

Summary
Cryptococcus neoformans is an opportunistic fungal pathogen that causes life-threatening meningitis most commonly in populations with impaired immunity. Here we resolved the transcriptome of the human brain endothelium challenged with C. neoformans to establish whether C. neoformans invades the CNS by co-opting particular signaling pathways as a means to promote its own entry. Among the 5 major pathways targeted by C. neoformans, the EPH-EphrinA1 (EphA2) tyrosine kinase receptor-signaling pathway was examined further. Silencing the EphA2 receptor transcript in a human brain endothelial cell line or blocking EphA2 activity with an antibody or chemical inhibitor prevented transmigration of C. neoformans in an in vitro model of the BBB. In contrast, treating brain endothelial cells with an EphA2 chemical agonist or an EphA2 ligand promoted greater migration of fungal cells across the BBB. C. neoformans activated the EPH-tyrosine kinase pathway through a CD44-dependent phosphorylation of EphA2, promoting clustering and internalization of EphA2 receptors. Moreover, HEK293T cells expressing EphA2 revealed an association between EphA2 and C. neoformans that boosted internalization of C. neoformans. Collectively the results suggest that C. neoformans promotes EphA2 activity via CD44 and this in turn creates a permeable barrier that facilitates the migration of C. neoformans across the BBB.
Keywords: Cryptococcus neoformans, EphA2 receptor tyrosine kinase, blood-brain barrier, cytoskeleton re-modeling, transcytosis of brain endothelial cells
Introduction
Infection of the central nervous system (CNS) causes severe morbidity and often high mortality.[1] Circulating eukaryotic pathogens use a complex variety of mechanisms to evade host immunity, traverse the blood-brain barrier (BBB) and ultimately invade the CNS.[1] Debilitating symptoms of CNS fungal infections can be difficult to treat partly because of the limited repertoire of non-toxic antifungal drugs. Unfortunately, even after effective treatment neurological sequelae can remain. It’s believed that the altered and/or damaged state of the BBB along with changes in homeostatic interactions between the BBB, astrocytes and neurons (i.e. the neurovascular unit) may contribute to long-term cognitive deficiencies.[2] In a recent study, fifteen autopsy cases of men and women with neuropathologically and microbiologically confirmed fungal infections of the brain found that compared to bacterial meningitis and septic encephalitis, fungal encephalitis was characterized by a strong microglial activation and astrocyte proliferation, more neuronal damage and less endogenous repair.[3] These findings indicated severe damage to the brain. Unlike in other infectious diseases of the CNS, where a co-existence of damage and repair was observed, fungal encephalitis was characterized by axonal injury, astrocytosis, microglial activation and minimal neuronal regeneration.[3] The notion that fungal brain infections can lead to significant neuronal damage was further supported by a recent set of studies suggesting that Alzheimer’s disease might be linked to fungal infection in the brain.[4,5] Studies suggested that amyloid-beta, the component of the amyloid plaques found in brains of Alzheimer’s patients, may function as an antimicrobial peptide that responds to the presence of the pathogen by forming and concentrating around the pathogen.[4–6]
Cryptococcus neoformans is the leading cause of fungal meningitis and accounts for 15% of AIDS-related deaths worldwide.[7] The global incidence of cryptococcal meningitis (CM) has been estimated at 223,100 resulting in 181,100 annual fatalities with approximately 73% of all cases occurring in sub-Saharan Africa.[7] CM continues to cause significant morbidity and mortality in the United States. Of the 30,840 hospitalizations attributed to CM between 1997 and 2009, approximately 3,440 deaths were reported.[8] Of all the CM cases, 21.6% occurred among HIV-uninfected patients and although a steady decline in the HIV-infected deaths was observed, the persistent burden of CM among HIV-uninfected patients is concerning.[8] Indeed without rapid intervention, CM is universally fatal regardless of the immune status of the host.
Several studies have shown that C. neoformans can move freely within the bloodstream, lodge within the lumen of the capillaries and cross the BBB directly via a transcellular mechanism.[9–14] Here cryptococci adhere to- and are internalized by the brain endothelium from the luminal (apical) side. Subsequently, fungal cells transmigrate through the endothelial cytoplasm, exit on the abluminal (basolateral) side of the BBB and invade the brain parenchyma. This is an extraordinary achievement given that a crucial function of the BBB is to protect the brain from harmful agents. Cryptococci can breach the endothelium through an active process via protein-mediated transcytosis events that require C. neoformans viability, several fungal and host gene products including a metalloprotease (Mpr1), urease, CD44 and cytoskeleton remodeling of brain endothelial cells.[10,11,15–18] More recent evidence suggests that C. neoformans can also breach the BBB through a stealth-like mechanism by co-opting monocytes.[19,20]
Despite the growing knowledge about fungal gene products that play a role in the trans-cellular crossing of the BBB, the identity and details of key signaling pathways in the brain endothelium that mediate the transcellular movement of cryptococci into the CNS are just beginning to be unraveled. In C. neoformans, capsule-bound hyaluronic acid serves as a ligand for the CD44 host receptor.[21–23] Knockdown of CD44 in human brain microvascular endothelial cells significantly reduced the adherence of C. neoformans demonstrating that CD44 acts as a receptor for hyaluronic acid in C. neoformans.[21,22] In addition, mice deficient in CD44 (CD44 knockout mice) showed improved survival and less brain fungal burden further supporting a role for CD44 in the attachment of cryptococci to the BBB.[22]
Invasion of C. neoformans into the brain endothelium requires the re-organization of the actin cytoskeleton.[9–11] Studies involving scanning electron microscopy have shown that following binding of C. neoformans, human brain microvascular endothelial cells produce microvilli that envelop and internalize C. neoformans by a zipper-like mechanism.[10] The rearrangement of actin filaments plays a crucial role during internalization since this produces the force required to generate the microvilli that engulf C. neoformans. Similar mechanisms have been shown to mediate the internalization of other pathogens.[24,25] The remodeling of actin filaments is mediated by the small GTPases – RhoA, Rac1 and Cdc42, many of which have been shown to play a role in the internalization of C. neoformans and other pathogens. [16,25]
Recent studies have demonstrated that some pathogens such as Chlamydia trachomatis, hepatitis C virus, KSHV, and Plasmodium sporozoites invade host cells by engaging EphA2, a tyrosine kinase receptor (TKR).[26–29] EphA2 belongs to the Ephrin (EPH) family of TKR and along with their membrane-tethered ligands (knows as ephrins), they make up the largest TKR subfamily.[27] The EphA2 receptor binds to GPI-linked ephrinA1 ligand and mediates diverse cellular activities including cytoskeleton re-modeling, endothelial cell barrier integrity, cell adhesion, motility and is also implicated in neovascularization and oncogenesis.[27,30,31] The binding of ephrinA1 to EphA2 induces dimerization and phosphorylation of EphA2 triggering signaling events mediated by PI3K, MAPK, Src kinases, Rac1 and Rho GTPases.[27] EphA2 is unique among other TKRs, in that ligand binding to EphA2 produces reverse signaling in cells where the ligand is bound to the cell membrane.[31] A crystal structure of the ligand-binding domain of EphA2 and ephrinA1 suggested that EphA2 undergoes only minor conformational changes upon ephrinA1 binding indicating the ephrinA1-binding pocket on EphA2 is formed prior to ligand binding, consistent with the “lock and key” model. [32,33] The extracellular portion of EphA2 consists of an N-terminal glycosylated ligand-binding domain, a region rich in cysteines and two fibronectin type III repeats. The cytosolic portion of EphA2 mediates protein-protein interactions through its SAM motif, a PDZ binding motif, and its kinase domain.[31]
In this study we characterized the transcriptome of human brain microvascular endothelial cells (referred to as hCMEC) challenged with C. neoformans in an in vitro model of the BBB. We mapped the transcriptome to known canonical signaling pathways according to the ratio of differentially expressed transcripts to the total number of genes attributed to each pathway. We identified the EPH-EphrinA1 (EphA2) tyrosine kinase receptor-signaling pathway and found that the EphA2 receptor mediated the migration of C. neoformans across the BBB in a CD44-dependent manner. Silencing the EphA2 transcript or inhibiting EphA2 activity with an antibody or an inhibitor (dasatinib) prevented C. neoformans from crossing the BBB while activation of EphA2 with the ephrinA1 ligand or an agonist (doxasozin) enhanced crossing of C. neoformans. The EphA2 receptor was phosphorylated during C. neoformans infection but phosphorylation was prevented by dasatinib, consistent with less cryptococci crossing brain endothelial cells when treated with dasatinib. Localization studies of C. neoformans and EphA2 in human brain endothelial cells, live-cell recording of HEK293T cells expressing EphA2 and protection assays demonstrated a clear association between cryptococci and EphA2 consistent with a role for EphA2 during internalization of C. neoformans. Collectively, the data suggest that C. neoformans engages the EphA2 receptor in order to cross the BBB.
Material and Methods
Brain microvascular endothelial cells and culture conditions for RNA sequencing analysis
A human brain capillary endothelial cell line (labeled as, hCMEC/D3) was obtained from Dr. Weksler (Cornell University) who developed and characterized this cell line as a model for the human blood-brain barrier [34,35]. hCMEC/D3 cells were maintained in a 25cm2 flask as previously described.[9,34] Briefly, 75cm2 tissue culture treated flasks were coated with rat tail collagen type I (Corning, Corning, NY), at a concentration of 0.15 mg/mL in 0.05M acetic acid for 30 minutes, before removal of coating solution, and washing with PBS. Confluent cultures of hCMEC/D3 cells were washed with PBS, then trypsinized for approximately 2–3 minutes at 37°C, or until visible detachment was observed. hCMEC/D3 cells were seeded into the new flasks based on the growth area of the flasks, or 1 mL of trypsinized cells were used to inoculate a second 75cm2 flask. Subsequently, the hCMEC/D3 cells were cultivated for approximately 2 weeks at 37°C and 5% CO2, with medium changes occurring every 3–4 days. hCMEC/D3 cells were induced to differentiate through the use of decreasing concentrations of supplements in the medium with each subsequent medium change after reaching confluence, down to a minimum of 0.25x supplements.
Infection of hCMEC/D3 cells with C. neoformans
A strain of C. neoformans var. grubii (H99) was grown in yeast peptone dextrose (YPD) medium at 30°C with gentle agitation. Fungal cells were washed with PBS and finally resuspended in YPD, before counting with a haemocytometer and diluting to a multiplicity of infection (MOI) of 5 in YPD before starting the assay. The diluted C. neoformans was added to PBS washed hCMEC/D3 cells (or YPD alone for a control), and incubated for 3h at 37°C with 5% CO2 in EGM-2 with 100 units of potassium penicillin and 100μg of streptomycin sulfate per ml of culture medium. After 3h incubation, both C. neoformans and hCMEC/D3 were washed with PBS and harvested for RNA isolation.
Total RNA isolation, cDNA library preparation and RNA-sequencing
The total RNA were isolated using RNeasy Mini Kit (Qiagen). The hCMEC/D3 was harvested, and washed 3 times. Total RNA from hCMEC/D3 samples was isolated, then subjected to sample quality assessments utilizing both Nanodrop and Agilent Biolanalyzer analysis. Samples were not submitted for mRNA isolation until the sample RNA integrity number (RIN) score was greater than 7 and the 28S and 18S rRNA peaks were observed. The mRNA samples were used to prepare cDNA libraries then sequenced on the Illumina HiSeq platform (RNA-Seq). The transcriptomic profiles were retrieved from mapping the short reads to the reference genome. In our study, human genome database Hg19 from HUGO was used as the reference genome. Statistics were run on all samples, using the Bioconductor package edgeR. Differentially expressed genes were sorted by adjusted P-values (P <0.01 as significance cutoff). Each directory also contained a Multi-Dimensional Scaling (MDS) plot, showing the distance relationship (i.e. similarity) between each sample based on gene expression. As shown from the MDS plot the samples from the same treatment of H99 with hCMEC/D3 cells (HB1-HB3) were clustered and the control samples of hCMEC/D3 cells alone (B1-B3) also formed a cluster different from the treated ones, suggesting that our experimental conditions were reproducible (S2_Fig 2). The DNA Technology and Expression Analysis Core of the University of California, Davis provided the raw data from the Illumina reads of the cDNA of the transcripts. The Illumina raw data was subjected to RNAseq analysis by the Bioinformatics core facility, U.C. Davis Genome Center.
