemic heart disease.
Results
We examined fatty acid epoxide levels and metabolism in tissues and body fluids from WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice. Ephx1−/− and Ephx2−/− mice were previously generated and maintained on a C57BL/6 background (11, 12). In-crossing of Ephx1+/−Ephx2+/− mice yielded pups with no overt phenotypes; pups were born in normal Mendelian ratios (Table S1), had similar body weights and organ/body weight ratios (Table S2), and exhibited no gross anatomical or histological abnormalities.
Mice showed expected alterations of gene expression (Fig. 1, A and B). Ephx1−/− mice have been reported to express low levels of a truncated, noncoding, Ephx1 mRNA transcript (11). Ephx1 mRNA levels were reduced in Ephx1−/− and Ephx1−/−Ephx2−/− mice relative to WT. Ephx2 mRNA levels were nearly absent in Ephx2−/− and Ephx1−/−Ephx2−/− mice. Likewise, EPHX1 protein was absent from Ephx1−/− and Ephx1−/−Ephx2−/− mice, and EPHX2 protein was absent from Ephx2−/− and Ephx1−/−Ephx2−/− mice. EPHX1 was detected in both the S9 fraction, which contains both cytosol and microsomes, and in microsomes. EPHX2 was detected in both S9 and microsomal fractions. This is consistent with previous findings of nearly equal distribution of EPHX2 between cytosolic and microsomal fractions (13–15). We observed no compensatory regulation of hepatic Ephx1, Ephx2, Ephx3, or Ephx4 mRNAs (Fig. 1A, Fig. S1). In addition, hepatic expression of Cyp2c and Cyp2j family members was also comparable across genotypes. Similar Ephx1, Ephx2, Ephx3, and Ephx4 expression profiles were observed in mouse heart (Fig. S2).
Figure 1.

Hepatic epoxide hydrolase expression in WT and Ephx-deficient mice. A, Ephx1 and Ephx2 mRNA levels in livers from WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice relative to Gapdh; n = 4 mice per genotype group. *, p < 0.05 versus WT; ∧, p < 0.05 versus Ephx1−/−; #, p < 0.05 versus Ephx2−/−. Results are expressed as mean ± S.E. B, expression of EPHX1, EPHX2, and β-actin proteins in liver S9 and microsomal fractions from WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice.
We quantified oxylipin levels in plasma from WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice using LC-MS/MS (Fig. 2, A and B, Table S3). Disruption of Ephx1 alone did not alter plasma 14,15-EET, 11,12-EET, 8,9-EET, or 5,6-EET levels compared with WT, nor did it alter levels of epoxides derived from linoleic acid (12,13-EpOME and 9,10-EpOME), docosahexaenoic acid (19,20-EpDPE), or eicosapentaenoic acid (17,18-EpETE). In contrast, Ephx1−/− plasma had significantly lower levels of 8,9-DHET, 5,6-DHET, 19,20-DiHDPA, and 17,18-DHET compared with WT. Plasma from Ephx2−/− mice had significantly higher levels of 14,15-EET, 11,12-EET, 8,9-EET, 17,18-EpETE, 12,13-EpOME, and 9,10-EpOME and significantly lower levels of 14,15-DHET, 11,12-DHET, 8,9-DHET, and 12,13-DiHOME compared with WT. Disruption of both Ephx1 and Ephx2 more dramatically altered plasma oxylipin levels. Compared with WT, Ephx1−/−Ephx2−/− mice had significantly higher levels of every CYP-derived epoxide measured except 5,6-EET. Importantly, in Ephx1−/−Ephx2−/− mouse plasma, levels of all the fatty acid diols were low or undetectable. Moreover, compared with Ephx2−/− mice, plasma from Ephx1−/−Ephx2−/− mice had significantly higher levels of 11,12-EET, 17,18-EpETE, and 19,20-EpDPE, and significantly lower levels of all diols measured except 12,13-DiHOME. These data suggest functional compensation of EPHX1 and EPHX2 with regard to fatty acid epoxide hydrolysis in vivo. Together, EPHX1 and EPHX2 account for hydrolysis of the nearly all circulating epoxides derived from arachidonic, linoleic, eicosapentaenoic, and docosahexaenoic acids.
Figure 2.

Plasma oxylipin levels in WT or Ephx-deficient mice. A, plasma levels of AA-derived epoxides and diols in WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice. B, plasma levels of docosahexaenoic-, eicosapentaenoic-, and linoleic acid–derived epoxides and diols in WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice; n = 9–11 per group. *, p < 0.05 versus WT; #, p < 0.05 versus Ephx2−/−. Results are expressed as mean ± S.E.
Our in vivo data suggest a substantial role of EPHX1 in hydrolysis of endogenous fatty acid epoxides that was not predicted by prior in vitro assays. In vitro kinetic assays, typically performed at supraphysiologic (μm) substrate concentrations (7, 8), may underestimate the capacity for EPHX1 to hydrolyze fatty acid epoxides in vivo. Indeed, intracellular EET levels are estimated to be in the low nm range (16, 17). To determine the relative contribution of EPHX1 and EPHX2 to EET hydrolysis at physiologically relevant concentrations, liver lysates from all four genotypes were incubated with exogenous 11,12-EET at 1–1000 nm concentration (Fig. 3). In this assay, EPHX2 was the dominant hydrolase. Ephx1−/− lysates showed no decrease in enzymatic activity compared with WT, whereas Ephx2−/− lysates show nearly complete reduction of hydrolysis (94–99%) at all 11,12-EET concentrations. Ephx1−/−Ephx2−/− lysates had a small additional decrease in 11,12-EET hydrolysis compared with Ephx2−/− lysates. These data suggest that EPHX1 is responsible for only a small fraction (1–4%) of 11,12-EET hydrolysis in the liver under these experimental conditions.
Figure 3.
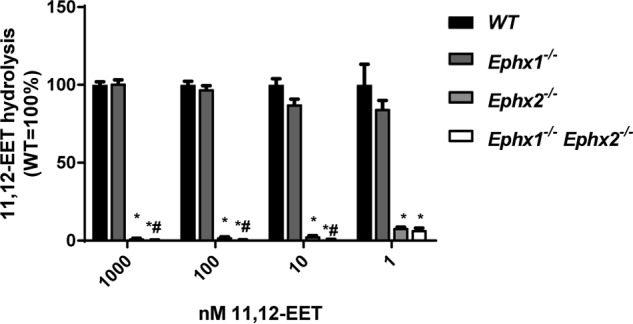
Relative contribution of EPHX1 and EPHX2 to EET hydrolysis in liver S9 fractions. Metabolism of 11,12-EET (1–1000 nm) by liver lysates from WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice. n = 4 per group. *, p < 0.05 versus WT; #, p < 0.05 versus Ephx2−/−. Results are expressed as mean ± S.E.
