

Abstract
HIV-Nef activates the myeloid cell-typical tyrosine kinase Hck, but its molecular role in the viral life cycle is not entirely understood. We found that HIV plasma extracellular vesicles (HIV pEV) containing/10 proteases and Nef also harbor Hck, and analyzed its role in the context of HIV pEV secretion. Myeloid cells required Hck for the vesicle-associated release of ADAM17. This could be induced by the introduction of Nef and implied that HIV targeted Hck for vesicle-associated ADAM17 secretion from a myeloid compartment. The other contents of HIV-pEV, however, including miRNA and effector protein profiles, as well as the presence of haptoglobin suggested hepatocytes as a possible cellular source. HIV liver tissue analysis supported this assumption, revealing induction of Hck translation, evidence for ADAM protease activation and HIV infection. Our findings suggest that HIV targets Hck to induce pro-inflammatory vesicles release and identifies hepatocytes as a possible host cell compartment.
Keywords: Hck, Chronic HIV infection, Nef, ADAM17, Plasma extracellular vesicles, Liver, Hepatocytes, Myeloid cells
Highlights
-
•
Hck is found along with HIV Nef and ADAM17 in plasma extracellular vesicles (pEV) from HIV-infected individuals.
-
•
Hck is required for the secretion of ADAM17 via pEV from myeloid cells and hepatocytes.
-
•
Liver tissue from HIV infected individuals revealed induction of Hck expression and presence of Nef.
1. Introduction
One of the well documented effects of HIV Nef is the activation of the tyrosine kinase Hck by interaction of its PxxP motif with the Hck SH3 domain (Saksela et al., 1995). This opens an intramolecular lock and constitutes the first step in the activation of the kinase (Lee et al., 1995, Lee et al., 1996, Moarefi et al., 1997). While the PxxP motif seemed required for disease progression in animal models (Khan et al., 1998) and humans (Trible et al., 2007), the molecular role of Hck in the viral life cycle remained unclear (Saksela, 2011). Hck is expressed predominantly in myeloid cells but not T cells, the main host cell of HIV. It was therefore speculated that in T cells Nef may recruit another SH3 domain-containing protein; however, the identity of this protein is still a matter of debate (Saksela, 2011).
In transient transfection systems Nef-induced Hck activation dysregulated signaling at the Golgi apparatus (Hassan et al., 2009, Hiyoshi et al., 2012), consistent with reports suggesting that secretory membrane trafficking from the Golgi is regulated by Src kinases (Sallese et al., 2009). In line with such a function, Hck augmented production and release of pro-inflammatory cytokines like TNF (English et al., 1993, Ernst et al., 2002). ProTNF is processed also in Golgi-derived compartments after PMA stimulation or Nef-induced ADAM17 activation, and is secreted in vesicles via membrane protrusions (Ostalecki et al., 2016). Such protrusions were also observed following Hck activation (Carreno et al., 2002).
A number of reports demonstrated HIV replication in liver cells in vitro and in vivo (Blackard and Sherman, 2008, Cao et al., 1992, Cao et al., 1990, Housset et al., 1993, Tuyama et al., 2010) and HIV infection is frequently associated with liver disorders (Mendeni et al., 2011, Price et al., 2012). HIV-induced immunosuppression and the risk of liver-related death correlate strongly, and liver-related morbidity is the most frequent cause of death in chronic infection (Towner et al., 2012, Weber et al., 2006). At present, however, HIV liver infection is not considered to be relevant for disease pathogenesis.
We recently reported that the number of ADAM17/Nef-containing plasma extracellular vesicles (pEV or HIV pEV) is strongly upregulated in HIV infection. Their number did not decline during therapy and their ADAM17/Nef content correlated inversely with CD4 T cell counts. Hence identifying the compartment shedding HIV pEV was of considerable importance. Preliminary findings had suggested that they did not derive from T cells (Lee et al., 2016). In this report we aimed at identifying their cellular origin, analyzing the factor content in HIV pEV. The detection of Hck not only explained a possible role of the tyrosine kinase in HIV biology, but also pointed at myeloid cells and hepatocytes as the likely cellular origin of HIV pEV.
2. Materials and Methods
2.1. Cell Lines
Liver cell lines Huh7 and Sk-Hep-1 (kindly provided by P. Knolle, Technische Universität München) were grown in DMEM (Sigma-Aldrich) supplemented with 10% Fetal calf serum (FCS, Sigma-Aldrich) and 1% penicillin-streptomycin (Lonza). Sk-Hep1 cells were additionally maintained in 40 μM β-mercaptoethanol (Carl Roth). LX-2 cells were provided by SL. Friedman (Icahn School of Medicine) and cultured in DMEM high glucose (Life Technologies) supplemented with 2% FCS, 1% penicillin-streptomycin. All cells were grown at 37 °C under 5% CO2.
2.2. EV Depletion of FCS and Human Serum for Cell Culture
To assure that EV generated from cell culture were not contaminated by outside sources, heat inactivated FCS and human serum for medium supplementation were depleted of bovine EV by ultracentrifugation for 18 h at 110,000g, 4 °C before use.
2.3. Isolation and Purification of EV
EV purification was performed essentially as described previously (Lee et al., 2016). Briefly, supernatants were collected after 48 h and centrifuged for 20 min at 2000g, 30 min at 10,000g and ultra-centrifuged for 1 h at 100,000g. Pellets were resuspended in 35 ml PBS and centrifuged at 100,000g for 1 h. Pellets were resuspended in 100 μl PBS and considered as EV preparations.
For EV purification from patient samples, 30 ml blood plasma was diluted with 30 ml PBS and centrifuged for 30 min at 2000g, 45 min at 12000g and ultra-centrifuged for 2 h at 110,000g. Pellets were resuspended in 30 ml PBS and centrifuged at 110,000g for 1 h. Pellets were again resuspended in 100 μl PBS and considered as EV preparations. For further purification, EV were diluted in 2 ml of 2.5 M sucrose, 20 mM Hepes/NaOH, pH 7.4 and a linear sucrose gradient (2–0,25 M sucrose, 20 mM Hepes/NaOH pH 7.4) was layered on top of the EV suspension. The samples were then centrifuged at 210,000g for 15 h. Gradient fractions were collected and the refractive index was determined. Each fraction was diluted in 10 ml PBS and ultra-centrifuged for 1 h at 110,000g. Pellets were solubilized in SDS sample buffer or resuspended in 100 μl PBS and analyzed by immunoblotting or Cytokine/Chemokine/soluble Factor (CCF) protein array (see Supplementary information).
To validate our centrifugation-based pEV isolation protocol, we generated an EV spike-in control (from a stable cell line producing EV), containing an EBV-derived miRNA (BHRF1-2*) that was not found in human pEV-miRNAs, but was detectable by the miRNA microarray (Agilent). After spike-in, BHRF1-2* miRNA was readily detected with comparable efficiency in 4 different plasma samples (data not shown).
2.4. Peripheral Blood Mononuclear Cell (PBMC) Preparation
Leukoreduction system chambers (LRSCs) (Pfeiffer et al., 2013) from healthy donors were acquired after plateletpheresis. The resulting platelet free cell sample was diluted 1:2 in PBS and the PBMC containing buffy coat was isolated after density gradient centrifugation on Lymphoprep (Axix Shield 1114544) at 500g for 30 min at room temperature. PBMCs were then washed 3 times in PBS/1 mM EDTA; 1. wash: 282 g, 15 min, 4 °C; 2. wash: 190 g, 10 min, 4 °C; 3. wash: 115 g, 12 min, 4 °C.