RNA-sequencing validation by quantitative PCR
The hCMEC/D3 cells were grown and infected with C. neoformans (H99) as described above protocol above but instead of the 75cm2 flask, the 25cm2 flask was used. hCMEC/D3 cells were harvested and the total RNA was isolated by using RNeasy kit as described above. Total RNA was used to generate cDNA with the Quantitect Reverse Transcription kit (Qiagen). Expression levels of twelve genes were analyzed using quantitative-PCR as a validation of RNA-seq (S1_Table 1). The cDNA samples were diluted to 20ng/μl. Gene-specific primers were used (S2_Table 2). All quantitative-PCR reactions were performed in 96 well plates using the ABI 7700 sequence detection system (Perkin-Elmer Applied Biosystems) and the amplifications were done using the SYBR Green PCR Master Mix (Applied Biosystems). The thermal cycling conditions were 50°C for 2 min followed by an initial denaturation step at 95°C for 10 min, 45 cycles at 95°C for 30s, 60°C for 30s and 72°C for 30s. The experiments were carried out in triplicate for each gene. The relative quantification in gene expression was determined using the 2-ΔΔCt method. Using this method, we obtained the fold-changes in gene expression normalized to an internal control gene (YWHAZ).
Western blot analysis, antibodies, inhibitors and recombinant proteins
hCMEC/D3 cells were washed with 1X phosphate buffered saline (PBS), disrupted with 1X lysis buffer (1XPBS-CM: 10XPBS, 9mM CaCl2, 4.9mM MgCl2, 3% w/v n-octyl-B-D-glucopyranoside). Supernatants were collected, centrifuged and protein concentrations determined by Bradford Assay. Supernatants were boiled at 100°C for 5 min with 2X Laemmli sample buffer (Bio-Rad). Approximately 50μg of total protein were subjected to 5%–10% sodium dodecyl sulfate polyacrylamide gel electrophoresis using Bio-Rad Mini-Protean Tetra system, followed by transfer onto immune-blot PVDF membrane (Bio-Rad). Non-specific binding sites were blocked by incubating with 5% non-fat dry milk freshly prepared in Tris-buffered saline with 0.5% Tween-20 (TBST) for 1 h at room temperature with shaking. The PVDF membranes were then incubated with primary antibodies (Rabbit polyclonal EphA2 IgG (Santa Cruz Technology) or phospho-EphA2 S897 (Cell Signaling) or histidine-6 (Neuromab, Inc) diluted at 1:1000 overnight at 4°C. After five washes (5 min each wash) with TBST, the membranes were incubated with secondary antibodies (Goat anti-rabbit (Abcam) or Goat anti-mouse (Abcam)) diluted 1:5000 conjugated to horseradish peroxidase for 1 h at room temperature. After washing five times (5 min each wash) in TBST, the signals were visualized using Supersignal West Pico enhanced chemiluminescence (ECL) detection reagents (Thermofisher Scientific). The polypeptide bands from the PVDF membrane were exposed on CL-XPosure Film (Thermo Scientific). Phalloidin stain, dasatinib, monoclonal antibody for EphA2 IgG and control IgG were purchased from Cell Signaling Technology. Doxazosin was purchased from Sigma Aldrich. Recombinant human ephrinA1 was purchased from Origene.
The in vitro blood-brain barrier (BBB) model and transcytosis assay
The in vitro static monolayer model of the human BBB consisted of a transwell apparatus with the lower chamber representing the abluminal (basolateral) side and upper chamber representing the luminal (apical) side separated by a porous membrane (8μM, Corning). The hCMEC/D3 cells used for the transcytosis assay were between passages 25 and 35. A confluent monolayer in a 25cm2 culture flask was trypsinized and re-suspended in 9 ml of cell culture medium. 1.25×105 cells of hCMEC/D3 cells were seeded in rich endothelial growth medium (EBM-2, Lonza) with growth factors and antibiotics (gentamicin and amphotericin B) on a collagen-coated transwell insert. Once added to the transwell apparatus, the hCMEC/D3 cells were cultured for approximately 2 weeks at 37°C and 5% CO2. During the 2-week incubation, the medium (concentration of supplements) was changed from 1x-strength on days 3 and 6, to 0.5x-strength on day 9, and 0.25x-strength on day 12, and used in the assay on days 12–14. The use of the lower-strength medium was required to reduce the growth factors in the medium in order to promote cell differentiation and tight junction formation. Integrity of the BBB monolayer was confirmed by routine measurement of the trans-endothelial electrical resistance (TEER) prior to the transcytosis assay using an endometer (World Precision Instruments). The transcytosis assays were performed by washing hCMEC/D3 cells with PBS and adding fresh EGM-2 with 100 units of potassium penicillin and 100μg of streptomycin sulfate per ml of culture medium, adding 2×105 cells of C. neoformans to the top chamber of the transwell and incubating at 37°C and 5% CO2 for pre-determined times before collecting cryptococci that had migrated to the lower chamber. Cryptococci were plated onto YPD agar medium and counted after 48h incubation at 30°C. A cp1Δ strain in the H99 alpha background was obtained from the 2008 Madhani partial deletion collection[36] H99 alpha was used as a control for all the experiments performed with the cps1Δ mutant.
Gene Knockdown By siRNA
For gene silencing 3.75×105 of hCMEC/D3 cells were seeded in each well of 12-well transwell plate to allow the cells become 70% confluent at transfection. The cells were incubated in growth medium with growth factors and antibiotics for 24h at 37°C, 5%CO2. Cells were transiently transfected with 10μM siRNA diluted in Opti-MEM medium (Gibco) and Lipofectamine RNAiMAX (Fisher Scientific) for 48h at 37°C, 5% CO2. This incubation was repeated with newly prepared siRNA mixture and growth medium with decreasing growth factors every 48 hours. After a total of 7 days hCMEC/D3 cells were harvested and the total RNA was isolated for gene expression by qPCR and the transcytosis assays were performed. The siRNA sequences for EFNA1, EphA2, and the negative control (Silencer Select negative control No. 1 siRNA) were all purchased from ThermoFisher Scientific.
FITC-Dextran permeability assay
Permeability across the hCMEC/D3 cells in the in vitro model of the BBB was assessed by the passage of FITC-dextran (average molecular mass, 70kDa). Following treatment of hCMEC/D3 cells, 1 mg/ml of FITC-labeled dextran was added to each transwell. Fluorescence of FITC-dextran was measured in samples taken from both the upper and lower wells of black bottom 96-well plates. Measurements were recorded at 538 nm (excitation wavelength, 485 nm) with a spectrophotometer (SpectraMax M5). Results were representative of five independent experiments.
Immunofluorescence (IF) confocal microscopy
hCMEC/D3 cells were grown to confluence (~2 weeks) on collagen coated 12-well transwell inserts (8μm, Corning). C. neoformans was labeled with fluorescein isothiocyanate (FITC) (ThermoFisher Scientific) for 10 minutes at 37°C. hCMEC/D3 cells were washed with 1X PBS twice and infected at an MOI of 5 with FITC-labeled C. neoformans for 1.5 hours at 37°C, 5% CO2. After infection, hCMEC/D3 cells were washed with 1X PBS and fixed with 4% paraformaldehyde for 5 minutes at RT. The cells were washed with ice-cold 1X PBS three times and air-dried. The transwell filter was excised and washed twice with Immunofluorescence Buffer (IF buffer, 0.15M NaCl, 5 mM EDTA, 20 mM HEPES, pH 7.5). The cells were incubated with EphA2 primary antibody (Abcam) 1:50 overnight at 4°C in PBS with 1% BSA. After washing three times for 15 minutes in IF buffer, the cells were secondary stained with anti-rabbit IgG (AF 568, Abcam) 1:1000 for 1 hour at RT in PBS with 1% BSA. For staining of phalloidin the PFA fixed cells were incubated with AF647 conjugate (Cell Signaling) at 1:20 for 15 minutes at RT and washed with 1X PBS. For staining of nuclei the PFA fixed cells were incubated with DAPI stain (Cell Signaling Technology) at 1:5000 for 2 minutes at RT and washed with 1X PBS three times. Samples were mounted with VectaMount HQ (Vector Labs, Inc) and imaged on a Leica TCS SP8 STED 3X confocal microscope.
Protein expression studies in HEK293T cells
HEK293T cells, purchased from American Type Culture Collection (ATCC, Manasass, VA) were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Life Technologies) and antibiotics (penicillin/streptomycin/gentamicin/amphotericin B).
Preparation of EphA2 expression vectors
EphA2-His6 was produced by cloning EphA2 cDNA (gift from Dr. Kalina Hristova) into pcDNA 3.1/V5-His TOPO according to the directions on the Topo TA cloning kit (ThermoFisher Scientific, Waltham, MA). EphA2 cDNA was amplified by PCR using primers EphA2Fwd_HindIII 5′-GGGCCCAAGCTTACCAGCAA CATGGAGCTCCAGGCAGCC-3′ and EphA2 Rvs_KpnI 5′ CCCGGGGGTACCTTTGATGGGGATCCCCACAGTGTTCAC-3′. These oligonucleotides include HindIII and KpnI restriction sites for cloning into HindII and KnpI sites of pcDNA 3.1/V5-His TOPO vector.
Transfection
Cells were grown in 25cm2 flasks using DMEM modified with 10%FBS, 1% penicillin/streptomycin, and 0. 1% gentamicin-amphotericin B. After reaching 90% confluence, cells were washed with 1x PBS, and trypsinized (Trypsin EDTA 0.05%) for 1 minute. Trypsinized cells were transferred to a new 25cm2 flask containing 5 ml modified DMEM. Cells were grown for approximately 24 hours at 37°C + 5% CO2, until 70%–90% confluence then transfected with pcDNA 3.1-EphA2/V5-His TOPO or empty vector with Lipofectamine 3000 reagents (ThermoFisher Scientific) according to the manufacturer’s protocol. After transient transfection for 48 hours, HEK293T cells were used for infection studies.
Western Blot Analysis
Transfected cells were lysed as previously described in the Western blot analysis section. The PVDF membranes were then incubated with primary antibodies (mouse monoclonal histidine-6 IgG, Neuromab) diluted at 1:1000 overnight at 4°C. Membranes were incubated with secondary antibodies (goat anti-mouse IgG, Abcam) diluted 1:5000 conjugated to horseradish peroxidase for 1 h at room temperature.
Live-cell recording
HEK293T cells were grown in 25cm2 flasks using DMEM as described above. After reaching 90% confluence, the cells were washed with 1x PBS, and trypsinized (Trypsin EDTA 0.05%) for 1 minute. Trypsinized cells were transferred to a chamber slide for live-cell recording (LabTek Inc.) containing 200 μl DMEM + 10% FBS. HEK293T cells were grown for approximately 24 hours at 37°C + 5% CO2, until reaching 70%–90% confluence then transfected with pcDNA 3.1-EphA2/V5-His TOPO or empty vector for 48 hours. Cells were washed with 1X PBS and stained with 5μm DRAQ5 (Cell Signaling) for 15 minutes in the dark at room temperature. Cells were washed and stained with Ni-NTA-ATTO 550 (Sigma) for 15 minutes in the dark at room temperature. At time zero FITC-labeled C. neoformans (H99 strain, MOI of 5) or green fluorescent microspheres (25μm average diameter, Sigma) were added to the cells. Cells were viewed with an environmental chamber (Ibidi, Inc.) and a Leica TCS SP8 STED 3X confocal microscope at the UC Davis Advanced Imaging Facility.
Statistical analysis
Statistical significance was determined by un-paired Student’s t-test with Welch’s correction or running ANOVA using GraphPad Prism Program (GraphPad Software Inc.). P values <0.05 were considered significant.
Results
The transcriptome of human brain capillary endothelial cells challenged with C. neoformans suggests a role for the EPH-EphrinA1 (EphA2) tyrosine kinase receptor-signaling pathway
We sought to define the transcriptional response of the human BBB when challenged with C. neoformans. In order to capture the transcriptome representing the initial interface between human brain capillary endothelial cells (a.k.a. the BBB) and fungal cells, a strain of C. neoformans and a human brain capillary endothelial cell line (hCMEC/D3, referred to as brain endothelial cells throughout the text) were co-incubated for 3h (S1_Fig 1). This cell line recapitulates many features of primary human brain capillary endothelial cells and thus the cell line has been used as a model of the human blood-brain barrier.[34,37] This time point was chosen based on previous studies demonstrating that attachment with concomitant changes in cell surface morphology of occurred by 3h.[9,10,12] We identified a total of 1525 transcripts with a log2 (fold-change) value >1.5 – this represented approximately 3% of the entire transcripts in brain endothelial cells. Among the differentially expressed genes, 56% were upregulated and 44% were downregulated (Fig 1A). Twelve genes identified by RNAseq were randomly selected and validated by quantitative PCR (S1_Table 1). We also found that gene expression profiles of brain endothelial cells grown on either transwells or on 25cm2 flasks were similar (data not shown).
Fig 1. The transcriptome of human brain capillary endothelial cells (a.k.a. BBB) challenged with C. neoformans.

(A) Among the differentially expressed transcripts identified in brain endothelial cells exposed to C. neoformans, 56% are upregulated and 44% are downregulated. These transcripts represent approximately 3% of all transcripts detected in the hCMEC/D3 cell line used here as an in vitro model of the human BBB. (B) The transcripts are grouped according to known or predicted function.