Our in vitro 11,12-EET hydrolysis assays do not reflect the pattern observed in vivo where EPHX1 contributes significantly to EET hydrolysis in the absence of EPHX2. EPHX1 is localized to the endoplasmic reticulum (ER) adjacent to the P450s that generate EETs. Although rarely studied for its role in EET hydrolysis, EPHX1 metabolism of xenobiotics has been extensively investigated (18). Indeed, EPHX1 can bind to xenobiotic metabolizing P450s, and this binding stimulates EPHX1 activity in what might be a coupled reaction (19–21). We hypothesized that EPHX1 may also participate in a coupled reaction with P450s that metabolize endogenous fatty acids. To test this, we incubated WT liver microsomes, which contain cytochromes P450, EPHX1, and EPHX2, with 10 μm arachidonic acid and increasing concentrations of NADPH. High NADPH concentrations induced rapid production of 11,12-EET (1900 pg/mg protein/min). Decreasing the concentration of NADPH progressively slowed the rate of 11,12-EET production to <100 pg/mg protein/min (Fig. 4, A and B). At the highest rate of EET production (200 μm NADPH), disruption of EPHX1 did not reduce hydrolysis, whereas disruption of EPHX2 reduced hydrolysis of 11,12-EET by 85%. Disruption of both EPHX1 and EPHX2 abolished 11,12-EET hydrolysis. Thus, EPHX1 appears to be responsible for 15% of EET hydrolysis under these conditions. Importantly, the relative contribution of EPHX1 to EET hydrolysis increased as the rate of EET formation decreased. Thus, at 0.2 μm and 0.02 μm NADPH, EPHX1 was capable of 61 and 86% of 11,12-EET hydrolysis, respectively. At the lowest rate of EET production (no added NADPH), EPHX1 was capable of hydrolyzing nearly all EETs as they formed. Formation and hydrolysis of 8,9- and 14,15-EET were similarly regulated (Fig. 4, C–E). The contribution of EPHX1 and EPHX2 to EET hydrolysis at low rates of EET production closely resembles the pattern observed in mouse plasma, suggesting that in vivo production of EETs by P450s occurs near the low end of enzymatic capacity.
Figure 4.

Relative contribution of EPHX1 and EPHX2 to hydrolysis during EET formation in liver microsomes. Microsomes made from livers of WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice were incubated with 10 μm arachidonic acid in the presence/absence of increasing concentration of NADPH. Relative formation of 11,12-DHET (A), 11,12-EET (B), 14,15-DHET (C), 14,15-EET (D), 8,9-DHET (E), and 8,9-EET (F) at 0–200 μm NADPH are shown as a percentage of WT (A, C, E) or Ephx1−/−Ephx2−/− (B, D, F). Formation rates at each NADPH concentration are shown above each group in pg/mg protein/min. n = 4 per group, *, p < 0.05 versus WT; #, p < 0.05 versus Ephx2−/−. Results are expressed as mean ± S.E.
To support the presence of a coupled reaction between P450s that metabolize endogenous fatty acids and EPHX1, we examined physical interactions between the enzymes using FRET (Fig. 5). EPHX1 N-terminal was labeled with cyan fluorescent protein (CFP), whereas CYP2C8 and CYP2C9 were labeled with YFP. FACS-FRET measurements showed a physical association between EPHX1 and both CYP2C8 or CYP2C9. This association was as robust as was observed with the positive control (CYP2J5 and cytochrome P450 reductase (CYPOR)). This direct interaction between the enzymes suggests an active transfer of the EET substrate that could have major implications for the balance between the epoxide as the intermediate and the diol as the final product of the enzymatic cascade.
Figure 5.

Direct interaction between CYP2C epoxygenases and EPHX1. A, schematic of membrane topology of the YFP-CYP and the CFP-EPHX1 fusion proteins used for the FRET analysis. Intracellular co-localization of the above fusion proteins in HEK-293 cells (1 = YFP (red), 2 = CFP (green), 3 = co-localization (yellow)). Scale bar = 10 μm. B, representative FACS analyses of FRET-positive cells for the negative control, CYP2C8-EPHX1, and CYP2C9-EPHX1 interactions. FRET signal intensity (abscissa) is plotted against CFP signal intensity (ordinate). Gated areas contain FRET-positive cells. C, percentage of FRET-positive cells calculated from three to five independent replicate experiments except n = 1 for the positive control (CYP2J5-cytochrome P450 reductase (CYPOR)). *, p < 0.0001 versus negative control (CYP2J5–ER-membrane-anchor). Results are expressed as mean ± S.E.
To further examine the relative roles of EPHX1 and EPHX2 in EET hydrolysis in an intact organ, hearts were isolated and perfused in retrograde fashion using the Langendorff method (4, 22). Perfusates from each genotype were collected during the last 20 min of baseline and the first 20 min of reperfusion after global, no-flow ischemia, and assayed for fatty acid epoxides and diols by LC-MS/MS (Fig. 6, Table S4). Levels of most of the fatty acid epoxides and diols in Ephx1−/− and Ephx2−/− heart perfusates were similar to those in WT at baseline and after reperfusion, although Ephx1−/− heart perfusates had significantly higher 19,20-EpDPE and significantly lower 19,20-DiHDPA levels, and Ephx2−/− heart perfusates had significantly higher 17,18-EpETE levels at baseline. In contrast, Ephx1−/−Ephx2−/− heart perfusates had significantly lower levels of 14,15-DHET, 11,12-DHET, 19,20-DiHDPA, and 17,18-DHET than both WT or Ephx2−/− hearts at baseline and during reperfusion. Of note, production of 11,12-EET plus 11,12-DHET was ∼800 pg/g heart tissue/min at baseline and increased to 2200 pg/g/min during reperfusion. Likewise, production of 14,15-EET plus 14,15-DHET levels increased after reperfusion. Importantly, EPHX1 contributed more to EET hydrolysis under conditions where EET production occurred at lower rates (i.e. at baseline).
Figure 6.

Role of EPHX1 and EPHX2 in regulating heart oxylipin levels. Shown are oxylipin levels in cardiac perfusates from WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− hearts expressed as pg/g heart tissue/min. n = 6–7 per genotype group; *, p < 0.05 versus WT; #, p < 0.05 versus Ephx2−/−.
Ephx2−/− hearts have improved functional recovery after ischemia (4). We hypothesized, that Ephx1−/−Ephx2−/− hearts, which have a more profound reduction in EET hydrolysis to DHETs, might have an additional improvement of postischemic functional recovery. Baseline cardiac parameters were similar in all four genotypes (Table S5). After 20 min of ischemia, recovery of left ventricular developed pressure (LVDP) at 40 min of reperfusion (R40) in WT hearts was 31% (Fig. 7). Disruption of EPHX1 alone had no effect on LVDP recovery (38%). Disruption of EPHX2 alone resulted in significantly improved LVDP recovery (51%) as we have reported previously (4). Remarkably, LVDP recovery in Ephx1−/−Ephx2−/− hearts was 71%, which was significantly higher than both WT and Ephx2−/− hearts. Consistent with these data, rate-pressure product (RPP) was also significantly higher in Ephx1−/−Ephx2−/− hearts compared with both WT and Ephx2−/− hearts. Thus, reduced EET hydrolysis in Ephx1−/−Ephx2−/− hearts has important functional consequences.