2.5. Generation of Immature/Mature Dendritic Cells (DC)
PBMCs were isolated from LRSCs as described above, resuspended in 1 × BD IMag Buffer (BD Biosciences 552362) and counted. Monocytes were then isolated from 1.5 × 107 PBMCs using BD IMag Anti-Human CD14 Magnetic Particles (BD Biosciences 557769) according to the manufacturer's instructions. 6.0 × 106 monocytes per well were then seeded in a 6 well plate in RPMI supplemented with 1% heat inactivated human serum from human male AB plasma (Sigma-Aldrich). Monocyte-derived DC were generated supplementing the medium with 800 IU/ml of recombinant GM-CSF and 250 IU/ml of recombinant IL-4 (both from CellGenix) on day 1 after isolation and 400 IU/ml of recombinant GM-CSF and 250 IU/ml of recombinant IL-4 on days 3, 5 and 6. For EV isolation from immature DC, cells were washed with PBS on day 7 and 10 ml RPMI containing 1% of EV-depleted, heat-inactivated human serum and 1% of penicillin/streptomycin was added. After 24 h the supernatant was harvested. For EV isolation from mature DC, immature DC cultures were supplemented for 24 h with a maturation cocktail 200 IU/ml IL-1ß, 1000 IU/ml IL-6 (both from CellGenix), 10 ng/ml TNF (beromun; Boehringer Ingelheim) and 1 μg ml− 1 Prostin E2 (PGE2, Pfizer). Subsequently cells were washed 1 time with PBS and seeded in 10 ml of RPMI supplemented with 1% of heat-inactivated and EV-depleted serum and 1% of penicillin/streptomycin. After additional 24 h the supernatant was harvested. EV from immature and mature DC were purified as described above.
2.6. Generation of Macrophages
PBMCs were isolated from LRSCs as described above. Monocytes were separated from the non-adherent fraction (NAF) by plastic adherence on cell culture flasks and cultured in RPMI supplemented with 1% human serum and 1% of penicillin/streptomycin. On days 1, 3, 5, 7 and 9 after seeding, medium was supplemented with 800 IU/ml of GM-CSF. On day 11, medium was removed, cells were washed with PBS and 20 ml of RPMI supplemented with 1% of EV depleted human serum and 1% of penicillin/streptomycin was added. After 24 h supernatant was harvested and EV were isolated as described above.
2.7. Generation of Primary Myeloid Cells (Adherent PBMC)
PBMCs were isolated from LRSCs as described above. Monocytes were separated from the non-adherent fraction (NAF) by plastic adherence on cell culture flasks and cultured in RPMI supplemented with 1% human serum and 1% of penicillin/streptomycin. On day 1 after seeding, medium was supplemented with 800 IU/ml of recombinant GM-CSF and 250 IU/ml of recombinant IL-4 (both from CellGenix). After 24 h supernatant was harvested and EV were isolated as described above.
2.8. Nef Antibodies and Detection Reagents
Different anti-Nef antibodies and reagents were used: (1) anti-Nef JR6, a mouse monoclonal antibody (Abcam ab42358); (2) anti-Nef 2A3, a mouse monoclonal antibody (Abcam ab77172); (3 and 4) anti-Nef sheep serum, either as a purified biotinylated polyclonal antibody or non-labeled (both from Targeted Affinity Oy, Helsinki); (5) anti-Nef polyclonal serum (provided by Mark Harris, Leed University). All Nef-antibodies were used to demonstrate the presence of Nef in pEV. For immunoblotting JR6 turned out to have the highest sensitivity and specificity as judged by the ratio of Nef vs. background staining. For detection in tissue we used the biotinylated anti-Nef sheep serum and the JR6 antibody.
2.9. Antibodies
The following antibodies were used for immunostaining or immunoblotting: anti-ADAM10 (mouse monoclonal, Abcam ab73402), anti-ADAM10 (mouse monoclonal, Helmholtz Zentrum Munich), anti-ADAM17 (rabbit polyclonal, Cell Signaling 3976), anti-HCK (rabbit polyclonal, Santa Cruz, sc-72), anti-Gag p24 (mouse monoclonal, Abcam ab9071), anti-Gag p24 (mouse monoclonal, Abcam ab9044), anti-CD63 (mouse monoclonal, BD Biosciences 556019), anti-CD81 (mouse monoclonal, BD Biosciences 555675), anti-Tsg101 (mouse monoclonal, Santa Cruz Cruz sc-7964), anti-Vpu (rabbit polyclonal, Biozol FBX-VPU-101AP-100), anti-Vpr (rabbit polyclonal, NIH, provided by U. Schubert), anti-phopho Src (rabbit polyclonal, Cell signaling 2101), anti-Vinculin (rabbit polyclonal, Millipore AB6039), anti-Erk1/2 (rabbit polyclonal, Cell signaling 4695), anti-Fyn (rabbit polyclonal, Genetex 109428) anti-c-Yes (rabbit polyclonal, Genetex 100616), anti- Fgr (rabbit polyclonal, Genetex 102947), anti-Blk (rabbit polyclonal, Genetex 111546), anti-Lck (mouse monoclonal, Santa cruz sc-433), anti-Lyn (rabbit polyclonal, Cell signaling 2732), anti-β-actin (mouse monoclonal, Sigma-Aldrich A5316), Propidium iodide (Genaxxon bioscience, M3181.0010), DAPI (4′,6-diamidino-2-phenylindole, Biomol ABD-17510).
Primary antibodies were used at 1–2 μg ml− 1 for immunoblotting, 2 μg ml− 1 for immunofluorescence and 5–10 μg ml− 1 for FACS analysis. The following secondary antibodies were used: Alexa Fluor 488 goat anti-mouse and Alexa Fluor 555 goat anti-rabbit IgG (both from Life Technologies) and anti-mouse IgG-Biotin conjugate and anti-rabbit IgG-Biotin conjugate (LSAB +, Dako REAL Detection Systems, HRP/AEC, Rabbit).
2.10. DNA Constructs, Transfections and Protein Assays
Expression plasmids for Nef and Nef-cofactors (hnRNPK, PKCδ, Lck) were described previously (Lee et al., 2013). The HIV-Δenv (pNL-4.3Δenv (Clavel et al., 1989)) expression plasmid was kindly provided by U. Schubert (Department of Virology, University of Erlangen). Hck expression plasmids were kindly provided by Kalle Saksela. The p59 isoform of human Hck was provided with a Kozak consensus sequence and a kinase-inactive (K269N) derivative or an activated version lacking the C-terminal inhibitory tyrosine phosphorylation site (Y501F) was generated by PCR mutagenesis. The constructs were inserted into the pEBB-PP expression vector as BamHI-NotI (K269N) or BamHI-KpnI (Y501F) fragments. In the latter case this creates an in-frame fusion with a 123 aa biotin acceptor domain from Propionibacterium shermanii transcarboxylase. The GFP-proTNF-RFP fusion protein was described previously (Lee et al., 2013). For immunoblotting experiments, plasmids were transfected with Lipofectamine® LTX with Plus™ Reagent (Invitrogen) according to the manufacturer's instructions, or using the classical calcium phosphate procedure. Cells were analyzed 24–72 h after transfection. Immunoprecipitations were performed as described previously (Lee et al., 2013). In general 20 μg of cellular protein lysate and 10 μg of EV lysate were loaded per lane. The latter corresponded to the secretion from 2 to 4 mio Huh7 cells within 48 h.
2.11. Human Cytokine/Chemokine/Soluble Factor (CCF) Array
Purified EV from sucrose gradient fractions corresponding to equal volume of cellular supernatant (in general 60 ml from 10 mio. Transfected cells) or equal plasma volume were applied to the RayBio Human Cytokine Array C-S (Hölzel Diagnostika, AAH-CYT-1000-2) according to the manufacturer's instructions. A minimum of 20 μg EV proteins was used per filter incubation (see Supplement table S1).
2.12. ADAM17/α-Secretase Acitivity Assay
The assay was performed essentially as described previously (Lee et al., 2016) using a commercial, SensoLyte®520 α-Secretase Activity Assay Kit (AnaSpec 72085), according to the manufacturer's instructions. Briefly, we placed sucrose gradient purified pEV (the equivalent of 1 ml plasma) on a 96-well, black, flat bottom plate (Greiner 655900) and added a 5-FAM (fluorophore) and QXL™ 520 (quencher) labeled FRET peptide substrate for continuous measurement of enzyme activity. Upon cleavage of the FRET peptide by the active enzyme, the fluorescence of 5-FAM is recovered and continuously monitored at excitation/emission = 490 nm/520 nm by a preheated (37 °C) TECAN infinite M200 Pro plate reader.