Of the transcripts identified only those with a log2 fold-change of 2 or greater were grouped according to their known or predicted function (Fig 1B, Table 1, hBMECs = brain endothelial cells). We found that the transcriptome of brain endothelial cells resulting from exposure to fungal cells included genes predicted to act within several different functional groups such as: the immune/inflammatory response, the stress-apoptosis response, metabolism and transcription. As expected we identified many cytoskeleton- and cell signaling-related transcripts that were differentially regulated. It is worth noting that although the movement of solutes and ions across brain endothelial cells is usually tightly regulated, several genes encoding transporters displayed altered expression. The SLC16 gene family of key transporters known as MCTs, like the monocarboxylate transporter (MCT2) that moves lactic acid across the plasma membrane was downregulated while the hexose (glucose & fructose) transporter (MCT14) was upregulated (Table 1). Several ion channels including ASIC3 acid sensing (proton-gated) channel, CACNG8 calcium channel, and KCNN2 potassium channel were also upregulated, while the CLIC4 chloride channel was downregulated (Table 1). Collectively, these changes were indicative of shifts in metabolism and pH regulation induced by interactions with fungal cells.
Table 1.
Differentially expressed genes in hBMECs challenged with C. neoformans.
| Accession# | ids | logFC | PValue | FDR |
|---|---|---|---|---|
| Transcription Factors/Regulation | ||||
| 21160 | LINC00473 -transcriptional regulator | 6.29 | 6.19E-58 | 1.31E-53 |
| 23355 | C10orf10 - activator of transcription factor ELK1 | 3.91 | 1.57E-50 | 1.66E-46 |
| 7180 | MN1 - is a transcription coregulator | 3.23 | 1.06E-43 | 4.48E-40 |
| 6782 | MAFK - transcription factor | 2.13 | 7.72E-14 | 7.02E-12 |
| 11281 | SRA1 - steroid receptor RNA activator 1 | 2.09 | 8.99E-14 | 8.03E-12 |
| 3238 | EGR1 - transcriptional regulator | −2.08 | 2.05E-21 | 8.70E-19 |
| 2350 | CREBL2 - regulator of CREB1 transcriptional activity | −2.64 | 1.97E-30 | 2.98E-27 |
| 11094 | SNAI2 - act as transcriptional repressors bind to E-box motifs | −2.13 | 2.24E-14 | 2.45E-12 |
| Immune/Inflammatory Response | ||||
| 7952 | NPTX1 - neuronal Pentraxins; pattern recognicition receptors | 4.42 | 8.66E-17 | 1.64E-14 |
| 23228 | CPAMD8, Complement components | 3.88 | 5.17E-23 | 2.54E-20 |
| 10672 | CXCL12 - chemokine protein activate leukocytes | 2.53 | 6.82E-23 | 3.28E-20 |
| 24182 | C1QL1 - complement component 1 | 2.12 | 1.54E-17 | 3.32E-15 |
| 7203 | CD200 - immunoglobulin superfamily | 2.04 | 4.20E-14 | 4.12E-12 |
| 2334 | CR1- complement component (3b/4b) receptor 1 | 2.47 | 0.00719469 | 0.02679599 |
| 1608 | CCR7-chemokine (C-C motif) receptor 7 | 3.73 | 3.77E-14 | 3.77E-12 |
| 11921 | TNFRSF6B - tumor necrosis factor receptor superfamily | 2.35 | 2.00E-06 | 2.43E-05 |
| 24182 | C1QL1- complement component 1, q subcomponent-like 1 | 2.12 | 1.54E-17 | 3.32E-15 |
| 6476 | LAG3 -lymphocyte-activation gene 3 | 2.09 | 4.40E-05 | 0.00036361 |
| 1695 | CD7-CD7 molecule | 7.78 | 1.16E-06 | 1.51E-05 |
| 15563 | IL37-interleukin 37 | 2.40 | 1.93E-05 | 0.00017705 |
| 6713 | LTB4R-leukotriene B4 receptor | 2.39 | 5.69E-10 | 1.88E-08 |
| 23228 | CPAMD8-C3 and PZP-like, alpha-2-macroglobulin domain | 3.88 | 5.17E-23 | 2.54E-20 |
| 2926 | DMBT1-encodes a protein involed in immune system | 3.23 | 7.69E-26 | 5.62E-23 |
| 18163 | IFNE-interferon, epsilon | 2.83 | 3.92E-08 | 7.60E-07 |
| 10729 | SEMA4A-sema domain, immunoglobulin domain (Ig) | 2.01 | 0.00154048 | 0.00758141 |
| 6879 | MAPK6-mitogen-activated protein kinase 6 | −2.30 | 1.45E-23 | 7.47E-21 |
| 31405 | EBF1-early B-cell factor 1 | −2.13 | 8.54E-05 | 0.00064213 |
| Cell Adhesion | ||||
| 1764 | CDH5 - Caherin 5 VE-caherin | 2.80 | 1.16E-28 | 1.30E-25 |
| 7521 | CDHR5 - cadherin-related family member 5 | 2.79 | 0.00169751 | 0.0082261 |
| 2035 | CLDN14-claudinl4 | 2.53 | 0.00526077 | 0.02078883 |
| 8661 | PCDH9-protocadherin 9 | −2.74 | 1.56E-07 | 2.61E-06 |
| 13404 | PCDH10-protocadhierin 10 | −3.66 | 5.02E-05 | 0.00040629 |
| Cytoskeleton | ||||
| 24071 | TUBA3D -tubulin, alpha 3d | 2.68 | 0.00400201 | 0.01657552 |
| 25517 | ANKZF1- ankyrin repeat and zinc finger domain containing 1 | 2.30 | 3.71E-20 | 1.38E-17 |
| 16467 | ACAP1- ArfGAP with coiled-coil, ankyrin repeat | 2.16 | 2.73E-07 | 4.25E-06 |
| 6136 | ITGA11- integrin, alpha 11 | 3.43 | 2.39E-07 | 3.76E-06 |
| 21446 | HAPLN3-hyaluronan and proteoglycan link protein 3 | 2.06 | 8.41E-08 | 1.53E-06 |
| 24352 | TNS4-tensin 4 | 2.30 | 3.12E-08 | 6.22E-07 |
| 11274 | SPTB-spectrin | 2.13 | 2.63E-05 | 0.00023217 |
| 14294 | SHANK3-SH3 and multiple ankyrin repeat domains 3 | 3.16 | 0.00102482 | 0.00538126 |
| 15846 | PALMD-palmdelphin, a lipid associated protein | 2.40 | 0.0090093 | 0.03217987 |
| 15756 | SDCBP2-syndecan binding protein (syntenin) 2 | 2.26 | 0.00812824 | 0.0296268 |
| 6140 | ITGA4-integrin, alpha 4 | −2.58 | 0.00094734 | 0.00506101 |
| 23576 | HOOK3-hook microtubule-tethering protein 3 | −2.04 | 2.33E-13 | 1.90E-11 |
| Receptors/Surface Proteins | ||||
| 11784 | THBD - thrombomodulin | 2.45 | 7.88E-29 | 9.27E-26 |
| 3540 | F2RL3 - Protease-activated receptor-like 3 | 4.37 | 7.44E-30 | 1.05E-26 |
| 3496 | MPZL2- myelin protein zero-like 2 | 2.46 | 1.05E-15 | 1.60E-13 |
| 7981 | NR4A2 - NURR1 nuclear receptor-related protein | 4.89 | 1.64E-34 | 3.47E-31 |
| 7982 | NR4A3 - nuclear receptor | 3.86 | 1.40E-28 | 1.48E-25 |
| 12694 | VIPR1 - a receptor for vasoactive intestinal peptide | 6.18 | 1.00E-23 | 5.30E-21 |
| 18628 | STAB1 - Stabilin-1; transmembrane receptor | 2.69 | 3.58E-12 | 2.13E-10 |
| 21446 | HAPLN3- hyaluronan and proteoglycan link protein 3 | 2.06 | 8.41E-08 | 1.53E-06 |
| 3025 | DRD4-dopamine receptor D4 | 3.34 | 0.00277866 | 0.01236211 |
| 3221 | EFNA1 - Ephrin A1 | 2.06 | 1.37E-14 | 1.62E-12 |
| 3396 | EPHB6-EPH receptor B6 | 2.17 | 5.39E-08 | 1.02E-06 |
| 727 | ARTN-artemin | 4.16 | 0.00071342 | 0.00397574 |
| 14911 | TAS2R4-taste receptor, type 2, member 4 | 3.16 | 4.16E-05 | 0.00034564 |
| Cell Signaling | ||||
| 14565 | SORBS1, sorbin and SH3 containing proteins | 2.89 | 2.18E-25 | 1.49E-22 |
| 20318 | SMOC1 - secreted modular calcium-binding protein | 2.61 | 2.77E-22 | 1.28E-19 |
| 684 | ARHGEF4-Rho guanine nucleotide exchange factor (GEF) 4 | 2.43 | 6.46E-11 | 2.74E-09 |
| 14130 | ARHGAP9 - Rho GTPase activating protein 9 | 2.56 | 0.00662294 | 0.02506775 |
| 14945 | PPP1R3G-protein phosphatase 1, regulatory subunit 3G | 3.27 | 7.58E-06 | 7.82E-05 |
| 3496 | MPZL2- myelin protein zero-like 2 | 2.46 | 1.05E-15 | 1.60E-13 |
| 9039 | PLA2G6-phospholipase A2, group VI | 2.01 | 4.60E-10 | 1.55E-08 |
| 29134 | DENND3-DENN/MADD domain containing 3 | 2.21 | 1.84E-16 | 3.22E-14 |
| 8792 | PDE7B-phosphodiesterase 7B | 2.33 | 6.89E-07 | 9.56E-06 |
| 12778 | WNT9A-wingless-type MMTV integration site family | 2.26 | 2.19E-12 | 1.40E-10 |
| 26036 | COMMD8-COMM domain containing 8 | −2.18 | 1.17E-09 | 3.53E-08 |
| 16015 | MOB1A-MOB kinase activator 1A | −2.04 | 1.15E-15 | 1.72E-13 |
| 29148 | FBXO45-F-box protein 45 | −2.07 | 1.02E-19 | 3.50E-17 |
| 3638 | FDX1-ferredoxin 1 | −2.17 | 4.72E-14 | 4.60E-12 |
| 16132 | SRXN1-sulfiredoxin 1 | −2.33 | 2.37E-15 | 3.32E-13 |
| 19903 | RRAGD-Ras-related GTP binding D | −2.04 | 6.96E-07 | 9.63E-06 |
| 2350 | CREBL2-cAMP responsive element binding protein-like 2 | −2.64 | 1.97E-30 | 2.98E-27 |
| 26545 | ARHGAP42-Rho GTPase activating protein 42 | −2.80 | 7.00E-13 | 5.04E-11 |
| 16924 | ARAP2-ArfGAP with RhoGAP domain, ankyrin repeat | −2.93 | 4.94E-32 | 9.51E-29 |
| 16924 | ARAP2 -a phosphatidylinositol (3,4,5)-binds RhoA-GTP | −2.93 | 4.94E-32 | 9.51E-29 |
| 16015 | MOB1A - kinase activator protein | −2.04 | 1.15E-15 | 1.72E-13 |
| 24463 | UBTD2 - ubiquitin domain containing protein | −2.25 | 1.60E-24 | 9.70E-22 |
| 10056 | RNF11 -Ring finger protien; mediates protein-protein interactions | −2.36 | 3.05E-24 | 1.75E-21 |
| 6879 | MAPK6 - MAP kinase 6 | −2.30 | 1.45E-23 | 7.47E-21 |
| 696 | ARL5A - ADP-ribosylation factor | −2.58 | 2.78E-26 | 2.26E-23 |
| 4381 | GNA13-Gprotein | −2.41 | 5.42E-26 | 4.25E-23 |
| 16967 | ARPP19 - cAMP regulated phosphoprotein | −2.27 | 5.86E-26 | 4.43E-23 |
| Proteases | ||||
| 16305 | ADAMTS15 - a metalloproteinase domain | 3.93 | 4.43E-31 | 7.82E-28 |
| 1383 | CA9, carbonic anhydrase, zinc metalloenzymes | 3.73 | 6.60E-22 | 2.98E-19 |
| 202 | ADAM23-ADAM metallopeptidase domain 23 | 2.38 | 0.00046464 | 0.00274772 |
| 1478 | CAPN11- calpain 11 | 2.27 | 2.69E-05 | 0.00023625 |
| 34407 | PRSS53-protease, serine, 53 | 2.03 | 0.00020773 | 0.00138617 |
| 24635 | PRSS48-protease, serine, 48 | 3.81 | 0.00317722 | 0.01378664 |
| Metabolism | ||||
| 2595 | CYP1A1-cytochrome P450, family 1, subfamily A, polypeptide 1 | 4.89 | 1.51E-17 | 3.31E-15 |