Figure 7.
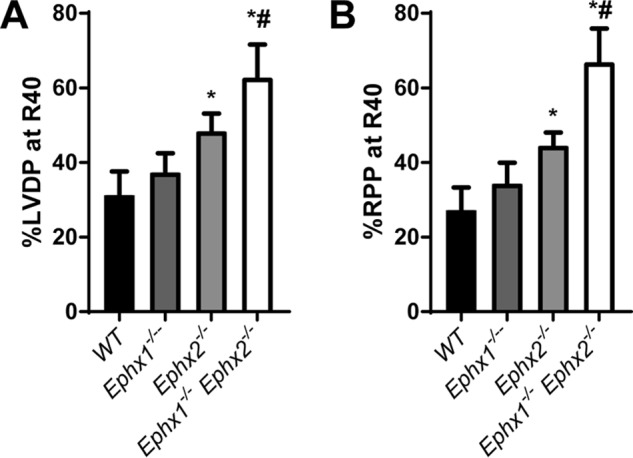
Role of EPHX1 and EPHX2 in in postischemic recovery of heart function. A and B, recovery of left ventricular developed pressure (LVDP) (A) and rate pressure product (RPP) (B) at 40 min of postischemic reperfusion (R40) as percent of baseline in WT, Ephx1−/−, Ephx2−/−, and Ephx1−/−Ephx2−/− mice. n = 8–10 per genotype group, *, p < 0.05 versus WT; #, p < 0.05 versus Ephx2−/−. Results are expressed as mean ± S.E.
Taken together, our data suggest a new paradigm for EET hydrolysis by epoxide hydrolases (Fig. 8). When EET formation rates are low (e.g. under basal conditions), EPHX1 can hydrolyze most of the EETs as they are formed through a coupled reaction with P450 enzymes; EPHX2 contributes minimally. However, when EET production rates are high (e.g. during postischemic reperfusion), EPHX1 capacity is overwhelmed and EPHX2 contributes significantly to EET hydrolysis. Thus, EPHX2 is an excellent EET scavenger; it readily clears EETs from the cytosol. By contrast, EPHX1 is a slow EET scavenger, but it contributes significantly to EET hydrolysis under basal conditions. The role of EPHX1 with regard to EET hydrolysis is unmasked in the absence of EPHX2.
Figure 8.
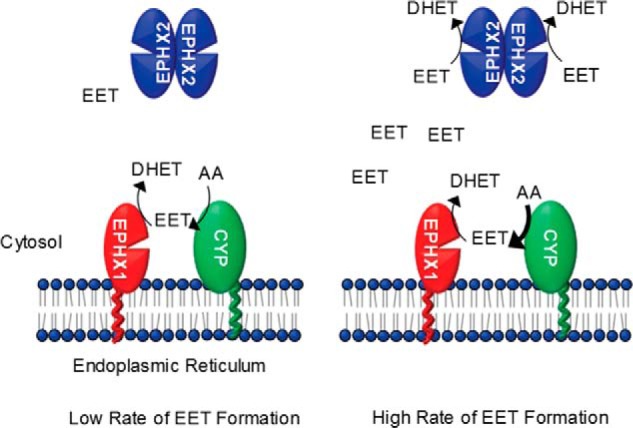
New paradigm for hydrolysis of EETs. Cytochrome P450 epoxygenases are localized to the cytoplasmic face of the endoplasmic reticulum adjacent to EPHX1. EPHX2 is predominantly cytoplasmic. When EET formation occurs at low rates (e.g. basal conditions), EPHX1 can hydrolyze most of the EETs as they are formed; EPHX2 contributes minimally. However, when EET production rates are high (e.g. postischemic reperfusion), EPHX1 capacity is overwhelmed and EPHX2 then contributes significantly to EET hydrolysis.
Discussion
In summary, this manuscript contains several important and novel findings regarding the functional role of epoxide hydrolases: 1) disruption of both EPHX1 and EPHX2 almost completely abolishes EET hydrolysis in mouse plasma; 2) in vitro kinetic assays demonstrate that, relative to EPHX2, EPHX1 is a poor EET scavenge; 3) EPHX1 plays a prominent role in EET hydrolysis when EET formation rates are low; 4) disruption of both EPHX1 and EPHX2 suppresses EET hydrolysis in mouse heart; and 5) Ephx1−/−Ephx2−/− hearts have significantly improved recovery of cardiac function after ischemia compared with both WT or Ephx2−/− hearts. Together, these findings dramatically alter our understanding of fatty acid epoxide hydrolysis and may have many important clinical and therapeutic implications.
The contribution of EPHX1 to EET hydrolysis is surprising in several respects. Our findings are in stark contrast to prior studies which suggest that EPHX1 is too slow to contribute significantly to EET hydrolysis and may only contribute to hydrolysis of a selective subset of fatty acid epoxides (7, 8). In vitro, EPHX2 shows higher catalytic activity than EPHX1 for most epoxides. These published in vitro studies poorly reflect the fact that EPHX1 significantly contributes to hydrolysis of a broad spectrum of fatty acid epoxides in vivo. EPHX2 likely maintains higher catalytic capability in vivo relative to EPHX1. Morisseau and co-workers (23) note that tissue concentrations of EPHX2 are 3 to 400 nm, whereas EET concentrations are in the low nm range. They conclude that, for EPHX2, substrate accessibility, not hydrolase activity, is the limiting factor for the conversion of epoxy fatty acids. Although EPHX1 may have lower catalytic capability than EPHX2, it likely has increased accessibility to epoxy fatty acids. For most epoxides, the role of EPHX1 only becomes apparent in Ephx1−/−Ephx2−/− mice or tissues. With the exception of 12,13-EpOME, EPHX1 significantly regulates every fatty acid epoxide examined. Ephx1−/−Ephx2−/− plasma contain low levels of both DiHOMEs. These data may suggest a minor role for additional hydrolases such EPHX3, which shows high catalytic activity toward 9,10-EpOME (7).
EPHX1 appears to act primarily as a “first-pass” hydrolase for fatty acid epoxides. It has little capacity to scavenge EETs out of the cytosol. Under normal physiologic conditions, our data suggest that P450s generate EETs at a fraction of their maximal capacity (∼5%). The reason that EPHX1 can significantly contribute to EET hydrolysis in vivo is that most epoxy fatty acids formed occur at low basal levels in most tissues, most of the time. Ischemia and other stimuli such as inflammation activate cytosolic phospholipase A2 which releases AA from membrane phospholipid stores. The free AA can then be rapidly metabolized by P450s to EETs. At these higher rates of EET formation, the limited capacity of EPHX1 is surpassed and significant EET-signal is produced; EPHX2 plays a predominate role under these stimulated conditions.