2.13. Patient Material
Patient material was obtained from patients of the HIV clinic (headed by T. Harrer) at the Department of Medicine 3, University Hospital Erlangen. Plasma was drawn from patients after informed consent. At the time of sampling, non-viremic HIV patients were under ART for prolonged periods without detectable viral load. None of the patients had an active HBV or HCV virus infection at the time of blood - or tissue sampling (see also Supplement table S2). CD4 and CD8 counts (cells/μl blood) were determined by the Department of Medicine 3. In general, 6–7 ml of plasma was obtained from each individual per visit. Liver FFPE tissue samples were obtained from 3 non-viremic clinically healthy individuals under ART (see Supplement table S2). HIV spleen tissue was obtained from a non-viremic HIV-infected individual after resection for a non HIV-related condition. Skin tissue was obtained from two non-viremic individuals who had punch biopsies because of non-HIV-related skin conditions. A fourth liver sample and gut tissue was obtained from a non-viremic individual who died shortly after admission in 2015 due to liver failure (see Supplement table S2).
2.14. Immunostaining and Confocal Microscopy
For immunohistochemistry staining, FFPE samples were deparaffinized and antigen retrieval was achieved in Tris-EDTA buffer pH 9 at 100 °C for 30 min. For antigen detection the LSAB method (Dako REAL Detection Systems) was used, performed by the Dako Autostainer Plus.
For ICC analyses, transfected cells were cultured on glass slides for 2 h at 37 °C and subsequently fixed by 3% PFA for 30 min at room temperature followed by three washes with PBS/1% BSA. Cells were then permeabilized with 0.1% Triton X-100/1% BSA and immunostained by standard procedures (primary and secondary antibodies). Finally, the cells were washed 30 min with PBS/1% BSA and mounted with Fluoromount-G (SouthernBiotech). Slides were analyzed on a Zeiss Laser Scanning Microscope LSM780 equipped with the ZEN software (Carl Zeiss AG, Oberkochen, Germany).
For confocal microscopy analyses, fixed samples were imaged with a laser scanning confocal microscope (LSM780; Carl Zeiss AG, Oberkochen, Germany) equipped with a 63 × objective. For Alexa488 the illumination was set at 488 nm and emissions were collected between 506 and 583 nm. For Alexa555 the illumination was at 561 nm and emission collected between 574 and 667 nm. Detecting DAPI, illumination was set to 405 nm and emission collected between 410 and 495 nm.
2.15. Multi-Epitope Ligand Cartography (MELC) Technology
The MELC technology and it application for SPPL3 and ADAM protease detection has been described recently (Schubert et al., 2006). Briefly, a slide with a tissue specimen was placed on an inverted wide-field fluorescence microscope (Leica DM IRE2, Leica Microsystems, Wetzlar, Germany; × 20 air lens; numerical aperture, 0.7) fitted with fluorescence filters for fluorescein isothiocyanate and phycoerythrin. Fluorochrome-conjugated antibodies and wash solutions were added and removed robotically under temperature control, avoiding any displacement of the sample and objective. The repetitive cyclic process of this method includes the following steps: (a) fluorescence tagging, (b) washing, (c) imaging and (d) photo bleaching; phase-contrast and fluorescence images were recorded by a high-sensitivity cooled CCD camera (Apogee KX4, Apogee Instruments, Roseville, CA; 2048 × 2048 pixels; 2 × binning results in images of 1024 × 1024 pixels; final pixel size was 900 × 900 nm). Data acquisition was fully automated.
2.16. Transmission Electron Microscopy
The EV sample was fixed in 2% (w/v) paraformaldehyde in PBS at 4 °C overnight. Fixed EV were spread on carbon-coated 400-square-mesh copper grids (Electron Microscopy Sciences, Hatfield, PA, USA). After 20 min of incubation grids were washed with PBS and post-fixed with 2% glutaraldehyde (w/v) in PBS for 5 min. After a series of washing steps using distilled water, grids were incubated in a 3% aqueous solution of uranyl acetate (pH 4.5) that had been filtered through a 0.22 μm filter for 5 min. Grids were dried at room temperature and examined with a transmission electron microscope (Leo 912; Zeiss, Oberkochen, Germany).
To analyze pEV and EV by electron microscopy, we have purified these vesicles by different means, including iodixanol gradient (Optiprep) and antibody-coupled bead isolation. In all cases the vesicles appeared to have a similar structure and size as demonstrated in Supplement Fig. 3C.
2.17. Immunoisolation of pEV/EV by Magnetic Beads
Antibodies were coupled to magnetic microbeads by a Miltenyi Biotec (Bergisch Gladbach, Germany). For isolation of pEV, 2 ml blood plasma was diluted with 2 ml PBS and 50 μl of antibody-coupled beads were added for 1 h and subsequently subjected to magnetic immunoisolation with MACS® Technology (Miltenyi Biotec) using MS columns. To purify EV from cells, cell cultured supernatants were collected and were purified by combining differential centrifugation and column-based bead isolation. The vesicles were finally eluted with 45 μl of hot (95 °C) SDS sample buffer and all of the vesicle lysate was subsequently analyzed by western blot. The column flow-through was collected and centrifuged at 110,000g (pEV) or 100,000g (EV from cells) for 1 h. Pellets were solubilized in SDS sample buffer and analyzed by western blot.
2.18. DNA/RNA Extraction and PCR Amplification
DNA extraction from paraffin embedded liver tissues DNA extraction was done with NucleoSpin Tissue: Cat. No: 740952.250, according to the manufacturer's instructions. Subsequently, whole genome amplification was performed via Degenerate Oligonucleotid-Primed PCR (DOP-PCR) as previously described (Telenius et al., 1992). Reverse transcription of extracted pEV RNA was performed using the commercially available QantiTect Reverse Transcription kit (Qiagen, Cat. No: 205311).
For amplification of HIV LTR and pol sequences, the following specific primers were used in nested PCRs: LTR, outer primers: 9148s (sense) (CAA GGC TAC TTC CCT GAT TGG CA), 9148sG (CAA GGA TTC TTC CCA GAT TGG CA), 9148sD (CAA GGC TTC TTC CCT GAT TGG CA) 9545as (antisense) (GAG ACC CAG TAC AGG CAA AAA GC), 9545asG (GAG ACC CAG TAC AGG CGA GAA GC), 9545asA (GAG ACC CAG TAC AGG CGA AAA GC). LTR, nested primers: 9172s (AAC TAC ACA CCA GGG CCA GGG), 9172sA (AAT TAC ACA CCA GGG CCA GGG), 9172sC (AAC TAC ACA CCG GGA CCA GGG), 9519as (TGC TTA TAT GCA GCA TCT GAG GG), 9519asG (TGC TTA TAT GCA GCT TCT GAG GG).
Pol, outer primer: pol2249s (ATC ACT CTT TGG CAA CGA CC) pol2589as (TGG CCA TCC ATT CCT GG). Pol, nested primers: pol2372s (AAA AAT GAT AGG GGG AAT TGG), pol2557as (CTG GTA CAG TTC AAT AGG AC).
The PCR was performed with the Peqlab PeqSTAR 2 × thermocycler system using AmpliTaq DNA polymerase and the included buffer (Applied Biosystems) according to the manufacturer's instructions. PCRs were performed with the following cycling conditions: LTR outer and nested: 95 °C 3′/95 °C 3′/60 °C 40″/72 °C 45″//95 °C 1′/60 °C 40″/72 °C 45″ + 2″/cycle//× 29/72 °C 5′/4 °C storage. Pol outer and nested: 95 °C 2′//95 °C 30″/52 °C 40″/72 °C 90″ + 1′/cycle//× 30/72 °C 5′/4 °C storage. PCR products were detected on a 1% agarose gel using the Amersham Imager 600 system.
2.19. Statistical Analysis
GraphPad Prism and Microsoft Excel software were used for statistical analysis. The standard deviation of the mean was calculated using the Student's t-test.
3. Results
3.1. HIV pEV Contain Hck
In vitro, Nef-induced extracellular vesicles (EV) upload proteins from lipid rafts (Lee et al., 2013). We therefore speculated that pEV from patients may harbor a tyrosine kinase that could help to identify their cellular source. Purified pEV from 8 non-viremic patients and 3 healthy controls were blotted for 7 tyrosine kinases. Hck was identified in all HIV samples but not in controls (Supplement Fig. S1a and Fig. 1a, red box). Only in one case (HIV.08) also Yes and Fyn were found (Fig. 1a). In addition, a phopho-Src (p-Src) staining was detected, suggesting that the pEV-associated tyrosine kinase was activated. Using the HIV.08 plasma sample and a bead-coupled anti-αvβ5 antibody that isolates HIV pEV (Lee et al., 2016), we found that Hck, but not Yes, co-isolated with Nef and ADAM17 (Fig. 1b, red box). These results suggested that Nef-induced Hck activation was potentially connected to the generation and/or function of HIV pEV.