| 4092 | GAD1 - glutamate decarboxylase 1 | 4.63 | 6.29E-20 | 2.26E-17 |
| 20244 | CYP4X1-cytochrome P450, family 4, subfamily X, polypeptide 1 | 2.34 | 4.33E-07 | 6.37E-06 |
| 33480 | CYP27C1-cytochrome P450, family 27, subfamily C, polypeptide 1 | 2.22 | 0.00882842 | 0.0316934 |
| 24140 | MGAT5B- mannosyl-glycoprotein-acetyl-glucosaminyltransferase | 2.11 | 4.51E-05 | 0.0003714 |
| 1383 | CA9-carbonic anhydrase IX | 3.73 | 6.60E-22 | 2.98E-19 |
| 20093 | ADSSL1-adenylosuccinate synthase like 1 | 2.67 | 1.48E-08 | 3.21E-07 |
| 30252 | NAT6-N-acetyltransferase 6 (GCN5-related) | 2.36 | 1.16E-11 | 5.87E-10 |
| 16431 | VNN3-vanin 3 | −2.04 | 0.00062452 | 0.00354502 |
| 19365 | PANK3 - pantothenic acid kinase; biosynthesis of CoA | −2.34 | 1.94E-24 | 1.14E-21 |
| 18170 | CMPK1 - cytidine monophosphate (UMP-CMP) kinase 1 | −2.24 | 2.99E-25 | 1.98E-22 |
| 6821 | MAN1A1-mannosidase, alpha, class 1A, member 1 | −2.35 | 0.00299247 | 0.01313127 |
| 696 | ARL5A-ADP-ribosylation factor-like 5A | −2.58 | 2.78E-26 | 2.26E-23 |
| Stress Response/Apoptosis | ||||
| 16039 | ANGPTL4 - acts as an apoptosis survival factor | 3.90 | 2.52E-44 | 1.33E-40 |
| 11373 | STC1 - Stanniocalcin-1 is a glycoprotein; hypoxia-responsive | 3.12 | 6.34E-25 | 4.07E-22 |
| 24944 | DDIT4 - DNA damage-inducible transcript 4 protein | 2.66 | 1.05E-22 | 4.95E-20 |
| 23841 | GRIP2- glutamate receptor interacting protein 2 | 2.00 | 0.00258596 | 0.01165435 |
| 4092 | GAD1- glutamate decarboxylase 1 | 4.63 | 6.29E-20 | 2.26E-17 |
| 14661 | EGLN3- egl-9 family hypoxia-inducible factor3 alpha subunit | 3.74 | 8.92 E-08 | 1.61E-06 |
| 38749 | SDIM1-stress responsive DNAJB4 interacting membrane protein 1 | 5.45 | 5.97E-06 | 6.35E-05 |
| 23172 | MYCT1-myc target 1 | 2.98 | 0.00598148 | 0.02315718 |
| 21 | AATK-apoptosis-associated tyrosine kinase | 2.59 | 1.34E-05 | 0.00012838 |
| 24944 | DDIT4-DNA-damage-inducible transcript 4 | 2.66 | 1.05E-22 | 4.95E-20 |
| 2726 | DDIT3-DNA-damage-inducible transcript 3 | −2.46 | 1.89E-26 | 1.60E-23 |
| 4373 | GMFB - glia maturation factor, beta | −2.42 | 3.66E-20 | 1.38E-17 |
| 14065 | HDAC9 - histone deacetylase 9 | −2.49 | 7.70E-12 | 4.09E-10 |
| Hormone secretion | ||||
| 9604 | PTGS1 - prostaglandin-endoperoxide synthase 1 | 2.52 | 1.56E-13 | 1.32E-11 |
| 5472 | IGFBP3- insulin-like growth factor binding protein 3 | 2.29 | 1.93E-10 | 7.18E-09 |
| 11281 | SRA1 - steroid receptor RNA activator 1 | 2.09 | 8.99E-14 | 8.03E-12 |
| 4263 | GHR-growth hormone receptor | 2.06 | 0.00811349 | 0.02958832 |
| 3179 | EDNRA - receptor for endothelin-1 | 4.22 | 1.84E-11 | 8.81E-10 |
| 3176 | EDN1-endothelin 1 | 2.48 | 0.00573075 | 0.02234169 |
| 3177 | EDN2 - endothelin 2 - promote myelination | 4.67 | 2.22E-36 | 7.84E-33 |
| 11374 | STC2-stanniocalcin 2 | 2.39 | 4.08E-27 | 3.75E-24 |
| 5472 | IGFBP3-insulin-like growth factor binding protein 3 | 2.29 | 1.93E-10 | 7.18E-09 |
| 16039 | ANGPTL4 - angiopoietin-like 4 | 3.90 | 2.52E-44 | 1.33E-40 |
| 7982 | NR4A3-nuclear receptor subfamily 4, group A, member 3 | 3.86 | 1.40E-28 | 1.48E-25 |
| 259 | ADM-adrenomedullin | 2.77 | 3.23E-35 | 8.56E-32 |
| 11330 | SSTRl-somatostatin receptor 1 | 2.26 | 0.00017148 | 0.00117183 |
| Transporters/Carriers | ||||
| 11063 | SLC7A5 - solute carrier family 7 member 5 | 2.03 | 1.37E-21 | 5.92E-19 |
| 10953 | SLCO4A1 - Solute carrier organic anion transporter | 3.07 | 5.26E-35 | 1.24E-31 |
| 29196 | SLC45A4, solute carrier family 45, member 4 | 2.35 | 5.83E-10 | 1.92E-08 |
| 18301 | SLC2A14, hexose (glucose & fructose) transporter | 2.23 | 5.92E-14 | 5.65E-12 |
| 11073 | SLC9A3, NHE3 cation proton antiporter 3 | 3.95 | 4.09E-05 | 0.00034161 |
| 44187 | SLC2A1-AS1, antisense RNA 1, affiliated with IncRNA class | 2.02 | 4.35E-05 | 0.00036006 |
| 15827 | SLC13A4, sodium/sulfate symporter | 2.18 | 0.00021499 | 0.00142809 |
| 13941 | SLC44A4, choline transporter-like protein 4 | 2.62 | 0.00418046 | 0.01719353 |
| 6291 | KCNN2 - potassium/calcium-activated channel | 2.23 | 0.00991974 | 0.03475682 |
| 29103 | ATP2C2-ATPase, Ca++ transporting, type 2C, member 2 | 2.40 | 0.00183352 | 0.00878075 |
| 13628 | CACNG8 - calcium channel, voltage-dependent | 2.08 | 1.35E-05 | 0.00012893 |
| 6291 | KCNN2-potassium/calcium-activated channel | 2.23 | 0.00991974 | 0.03475682 |
| 7060 | MGP-matrix Gla protein, calcium binding protein | 3.55 | 1.01E-09 | 3.09E-08 |
| 101 | ASIC3-acid-sensing (proton-gated) ion channel 3 | 2.03 | 0.00026748 | 0.00172908 |
| 21353 | KLHL31-kelch-like family member 31 | 4.32 | 0.00356567 | 0.01511412 |
| 13533 | ATP8A2-ATPase, aminophospholipid transporter, class I | −2.33 | 0.00695826 | 0.02611289 |
| 13621 | SLC6A15, amino acid transporter, member 15 of the solute carrier | −2.16 | 8.55E-08 | 1.55E-06 |
| 29681 | SLC25A33, pyrimidine nucleotide carrier | −2.09 | 4.05E-11 | 1.79E-09 |
| 22082 | VMA21 - vacuolar ATPase | −2.49 | 5.52E-24 | 3.00E-21 |
| 13518 | CLIC4 - chloride intracellular channel | −2.04 | 2.49E-20 | 9.57E-18 |
| 10928 | SLC16A7, monocarboxylate transproter | −2.78 | 0.00048093 | 0.00283142 |
| 23576 | HOOK3-hook microtubule-tethering protein 3 | −2.04 | 2.33E-13 | 1.90E-11 |
| 20983 | NECAB1-N-terminal EF-hand calcium binding protein 1 | −2.04 | 2.95E-09 | 7.95E-08 |
| 19008 | KLHL11-kelch-like family member 11, proteosomal degradation | −2.38 | 3.46E-10 | 1.22E-08 |
| RNA processing | ||||
| 10130 | RNU1-3-RNA, U1 small nuclear 3 | 3.32 | 0.00152132 | 0.00749929 |
| 40645 | ATP11A-AS1-ATP11A antisense RNA 1 | 3.26 | 0.00038146 | 0.00233068 |
| 16416 | LINC00313-long intergenic non-protein coding RNA 313 | 3.24 | 3.81E-11 | 1.70E-09 |
| 42742 | LINC00410-long intergenic non-protein coding RNA 410 | 2.76 | 0.00067119 | 0.00377125 |
| 40364 | MATN1-AS1-MATN1 antisense RNA 1 | 2.54 | 0.00546353 | 0.02146989 |
| 25810 | HEXA-AS1-HEXA antisense RNA 1 | 2.36 | 0.00380048 | 0.01587745 |
| 10031 | RMRP-RNA component of mitochondrial RNA processing | 2.15 | 0.00166202 | 0.0080893 |
| 32591 | SNORA6-small nucleolar RNA, H/ACA box 6 | 2.05 | 0.00185458 | 0.00885758 |
| 40705 | LENG8-AS1-LENG8 antisense RNA 1 | 2.04 | 3.05E-05 | 0.00026381 |
| 10116 | SNORA73B-small nucleolar RNA, H/ACA box 73B | 2.04 | 0.00023708 | 0.00155775 |
| 33791 | LINC00173-long intergenic non-protein coding RNA 173 | 2.03 | 0.00821902 | 0.02989594 |
| 44187 | SLC2A1-AS1-SLC2A1 antisense RNA 1 | 2.02 | 4.35E-05 | 0.00036006 |
| 40209 | EIF2B5-AS1-EIF2B5 antisense RNA 1 | 2.01 | 0.00216837 | 0.0101011 |
| 31955 | PABPC4L-poly(A) binding protein, cytoplasmic 4-like | −3.77 | 0.00011564 | 0.00083691 |
| Diseases | ||||
| 7180 | MN1-meningioma (disrupted in balanced translocation) 1 | 3.23 | 1.06E-43 | 4.48E-40 |
| 20708 | APBB3-amyloid beta (A4) precursor protein-binding | 2.75 | 7.67E-25 | 4.78E-22 |
| 15955 | SEZ6-seizure related 6 homolog (mouse) | 2.72 | 0.00120819 | 0.00618167 |
| Cell cycle | ||||
| 25097 | CABLES1-Cdk5 and Abl enzyme substrate 1 | 2.35 | 5.41E-17 | 1.07E-14 |
| 28600 | PROCA1-protein interacting with cyclin A1 | 2.10 | 1.35E-05 | 0.00012894 |
| 14065 | HDAC9-histone deacetylase 9 | −2.49 | 7.70E-12 | 4.09E-10 |
Genes are grouped according to known or predicted function. Changes in gene expression are represented by log2FC (fold-change). FC values are ≥ 2 and ≤ −2. FDR represents the false discovery rate. Details of the RNA-Seq analysis are described in the material & methods.
Mapping the transcriptome to known canonical signaling pathways using Ingenuity Pathway Analysis (IPA, Qiagen, Inc.) revealed that 5 unique pathways dominated the transcriptome (Table 2). These included the EPH-ephrinA1 (EphA2) tyrosine kinase receptor pathway, axonal guidance pathway involving MAPK signaling and cytoskeleton regulation, RhoGDI (Rho GDP-dissociation inhibitors are important regulators of Rho-GTPases in cytoskeleton-remodeling, vesicular trafficking and gene expression), CXCR4-pathway (immune/inflammatory response), and IL-8 signaling pathway (cell proliferation, survival and invasion) (Table 2). Of the 5 pathways identified, approximately 11.49% (20 genes) of the 174 genes known to function within the EphA2-signaling pathway were differentially regulated in brain endothelial cells exposed to C. neoformans (Table 2).
Table 2.
The 5 dominant canonical signaling pathways in hBMECs challenged with C. neoformans.
| # | Pathway | Number of DEGs | Number of total genes | Ratio | P-value |
|---|---|---|---|---|---|
| 1 | Ephrin Receptor Signaling | 20 | 174 | 0.115 | 5.17E-05 |
| 2 | Axonal Guidance Signaling | 36 | 433 | 0.083 | 1.01E-04 |
| 3 | RhoGDI Signaling | 19 | 173 | 0.11 | 1.45E-04 |
| 4 | CXCR4 Signaling | 17 | 152 | 0.112 | 2.56E-04 |
| 5 | IL-8 Signaling | 19 | 183 | 0.104 | 3.01E-04 |
The top canonical signaling pathways are mapped according to the ratios of differentially expressed genes (DEGs) to the total number of genes attributed to the pathway and the corresponding P-value.