We propose that P450 epoxygenases and EPHX1 biosynthesize and hydrolyze EETs in a manner analogous to xenobiotics (24, 25). For xenobiotic metabolism, a coupled reaction prevents the release of highly reactive epoxide intermediates into membranes or cytosol. One alternative is that P450-derived epoxides may not be immediately hydrolyzed but rather remain associated with the ER membrane similar to other lipophilic molecules (26, 27). EPHX1, tethered to the ER, may effectively scavenge these EETs. It is possible that EPHX2, which is known to be “trapped” or bound to microsomes (Fig. 1) (14, 28), might also hydrolyze EETs off membranes in a similar fashion.
In vitro kinetic assays using recombinant proteins dramatically underestimated the potential of EPHX1 as a relevant hydrolase for a wide panel of fatty acid epoxides. The cytoplasmic enzyme EPHX2 excels at high throughput metabolism of exogenous EETs in solution. It should not be surprising that EPHX1 performs slowly under conditions that poorly reflect its normal cellular environment. EPHX1 may have much higher hydrolase activity in vivo for several reasons: 1) EPHX1/P450 binding might stimulate hydrolase activity (19), 2) EPHX1 might benefit from direct substrate handoff in a coupled reaction with P450s (24), 3) EPHX1 might have higher activity when embedded in ER membranes, and 4) EPHX1 may have an advantage in scavenging free EETs that remain associated with ER membranes. Previous findings obtained in highly dilute solubilized solutions may not translate well to the complex, concentrated, membrane-containing environments found in vivo. Our findings represent another cautionary tale of the perils of estimating metabolism of lipophilic molecules by membrane-bound enzymes that have been taken out of their native environment (26, 27, 29).
A coupled reaction between P450s and EPHX1 suggests tight regulation of basal EET concentrations; however, this reaction may also suggest the possibility of a concerted mechanism for producing bioactive diols. Although fatty acid epoxides are commonly thought of as the more active oxylipin, fatty acid diols do retain some selective bioactivity. For example, 11,12-DHET lacks anti-inflammatory or mitogenic effects, but is vasodilatory in some vascular beds (3, 5, 6) and attenuates cAMP production in forskolin-stimulated cells (30). The AA-derived diols have little effect on angiogenesis; however, the DHA-derived diol 19,20-DiHDPA is reported to promote retinal angiogenesis (31–33). Thus, EPHX1 may play a role in both limiting basal epoxide signaling and promoting physiologic responses through production of bioactive diols.
Our data in the various Ephx−/− mice and tissues shed light on regulation of EETs in cells that lack either EPHX1 or EPHX2. EPHX1 can hydrolyze a significant proportion of EETs in EPHX2-deficient cells; likewise, EPHX2 can hydrolyze a significant proportion of EETs in cells with low EPHX1 expression. Ephx2 is abundantly expressed in several tissues including liver, ovary, kidney, and intestinal tissues, but is expressed at much lower levels in the lung, brain, skin, and testes (Fig. S3A). In contrast, Ephx1 is abundant in liver, lung, ovary, and testes, but is expressed at lower levels in other tissues including the heart (Fig. S3B). Moreover, within a given tissue, expression of EPHX1 and EPHX2 varies by cell type. For example, in the heart, EPHX2 is abundant in cardiomyocytes and endothelial cells, but expressed at low levels in smooth muscle cells and fibroblasts; in the liver, EPHX2 is abundant in hepatocytes, but expressed at low levels in endothelial cells (Fig. S3C). It is remarkable that despite its low level of expression in the heart, EPHX1 contributes significantly to EET hydrolysis in this tissue under basal conditions. EPHX1 may also play a more prominent role in hydrolysis of fatty acid epoxides in tissues or cells where EPHX2 expression is low or absent. EPHX2 also contains a functional lipid phosphatase domain (34). Although endogenous substrates for this domain have not been identified, its presence suggests that EPHX2 may have important physiologic roles aside from fatty acid epoxide hydrolysis.
There is substantial variation in the human EPHX1 gene. There are over 140 single nucleotide polymorphisms (SNPs) listed in the National Cancer Institute dbSNP database (https://www.ncbi.nlm.nih.gov/snp/).3 The most frequently studied SNPs are Y113H (rs1051740) and H139R (rs2234922), both of which are believed to reduce EPHX1 activity (18). EPHX1 has been widely studied for its role in bioactivation or detoxification of carcinogens (35) and associations of EPHX1 polymorphisms with cancer have been found (18). EETs are mitogenic, anti-apoptotic, and pro-angiogenic (3) and promote tumor growth and metastasis (32). Thus, EPHX1, via its regulation of EET-mediated effects, may also modulate tumor growth independent of its role in carcinogen metabolism. Although EPHX1 polymorphisms have not yet been examined with respect to cardiovascular disease outcomes, our findings suggest a role for EPHX1 in diseases such as atherosclerosis, sepsis, stroke, and myocardial infarction (MI), independent of their role in xenobiotic metabolism.
Our findings may also have important clinical implications. Preclinical studies have demonstrated that EPHX2 disruption or inhibition improves postischemic cardiac functional recovery and reduces MI (4, 36–38). EPHX2 inhibitors, which have completed phase I clinical trials (39), have been proposed as treatments for patients suffering MI (40, 41). Our data suggest that dual inhibition of EPHX1 and EPHX2 might offer additional benefit to MI patients. EPHX1 appears to play a particularly important role in omega-3 fatty acid epoxide hydrolysis. Although less well studied, EpETEs and EpDPEs appear to have potent EET-like properties (42). In addition, 17,18-EpETE and 19,20-EpDPE have potent antiarrhythmic effects in the heart (43). Given the important role of EPHX1 in xenobiotic metabolism, long term use of EPHX1 inhibitors may be unwise; however, our data suggest a possible benefit for dual inhibition of EPHX1 and EPHX2 in acute treatment of cardiac arrhythmia and sudden cardiac death.
Our data suggest a new paradigm for hydrolysis of EETs; however, several important questions remain. Although EPHX1 and EPHX2 together account for nearly all EET hydrolysis observed in mouse plasma in vivo, Ephx1−/−Ephx2−/− mice exhibit appreciable hydrolysis of 9,10- and 11,12-EpOME (∼20% that of WT mice). Moreover, Ephx1−/−Ephx2−/− hearts produce significant amounts of DHETs during reperfusion. EPHX3 has been shown to have high catalytic activity against EETs and 9,10-EpOME in vitro (7); however, Ephx3−/− mice exhibit normal fatty acid epoxide hydrolase activity in multiple tissues, both in vitro and in vivo (10). Whether the residual fatty acid epoxide hydrolysis in Ephx1−/−Ephx2−/− mice is because of EPHX3 remains unknown. In addition, although Ephx1−/−Ephx2−/− mice have dramatically reduced plasma levels of fatty acid diols, this does not always correspond to increases in plasma levels of the corresponding fatty acid epoxides. In this regard, EETs may be shunted toward other metabolic pathways in cells lacking EPHX2 activity. Inhibition of EPHX2 increases EET esterification into plasma membranes and formation of chain-shortened epoxides (44–46). These chain-shortened epoxides can maintain potent biological activities (47), which may explain why Ephx2−/− and Ephx1−/−Ephx2−/− hearts show improved functional recovery despite minimal increases in EETs.