Fig. 1.

Hck is required for extracellular vesicle (EV)-associated secretion of ADAM17. (a–b) Hck is present in HIV pEV. (A) Blotting of Hck and indicated tyrosine kinases in pEV purified by differential centrifugation (from 2 ml plasma) from 2 non-viremic HIV patients (HIV.07/0.08) and one healthy control (cont.02). For blotting control the tyrosine kinases were transiently expressed in 293T cells. (b) Blotting of Hck, Yes and Nef in pEV from HIV.08 and a healthy donor (cont.03) isolated by anti-αvβ3-coupled magnetic beads. For blotting control lysates from NIH3T3 and 293T cells transfected with ADAM17 and Nef were used. Beads: bead-isolated pEV; FT: flow through. (c–e) Nef-activated Hck correlates with ADAM17 activation. (c) Blotting of indicated factors in lysates of transiently transfected 293T cells. NAKC: Nef-associated kinase complex (hnRNPK, Lck, PKCδ). (d) Immunoprecipitation of Hck from transiently transfected 293T cells as indicated, and blotting for p-Src. (e) Blotting of indicated factors in lysates of 293T cells stably expressing (stbl. exp.) p59 Hck, and transiently transfected (trans. transf.) as indicated (lower four panels). EV purified from the supernatants were blotted for ADAM17 and Tsg101 (upper two panels). (f–m) EV-associated ADAM17 secretion from myeloid cells requires Hck. (f) Blotting of indicated factors in lysates of immature and mature dendritic cells (iDC/mDC). (g) Blotting of indicated factors in lysates of iDC and mDC, treated/not treated with the tyrosine kinase inhibitor PP2. (h) Blotting of indicated factors in lysates of iDC, mDC, Jurkat and 293T cells, transiently transfected as indicated, or untreated (mDC). (j) Immunoprecipitation of Hck from Nef-transfected iDC and mDC and blotting for indicated factors. (k) Same experiment as shown in (H), blotting EV purified from culture supernatants for indicated factors. (l) Blotting of indicated factors in lysates of Nef-transfected macrophages. (m) Same experiment as shown in (l), blotting EV purified from culture supernatants. Colored boxes in all panels depict results that are described in the text.
3.2. Nef-Induced Hck Activation Correlates With ADAM17 Activation and Secretion
In view of its role in TNF secretion and Golgi signaling, we assumed an involvement of Hck in ADAM17 activation, the sheddase of TNF, and/or pEV-associated release. As expected, co-transfection of Hck with Nef activated the tyrosine kinase, evidenced by a shift to a slower migrating protein band (Fig. 1c, red box) and binding of the phospho-specific anti-Src family antibody (Fig. 1d, red box). This effect correlated with the appearance of an activated form of ADAM17 (Fig. 1c, blue box), which we previously observed only after co-expression of Nef and associated factors (Nef-associated kinase complex or NAKC; Fig. 1c, green boxes) (Lee et al., 2013).
Nef transfection into Hck-expressing 293T cells (p59 stable transfection) induced the uploading of activated ADAM17 into EV (Fig. 1e, red box). Conversely, a mutation in the Hck-interacting PxxP domain of Nef (P76/78A: Nef.AxxA) abolished this effect (blue box). Taken together, Hck activation by Nef correlated with ADAM17 activation and its pEV-associated release.
3.3. Myeloid Cells Require Hck for ADAM17 Activation and Secretion
We asked whether cells of the myeloid lineage, which express Hck, require the tyrosine kinase for ADAM17 activation and/or EV release. In mature DC (mDC), but not immature DC (iDC) (Supplement Fig. S2a and b), a p-Src reactive and shifted Hck protein band was detected, as well as activated ADAM17 and proTNF cleavage (Fig. 1f, red box). These effects were abolished in the presence of a tyrosine kinase inhibitor (PP2) (Fig. 1g, red box). An introduction of Nef into iDC induced ADAM17 activation and proTNF cleavage, while Jurkat T cells were non-responsive and contained no proTNF (Fig. 1h, compare red and blue box). In the same Nef-transfected iDC lysates an activation of Hck was verified, evidenced by protein band shift and p-Src reactivity (Fig. 1j, red box). Extracellular vesicles (EV) purified from Nef-transfected, but not vector-transfected iDC culture supernatants contained activated ADAM17 (Fig. 1k, red box), similar as EV from mDC without Nef transfection (blue box). No ADAM17 was found in Jurkat-derived EV (green box).
Similar observations were made in primary macrophages (Supplement Fig. S2c). Transfection of Nef activated ADAM17 (Fig. 1l, red box) and lead to the secretion of ADAM17-containig EV (Fig. 1m, red box), although the EV secretion activity of macrophages was rather low as judged by the (volume-adjusted) EV protein abundance from 293T cells and macrophages. Taken together, in DC and macrophages Hck activation correlated with ADAM17 activation and secretion, and the presence of Nef induced this mechanism.
3.4. HIV pEV Marker Profiles Implicate Hepatocytes as Their Cellular Source
To identify a putative myeloid compartment shedding HIV pEV, we compared the cytokine, chemokine and soluble factor (CCF) profile of EV secreted by primary myeloid cells from infected individuals with that from HIV pEV, which we described recently (Lee et al., 2016). Plastic adherent PBMC (mostly monocytes and iDC) from 5 non-viremic individuals cultured in vitro (72 h) secreted EV with a CCF pattern lacking almost all the factors found in HIV pEV (Fig. 2a, red box). Hence it seemed unlikely that HIV pEV derived from peripheral myeloid cells.
Fig. 2.
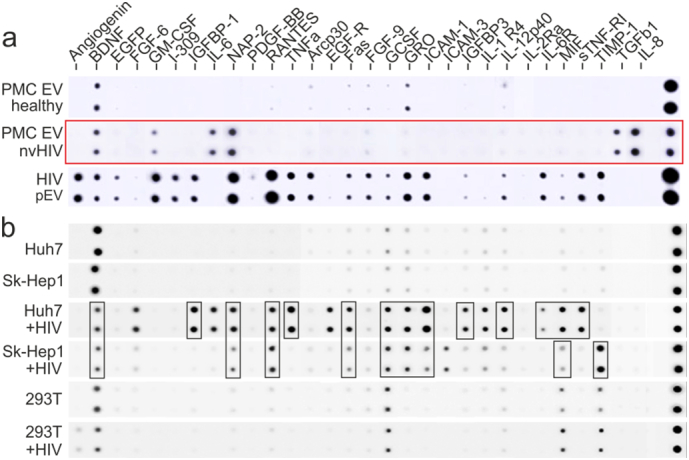
Marker profiles imply hepatocytes as a possible source of HIV pEV. (a) Protein array (RayBiotech) analyzing the content of cytokines, chemokines and soluble factors (CCF) in EV secreted by primary myeloid cells (PMC EV: plastic-adherent PBMC) from 5 infected non-viremic individuals and 5 healthy donors, cultured for 72 h (cultured separately, pooled for analysis). For comparison HIV pEV (15 ml plasma) from one non-viremic HIV patient was assessed as described recently 24. (b) CCF protein array analysis as in (B). EV were purified from 2 liver lines (Huh7, SkHep-1), and 293T cells. The cells were transfected for 72 h with a HIVΔenv construct or vector control (expression control in Supplement Fig. 4b). Assay input details are summarized in Supplement table S1. Colored boxes in panels depict results that are described in the text.
We noticed that the CCF content of HIV pEV contained many hepatocyte-typical inflammatory factors (Rowell et al., 1997), or factors predominantly secreted by liver cells (e.g. IGFBP-1 and 3 (Arany et al., 1994) and IL6-R (Desgeorges et al., 1997)) (Fig. 2a and (Lee et al., 2016)). We therefore compared the CCF profiles of EV derived from Huh7 and SK-Hep1 cells transfected with HIVΔenv or vector control. While vector-transfected cells produced EV with BDNF only, HIVΔenv-transfected cells secreted EV containing 20 different CCF, of which 15 matched those in HIV pEV (Fig. 2b, black boxes). For control, 293 T cells were transfected with HIVΔenv and produced EV with a greatly reduced CCF pattern. Together these result supported the assumption that liver cells were one possible source of HIV pEV.