The transmigration of C. neoformans is dependent on the expression of the EphA2-tyrosine kinase receptor in brain endothelial cells
The transcriptome analysis indicated a likely role for EphA2-signaling pathway in mediating the fungal-brain endothelium interaction. EphrinA1, a ligand for the EphA2 tyrosine kinase receptor, was one of the 174 transcripts involved in EPH-mediated signaling. To characterize the function of EphA2 during fungal crossing of the BBB, we used siRNA in brain endothelial cells (hCMEC/D3 cell line) to knockdown the EphA2 transcript (Fig 2A). Following siRNA, approximately 51% of the EphA2 transcript was repressed (Fig 2A). We then used the siRNA-EphA2 cells in the in vitro model of the human BBB to perform transcytosis assays in order to determine whether C. neoformans was able to cross the endothelial barrier (Fig 2, lower left panel). In this static model of the BBB, the hCMEC/D3 (human brain capillary endothelial cells) cell line was seeded onto a transwell suspended in appropriate media. The brain endothelial cells were grown to confluency and allowed to fully differentiate into a tight, intact barrier [9,34]. The chamber above the endothelial cells represented the apical (luminal) side of the BBB and the chamber below represented the basolateral (abluminal) side of the BBB. In the transcytosis assays, fungal cells were added to the luminal side of the barrier, collected from the abluminal side following transcytosis and plated for CFU determination (Fig 2, lower left panel).
Fig 2. siRNA knockdown of EphA2 tyrosine kinase receptor prevents the migration of C. neoformans across brain endothelial cells in an in vitro model of the BBB.
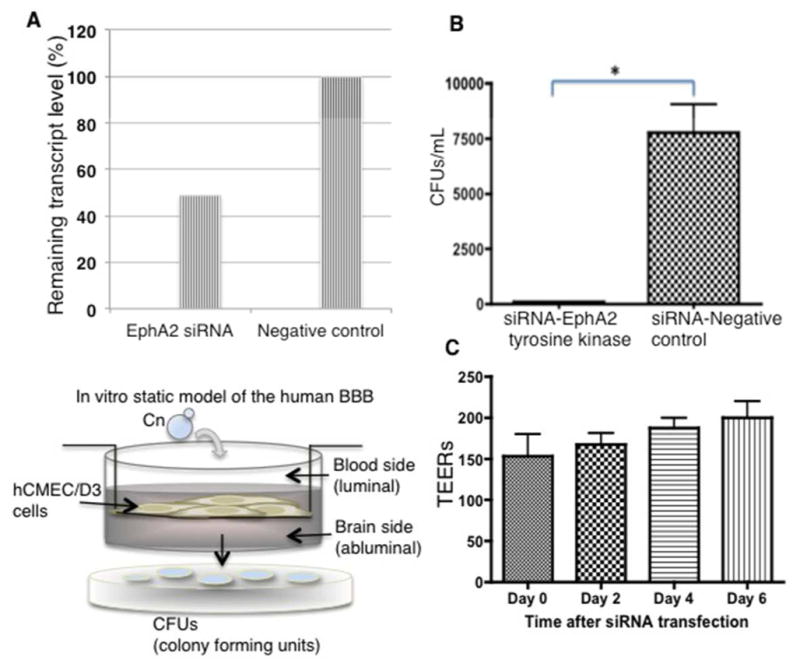
(A) The EphA2 transcript was repressed by siRNA in brain endothelial cells. Quantitative PCR revealed ~51% repression of EphA2 transcript in contrast to the siRNA control. (B) Transcytosis assays in the in vitro BBB model show a significant reduction in the movement of C. neoformans across endothelial cells when EphA2 transcript is repressed. Transmigration of C. neoformans was not affected when endothelial cells were transformed with an siRNA negative control. Transcytosis assays were performed as described below. Fungal cells were collected from bottom well following 24h co-incubation (n=5, *P<0.05). (C) Following siRNA of EphA2 transendothelial electrical resistance measurements (TEERs) were performed at the indicated times with an endometer. The TEER measurements indicate an intact barrier. The modest upward trend of TEERs suggests a tightening of the barrier. (Lower left panel) A schematic diagram depicting an in vitro, static monolayer model of the human BBB. Immortalized human brain endothelial cells (hCMEC/D3 cell line) are grown and differentiated on a transwell. Barrier integrity is confirmed by monitoring tightness of barrier junctions with TEERs and dextran permeability. Transcytosis assays are performed as follows: Fungal cells are added to the luminal side (top of transwell), collected from the bottom well (abluminal side of BBB) at indicated times and plated onto agar plates for CFU determination.
We found that repression of the EphA2 transcript prevented the migration of C. neoformans across the in vitro BBB model (Fig 2B). The reduced expression of EphA2 did not appear to alter the integrity of the barrier since the TEER (transendothelial electrical resistance) values of the endothelial cells did not change significantly, thus suggesting that the intercellular junctions were intact; however an upward trend of higher TEER values was observed following siRNA transfection indicating a tighter barrier (Fig 2C). Interestingly, we found that silencing the ephrinA1 ligand of EphA2 did not alter the migration of C. neoformans across the BBB (S3_Fig 3).
The role of EphA2 was further examined by blocking EphA2 activity with a monoclonal antibody (mAb) raised against EphA2 or by the addition of an inhibitor of the EPH class of tyrosine kinase receptors. Brain endothelial cells were treated with EphA2-mAb for 45mins[38] and subsequently challenged with C. neoformans. Following a 3h co-incubation of EphA2-mAb-treated endothelial cells with C. neoformans in the in vitro model of the BBB, significantly fewer fungal cells crossed the BBB (Fig 3A). In contrast, the addition of a control antibody (IgG) to the brain endothelial cells did not alter the transmigration of fungal cells in the transcytosis assay (Fig 3A). Treatment with either antibody did not appear to affect dextran permeability across the BBB suggesting an intact barrier (Fig 3B). Transcytosis assays performed in the in vitro BBB model with brain endothelial cells treated with dasatinib, a well-characterized inhibitor of tyrosine kinase receptors within the EPH class[38,39] demonstrated a similar reduction in the migration of fungal cells across the endothelial barrier (Fig 3C). As observed above with the addition of EphA2-mAb, the barrier integrity was not altered during treatment with dasatinib (Fig 3D). Taken together, the results suggested that migration of C. neoformans across the BBB is dependent on the functional expression of EphA2 in brain endothelial cells.
Fig 3. Blocking EphA2 activity diminishes the crossing of C. neoformans.

(A) Addition of peptide monoclonal antibody (mAb) against the extracellular N-terminal region of EphA2 blocks activity of EphA2. Brain endothelial cells were treated with EphA2-mAb (125μg) for 45mins and subsequently challenged with C. neoformans. Following a 3h co-incubation in the in vitro BBB model, transcytosis assays showed reduced fungal crossing, in contrast to the control antibody (IgG). (B) Treatment of endothelial cells with the antibodies did not affect dextran permeability suggesting an intact barrier. (C) & (D) Similarly, addition of 10μm or 20μm dasatinib (an inhibitor of EphA2) to brain endothelial cells in the in vitro BBB model, reduced fungal crossing but did not appear to alter dextran permeability. The DMSO solvent control had no effect on fungal crossing. Transcytosis assays were performed as described above. n=8, *P<0.05, ***P<0.001, n/s = not significant.
C. neoformans activates EphA2 in brain endothelial cells by promoting its phosphorylation
We next examined whether the association of C. neoformans with brain endothelial cells could stimulate EphA2 activity. It is well established that EphA2 is activated through phosphorylation - this is a key step for receptor signaling and internalization.[28,40] To test this, brain endothelial cells were co-incubated with fungal cells and lysates were examined by Western blot analysis with an antibody specific to the phosphorylated serine897 residue in EphA2. We detected a polypeptide corresponding to phosphorylated-EphA2 in brain endothelial cells that were previously challenged with fungal cells for 15, 30 or 60 minutes (Fig 4A). In contrast, the co-incubation of S. cerevisiae and brain endothelial cells did not promote EphA2 phosphorylation (Figure 4B). Upon further analysis of EphA2, we found that the presence of dasatinib blocked the phosphorylation of EphA2 that was otherwise detected upon exposure to C. neoformans (Fig 4C, D). This result is consistent with the significant reduction in fungal cells crossing the BBB where the brain endothelial cells had been treated with dasatinib (Fig 3C).
Fig 4. C. neoformans induces the phosphorylation of EphA2.

(A) Western blot analysis demonstrated the phosphorylation of EphA2 in brain endothelial cells when cells were challenged with C. neoformans (B) S. cerevisiae or (E) cps1Δ for 15min, 30min or 1h (middle panel). A polyclonal anti-phospho antibody to EphA2 was used to detect the phosphorylated form of EphA2. GAPDH was used as a loading control. (C) Endothelial cells treated with dasatinib and co-incubated with C. neoformans revealed a lack of EphA2 phosphorylation, however DMSO control clearly indicated phosphorylated EphA2, thus consistent with the notion that C. neoformans activates EphA2. (D, F) Relative band intensity of phosphorylated EphA2.
Collectively, the data support the notion that C. neoformans activates EphA2 by promoting its phosphorylation and that EphA2 phosphorylation contributes to the transmigration of C. neoformans; however, we asked whether the activity of EphA2 was dependent on the association of cryptococci with CD44 - the receptor for hyaluronic acid. Western blot analysis revealed that when a strain lacking hyaluronic acid (cps1Δ) was co-incubated with brain endothelial cells for 15, 30 or 60 minutes, EphA2 was not phosphorylated suggesting a lack of activity (Fig 4E, F). The aforementioned result is consistent with the notion that C. neoformans promotes EphA2 activity through CD44.
Activation of EphA2 enhances the transcellular migration of C. neoformans across brain endothelial cells in the in vitro model of the BBB
EphrinA1 can serve as a ligand for EphA2 – their association promotes the phosphorylation and internalization of EphA2. We asked whether the addition of ephrinA1 could promote the internalization and transmigration of C. neoformans via EphA2. To test this, recombinant ephrinA1 was added to brain endothelial cells while the movement of C. neoformans across the brain endothelial cells in the in vitro BBB model was monitored. We found that the addition of ephrinA1 significantly enhanced the migration of C. neoformans across the BBB (Fig 5A). The data indicated a dose-dependent trend although no significance was detected between concentrations of ephrinA1 used here (Fig 5A).
Fig 5. Activation of EphA2 promotes crossing of C. neoformans in the in vitro model of the BBB.
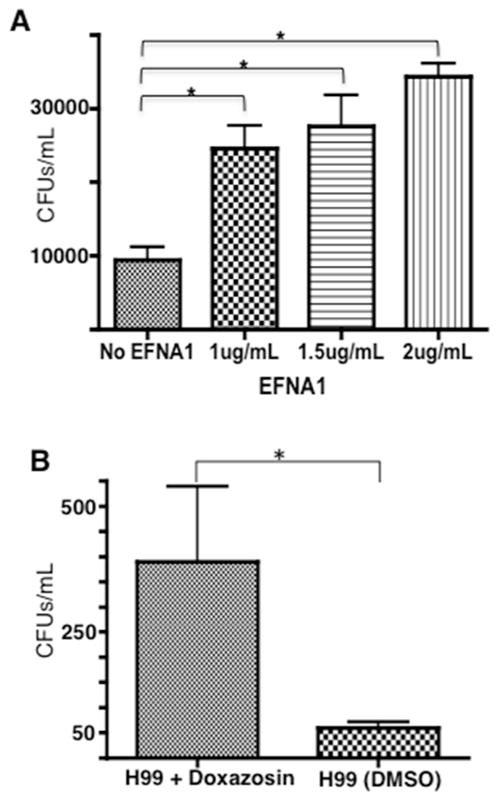
(A) Addition of human recombinant ephrinA1 (EFNA1, Origene), a ligand of EphA2, to brain endothelial cells enhanced crossing of C. neoformans in the in vitro BBB model. Three concentrations of EFNA1 (1, 1.5, and 2 μg/ml) were co-incubated with 2×105 cells of C. neoformans cells and added to the top chamber of the transwell in the in vitro BBB model. Following 24h, fungal cells were collected from the bottom chamber and placed onto agar plates for CFU determination. (B) Transcytosis assay revealed that the EphA2 agonist, doxazosin, facilitated migration of C. neoformans across brain endothelial cells. DMSO, a solvent for doxazosin and used here as a negative control, had no effect. Brain endothelial cells were exposed to 100μM doxazosin and co-incubated with 2×105 cells of C. neoformans. Following 24h, fungal cells were collected from bottom chamber placed onto agar plates for CFU determination *P<0.05, n=8.
We further examined whether an active EphA2 in brain endothelial cells enhanced the transmigration of C. neoformans by using a small molecule agonist (doxazosin) of EphA2. Endothelial cells were treated with doxazosin in the in vitro BBB model and transcytosis assays were performed following a 3h co-incubation with C. neoformans. Doxazosin is a potent agonist of EphA2 and interacts directly with the ligand-binding domain on the extracellular N-terminal region of EphA2.[32] Based on our observations above, we would predict a similar increase in fungal crossing. As expected the migration of fungal cells across the BBB was significantly enhanced in the presence of doxazosin (Fig 5B). Thus while inhibiting EphA2 activity prevented the migration of C. neoformans, we found that stimulating EphA2 activity with a ligand or an agonist enhanced crossing.