In conclusion, our findings represent a significant advance in the eicosanoid field. The discovery that EPHX1 contributes significantly to EET hydrolysis in vivo explains the enigmatic residual hydrolysis observed after EPHX2 genetic disruption or pharmacological inhibition, and represents a new paradigm for the role of EPHXs in EET metabolism. The findings suggest new avenues of research regarding the role of EPHX1 polymorphisms in human disease, and offer a novel therapeutic target for the treatment of ischemic heart disease.
Experimental procedures
Animals
Mice with constitutive global disruption of Ephx1 (Ephx1−/−) and Ephx2 (Ephx2−/−) were generated as described previously (11, 12) and generously provided by Dr. Frank Gonzalez (NCI, National Institutes of Health). Mice were back-crossed for more than 10 generations onto a pure C57BL/6 background. Ephx1+/−Ephx2+/− mice were in-crossed to produce Ephx1+/+Ephx2+/+ (WT), Ephx1−/−Ephx2+/+ (Ephx1−/−), Ephx1+/+Ephx2−/− (Ephx2−/−), and Ephx1−/−Ephx2−/− mice for study. Relatively equal proportions of male and female mice, age 10–20 weeks, were used in each study. Mice were maintained in cages with a 12:12 h light-dark cycle and free access to NIH 31 chow (Envigo, Madison, WI) and water. All procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the NIEHS Animal Care and Use Committee.
Langendorff isolated perfusion
Mice were anesthetized with pentobarbital, hearts were removed, cannulated, and perfused in the Langendorff mode as described previously (4, 22). Hearts were perfused with modified Krebs-Henseleit buffer (120 mm NaCl, 25 mm NaHCO3, 4.7 mm KCl, 1.2 mm KH2PO4, 1.20 mm MgSO4, 11 mm glucose, and 1.8 mm CaCl2) bubbled with 95% air and 5% CO2. A water-filled balloon connected to a pressure transducer was inserted into the left ventricle to monitor cardiac function. Hearts were equilibrated for 40 min and then subjected to 20 min of global, no-flow ischemia, followed by 40 min of reperfusion. Recovery of contractile function was measured as left ventricular developed pressure at the end of reperfusion expressed as a percentage of preischemic LVDP. Rate pressure product was calculated as LVDP × heart rate. n = 8–10 per group with roughly equal proportions of males and females in each genotype group. In separate experiments to collect heart perfusates for LC-MS/MS analysis, hearts were cannulated and perfused as above but without balloon insertion. Heart perfusates were collected during the last 20 min of equilibration (Baseline) and during the first 20 min of reperfusion into 50-ml conical tubes containing 5 μl of 10 mm trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) (kindly provided by Bruce Hammock, UC Davis) on dry ice and stored at −80 °C prior to extraction and analysis. n = 6–7 per group with roughly equal proportion of males and females in each genotype group.
Cell fractionation and protein immunoblotting
All chemicals were from Sigma unless otherwise noted. Cell fractionation was performed similarly to previously published methods (48). 100 mg liver tissue was added to 900 μl ice-cold lysis buffer (0.25 m sucrose, 10 mm Tris, pH 7.5, 0.2 μg/ml leupeptin, 0.04 units/ml aprotinin, 0.25 μm PMSF) and homogenized using a TissueLyser II (single stainless steel beads, 4 °C, 10 min at 30 Hz). The homogenate was centrifuged three times at 9000 × g for 10 min. The 9000 × g supernatant, which contained both cytosol and microsomes, was considered the S9 fraction. The 9000 × g supernatant was centrifuged at 100,000 × g for 90 min, washed with 0.15 m KCl, and centrifuged again at 100,000 × g. Microsomal pellets were resuspended in 50 mm Tris, pH 7.5, containing 1 mm DTT, 1 mm EDTA, and 20% glycerol. S9 fraction and microsomal protein concentrations were determined by BCA Assay (Bio-Rad).
20 μg of microsomes or S9 fractions were separated by 10% SDS-PAGE and transferred to nitrocellulose. Membranes were blocked overnight at 4 °C in PBS containing 0.1% Tween 20 and 5% nonfat milk (PBS-T/NFM). Membranes were probed with antibodies to EPHX1 (sc-135984 at 1:200) (Santa Cruz Biotechnology, Dallas, TX) or EPHX2 (sc-25797 at 1:1000) (Santa Cruz Biotechnology) in PBS-T/NFM for 2 h at room temperature. Blots were stripped and reprobed with β-actin (clone AC-74 at 1:5000) (Sigma). Staining was detected using HRP-conjugated secondary antibodies from Calbiochem (1:5000 dilution) and enhanced chemiluminescence.
mRNA analysis
mRNA was isolated from liver and heart tissues using RNeasy Mini Kits from Qiagen (Valencia, CA) and converted to cDNA using High Capacity cDNA Reverse Transcription Kit from Life Technologies (Carlsbad, CA). Ephx1 (Mm00468752_m1), Ephx2 (#Mm00514706_m1), Ephx4 (#Mm01203346_m1), and Gapdh (Mm99999915_g1) were detected using TaqMan probes according to manufacturer's instructions. Ephx3, Cyp2c, and Cyp2j expression was detected by SYBR Green using established methods (7, 49). n = 5–8 per group with roughly equal proportions of male and female livers and hearts.
EET hydrolysis assay
200 μg liver lysates were mixed with ice-cold solutions containing 0, 1, 10, 100, or 1000 nm 11,12-EET (Cayman Chemical, Detroit, MI) in a total of 100 μl PBS containing 0.1% BSA. Samples were incubated at 37 °C for 5 min before reactions were stopped by the addition of 1 ml ice-cold PBS. 10 μl of internal standard (30 ng each of PGE2-d4, 11,12-EET-d11, and 11,12-DHET-d11) (Cayman) was then added to each sample and lipids were extracted with 2 ml ethyl acetate, placed into tubes containing 6 μl of 30% glycerol in methanol, dried under vacuum centrifugation, covered with argon gas, and stored at −80 °C. n = 4 per group; liver lysates were prepared from 2 males and 2 females for each genotype.
AA metabolism assay
50 μg liver microsomes were added to a reaction buffer (50 mm Tris-Cl, pH 7.4, 150 mm KCl, 10 mm MgCl2) containing 10 μm arachidonic acid (Cayman Chemical) and 0, 0.02, 0.2, 2, 20, or 200 μm NADPH (Sigma). Samples were incubated for 10 min at 37 °C. A control reaction (10 μm arachidonic acid, 0 μm NADPH) was kept on ice for 10 mins. Rates of lipid formation were corrected for metabolites found in this control reaction. Reactions were stopped by the addition of 2 ml ethyl acetate and lipids were extracted as described above. n = 4; microsomes were prepared from livers of 2 male and 2 female mice of each genotype.