3.5. HIV pEV Contain Liver-Typical Factors
For further clarification, we screened lysates of HIV pEV for liver-typical factors. We detected haptoglobin, an acute phase protein mainly secreted by liver cells (Yang et al., 2013). The factor was found in sucrose gradient fractions along with activated Hck and Nef (Fig. 3a, red box), but not in respective fractions of controls. Using anti-αvβ5 coupled beads, haptoglobin was co-isolated with ADAM17 and Nef from HIV pEV of 2 non-viremic patients with high and low CD4 count (Fig. 3b). Haptoglobin correlated in abundance with Nef (red boxes) and was not found in the flow through. Resting Huh7 cells did not express haptoglobin, however expression was induced after transfection of the HIVΔenv construct (Fig. 3c, red box). In addition, haptoglobin was then found in EV purified from these supernatants (blue box).
Fig. 3.

HIV pEV contain liver-typical factors. (a–b) Presence of haptoglobin in HIV pEV. (a) Western blot analysis of pEV purified by sucrose gradient from a non-viremic HIV patient (non-vir. HIV) and a healthy control. The individual fractions were blotted for indicated factors including haptoglobin (red box). Lys.: lysates of transfected 293T cells serving as positive control. (b) HIV pEV isolation from plasma of 2 non-viremic patients with a high and low CD4 count using anti-αvβ3-coupled magnetic beads. Bead isolated pEV (bead) and flow through (FT) were analyzed for the indicated markers. (c) Lysates of Huh7 cells transfected with HIVΔenv or vector (vec.), and EV derived from these cells were blotted for markers as indicated. Nef-transfected HeLa cells that express haptoglobin served as positive control. (d–g) HIV pEV and Huh7-derived marker profiles and morphology are identical. (d) Blotting of anti-αvβ3 antibody-isolated HIV pEV from one non-viremic patient. Bead isolated pEV (bead) and flow through (FT) were analyzed for the indicated markers. (e–g) Same experimental set up as in (d); however, EV were obtained from different cell culture supernatants as indicated. Cells were either transfected for 72 h with a HIVΔenv viral construct (HIV) (expression control in Supplement Fig. S4a) or an empty vector (vc.). (h) Electron micrographs of anti-αvβ3 antibody-isolated HIV pEV and Huh7-derived EV that were transfected with HIVΔenv. Colored boxes in all panels depict results described in the text.
Next we compared receptor surface pattern and morphology of HIV pEV with that of liver cell-derived EV. Anti-αvβ5 bead-isolated HIV pEV contain ADAM17, Vpu, Nef (Fig. 3d, red box) and lack Tsg101 and CD81 (green boxes) (Lee et al., 2016). The latter discriminates them from classical multivesicular body (MVB)-derived exosomes. Electron micrographs supported this conclusion, revealing vesicles of 100–150 nm size, composed of a membranous coat and a spherical core, structures not described for exosomes (Thery et al., 2009) (Fig. 3h). Notably, pEV purified by iodixanol gradient (Optiprep®) from cancer patients had a similar appearance, structure and size as HIV pEV, indicating that this type of vesicle may develop not only in the course of HIV infection (Supplement Fig. S3a).
Anti-αvβ5-isolated EV from HIVΔenv-transfected Huh7 cells had the same protein profile (Fig. 3e, red and green boxes) and identical shape, structure and size as HIV pEV (Fig. 3h). Notably, EV with similar appearance, structure and size were also secreted by HIV-transfected 293T cells and mature dendritic cells (purification by Optiprep® gradient; Supplement Fig. 3a). The latter have, like HIV EV and pEV, a rich content of effector molecules (see below Fig. 3, and data not shown). Hence, the EV secretion induced by HIV is not specific for Huh7 hepatocytes.
Sk-Hep1-derived EV gave a comparable profile (Fig. 3f), but lacked activated ADAM17 (red box). Bead-isolated EV from HIVΔenv-transfected Jurkat cells did not harbor Nef or ADAM17, but Tsg101 and CD81. Instead, Nef was found in the flow through. In addition, Vpu was not uploaded into EV, although it was present in the cell lysate (Fig. 3g; expression control in Supplement Fig. S4a). These data suggested that hepatocyte-derived EV matched the protein profile and morphological appearance of HIV pEV.
3.6. HIV Liver Tissue Expresses Hck, SPPL3 and Viral Gene Products
In healthy liver Hck is present only in bile duct cells (www.proteinatlas.org/). Also the hepatocyte-like Huh7 and liver sinus endothelial cells (SkHep1) revealed only low Hck abundance; however, transfection of HIVΔenv upregulated Hck protein levels in both cell types. In addition, Hck was activated in Huh7 cells as demonstrated by p-Src staining and protein band shift (Supplement Fig. S3b). In line with this result, ADAM17 was activated and secreted in/by Huh7 but not in/by SkHep1 cells (Supplement Fig. S3c).
We analyzed liver tissue from a HIV-infected non-viremic individual who died of liver cirrhosis-associated complications (Supplement table S2). Almost all hepatocytes stained strongly for Hck, while in a liver sample from a healthy control only spindle-shaped endothelial cells gave positive signals (Fig. 4a, images 1). For control, gut tissue of the patient was stained for Hck, but no difference was recorded in comparison to the control (images 2).
Fig. 4.

Overexpression of Hck, SPPL3 and Nef in HIV liver tissue. (a) Immunohistochemistry analysis of liver and gut tissue sections (fresh frozen tissue) from a non-viremic HIV patient (P04, see Supplement table S2), and two healthy individuals. Different sections were stained by different cellular markers and Nef as indicated. To demonstrate Nef subcellular localization one section was stained with a second anti-Nef antibody (JR6). Inserts serve to magnify cells (images 1 and 4). Arrows in images 5 indicate cells positive or negative for Nef. (b) Analysis of HIVΔenv-transfected Huh7 cells by multi-epitope-ligand-cartography (MELC). Huh7 cells were transfected with HIVΔenv and 48 h later analyzed by MELC-technology 30 for the co-expression of the indicated proteins. A17: ADAM17; Vim.: Vimentin. (c) Immunohistochemistry analysis of Nef expression in two FFPE liver sections (biopsies) from non-viremic HIV patients (P03 and P04, see Supplement table S2) under ART and two healthy individuals. (d) Nef expression in spleen, skin and lymph node (fresh frozen tissue). Boxes depict areas that were magnified. * Non-specific staining of epidermal melanin. Quantification of positive cells in (A, C and D) is shown in Supplement table S3.
ADAM17 (data not shown) and ADAM10 expression did not differ in liver tissues from patient and control (Fig. 4a, images 3). Thus we looked for alternative evidence suggesting ADAM protease activation. We recently described that the expression of the endopeptidase SPPL3 activates ADAM10 (Ostalecki et al., 2017). This protease is also activated by HIV and shuttled into EV (Supplement Fig. S3c, and (Lee et al., 2016)). In healthy liver tissue, spindle-shaped cells, but not hepatocytes, expressed SPPL3. Conversely, in the liver tissue a prominent staining of SPPL3 was found in hepatocytes predominantly in a perinuclear compartment (images 4, see inserts). To determine whether these effects could have been caused by Nef, we stained the tissue with different Nef antibodies. Most but not all hepatocytes gave a signal for Nef (images 5, upper panels). Using the Nef JR6 antibody, we found that the viral protein, like SPPL3, localized to a perinuclear compartment (images 5, lower panels). No staining for Nef was seen with control tissue.
For confirmation we transfected Huh7 hepatocytes with HIVΔenv and analyzed the cells for co-expression of Hck, SPPL3, ADAM17 and viral proteins using the MELC technology (Ostalecki et al., 2017). This technology allows the assessment of multiple proteins in one cell layer. The results confirmed that protein abundance and colocalization of Hck, SPPL3 and ADAM17 increased notably in the presence of Nef and Vpu (Fig. 4b). Thus it seemed plausible that HIV infection of liver tissue induced Hck expression and ADAM protease activation.