C. neoformans co-localizes with EphA2 and F-actin in brain endothelial cells
In order to assess the localization of EphA2 and C. neoformans in brain endothelial cells, confocal microscopy was used. Here, endothelial cells were initially challenged with C. neoformans and subsequently fixed. Phalloidin staining, used to delineate F-actin, revealed the presence of F-actin throughout brain endothelial cells suggesting cytoskeleton involvement as expected (Fig 6A, B, C, indicated by arrows). The punctate localization pattern of EphA2 in the endothelial cells (shown in red) revealed clusters of EphA2 along the cell surface and along F-actin fibers (Fig 6A, B). The presence of C. neoformans was detected as FITC-labeled cryptococci (Fig 6F). Interestingly, cryptococci were closely associated with F-actin and EphA2 in structures that embraced fungal cells (Fig 6). In addition clusters of the EphA2 receptor were observed in adjacent cells that were in close proximity to C. neoformans (Fig 6, B, D, indicated by star).
Fig 6. Clustering of EphA2 receptors co-localize with F-actin and C. neoformans in brain endothelial cells.

The EphA2 receptor co-localized with F-actin and both surrounded C. neoformans (indicated by arrows). In addition, clustering of the EphA2 receptor was observed on adjacent brain endothelial cells in close proximity to fungal cells (indicated by star). Panels (A) & (B) represent merged confocal images of immunofluorescence of endothelial cells exposed to C. neoformans; (C) F-actin was detected by phalloidin (yellow); (D) The EphA2 receptor is shown in red, (E) nuclei are shown in blue with DAPI stain and (F) C. neoformans was detected by FITC (shown in green).
Further analysis revealed that cryptococci were enveloped by F-actin that resembled microvilli-like structures on what appeared to be a ruffled surface of brain endothelial cells (Fig 7, arrows). This was consistent with morphological changes indicative of actin-recruitment and cytoskeleton re-modeling as previously described.[10,11] The EphA2 receptor appeared to co-localize with the F-actin structures and clustered around C. neoformans. EphA2 clustering was also observed in adjacent brain endothelial cells that were near cryptococci (Fig 7, indicated by star).
Fig 7. Microvilli-like structures embrace C. neoformans.

Confocal microscopy revealed that brain endothelial cells exposed to C. neoformans produced microvilli-like structures of F-actin that embraced C. neoformans. EphA2 displayed a punctate localization pattern and co-localized with F-actin in association with C. neoformans (indicated by arrows). As noted above, a cluster of EphA2 receptors was clearly observed on the surface of brain endothelial cells that were very near to fungal cells (indicated by star). Panels (A) & (B) represent merged confocal images of immunofluorescence of endothelial cells exposed to C. neoformans. (C) Illustrates F-actin as detected by phalloidin (yellow); (D) The EphA2 receptor is shown in red; (E) nuclei are shown with DAPI stain (blue) and (F) C. neoformans was detected by FITC (shown in green).
The overexpression of EphA2 in HEK293T cells engages C. neoformans by promoting a direct interaction
Next, we asked whether the overexpression of EphA2 in a different mammalian cell type could promote association and internalization of C. neoformans. To test this, a HIS-tagged EphA2-cDNA was transformed into a HEK293T cell line. Following Western blot analysis that confirmed protein expression of EphA2 (Fig 8F), live-cell microscopy was performed in order to capture direct interactions between EphA2 and C. neoformans in real-time by acquiring images approximately every 2 min for 1h (S1_Movie). The live-cell recording studies revealed that the HEK293T cells overexpressing EphA2 produced cellular protrusions that directly associated and enveloped C. neoformans following 35 min of co-incubation (S1_Movie, panel A &C). The green colored, FITC-stained fungal cells appeared to hover next to HEK293T cells and EphA2 (in red) was predominately localized to the cellular protrusions that interacted with C. neoformans. The aforementioned observations were confirmed upon closer inspection of snap shots captured at various time points from the live-cell recording (Fig 8). The association between EphA2 and C. neoformans appeared to be specific since similar live-cell recording studies that replaced C. neoformans with fluorescent beads revealed no association between the beads and EphA2 (S3_Movie). Furthermore, following a 1h co-incubation, fungal cells appeared significantly less engaged with HEK293T that had not been transformed with EphA2-cDNA (S2_Movie). In both the bright-field and fluorescence images, fungal cells did not appear to be engulfed in the absence of EphA2. Collectively, these results strongly support the notion that EphA2 interacted directly with C. neoformans.
Fig 8. HEK293T cells overexpressing EphA2 display a direct interaction with C. neoformans.

(A)–(E) Immunofluorescence images represent still images captured at indicated time points from live-cell imaging of HEK293T cells expressing EphA2-cDNA and challenged with C. neoformans (S6_movie & S7_movie). Images display a clear association between EphA2 and C. neoformans as indicated by cell protrusions engulfing fungal cells (white arrows). Fungal cells were stained with FITC (green); Expressed EphA2 in HEK293T cells is shown in red; Nuclei are stained with DAPI and appear blue. (F) Western blot analysis detected the presence of the EphA2 in HEK293T cells (indicated by black arrow). The polypeptide band corresponding to EphA2HIS was not observed in non-transfected cells. GAPDH was used as a loading control (indicated by bottom black arrow).
EphA2 actively stimulates the internalization of C. neoformans
Collectively the data supported a central role for EphA2 in mediating an association of C. neoformans with the host. However, in order to establish whether EphA2 acted directly to internalize C. neoformans, a cell protection assay was performed (Fig 9). In this assay, HEK293T cells overexpressing EphA2 were exposed to C. neoformans for 1.5h, subsequently washed away and replaced with fluconazole, a static antifungal drug. Prior to this step, fluconazole activity was monitored to ensure HEK293T cells remained viable while fungal cells were susceptible (Fig 9C, 9D). If fungal cells had been internalized by the HEK293T they would not be exposed to fluconazole and therefore would be free to replicate. Eventually the HEK293T cells-containing C. neoformans could be lysed and the contents plated for CFU determination (Fig 9A). We detected significantly more CFUs from HEK293T overexpressing EphA2 than HEK293T cells alone (transformed with an empty plasmid), suggesting that EphA2 was directly responsible for the internalization of C. neoformans (Fig 9B).
Fig 9. EphA2 is responsible for the internalization of C. neoformans in HEK293T that overexpress EphA2.

(A) To establish whether EphA2 acted directly to internalize C. neoformans a cell protection assay was performed. HEK293T cells overexpressing EphA2 were exposed to C. neoformans for 1.5h, subsequently washed away and replaced with fluconazole (15μg/ml), a static antifungal drug. Following a further 48h co-incubation where internalized C. neoformans was protected from fluconazole and allowed to replicate, HEK293T cells were lysed and plated for CFU determination (B) Significantly more CFUs from HEK293T overexpressing EphA2 than HEK293T cells alone (transformed with an empty plasmid) were detected, suggesting that EphA2 was directly responsible for the internalization of C. neoformans. (C & D) Prior to the assay, fluconazole activity was monitored to ensure HEK293T cells remained viable and fungal cells were susceptible.
EphA2-mediated transmigration of C. neoformans may be dependent on CD44
It was previously shown that the down regulation of the CD44 receptor reduces the migration of C. neoformans across brain endothelial cells. [[21]22] To investigate the role of CD44 in the EphA2-mediated transmigration of C. neoformans, the cps1Δ deletion strain [70] was co-incubated with recombinant ephrinA1 or doxazosin for 6 h in transcytosis assays. The Cps1 gene encodes hyaluronic acid synthase and deletion of Cps1 produces several phenotypes including a decreased ability to associate with brain endothelial cells. This defect is likely due to reduced hyaluronic acid, which serves as a ligand for CD44.[23] We found that in contrast to the wild type H99 strain, significantly less cps1Δ cryptococci crossed the BBB as previously reported, (Fig 10A–D)[23] and the transmigration of cps1Δ was not enhanced by the addition of ephrinA1 or doxazosin (Fig 10A, B). Interestingly, blocking the EphA2 receptor with a monoclonal antibody or the inhibitor dasatinb, completely prevented all crossing of cps1Δ cryptococci (Fig 10C, D). We confirmed that the reduced migration of the cps1Δ strain was not due to a growth defect of the cps1Δ strain in endothelial medium (Fig 10E) and we established that the integrity of the in vitro BBB was maintained throughout the transcytosis assays (Fig 10F, G, H). Collectively the data is consistent with the established role of CD44[21,22] but further implicates CD44 in the EphA2-mediated transmigration and supports the notion that CD44 must be engaged to promote EphA2 activity.
Fig 10. EphA2-mediated transmigration of C. neoformans may involve CD44.

(A) Addition of human recombinant ephrinA1 (EFNA1, Origene) to brain endothelial cells did not rescue the crossing defect of the C. neoformans cps1Δ deletion strain. Two concentrations of EFNA1 (1 and 2 μg/mL) were co-incubated with 2×105 cells of C. neoformans cells and added to the top chamber of the transwell in the in vitro BBB model. Following 6h co-incubation, fungal cells were collected from the bottom chamber and placed onto agar plates for CFU determination, n=8 (B) Transcytosis assays with the EphA2 agonist, doxazosin, did not enhance crossing of the cps1Δ strain. DMSO is the solvent control. Brain endothelial cells were exposed to 100μM doxazosin and co-incubated with 2×105 cells of C. neoformans. Following 6h, fungal cells were collected from bottom chamber placed onto agar plates for CFU determination, n=5. (C) Brain endothelial cells were treated with EphA2-mAb (125μg) for 45mins and subsequently challenged with C. neoformans. Following a 3h co-incubation in the in vitro BBB model, transcytosis assays showed reduced crossing of WT and cps1Δ cryptococci, n= 8. (D) Addition of 10μm dasatinib to brain endothelial cells in the in vitro BBB model reduced crossing of WT and cps1Δ cryptococci, n= 5. (F, G, H) Barrier integrity was monitored by dextran permeability during treatments of brain endothelial cells with the antibodies (E) The defect in the transmigration of the cps1Δ strain is not due to growth inhibition in the endothelial cell culture medium. *P<0.05, **P<0.01, ***P<0.0001, n/s = not significant
Discussion
We have identified 5 signal transduction pathways in human brain endothelial cells that are targeted by C. neoformans. The EPH-ephrinA1 receptor, axonal guidance (related to EPH-signaling)[41], RhoGDI, CXCR4 and IL-8 (CXCL8) signaling pathways are significant because they contribute to an altered permeability of the BBB that can be transient or long-lasting.[42] CXCR4 is a GPCR (G-protein-coupled receptor) that selectively binds the chemokine ligand, CXCL12 (also known as Stromal Cell-Derived Factor 1 (SDF-1)), and subsequently activates signaling events that produce several biological responses including gene transcription, cell adhesion and cell migration.[43] Studies have indicated that the brain endothelium serves as a source for CXCL12 and its receptor (CXCR4) suggesting that CXCL12 can recruit CXCR4+ circulating lymphocytes and also act as a potential feedback mechanism for CXCR4 expression at the BBB.[44] This is consistent with our results where C. neoformans induced the expression of the CXCL12 transcript and promoted CXCR4-mediated signaling in brain endothelial cells. This finding is particularly interesting because it further supports the notion that C. neoformans might also transmigrate the BBB by a Trojan horse mechanism via infected phagocytes.[19,20,45]
IL-8 (interleukin-8), found pre-stored in microvascular endothelial cells, also stimulates G-protein-coupled signal transduction pathways that promote integrin-mediated adhesion of neutrophils.[46] High levels of IL-8 were found in CSF of patients with cryptococcal meningitis (CM) and some evidence suggested the cryptococcal capsular polysaccharide glucuronoxylomannan (GXM) induced IL-8 production in the brain.[47] A recent study found that endothelin-1, a potent vasoconstrictor, induced the production of IL-8 in human brain-derived endothelial cells and both altered BBB permeability.[42,48] We found that C. neoformans increased transcript levels of endothelin-1 & 2 and EDNRA (receptor for endothelin-1) consistent with the induction of IL-8-mediated signaling pathway.