Mouse plasma
200 μl mouse plasma was spiked with internal standard, mixed with 1 volume of 0.1% acetic acid in 5% methanol, and extracted with 3 ml ethyl acetate. Ethyl acetate was passed through Maestro A columns (Tecan, Mannedorf, Switzerland) under gravity flow into glass tubes containing 6 μl of 30% glycerol in methanol. Columns were washed with 1 ml acetonitrile (Sigma) and samples were dried and stored as above.
Cardiac perfusates
Samples were spiked with internal standards, mixed with 0.1 volume of 1% acetic acid in 50% methanol, and extracted by serial passage through Oasis HLB C18 3-ml columns (Waters, Milford, MA). Columns were washed twice with 3 ml 0.1% acetic acid in 5% methanol and eluted with methanol into glass tubes containing 6 μl of 30% glycerol in methanol. Samples were dried and stored as above.
Liquid chromatography tandem mass spectroscopy
All samples were reconstituted in 50 μl of 30% ethanol. Online liquid chromatography was performed with an Agilent 1200 Series capillary HPLC (Agilent Technologies, Santa Clara, CA). Separations were achieved using a Halo C18 column (2.7 μm, 100 × 2.1 mm) (MAC-MOD Analytical, Chadds Ford, PA), which was held at 50 °C. Mobile phase A was 85:15:0.1 water/acetonitrile/acetic acid. Mobile phase B was 70:30:0.1 acetonitrile/methanol/acetic acid. Flow rate was 400 μl/min; Gradient elution was used. Mobile phase percentage B and flow rate were varied as follows: 20% B at 0 min, ramp from 0 to 5 min to 40% B, ramp from 5 to 7 min to 55% B, ramp from 7 to 13 min to 64% B. From 13 to 19 min the column was flushed with 100% B at a flow rate of 550 μl/min. Samples were solvated in 50 μl of 30% ethanol. The injection volume was 10 μl. Samples were analyzed in duplicate. Analyses were performed on an MDS Sciex API 3000 equipped with a TurboIonSpray source (Applied Biosystems). Turbo desolvation gas was heated to 425 °C at a flow rate of 6 liters/min. Negative ion electrospray ionization tandem mass spectrometry with multiple reaction monitoring was used for detection. Analyte quantification was performed using Analyst 1.5.1 software (AB Sciex, Ontario, Canada). Relative response ratios of analytes and respective internal standards were compared with a standard curve of response ratios for each analyte. Lipid standards were stored in 100% ethanol under argon and used within 1 year of purchase from Cayman Chemical.
FRET-based FACS analysis of CYP-EPHX1 interaction
CYP and EPHX1 proteins were N-terminally fused to YFP and CFP, respectively. The resulting ER luminal localization of these fluorescent proteins results in a positive FRET signal if the two attached proteins interact on the other side of the membrane. Details and validation of this procedure, called FRET analysis of membrane protein interaction in the endoplasmic reticulum (FAMPIR), have been described previously (50). The negative control pair was an ER membrane–anchored YFP with CYP2J5-CFP. The positive control pair was CYP2J5-YFP with cytochrome P450 reductase–CFP. In brief, transient transfection of HEK-293 cells with a vector affording the expression of the two above fusion proteins was carried out using jetPEI® (Polyplus Transfection, Illkirch, France), according to the suppliers instructions. After 48 h, cells were trypsinized and subjected to FACS analyses on a LSR II Fortessa (BD Biosciences), using the sensitized emission method. The following filter settings were employed: YFP channel (488 nm, 530/30 filter, 505 LP), CFP channel (405 nm, 450/50 filter), FRET channel (405 nm 525/50 filter, 505 LP). Five × 104 cells were analyzed per sample. Bleed-through of the CFP and YFP signals into the FRET channel was compensated for by adjusting the respective recording parameters, using cells expressing only CFP or YFP chimeras. During analysis, the FRET intensity in the subpopulation displaying significant YFP and CFP expression was recorded and plotted against the intensity of the CFP fluorescence. Individual replicates were produced by separate transfection and FACS analyses for each data point. Typically, the different combinations of potential interaction partners were run in parallel.
Statistical analyses
Analysis of significance was determined by one-way analysis of variance (ANOVA) followed by post hoc t-tests using GraphPad Prism and Microsoft Excel software. For plasma analytes and biochemical assays, the only tests considered were as follows: WT versus Ephx1−/−, WT versus Ephx2−/−, WT versus Ephx1−/−Ephx2−/−, and Ephx2−/− versus Ephx1−/−Ephx2−/− mice. For cardiac perfusates, in addition to these comparisons, we conducted paired Student's t-tests to assess changes between baseline and postischemic analyte levels. p values less than 0.05 were considered significant.
Author contributions
M. L. E. and D. Z. conceptualization; M. L. E., A. G., J. P. G., F. B. L., A. C. O. L., J. A. B., and S. L. H. formal analysis; M. L. E., B. G. H., A. G., J. P. G., F. B. L., S. J. A., R. S., A. C. O. L., J. A. B., L. M. D., and S. L. H. investigation; M. L. E., J. P. G., F. B. L., R. S., A. C. O. L., and M. A. methodology; M. L. E. writing-original draft; M. L. E., A. G., M. A., and D. Z. writing-review and editing; M. A. and D. Z. supervision; M. A. and D. Z. funding acquisition; D. Z. resources.
Supplementary Material
Acknowledgments
We gratefully acknowledge Frank Gonzalez (NCI, National Institutes of Health) for providing EPHX1- and EPHX2-deficicient mice. We cordially thank Bruce Hammock and Art Spector for many insightful comments and recommendations.
This work was supported, in part, by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01 ES025034 to D. C. Z.) and by the Swiss National Foundation Grant PDFMP3_127330 (to M. A.). The authors declare that they have no conflicts of interest with the contents of this article.The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3 and Tables S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- AA
- arachidonic acid
- EETs
- epoxyeicosatrienoic acids
- DHETs
- dihydroxyeicosatrienoic acids
- ER
- endoplasmic reticulum
- CFP
- cyan fluorescent protein
- LVDP
- left ventricular developed pressure
- SNPs
- single nucleotide polymorphisms
- MI
- myocardial infarction.