Our findings in this liver tissue sample could have been the consequence of the advanced stage of the chronic infection and/or the liver cirrhosis. We therefore analyzed FFPE (formalin fixed paraffin embedded) liver biopsy sections from 3 non-viremic HIV patients (Supplement table S2) and healthy controls. About 25% of liver cells from one infected individual gave a Nef-specific staining (Fig. 4c, left images; summary in Supplement table S3). The result was confirmed by staining of a second - (Fig. 4c, right images) and a third liver tissue sample (data not shown). Nef expression was accentuated in endothelial cells (Fig. 4c, inserts 1, 2). However, hepatocytes were the predominant Nef-expressing cell population (Fig. 4c, insert 3). For comparison, other organ tissue sections from different non-viremic HIV patients were stained for Nef and positive results were found in spleen (1.7% of all cells), skin (0.8%) and in a lymph node (4.5%) (Fig. 4c, summary in Supplement table S3). Thus Nef was expressed in hepatocytes.
3.7. Expression of Gag p24 in HIV Liver Tissue
The expression of Nef implied that liver cells were infected by HIV as suggested previously (Cao et al., 1992, Housset et al., 1993). The liver sample described in Fig. 4a and all three liver biopsies were stained for p24. The results revealed a demarcated staining, predominantly in hepatocytes (Fig. 5a and b; third biopsy not shown). Quantification of p24 in one biopsy revealed 18% p24-positive cells (Supplement table S3).
Fig. 5.

HIV infection of the liver. (a–b) Immunohistochemistry analysis of Gag p24 in liver biopsies and tissue sample described in Fig. 4A and B. (a) Staining of p24 in the FFPE liver biopsies of Patient P01 and P02. Boxed areas were magnified (images 1–3). Quantification of positive cells (in %) is shown in Supplement table S3. (b) Staining of p24 in the liver sample (patient P04) shown in Fig. 4A and in a healthy control. Box and arrows indicate cells positive and negative for p24. (c–d) HIV DNA/RNA analysis by PCR in tissue and non-viremic pEV. (c) Two-round PCR amplification of HIV pol and LTR segments using extracted genomic DNA from liver samples shown above, applying a DOP amplification step. (d) Nested PCR amplification of pol and LTR segments using reverse transcribed RNA from gradient-purified pEV as templates. Included is a limiting dilution analysis using template amounts that correspond to indicated plasma volumes (right panel). (e) Detection of Gag p24 in plasma of non-viremic patients. Plasma (10 ml) from a healthy control and a non-viremic, low CD4 patient (98 CD4 cells/μl) were ultracentrifugated overnight. The pellet was resuspended in PBS and 1.5 ml plasma equivalents were blotted as indicated (dot blot technique). The red box depicts the p24 signal.
For confirmation, DNA was extracted from two tissue samples, amplified by DOP PCR and subjected to a second PCR using HIV pol- and LTR- specific primers. Both biopsy samples gave a positive reaction with both probes (Fig. 5c). When RNA from gradient-purified HIV pEV was analyzed for HIV transcripts, only LTR- but not pol-amplifications were readily detected after nested PCR. A limiting dilution revealed positive PCR signals from as few as 30 μl of pEV plasma equivalent (Fig. 5d). The results were consistent with a previous publication discovering TAR RNA in EV from infected individuals (Narayanan et al., 2013).
In light of these results we speculated that non-infectious Gag particles could be secreted by the liver. Plasma EV were pelleted from 10 ml of a non-viremic plasma sample and 1.5 ml pEV plasma equivalents were analyzed by dot blot. After prolonged membrane exposure, a faint p24 signal was detected (Fig. 5e, red box). Since we did not detect Gag p24 in HIV pEV (Lee et al., 2016) and since in chronic infection infected T cells release no or only few virus particles, this Gag p24 could have originated from a different reservoir, as for example infected liver cells. In summary all our results are consistent with the conclusion that HIV pEV were secreted at least in part by infected liver cells.
4. Discussion
We provide evidence that hepatocytes at least contribute to HIV pEV secretion. Relevant for this conclusion were the detection of liver-typical factors in bead-isolated HIV pEV, and the demonstration of viral proteins and Hck expression in liver tissue. It is possible that tissue myeloid cells, e.g. macrophages or kupfer cells, also secrete HIV pEV. However, since the EV secretion activity was low in macrophages (Fig. 1m) and the CCF profile from peripheral monocytes/iDC differed considerably from that of HIV pEV (Fig. 2b), we assume that myeloid cells may have only a minor role in maintaining the high plasma concentration levels of pEV.
We further demonstrate that Hck and Nef are linked to a particular secretion mechanism, potentially required for the release of pro-inflammatory EV. The biological relevance of HIV pEV is not entirely clear. Based on results in this and our previous report we would speculate that they stimulate the resting T cell compartment as implied by recent results (Arenaccio et al., 2014, Arenaccio et al., 2015). By side effect HIV pEV may contribute to HIV comorbidities and altered immunological parameters in chronic infection, for example through ingestion by monocytes (Lee et al., 2016).
We describe that HIV pEV, aside from lacking the exosome markers CD81 and Tsg101, have a two-layered composition, possibly a plasma membrane coat and a spherical core. This structure seems more complex than described for exosomes, which are generated by invagination of endosomal membranes into multivesicular body (MVB) (Thery et al., 2009). While this observation requires further ultrastructural analysis and conformation, it would support our assumption that HIV pEV are induced de novo by the activation of a specific secretion mechanism. We termed this mechanism vesicular or endosomal secretion; however, it could represent a variant of a non-conventional secretion mechanism (Nickel and Rabouille, 2009). In line with a role of Hck in pro-inflammatory factor release, cytoskeleton reorganization and plasma membrane configuration (Poh et al., 2015, Saksela, 2011), our results imply that the kinase is required to induce this secretion form. An important effector in this context could be dynamin-2 (Weller et al., 2010), interestingly a Nef-interacting molecule (Pizzato et al., 2007) that is directly involved in the formation of vesicles from the Golgi (Kessels et al., 2006).
Although independent laboratories, including ours, came to the conclusion that liver cells are infected in patients (Cao et al., 1992, Housset et al., 1993), liver infection is not considered to be relevant for HIV biology or pathogenesis. There are at least two reasons for this conviction. First, the viral entry mechanism into liver cells is not clear, although several groups described a CD4-independent mechanism that seemingly involves HIV co-receptors including CXCR4, CCR5 and a DC-SIGN-like protein (Bashirova et al., 2001, Cao et al., 1990, Iser et al., 2010). Second, replication in liver cells, like in DC or monocytes, appears to be too inefficient (Kong et al., 2012) to have a relevant role in pathogenesis. Indeed, we observed no correlation between viral copy number or p24 plasma levels in viremic patients and Nef abundance in HIV pEV (Lee et al., 2016). Hence the bulk of viral particles are produced by the T cell compartment. The relevance of liver infection for HIV pathogenesis may lie in the persistence of the infection, the activation of the resting T cell compartment by pEV and the transfer of virus to T cells by cell to cell contact. The latter could explain the rapid viral resurgence when treatment is interrupted, including the appearance of archival virus subtypes after prolonged treatment (Shen and Siliciano, 2008, Ruff et al., 2002).
Evidence for the relevance of HIV liver infection comes from clinical findings. Chronic infection is associated with a number of hepatic disorders ranging from elevated liver enzymes to liver cirrhosis (Ingiliz et al., 2009, Weber et al., 2006). Currently these clinical symptoms are attributed to HBV/HCV co-infections, other co-morbidities and toxic drug effects (Blackard and Sherman, 2008, Weber et al., 2006). However, a surprisingly strong but unexplained correlation was found between low CD4 cell counts and liver-related deaths (Weber et al., 2006). We described a correlation between low CD4 counts and ADAM17/Nef levels in pEV, which likely correlates with the number of infected hepatocytes. This would imply a connection between HIV infection of liver cells, immunodeficiency and clinical symptoms. Hence targeting HIV in the liver could be a new and promising avenue in treating the disease.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
Acknowledgement
We would like to thank Abbas Agaimy for providing HIV liver tissue sections, SL Friedmann for providing the LX-2 cells, U. Schubert for providing HIVΔenv construct.