The RNAseq analysis revealed several interesting aspects of how brain endothelial cells perceive and respond to fungal challenge. Our data points to changes in transcripts that would suggest significant endothelial cell stress such as the upregulation of the amyloid-beta precursor protein. A recent study demonstrated that amyloid-beta may function as an antimicrobial peptide and thus accumulate in the brain parenchyma in response to microbial infection.[6] We also found that several transcripts related to the immune response were also upregulated. For example, NPTX1, localized exclusively to the nervous system, belongs to the pentraxins, which are a class of multifunctional pattern recognition receptors involved in acute immunological responses.[49] Collectively, the set of defense-related transcripts we observed may suggest that that the BBB itself might contribute to a neuroimmune response to fungal cells by the production of inflammatory mediators or the expression of adhesion molecules – this would likely lead to endothelial cell stress and ultimately a weakened BBB.[50]
In this study we made several compelling observations that support the notion that transcytosis of C. neoformans across the BBB, is dependent on the EphA2-mediated internalization of C. neoformans by the BBB: 1) silencing the EphA2 transcript or blocking EphA2 activity with an antibody or chemical inhibitor prevented the transmigration of C. neoformans in the in vitro model of the BBB; 2) treating brain endothelial cells with an EphA2 chemical agonist (doxazosin) or an EphA2 ligand (ephrinA1) promoted greater migration of fungal cells across the BBB; 3) co-incubation of C. neoformans with brain endothelial cells led to the phosphorylation of EphA2 - this activation promoted clustering and internalization of EphA2 receptors that co-localized with F-actin and Cryptococci; and 4) expression of EphA2 in a different mammalian cell type, HEK293T, resulted in a direct interaction between EphA2 and C. neoformans that led to an increase in the internalization of C. neoformans and 5) EphA2-mediated transmigration of C. neoformans involves CD44. Collectively, the results suggest that C. neoformans promotes EphA2 activity via CD44, and this in turn facilitates the migration of C. neoformans across the BBB via EphA2-mediated signaling events that subsequently lead to a permeable barrier.
The binding of EphA2 to its ephrinA1 ligand induces receptor dimerization and subsequent trans-phosphorylation through the cytoplasmic domains of the receptors.[27] Interestingly, we found that silencing the ephrinA1 ligand in brain endothelial cells did not affect the transmigration of C. neoformans suggesting that the effect of EphA2 on fungal entry might be ligand-independent; however, we also observed significant increases in the traversal of cryptococci upon the addition of the ligand ephrinA1 and the agonist, doxasozin. Based on these results it would appear that the EphA2-dependent entry of cryptococci is both ligand independent and ligand-dependent. Although this notion is consistent with the role of EphA2 in the entry of hepatitis C virus as well as other defined functions of EphA2, we cannot rule out the possibility that other ephrin ligands may have bound EphA2 in the absence of ephrinA1 due to their promiscuous nature. [51,52]
EphA2 activity has been implicated in the reorganization of the actin cytoskeleton through activities of GTPases such as Rho, Rac and Cdc42.[52] Rho and Rac, both members of Ras-related superfamily of small GTPases, regulate actin polymerization to produce stress fibers and lamellipodia, respectively.[53,54] Cdc42, another member of the Rho family, triggers formation of actin-based protrusions called filopodia. [53,54] Similar membrane structures stemming from brain endothelial cells and embracing cryptococci have been observed and are likely required for internalization of C. neoformans. [9–11,18] Studies have demonstrated that internalization and transcellular movement of C. neoformans across brain endothelial cells depends on actin reorganization that is regulated by the activities of Rho-GTPases (RhoA, Rac1, and Cdc42) FAK, ezrin, and PKCα in the host.[16]
In the RNAseq analysis we identified 27 transcripts related to cell signaling, including a Rho-GTPase activating protein 9 (Rho-GAP9), Rho-GEF4 and phospholipase A2. Interestingly, the RhoGAP 9 has substantial GAP activity towards Cdc42, Rac1 and RhoA[55,56] thus it may serve as a master regulator in brain endothelial cells challenged with C. neoformans. Once activated, downstream events would include cytoskeleton remodeling[57] which is consistent with our observations of altered cytoskeleton-related transcripts. Thus our data strongly support the involvement of EphA2-mediated signaling events during the internalization and transcytosis of C. neoformans by brain endothelial cells. Based on these studies we propose that by activating EphA2-mediated signaling, C. neoformans can take advantage of the increased permeability of the BBB that results from changes in the cytoskeleton and the concomitant increase in endocytotic events.
Interestingly, some reports have suggested that EphA2 activation can increase paracellular permeability in endothelial cells by altering both tight and adherens junctions. [58–61] Although the traversal of cryptococci in the in vitro model of the BBB did not appear to breakdown intercellular junctions of the barrier, we detected changes in expression of transcripts that mediate endothelial cell connections (claudin, cadherin) including vasogenic agents (endothelin-1 & 2), thus suggesting an altered BBB permeability. We have found that longer co-incubation times produce lower TEER values and greater dextran permeability suggesting that the observed hyper-permeability of the BBB may be a result of a weakened barrier. These observations are consistent with a previous study where we demonstrated that human brain endothelial cells challenged with C. neoformans can undergo significant molecular and cellular changes indicative of BBB injury.[15] These changes included the translocation of HMGB1, a marker of cell injury, the upregulation of cyclophilinA, and the downregulation of proteins that function in energy production, protein processing and transcription.[15] In addition, studies have shown that cryptococcal infection can cause disruption and remodeling of tight junction proteins, ZO-1 and occludin.[23,62] Furthermore, histological analysis of blood-to-brain invasion of cryptococci demonstrated capillary damage where the presence of fungal cells in small capillaries led to structural changes in microvessel walls next to ruptured microcapillaries.[63] This is consistent with in vivo studies that found the BBB significantly breached by 24h post-infection as demonstrated by leakage of horseradish peroxidase.[64] The crossing of the BBB occurred in the cortical capillaries and caused severe damage to the microvessels.[64]
A recent in vivo study demonstrated that EphA2 receptor activation worsened ischemic brain injury in mice by promoting BBB damage. [65] Mice lacking EphA2 faired much better suggesting that the absence or inactivation of EphA2 activity may serve a protective role thus making EphA2 an attractive drug target. [65–67] In the case of C. neoformans, prolonged activity of EphA2 could conceivably alter and/or weaken endothelial cell junctions and thereby promote paracellular permeability. Given that patients with meningoencephalitis caused by C. neoformans eventually experience significant brain edema, the movement of excess fluid into the brain is likely occurring via disrupted tight junctions in a weakened barrier and thus fungal cells may ultimately take advantage of this opening into the brain. The notion that EphA2 could function to couple an initial transcytosis event with an eventual paracellular opening is intriguing and worth further exploration.
The roles of EphA2 and CD44 in promoting the transcellular migration of C. neoformans across the BBB may not be incongruous since it is conceivable that both receptors may work in unison. Indeed both CD44 and EphA2 have been implicated in regulating the endothelial cell barrier and angiogenesis.[68] A recent study found that treating endothelial cells with low molecular weight hyaluronan induced the association of CD44v10 (a variant of CD44) with EphA2 and the concomitant activation of EphA2 via phosphorylation thus suggesting a crosstalk between EphA2 and CD44.[68] Furthermore, the CD44-EphA2 complex facilitated recruitment of several proteins needed to induce RhoA activation during angiogenesis.[68]
In the case of C. neoformans, the inability of the cps1Δ strain to induce the phosphorylation of EphA2 or to cross the BBB upon stimulating EphA2 supports the notion that CD44 must be engaged in order to promote the EphA2-mediated transmigration of cryptococci. The working model (Fig 11) proposes that C. neoformans associates with CD44 on the luminal (apical) side of the brain endothelium and induces EphA2 phosphorylation via CD44-mediated transactivation of EphA2. Interestingly, despite the lack of Cps1, some transmigration of cps1Δ cryptococci still occurred, albeit to a lesser extent. This may reflect an incomplete lack of hyaluronan in the cps1Δ strain or alternatively a CD44-independent transmigration path may also exist. Once activated, EphA2 promotes GTPase-dependent signaling that reorganizes the actin cytoskeleton and internalizes C. neoformans via endocytosis. Subsequently the transcellular movement of C. neoformans across brain endothelial cells would be dependent on the activity of Annexin A2 (AnxA2) and other signaling events.[69] Sustained EphA2 activation could alter the integrity of the BBB by weakening intercellular junctions thereby boosting further entry of C. neoformans and fluid, resulting in brain edema.[59–61,65]
Fig 11. Working model of EphA2-dependent migration of C. neoformans across the BBB.
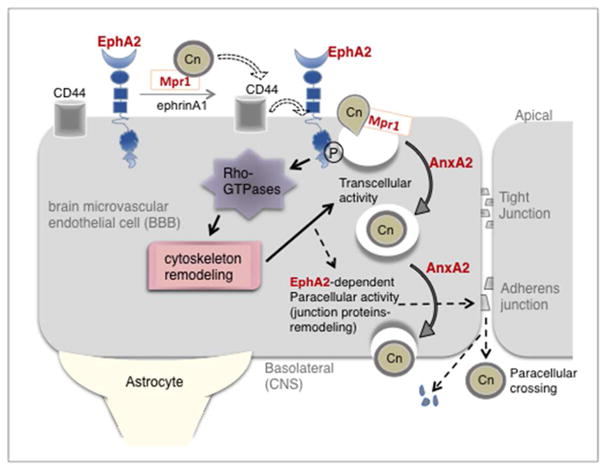
C. neoformans associates directly with the luminal side of the brain endothelium and induces EphA2 phosphorylation through transactivation of CD44 bound to C. neoformans. This promotes GTPase-dependent signaling that reorganizes the actin cytoskeleton and internalizes C. neoformans via endocytotic and transcellular mechanisms that require Mpr1 and Annexin A2 (AnxA2).[18,69] Sustained EphA2 activation could weaken intercellular junctions thereby increase paracellular permeability and boost further entry of C. neoformans along with excess fluid that would lead to brain edema (indicated by dashed arrows).
In summary, this study makes a significant contribution to resolving the complicated [60]tactics used by fungal pathogens to breach the BBB; it builds on what we understand so far and also opens up new lines of investigation that include a potential translational application. It is worth noting that EphA2 might serve as a potential target for small molecules or drugs that could be used to prevent fungal meningoencephalitis. Given the use of the lateral flow assay for Cryptococcal antigen detection in under 10 min in a symptom-free HIV+ population, drugs given to prevent fungal crossing into the brain could significantly reduce CM-related deaths.
Supplementary Material
Acknowledgments
We are grateful to members of the Gelli lab – in particular Dr. K. Vu who assisted with the quantitative PCR studies. We are also grateful to the DNA Technologies & Expression Analysis Core and the Advanced Imaging Facility at UC Davis. We are grateful to the National Institutes of Health for providing funding.
Footnotes
Conflict of Interest
The authors have no conflict of interest to declare.