References
- 1. Morisseau C., and Hammock B. D. (2013) Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 53, 37–58 10.1146/annurev-pharmtox-011112-140244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Imig J. D., and Hammock B. D. (2009) Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 8, 794–805 10.1038/nrd2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spector A. A., and Norris A. W. (2007) Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 292, C996–C1012 10.1152/ajpcell.00402.2006 [DOI] [PubMed] [Google Scholar]
- 4. Seubert J. M., Sinal C. J., Graves J., DeGraff L. M., Bradbury J. A., Lee C. R., Goralski K., Carey M. A., Luria A., Newman J. W., Hammock B. D., Falck J. R., Roberts H., Rockman H. A., Murphy E., and Zeldin D. C. (2006) Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ. Res. 99, 442–450 10.1161/01.RES.0000237390.92932.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larsen B. T., Miura H., Hatoum O. A., Campbell W. B., Hammock B. D., Zeldin D. C., Falck J. R., and Gutterman D. D. (2006) Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BK(Ca) channels: Implications for soluble epoxide hydrolase inhibition. Am. J. Physiol. Heart Circ. Physiol. 290, H491–H499 10.1152/ajpheart.00927.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Node K., Huo Y., Ruan X., Yang B., Spiecker M., Ley K., Zeldin D. C., and Liao J. K. (1999) Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285, 1276–1279 10.1126/science.285.5431.1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Decker M., Adamska M., Cronin A., Di Giallonardo F., Burgener J., Marowsky A., Falck J. R., Morisseau C., Hammock B. D., Gruzdev A., Zeldin D. C., and Arand M. (2012) EH3 (ABHD9): The first member of a new epoxide hydrolase family with high activity for fatty acid epoxides. J. Lipid Res. 53, 2038–2045 10.1194/jlr.M024448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marowsky A., Burgener J., Falck J. R., Fritschy J. M., and Arand M. (2009) Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience 163, 646–661 10.1016/j.neuroscience.2009.06.033 [DOI] [PubMed] [Google Scholar]
- 9. Marowsky A., Meyer I., Erismann-Ebner K., Pellegrini G., Mule N., and Arand M. (2017) Beyond detoxification: A role for mouse mEH in the hepatic metabolism of endogenous lipids. Arch. Toxicol. 91, 3571–3585 10.1007/s00204-017-2060-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoopes S. L., Gruzdev A., Edin M. L., Graves J. P., Bradbury J. A., Flake G. P., Lih F. B., DeGraff L. M., and Zeldin D. C. (2017) Generation and characterization of epoxide hydrolase 3 (EPHX3)-deficient mice. PLoS One 12, e0175348 10.1371/journal.pone.0175348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miyata M., Kudo G., Lee Y. H., Yang T. J., Gelboin H. V., Fernandez-Salguero P., Kimura S., and Gonzalez F. J. (1999) Targeted disruption of the microsomal epoxide hydrolase gene. Microsomal epoxide hydrolase is required for the carcinogenic activity of 7,12-dimethylbenz[a]anthracene. J. Biol. Chem. 274, 23963–23968 10.1074/jbc.274.34.23963 [DOI] [PubMed] [Google Scholar]
- 12. Sinal C. J., Miyata M., Tohkin M., Nagata K., Bend J. R., and Gonzalez F. J. (2000) Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J. Biol. Chem. 275, 40504–40510 10.1074/jbc.M008106200 [DOI] [PubMed] [Google Scholar]
- 13. Gill S. S., and Hammock B. D. (1980) Distribution and properties of a mammalian soluble epoxide hydrase. Biochem. Pharmacol. 29, 389–395 10.1016/0006-2952(80)90518-3 [DOI] [PubMed] [Google Scholar]
- 14. Gill S. S., and Hammock B. D. (1981) Epoxide hydrolase activity in the mitochondrial fraction of mouse liver. Nature 291, 167–168 10.1038/291167a0 [DOI] [PubMed] [Google Scholar]
- 15. Guenthner T. M., Hammock B. D., Vogel U., and Oesch F. (1981) Cytosolic and microsomal epoxide hydrolases are immunologically distinguishable from each other in the rat and mouse. J. Biol. Chem. 256, 3163–3166 [PubMed] [Google Scholar]
- 16. Morisseau C., Inceoglu B., Schmelzer K., Tsai H. J., Jinks S. L., Hegedus C. M., and Hammock B. D. (2010) Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J. Lipid Res. 51, 3481–3490 10.1194/jlr.M006007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ulu A., Harris T. R., Morisseau C., Miyabe C., Inoue H., Schuster G., Dong H., Iosif A. M., Liu J. Y., Weiss R. H., Chiamvimonvat N., Imig J. D., and Hammock B. D. (2013) Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J. Cardiovasc. Pharmacol. 62, 285–297 10.1097/FJC.0b013e318298e460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Václavíková R., Hughes D. J., and Souček P. (2015) Microsomal epoxide hydrolase 1 (EPHX1): Gene, structure, function, and role in human disease. Gene 571, 1–8 10.1016/j.gene.2015.07.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taura Ki K., Yamada H., Naito E., Ariyoshi N., Mori Ma M. A., and Oguri K. (2002) Activation of microsomal epoxide hydrolase by interaction with cytochromes P450: kinetic analysis of the association and substrate-specific activation of epoxide hydrolase function. Arch. Biochem. Biophys. 402, 275–280 10.1016/S0003-9861(02)00079-6 [DOI] [PubMed] [Google Scholar]
- 20. Kaminsky L. S., Kennedy M. W., and Guengerich F. P. (1981) Differences in the functional interaction of two purified cytochrome P-450 isozymes with epoxide hydrolase. J. Biol. Chem. 256, 6359–6362 [PubMed] [Google Scholar]
- 21. Kohn M. C., and Melnick R. L. (2001) Physiological modeling of butadiene disposition in mice and rats. Chem. Biol. Interact. 135–136, 285–301 [DOI] [PubMed] [Google Scholar]
- 22. Edin M. L., Wang Z., Bradbury J. A., Graves J. P., Lih F. B., DeGraff L. M., Foley J. F., Torphy R., Ronnekleiv O. K., Tomer K. B., Lee C. R., and Zeldin D. C. (2011) Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 25, 3436–3447 10.1096/fj.11-188300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morisseau C., Wecksler A. T., Deng C., Dong H., Yang J., Lee K. S., Kodani S. D., and Hammock B. D. (2014) Effect of soluble epoxide hydrolase polymorphism on substrate and inhibitor selectivity and dimer formation. J. Lipid Res. 55, 1131–1138 10.1194/jlr.M049718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gautier J. C., Urban P., Beaune P., and Pompon D. (1996) Simulation of human benzo[a]pyrene metabolism deduced from the analysis of individual kinetic steps in recombinant yeast. Chem. Res. Toxicol. 9, 418–425 10.1021/tx9500944 [DOI] [PubMed] [Google Scholar]
- 25. Carlson G. P. (2010) Metabolism and toxicity of styrene in microsomal epoxide hydrolase-deficient mice. J. Toxicol. Environ. Health Part A 73, 1689–1699 10.1080/15287394.2010.516240 [DOI] [PubMed] [Google Scholar]