Funding Sources
This work was supported by funds from the German Science Foundation (DFG): SFB 643 (Teilprojekt A9 2012-2015) and Ministry of Education and Research (BMBF: 031L0073A) (JH.L.). C.O. was supported by the Comprehensive Cancer Center (CCC) Erlangen. T.H. was supported by the Emerging Fields Program of the Friedrich-Alexander-University of Erlangen-Nürnberg.
Author Contributions
The project was designed and coordinated by A.S.B. Substantial contributions to the work were made by J-H.L. (pEV purification, transfections, Western blots and protein array); C.O. and B.S. (tissue staining and analysis); E.A. (transfections and confocal imaging); Z.Z. and T.V. (transfections and Hck stable cell line); H.B. (electron micrographs); T.H. (plasma and tissue sample analysis); K.S. (Nef antibodies and reagents, production and advice).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2018.01.004.
Appendix A. Supplementary Data
Supplementary material
References
- Arany E., Afford S., Strain A.J., Winwood P.J., Arthur M.J., Hill D.J. Differential cellular synthesis of insulin-like growth factor binding protein-1 (IGFBP-1) and IGFBP-3 within human liver. J. Clin. Endocrinol. Metab. 1994;79:1871–1876. doi: 10.1210/jcem.79.6.7527416. [DOI] [PubMed] [Google Scholar]
- Arenaccio C., Chiozzini C., Columba-Cabezas S., Manfredi F., Affabris E., Baur A., Federico M. Exosomes from human immunodeficiency virus type 1 (HIV-1)-infected cells license quiescent CD4 + T lymphocytes to replicate HIV-1 through a Nef- and ADAM17-dependent mechanism. J. Virol. 2014;88:11529–11539. doi: 10.1128/JVI.01712-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenaccio C., Anticoli S., Manfredi F., Chiozzini C., Olivetta E., Federico M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology. 2015;12:87. doi: 10.1186/s12977-015-0216-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashirova A.A., Geijtenbeek T.B., van Duijnhoven G.C., van Vliet S.J., Eilering J.B., Martin M.P., Wu L., Martin T.D., Viebig N., Knolle P.A., KewalRamani V.N., van Kooyk Y., Carrington M. A dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN)-related protein is highly expressed on human liver sinusoidal endothelial cells and promotes HIV-1 infection. J. Exp. Med. 2001;193:671–678. doi: 10.1084/jem.193.6.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackard J.T., Sherman K.E. HCV/ HIV co-infection: time to re-evaluate the role of HIV in the liver? J. Viral Hepat. 2008;15:323–330. doi: 10.1111/j.1365-2893.2008.00970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.Z., Friedman-Kien A.E., Huang Y.X., Li X.L., Mirabile M., Moudgil T., Zucker-Franklin D., Ho D.D. CD4-independent, productive human immunodeficiency virus type 1 infection of hepatoma cell lines in vitro. J. Virol. 1990;64:2553–2559. doi: 10.1128/jvi.64.6.2553-2559.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.Z., Dieterich D., Thomas P.A., Huang Y.X., Mirabile M., Ho D.D. Identification and quantitation of HIV-1 in the liver of patients with AIDS. AIDS. 1992;6:65–70. doi: 10.1097/00002030-199201000-00008. [DOI] [PubMed] [Google Scholar]
- Carreno S., Caron E., Cougoule C., Emorine L.J., Maridonneau-Parini I. p59Hck isoform induces F-actin reorganization to form protrusions of the plasma membrane in a Cdc42- and Rac-dependent manner. J. Biol. Chem. 2002;277:21007–21016. doi: 10.1074/jbc.M201212200. [DOI] [PubMed] [Google Scholar]
- Clavel F., Hoggan M.D., Willey R.L., Strebel K., Martin M.A., Repaske R. Genetic recombination of human immunodeficiency virus. J. Virol. 1989;63:1455–1459. doi: 10.1128/jvi.63.3.1455-1459.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgeorges A., Gabay C., Silacci P., Novick D., Roux-Lombard P., Grau G., Dayer J.M., Vischer T., Guerne P.A. Concentrations and origins of soluble interleukin 6 receptor-alpha in serum and synovial fluid. J. Rheumatol. 1997;24:1510–1516. [PubMed] [Google Scholar]
- English B.K., Ihle J.N., Myracle A., Yi T. Hck tyrosine kinase activity modulates tumor necrosis factor production by murine macrophages. J. Exp. Med. 1993;178:1017–1022. doi: 10.1084/jem.178.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M., Inglese M., Scholz G.M., Harder K.W., Clay F.J., Bozinovski S., Waring P., Darwiche R., Kay T., Sly P., Collins R., Turner D., Hibbs M.L., Anderson G.P., Dunn A.R. Constitutive activation of the SRC family kinase Hck results in spontaneous pulmonary inflammation and an enhanced innate immune response. J. Exp. Med. 2002;196:589–604. doi: 10.1084/jem.20020873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan R., Suzu S., Hiyoshi M., Takahashi-Makise N., Ueno T., Agatsuma T., Akari H., Komano J., Takebe Y., Motoyoshi K., Okada S. Dys-regulated activation of a Src tyroine kinase Hck at the Golgi disturbs N-glycosylation of a cytokine receptor Fms. J. Cell. Physiol. 2009;221:458–468. doi: 10.1002/jcp.21878. [DOI] [PubMed] [Google Scholar]
- Hiyoshi M., Takahashi-Makise N., Yoshidomi Y., Chutiwitoonchai N., Chihara T., Okada M., Nakamura N., Okada S., Suzu S. HIV-1 Nef perturbs the function, structure, and signaling of the Golgi through the Src kinase Hck. J. Cell. Physiol. 2012;227:1090–1097. doi: 10.1002/jcp.22825. [DOI] [PubMed] [Google Scholar]
- Housset C., Lamas E., Courgnaud V., Boucher O., Girard P.M., Marche C., Brechot C. Presence of HIV-1 in human parenchymal and non-parenchymal liver cells in vivo. J. Hepatol. 1993;19:252–258. doi: 10.1016/s0168-8278(05)80579-3. [DOI] [PubMed] [Google Scholar]
- Ingiliz P., Valantin M.A., Duvivier C., Medja F., Dominguez S., Charlotte F., Tubiana R., Poynard T., Katlama C., Lombes A., Benhamou Y. Liver damage underlying unexplained transaminase elevation in human immunodeficiency virus-1 mono-infected patients on antiretroviral therapy. Hepatology. 2009;49:436–442. doi: 10.1002/hep.22665. [DOI] [PubMed] [Google Scholar]
- Iser D.M., Warner N., Revill P.A., Solomon A., Wightman F., Saleh S., Crane M., Cameron P.U., Bowden S., Nguyen T., Pereira C.F., Desmond P.V., Locarnini S.A., Lewin S.R. Coinfection of hepatic cell lines with human immunodeficiency virus and hepatitis B virus leads to an increase in intracellular hepatitis B surface antigen. J. Virol. 2010;84:5860–5867. doi: 10.1128/JVI.02594-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels M.M., Dong J., Leibig W., Westermann P., Qualmann B. Complexes of syndapin II with dynamin II promote vesicle formation at the trans-Golgi network. J. Cell Sci. 2006;119:1504–1516. doi: 10.1242/jcs.02877. [DOI] [PubMed] [Google Scholar]
- Khan I.H., Sawai E.T., Antonio E., Weber C.J., Mandell C.P., Montbriand P., Luciw P.A. Role of the SH3-ligand domain of simian immunodeficiency virus Nef in interaction with Nef-associated kinase and simian AIDS in rhesus macaques. J. Virol. 1998;72:5820–5830. doi: 10.1128/jvi.72.7.5820-5830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Cardona M.W., Moreno-Fernandez M.E., Ma G., Shata M.T., Sherman K.E., Chougnet C., Blackard J.T. Low-level HIV infection of hepatocytes. Virol. J. 2012;9:157. doi: 10.1186/1743-422X-9-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.H., Leung B., Lemmon M.A., Zheng J., Cowburn D., Kuriyan J., Saksela K. A single amino acid in the SH3 domain of Hck determines its high affinity and specificity in binding to HIV-1 Nef protein. EMBO J. 1995;14:5006–5015. doi: 10.1002/j.1460-2075.1995.tb00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.H., Saksela K., Mirza U.A., Chait B.T., Kuriyan J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell. 1996;85:931–942. doi: 10.1016/s0092-8674(00)81276-3. [DOI] [PubMed] [Google Scholar]