References
- 1.Ueno N, Lodoen MB. From the blood to the brain: avenues of eukaryotic pathogen dissemination to the central nervous system. Curr Opin Microbiol. 2015;26:53–59. doi: 10.1016/j.mib.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grab DJ, Chakravorty SJ, van der Heyde H, Stins MF. How can microbial interactions with the blood-brain barrier modulate astroglial and neuronal function? Cell Microbiol. 2011;13:1470–1478. doi: 10.1111/j.1462-5822.2011.01661.x. [DOI] [PubMed] [Google Scholar]
- 3.Tauber SC, Eiffert H, Kellner S, Lugert R, Bunkowski S, et al. Fungal encephalitis in human autopsy cases is associated with extensive neuronal damage but only minimal repair. Neuropath Appl Neuro. 2014;40:610–627. doi: 10.1111/nan.12044. [DOI] [PubMed] [Google Scholar]
- 4.Pisa D, Alonso R, Rabano A, Rodal I, Carrasco L. Different Brain Regions are Infected with Fungi in Alzheimer’s Disease. Sci Rep. 2015;5:15015. doi: 10.1038/srep15015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, et al. The Alzheimer’s Disease-Associated Amyloid beta-Protein Is an Antimicrobial Peptide. PloS one. 2010:5. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8:340ra372. doi: 10.1126/scitranslmed.aaf1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, et al. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis. 2017;17:873–881. doi: 10.1016/S1473-3099(17)30243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyrgos V, Seitz AE, Steiner CA, Prevots DR, Williamson PR. Epidemiology of cryptococcal meningitis in the US: 1997–2009. PloS one. 2013;8:e56269. doi: 10.1371/journal.pone.0056269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vu K, Weksler B, Romero I, Couraud PO, Gelli A. Immortalized human brain endothelial cell line HCMEC/D3 as a model of the blood-brain barrier facilitates in vitro studies of central nervous system infection by Cryptococcus neoformans. Eukaryotic cell. 2009;8:1803–1807. doi: 10.1128/EC.00240-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang YC, Stins MF, McCaffery MJ, Miller GF, Pare DR, et al. Cryptococcal yeast cells invade the central nervous system via transcellular penetration of the blood-brain barrier. Infect Immun. 2004;72:4985–4995. doi: 10.1128/IAI.72.9.4985-4995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen SH, Stins MF, Huang SH, Chen YH, Kwon-Chung KJ, et al. Cryptococcus neoformans induces alterations in the cytoskeleton of human brain microvascular endothelial cells. J Med Microbiol. 2003;52:961–970. doi: 10.1099/jmm.0.05230-0. [DOI] [PubMed] [Google Scholar]
- 12.Chretien F, Lortholary O, Kansau I, Neuville S, Gray F, et al. Pathogenesis of cerebral Cryptococcus neoformans infection after fungemia. J Infect Dis. 2002;186:522–530. doi: 10.1086/341564. [DOI] [PubMed] [Google Scholar]
- 13.Jong A, Wu CH, Prasadarao NV, Kwon-Chung KJ, Chang YC, et al. Invasion of Cryptococcus neoformans into human brain microvascular endothelial cells requires protein kinase C-alpha activation. Cell Microbiol. 2008;10:1854–1865. doi: 10.1111/j.1462-5822.2008.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi M, Li SS, Zheng C, Jones GJ, Kim KS, et al. Real-time imaging of trapping and urease-dependent transmigration of Cryptococcus neoformans in mouse brain. J Clin Invest. 2010;120:1683–1693. doi: 10.1172/JCI41963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vu K, Eigenheer RA, Phinney BS, Gelli A. Cryptococcus neoformans Promotes Its Transmigration into the Central Nervous System by Inducing Molecular and Cellular Changes in Brain Endothelial Cells. Infection and Immunity. 2013;81:3139–3147. doi: 10.1128/IAI.00554-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JC, Crary B, Chang YC, Kwon-Chung KJ, Kim KJ. Cryptococcus neoformans activates RhoGTPase proteins followed by protein kinase C, focal adhesion kinase, and ezrin to promote traversal across the blood-brain barrier. The Journal of biological chemistry. 2012;287:36147–36157. doi: 10.1074/jbc.M112.389676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu TB, Kim JC, Wang Y, Toffaletti DL, Eugenin E, et al. Brain inositol is a novel stimulator for promoting Cryptococcus penetration of the blood-brain barrier. PLoS pathogens. 2013;9:e1003247. doi: 10.1371/journal.ppat.1003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vu K, Tham R, Uhrig JP, Thompson GR, 3rd, Na Pombejra S, et al. Invasion of the central nervous system by Cryptococcus neoformans requires a secreted fungal metalloprotease. mBio. 2014;5:e01101–01114. doi: 10.1128/mBio.01101-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santiago-Tirado FH, Onken MD, Cooper JA, Klein RS, Doering TL. Trojan Horse Transit Contributes to Blood-Brain Barrier Crossing of a Eukaryotic Pathogen. mBio. 2017:8. doi: 10.1128/mBio.02183-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charlier C, Nielsen K, Daou S, Brigitte M, Chretien F, et al. Evidence of a role for monocytes in dissemination and brain invasion by Cryptococcus neoformans. Infect Immun. 2009;77:120–127. doi: 10.1128/IAI.01065-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jong A, Wu CH, Gonzales-Gomez I, Kwon-Chung KJ, Chang YC, et al. Hyaluronic acid receptor CD44 deficiency is associated with decreased Cryptococcus neoformans brain infection. The Journal of biological chemistry. 2012;287:15298–15306. doi: 10.1074/jbc.M112.353375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jong A, Wu CH, Shackleford GM, Kwon-Chung KJ, Chang YC, et al. Involvement of human CD44 during Cryptococcus neoformans infection of brain microvascular endothelial cells. Cell Microbiol. 2008;10:1313–1326. doi: 10.1111/j.1462-5822.2008.01128.x. [DOI] [PubMed] [Google Scholar]
- 23.Jong A, Wu CH, Chen HM, Luo F, Kwon-Chung KJ, et al. Identification and characterization of CPS1 as a hyaluronic acid synthase contributing to the pathogenesis of Cryptococcus neoformans infection. Eukaryotic cell. 2007;6:1486–1496. doi: 10.1128/EC.00120-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nassif X, Bourdoulous S, Eugene E, Couraud PO. How do extracellular pathogens cross the blood-brain barrier? Trends Microbiol. 2002;10:227–232. doi: 10.1016/s0966-842x(02)02349-1. [DOI] [PubMed] [Google Scholar]
- 25.Eugene E, Hoffmann I, Pujol C, Couraud PO, Bourdoulous S, et al. Microvilli-like structures are associated with the internalization of virulent capsulated Neisseria meningitidis into vascular endothelial cells. J Cell Sci. 2002;115:1231–1241. doi: 10.1242/jcs.115.6.1231. [DOI] [PubMed] [Google Scholar]
- 26.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 28.Subbarayal P, Karunakaran K, Winkler AC, Rother M, Gonzalez E, et al. EphrinA2 Receptor (EphA2) Is an Invasion and Intracellular Signaling Receptor for Chlamydia trachomatis. PLoS pathogens. 2015:11. doi: 10.1371/journal.ppat.1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dutta D, Chakraborty S, Bandyopadhyay C, Valiya Veettil M, Ansari MA, et al. EphrinA2 regulates clathrin mediated KSHV endocytosis in fibroblast cells by coordinating integrin-associated signaling and c-Cbl directed polyubiquitination. PLoS pathogens. 2013;9:e1003510. doi: 10.1371/journal.ppat.1003510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carter N, Nakamoto T, Hirai H, Hunter T. EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas) Nat Cell Biol. 2002;4:565–573. doi: 10.1038/ncb823. [DOI] [PubMed] [Google Scholar]
- 31.Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 32.Petty A, Myshkin E, Qin H, Guo H, Miao H, et al. A small molecule agonist of EphA2 receptor tyrosine kinase inhibits tumor cell migration in vitro and prostate cancer metastasis in vivo. PloS one. 2012;7:e42120. doi: 10.1371/journal.pone.0042120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, et al. Ligand recognition by A-class Eph receptors: crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex. EMBO Rep. 2009;10:722–728. doi: 10.1038/embor.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 35.Weksler B, Romero IA, Couraud PO. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS. 2013;10:16. doi: 10.1186/2045-8118-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu OW, Chun CD, Chow ED, Chen C, Madhani HD, et al. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 2008;135:174–188. doi: 10.1016/j.cell.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poller B, Gutmann H, Krahenbuhl S, Weksler B, Romero I, et al. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. Journal of neurochemistry. 2008;107:1358–1368. doi: 10.1111/j.1471-4159.2008.05730.x. [DOI] [PubMed] [Google Scholar]
- 38.Chang Q, Jorgensen C, Pawson T, Hedley DW. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br J Cancer. 2008;99:1074–1082. doi: 10.1038/sj.bjc.6604676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang XD, Reeves K, Luo FR, Xu LA, Lee F, et al. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: rationale for patient selection and efficacy monitoring. Genome Biol. 2007;8:R255. doi: 10.1186/gb-2007-8-11-r255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fang WB, Brantley-Sieders DM, Hwang Y, Ham AJ, Chen J. Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. The Journal of biological chemistry. 2008;283:16017–16026. doi: 10.1074/jbc.M709934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein R. Eph/ephrin signalling during development. Development. 2012;139:4105–4109. doi: 10.1242/dev.074997. [DOI] [PubMed] [Google Scholar]
- 42.Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol. 2008;6:179–192. doi: 10.2174/157015908785777210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. 2007;1768:952–963. doi: 10.1016/j.bbamem.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. 2011;1812:220–230. doi: 10.1016/j.bbadis.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorrell TC, Juillard PG, Djordjevic JT, Kaufman-Francis K, Dietmann A, et al. Cryptococcal transmigration across a model brain blood-barrier: evidence of the Trojan horse mechanism and differences between Cryptococcus neoformans var. grubii strain H99 and Cryptococcus gattii strain R265. Microbes Infect. 2016;18:57–67. doi: 10.1016/j.micinf.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 46.Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol. 2014;10:593–619. doi: 10.1586/1744666X.2014.894886. [DOI] [PubMed] [Google Scholar]
- 47.Lipovsky MM, Gekker G, Hu S, Ehrlich LC, Hoepelman AI, et al. Cryptococcal glucuronoxylomannan induces interleukin (IL)-8 production by human microglia but inhibits neutrophil migration toward IL-8. J Infect Dis. 1998;177:260–263. doi: 10.1086/517368. [DOI] [PubMed] [Google Scholar]
- 48.Hofman FM, Chen P, Jeyaseelan R, Incardona F, Fisher M, et al. Endothelin-1 induces production of the neutrophil chemotactic factor interleukin-8 by human brain-derived endothelial cells. Blood. 1998;92:3064–3072. [PubMed] [Google Scholar]
- 49.Foo SS, Reading PC, Jaillon S, Mantovani A, Mahalingam S. Pentraxins and Collectins: Friend or Foe during Pathogen Invasion? Trends Microbiol. 2015;23:799–811. doi: 10.1016/j.tim.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Vries HE, Kuiper J, de Boer AG, Van Berkel TJ, Breimer DD. The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev. 1997;49:143–155. [PubMed] [Google Scholar]
- 51.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–U109. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitulescu ME, Adams RH. Eph/ephrin molecules--a hub for signaling and endocytosis. Genes Dev. 2010;24:2480–2492. doi: 10.1101/gad.1973910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 54.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 55.Peck J, Douglas Gt, Wu CH, Burbelo PD. Human RhoGAP domain-containing proteins: structure, function and evolutionary relationships. FEBS Lett. 2002;528:27–34. doi: 10.1016/s0014-5793(02)03331-8. [DOI] [PubMed] [Google Scholar]
- 56.Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- 57.Furukawa Y, Kawasoe T, Daigo Y, Nishiwaki T, Ishiguro H, et al. Isolation of a novel human gene, ARHGAP9, encoding a rho-GTPase activating protein. Biochem Biophys Res Commun. 2001;284:643–649. doi: 10.1006/bbrc.2001.5022. [DOI] [PubMed] [Google Scholar]
- 58.Larson J, Schomberg S, Schroeder W, Carpenter TC. Endothelial EphA receptor stimulation increases lung vascular permeability. Am J Physiol Lung Cell Mol Physiol. 2008;295:L431–439. doi: 10.1152/ajplung.90256.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miao Z, Dong Y, Fang W, Shang D, Liu D, et al. VEGF increases paracellular permeability in brain endothelial cells via upregulation of EphA2. Anat Rec (Hoboken) 2014;297:964–972. doi: 10.1002/ar.22878. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka M, Kamata R, Sakai R. EphA2 phosphorylates the cytoplasmic tail of Claudin-4 and mediates paracellular permeability. The Journal of biological chemistry. 2005;280:42375–42382. doi: 10.1074/jbc.M503786200. [DOI] [PubMed] [Google Scholar]
- 61.Fang WB, Ireton RC, Zhuang G, Takahashi T, Reynolds A, et al. Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J Cell Sci. 2008;121:358–368. doi: 10.1242/jcs.017145. [DOI] [PubMed] [Google Scholar]
- 62.Liu T-B, Kim J-C, Wang Y, Toffaletti DL, Eugenin E, et al. Brain inositol is a novel stimulator for promoting Cryptococcus penetration of the blood-brain barrier. PLoS pathogens. 2013;9:e1003247. doi: 10.1371/journal.ppat.1003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olszewski MA, Noverr MC, Chen GH, Toews GB, Cox GM, et al. Urease expression by Cryptococcus neoformans promotes microvascular sequestration, thereby enhancing central nervous system invasion. Am J Pathol. 2004;164:1761–1771. doi: 10.1016/S0002-9440(10)63734-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charlier C, Chretien F, Baudrimont M, Mordelet E, Lortholary O, et al. Capsule structure changes associated with Cryptococcus neoformans crossing of the blood-brain barrier. Am J Pathol. 2005;166:421–432. doi: 10.1016/S0002-9440(10)62265-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thundyil J, Manzanero S, Pavlovski D, Cully TR, Lok KZ, et al. Evidence that the EphA2 receptor exacerbates ischemic brain injury. PloS one. 2013;8:e53528. doi: 10.1371/journal.pone.0053528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou N, Zhao WD, Liu DX, Liang Y, Fang WG, et al. Inactivation of EphA2 promotes tight junction formation and impairs angiogenesis in brain endothelial cells. Microvasc Res. 2011;82:113–121. doi: 10.1016/j.mvr.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 67.Barquilla A, Pasquale EB. Eph receptors and ephrins: therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2015;55:465–487. doi: 10.1146/annurev-pharmtox-011112-140226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lennon FE, Mirzapoiazova T, Mambetsariev N, Mambetsariev B, Salgia R, et al. Transactivation of the receptor-tyrosine kinase ephrin receptor A2 is required for the low molecular weight hyaluronan-mediated angiogenesis that is implicated in tumor progression. The Journal of biological chemistry. 2014;289:24043–24058. doi: 10.1074/jbc.M114.554766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Na Pombejra S, Salemi M, Phinney BS, Gelli A. The Metalloprotease, Mpr1, Engages AnnexinA2 to Promote the Transcytosis of Fungal Cells across the Blood-Brain Barrier. Front Cell Infect Microbiol. 2017;7:296. doi: 10.3389/fcimb.2017.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.