- 26. Glatt H., and Oesch F. (1977) Inactivation of electrophilic metabolites by glutathione S-transferases and limitation of the system due to subcellular localization. Arch. Toxicol. 39, 87–96 [DOI] [PubMed] [Google Scholar]
- 27. Oesch F. (1987) Significance of various enzymes in the control of reactive metabolites. Arch. Toxicol. 60, 174–178 10.1007/BF00296975 [DOI] [PubMed] [Google Scholar]
- 28. Gill S. S., and Hammock B. D. (1981) Epoxide hydrolase activity in the mitochondrial and submitochondrial fractions of mouse liver. Biochem. Pharmacol. 30, 2111–2120 10.1016/0006-2952(81)90230-6 [DOI] [PubMed] [Google Scholar]
- 29. Dansette P. M., Rosi J., Bertho G., and Mansuy D. (2012) Cytochromes P450 catalyze both steps of the major pathway of clopidogrel bioactivation, whereas paraoxonase catalyzes the formation of a minor thiol metabolite isomer. Chem. Res. Toxicol. 25, 348–356 10.1021/tx2004085 [DOI] [PubMed] [Google Scholar]
- 30. Abukhashim M., Wiebe G. J., and Seubert J. M. (2011) Regulation of forskolin-induced cAMP production by cytochrome P450 epoxygenase metabolites of arachidonic acid in HEK293 cells. Cell Biol. Toxicol. 27, 321–332 10.1007/s10565-011-9190-x [DOI] [PubMed] [Google Scholar]
- 31. Hu J., Popp R., Frömel T., Ehling M., Awwad K., Adams R. H., Hammes H. P., and Fleming I. (2014) Muller glia cells regulate Notch signaling and retinal angiogenesis via the generation of 19,20-dihydroxydocosapentaenoic acid. J. Exp. Med. 211, 281–295 10.1084/jem.20131494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Panigrahy D., Edin M. L., Lee C. R., Huang S., Bielenberg D. R., Butterfield C. E., Barnés C. M., Mammoto A., Mammoto T., Luria A., Benny O., Chaponis D. M., Dudley A. C., Greene E. R., Vergilio J. A., et al. (2012) Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Invest. 122, 178–191 10.1172/JCI58128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ding Y., Frömel T., Popp R., Falck J. R., Schunck W. H., and Fleming I. (2014) The biological actions of 11,12-epoxyeicosatrienoic acid in endothelial cells are specific to the R/S-enantiomer and require the G(s) protein. J. Pharmacol. Exp. Ther. 350, 14–21 10.1124/jpet.114.214254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Newman J. W., Morisseau C., Harris T. R., and Hammock B. D. (2003) The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc. Natl. Acad. Sci. U.S.A. 100, 1558–1563 10.1073/pnas.0437724100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Decker M., Arand M., and Cronin A. (2009) Mammalian epoxide hydrolases in xenobiotic metabolism and signalling. Arch. Toxicol. 83, 297–318 10.1007/s00204-009-0416-0 [DOI] [PubMed] [Google Scholar]
- 36. Chaudhary K. R., Abukhashim M., Hwang S. H., Hammock B. D., and Seubert J. M. (2010) Inhibition of soluble epoxide hydrolase by trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is protective against ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. 55, 67–73 10.1097/FJC.0b013e3181c37d69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oni-Orisan A., Alsaleh N., Lee C. R., and Seubert J. M. (2014) Epoxyeicosatrienoic acids and cardioprotection: The road to translation. J. Mol. Cell. Cardiol. 74, 199–208 10.1016/j.yjmcc.2014.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jamieson K. L., Endo T., Darwesh A. M., Samokhvalov V., and Seubert J. M. (2017) Cytochrome P450-derived eicosanoids and heart function. Pharmacol. Ther. 179, 47–83 10.1016/j.pharmthera.2017.05.005 [DOI] [PubMed] [Google Scholar]
- 39. Chen D., Whitcomb R., MacIntyre E., Tran V., Do Z. N., Sabry J., Patel D. V., Anandan S. K., Gless R., and Webb H. K. (2012) Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol. 52, 319–328 10.1177/0091270010397049 [DOI] [PubMed] [Google Scholar]
- 40. Seubert J. M., Zeldin D. C., Nithipatikom K., and Gross G. J. (2007) Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat. 82, 50–59 10.1016/j.prostaglandins.2006.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nithipatikom K., and Gross G. J. (2010) Review article: epoxyeicosatrienoic acids: novel mediators of cardioprotection. J. Cardiovasc. Pharmacol. Ther. 15, 112–119 10.1177/1074248409358408 [DOI] [PubMed] [Google Scholar]
- 42. Konkel A., and Schunck W. H. (2011) Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim. Biophys. Acta 1814, 210–222 10.1016/j.bbapap.2010.09.009 [DOI] [PubMed] [Google Scholar]
- 43. Westphal C., Konkel A., and Schunck W. H. (2011) CYP-eicosanoids—a new link between omega-3 fatty acids and cardiac disease? Prostaglandins Other Lipid Mediat. 96, 99–108 10.1016/j.prostaglandins.2011.09.001 [DOI] [PubMed] [Google Scholar]
- 44. Weintraub N. L., Fang X., Kaduce T. L., VanRollins M., Chatterjee P., and Spector A. A. (1999) Epoxide hydrolases regulate epoxyeicosatrienoic acid incorporation into coronary endothelial phospholipids. Am. J. Physiol. 277, H2098–H2108 [DOI] [PubMed] [Google Scholar]
- 45. Fang X., Kaduce T. L., VanRollins M., Weintraub N. L., and Spector A. A. (2000) Conversion of epoxyeicosatrienoic acids (EETs) to chain-shortened epoxy fatty acids by human skin fibroblasts. J. Lipid Res. 41, 66–74 [PubMed] [Google Scholar]
- 46. Fang X., Kaduce T. L., Weintraub N. L., Harmon S., Teesch L. M., Morisseau C., Thompson D. A., Hammock B. D., and Spector A. A. (2001) Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J. Biol. Chem. 276, 14867–14874 10.1074/jbc.M011761200 [DOI] [PubMed] [Google Scholar]
- 47. Fang X., Weintraub N. L., Oltman C. L., Stoll L. L., Kaduce T. L., Harmon S., Dellsperger K. C., Morisseau C., Hammock B. D., and Spector A. A. (2002) Human coronary endothelial cells convert 14,15-EET to a biologically active chain-shortened epoxide. Am. J. Physiol. Heart Circ. Physiol. 283, H2306–H2314 10.1152/ajpheart.00448.2002 [DOI] [PubMed] [Google Scholar]
- 48. Graves J. P., Edin M. L., Bradbury J. A., Gruzdev A., Cheng J., Lih F. B., Masinde T. A., Qu W., Clayton N. P., Morrison J. P., Tomer K. B., and Zeldin D. C. (2013) Characterization of four new mouse cytochrome P450 enzymes of the CYP2J subfamily. Drug Metab. Dispos. 41, 763–773 10.1124/dmd.112.049429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Graves J. P., Gruzdev A., Bradbury J. A., DeGraff L. M., Li H., House J. S., Hoopes S. L., Edin M. L., and Zeldin D. C. (2015) Quantitative polymerase chain reaction analysis of the mouse Cyp2j subfamily: Tissue distribution and regulation. Drug Metab. Dispos. 43, 1169–1180 10.1124/dmd.115.064139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Orjuela Leon A. C., Marwosky A., and Arand M. (2017) Evidence for a complex formation between CYP2J5 and mEH in living cells by FRET analysis of membrane protein interaction in the endoplasmic reticulum (FAMPIR). Arch. Toxicol. 91, 3561–3570 10.1007/s00204-017-2072-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.