- Lee J.H., Wittki S., Brau T., Dreyer F.S., Kratzel K., Dindorf J., Johnston I.C., Gross S., Kremmer E., Zeidler R., Schlotzer-Schrehardt U., Lichtenheld M., Saksela K., Harrer T., Schuler G., Federico M., Baur A.S. HIV Nef, paxillin, and Pak1/2 regulate activation and secretion of TACE/ADAM10 proteases. Mol. Cell. 2013;49:668–679. doi: 10.1016/j.molcel.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Lee J.H., Schierer S., Blume K., Dindorf J., Wittki S., Xiang W., Ostalecki C., Koliha N., Wild S., Schuler G., Fackler O.T., Saksela K., Harrer T., Baur A.S. HIV-Nef and ADAM17-containing plasma extracellular vesicles induce and correlate with immune pathogenesis in chronic HIV infection. EBioMedicine. 2016;6:103–113. doi: 10.1016/j.ebiom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendeni M., Foca E., Gotti D., Ladisa N., Angarano G., Albini L., Castelnuovo F., Carosi G., Quiros-Roldan E., Torti C. Evaluation of liver fibrosis: concordance analysis between noninvasive scores (APRI and FIB-4) evolution and predictors in a cohort of HIV-infected patients without hepatitis C and B infection. Clin. Infect. Dis. 2011;52:1164–1173. doi: 10.1093/cid/cir071. [DOI] [PubMed] [Google Scholar]
- Moarefi I., LaFevre-Bernt M., Sicheri F., Huse M., Lee C.H., Kuriyan J., Miller W.T. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- Narayanan A., Iordanskiy S., Das R., Van D.R., Santos S., Jaworski E., Guendel I., Sampey G., Dalby E., Iglesias-Ussel M., Popratiloff A., Hakami R., Kehn-Hall K., Young M., Subra C., Gilbert C., Bailey C., Romerio F., Kashanchi F. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 2013;288:20014–20033. doi: 10.1074/jbc.M112.438895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel W., Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009;10:148–155. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- Ostalecki C., Wittki S., Lee J.H., Geist M.M., Tibroni N., Harrer T., Schuler G., Fackler O.T., Baur A.S. HIV Nef- and Notch1-dependent endocytosis of ADAM17 induces vesicular tnf secretion in chronic HIV infection. EBioMedicine. 2016;13:294–304. doi: 10.1016/j.ebiom.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostalecki C., Lee J.H., Dindorf J., Collenburg L., Schierer S., Simon B., Schliep S., Kremmer E., Schuler G., Baur A.S. Multiepitope tissue analysis reveals SPPL3-mediated ADAM10 activation as a key step in the transformation of melanocytes. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aai8288. [DOI] [PubMed] [Google Scholar]
- Pfeiffer I.A., Zinser E., Strasser E., Stein M.F., Dorrie J., Schaft N., Steinkasserer A., Knippertz I. Leukoreduction system chambers are an efficient, valid, and economic source of functional monocyte-derived dendritic cells and lymphocytes. Immunobiology. 2013;218:1392–1401. doi: 10.1016/j.imbio.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Pizzato M., Helander A., Popova E., Calistri A., Zamborlini A., Palu G., Gottlinger H.G. Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc. Natl. Acad. Sci. U. S. A. 2007;104:6812–6817. doi: 10.1073/pnas.0607622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh A.R., O'Donoghue R.J., Ernst M. Hematopoietic cell kinase (HCK) as a therapeutic target in immune and cancer cells. Oncotarget. 2015;6:15752–15771. doi: 10.18632/oncotarget.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J.C., Seaberg E.C., Badri S., Witt M.D., D'Acunto K., Thio C.L. HIV monoinfection is associated with increased aspartate aminotransferase-to-platelet ratio index, a surrogate marker for hepatic fibrosis. J. Infect. Dis. 2012;205:1005–1013. doi: 10.1093/infdis/jir885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell D.L., Eckmann L., Dwinell M.B., Carpenter S.P., Raucy J.L., Yang S.K., Kagnoff M.F. Human hepatocytes express an array of proinflammatory cytokines after agonist stimulation or bacterial invasion. Am. J. Phys. 1997;273:G322–G332. doi: 10.1152/ajpgi.1997.273.2.G322. [DOI] [PubMed] [Google Scholar]
- Ruff C.T., Ray S.C., Kwon P., Zinn R., Pendleton A., Hutton N., Ashworth R., Gange S., Quinn T.C., Siliciano R.F., Persaud D. Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure. J. Virol. 2002;76:9481–9492. doi: 10.1128/JVI.76.18.9481-9492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksela K. Interactions of the HIV/SIV pathogenicity factor Nef with SH3 domain-containing host cell proteins. Curr. HIV Res. 2011;9:531–542. doi: 10.2174/157016211798842107. [DOI] [PubMed] [Google Scholar]
- Saksela K., Cheng G., Baltimore D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef + viruses but not for down-regulation of CD4. EMBO J. 1995;14:484–491. doi: 10.1002/j.1460-2075.1995.tb07024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallese M., Giannotta M., Luini A. Coordination of the secretory compartments via inter-organelle signalling. Semin. Cell Dev. Biol. 2009;20:801–809. doi: 10.1016/j.semcdb.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Schubert W., Bonnekoh B., Pommer A.J., Philipsen L., Bockelmann R., Malykh Y., Gollnick H., Friedenberger M., Bode M., Dress A.W. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006;24:1270–1278. doi: 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- Shen L., Siliciano R.F. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J. Allergy Clin. Immunol. 2008;122:22–28. doi: 10.1016/j.jaci.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenius H., Carter N.P., Bebb C.E., Nordenskjold M., Ponder B.A., Tunnacliffe A. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718–725. doi: 10.1016/0888-7543(92)90147-k. [DOI] [PubMed] [Google Scholar]
- Thery C., Ostrowski M., Segura E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- Towner W.J., Xu L., Leyden W.A., Horberg M.A., Chao C.R., Tang B., Klein D.B., Hurley L.B., Quesenberry C.P., Jr., Silverberg M.J. The effect of HIV infection, immunodeficiency, and antiretroviral therapy on the risk of hepatic dysfunction. J. Acquir. Immune Defic. Syndr. 2012;60:321–327. doi: 10.1097/QAI.0b013e31824e9ef2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trible R.P., Emert-Sedlak L., Wales T.E., Ayyavoo V., Engen J.R., Smithgall T.E. Allosteric loss-of-function mutations in HIV-1 Nef from a long-term non-progressor. J. Mol. Biol. 2007;374:121–129. doi: 10.1016/j.jmb.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuyama A.C., Hong F., Saiman Y., Wang C., Ozkok D., Mosoian A., Chen P., Chen B.K., Klotman M.E., Bansal M.B. Human immunodeficiency virus (HIV)-1 infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: implications for the pathogenesis of HIV/hepatitis C virus-induced liver fibrosis. Hepatology. 2010;52:612–622. doi: 10.1002/hep.23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber R., Sabin C.A., Friis-Moller N., Reiss P., El-Sadr W.M., Kirk O., Dabis F., Law M.G., Pradier C., De W.S., Akerlund B., Calvo G., Monforte A., Rickenbach M., Ledergerber B., Phillips A.N., Lundgren J.D. Liver-related deaths in persons infected with the human immunodeficiency virus: the D: a:D study. Arch. Intern. Med. 2006;166:1632–1641. doi: 10.1001/archinte.166.15.1632. [DOI] [PubMed] [Google Scholar]
- Weller S.G., Capitani M., Cao H., Micaroni M., Luini A., Sallese M., McNiven M.A. Src kinase regulates the integrity and function of the Golgi apparatus via activation of dynamin 2. Proc. Natl. Acad. Sci. U. S. A. 2010;107:5863–5868. doi: 10.1073/pnas.0915123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Ma Y., Liu Y., Que H., Zhu C., Liu S. Elevated serum haptoglobin after traumatic brain injury is synthesized mainly in liver. J. Neurosci. Res. 2013;91:230–239. doi: 10.1002/jnr.23159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material