

SUMMARY
SCF ubiquitin ligase assembly is regulated by the interplay of substrate binding, reversible Nedd8 conjugation on Cul1, and the F-box protein (FBP) exchange factors Cand1 and Cand2. Detailed investigations into SCF assembly and function in reconstituted systems and Cand1,2 knockout cells informed the development of a mathematical model for how dynamical assembly of SCF complexes is controlled, and how this cycle is coupled to degradation of an SCF substrate. Simulations predicted an unanticipated hypersensitivity of Cand1/2-deficient cells to FBP expression levels, which was experimentally validated. Together, these and prior observations lead us to propose the adaptive exchange hypothesis, which posits that regulation of the koff of an FBP from SCF by the actions of substrate, Nedd8, and Cand1 molds the cellular repertoire of SCF complexes, and that the plasticity afforded by this exchange mechanism may enable large variations in FBP expression during development and in FBP gene number during evolution.
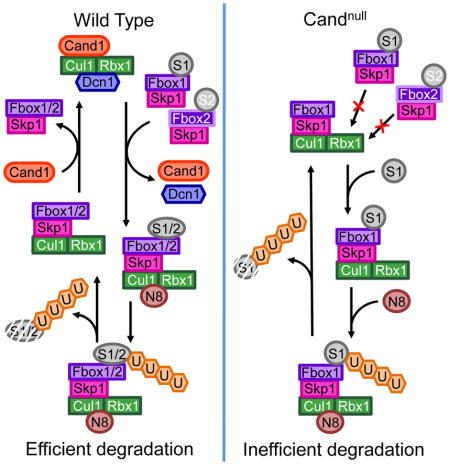
Graphical Abstract
INTRODUCTION
Ubiquitination plays an essential role in cells and organisms, and is achieved by a cascade of enzymes that activate ubiquitin and promote its conjugation to substrate proteins. Cullin-RING ubiquitin ligases (CRLs) comprise the largest family of E3/ubiquitin ligase enzymes that promote the conjugation step and are typified by the Skp1•Cul1•F-box (SCF) complexes (Deshaies and Joazeiro, 2009; Lydeard et al., 2013). SCFs are modular multisubunit complexes composed of the cullin Cul1, the RING domain protein Rbx1/Roc1/Hrt1, the adapter protein Skp1, and an interchangeable substrate receptor protein containing an F-box motif that binds Skp1. The human genome encodes 69 F-box proteins (FBPs) that can potentially form distinct SCFs, at least 54 of which have been detected (Katayama et al., 2013; Pierce et al., 2013; Chen et al., 2015; Jiang et al., 2016; Kamran et al., 2017; Reitsma et al., 2017). Since the substrate specificity of an SCF is determined by which one of the FBPs is recruited to the Cul1 scaffold, it is critical for cells to assemble and activate a specific SCF when its substrates are present.
SCFs are activated by covalent modification of Cul1 with Nedd8, which is mediated by a dedicated set of conjugation enzymes (Lydeard et al., 2013; Enchev et al., 2015). Nedd8 is removed by the COP9 signalosome (CSN), allowing Cul1 to bind the paralogous regulatory factors Cand1 and Cand2. Upon binding, Cand1 disrupts FBP•Skp1 association and inhibits Nedd8 conjugation (Duda et al., 2011). These features imply that Cand1 and Cand2 are negative regulators of SCFs, but studies of Cand1-deficient cells and organisms suggest it plays a positive role (Bosu et al., 2010; Lo and Hannink, 2006; Feng et al., 2004). To explain this paradox, it was hypothesized that Cand1-mediated recycling of SCF is required for optimal SCF function (Schmidt et al., 2009). Through quantitative kinetic studies of SCF subunit interactions, we previously found that the extremely low dissociation rate of an SCF complex was dramatically increased by Cand1 (Pierce et al., 2013), and Cand1 acts as a protein exchange factor that accelerates the equilibration of Cul1 with multiple FBP•Skp1 modules (Pierce et al., 2013; Zemla et al., 2013; Wu et al., 2013). In a recent study, we showed that the Cand proteins promote assembly of a specific SCF complex in response to generation of its cognate substrate (Reitsma et al., 2017). Despite this progress, there remain important gaps in our knowledge of FBP exchange and its role in substrate degradation, and essentially nothing is known about why such a complex system evolved.
Here, using biophysical methods coupled with phenotypic analysis of Cand-deficient cells and mathematical modeling, we develop a quantitative model for the Cand-fueled exchange cycle and pinpoint the defect in SCF substrate degradation in Cand1/2 double knockout cells. We show that mutant cells could not tolerate overexpression of individual FBPs, providing a simple rationale for the evolution of the exchange mechanism.
RESULTS
Quantitative Characterization of Cul1•Cand1 Assembly and Disassembly
We established a fluorescence resonance energy transfer (FRET) assay to directly measure the kinetic parameters of Cand1•Cul1, which is an essential prerequisite to modeling the SCF assembly/disassembly cycle in living cells–a major goal of this study. We employed Cul1 sortase-tagged at its C-terminus with AMC (Cul1AMC), and Cand1 lacking the first alpha helix and labeled with FlAsH via an N-terminal tetracysteine tag (FlAsHΔH1Cand1) (Fig 1A). Quenching of AMC fluorescence upon binding of FlAsHΔH1Cand1 was chased by unlabeled Cand1 (Fig 1A), which confirmed that the FRET signal depended on FlAsHΔH1Cand1 binding to Cul1AMC•Rbx1.
Figure 1. Properties of interactions among Cul1, Cand1 and Skp1•F-box protein revealed by FRET.

(A) FRET assay for Cand1•Cul1 complex formation. Fluorescence emission spectra from excitation at 350 nm of 70 nM Cul1AMC•Rbx1, 70 nM FlAsHΔH1Cand1, a mixture of the two (FRET), chase control for FRET, or buffer alone. +Chase indicates 700 nM Cand1. Proteins were added in the indicated order. Addition of chase to Cul1+Cand1 had a negligible effect on FRET due to the long t1/2 of the Cand1•Cul1 complex as shown previously (Pierce et al., 2013).
(B) kon for Cand1 binding to Cul1. The observed rates of Cand1•Cul1 assembly at different concentrations of Cand1 are plotted. Linear slope gives kon of 1.7 × 107 M−1s−1. Error bars, ± SEM, n = 5 (see also Fig S1A).
(C) kon for Cand1 binding to Cul1•Rbx1 preassembled with FBP. Similar to Fig 1B, except with 100 nM Skp1•Skp2 preincubated with 50 nM Cul1AMC•Rbx1. Linear slope gives kon of 2.0 × 106 M−1s−1. Error bars, ± SEM, n ≥ 4 (see also Fig S1B).
(D) Disruption of Cand1•Cul1 by Skp1•Skp2. The change in donor fluorescence versus time was measured following addition of 75 nM Skp1•Skp2 or 75 nM Skp1 to 25 nM FlAsHΔH1Cand1•Cul1AMC•Rbx1.
(E) koff of Cand1 from ternary exchange intermediate. The single exponential observed rates of Cand1 dissociation from 10 nM FlAsHΔH1Cand1•Cul1AMC•Rbx1 in the presence of increasing concentrations of Skp1•Skp2 were measured (see Fig S1C) and plotted. Fitting of the curve predicts a rate plateau at 67 sec−1. Error bars, ± SEM, n ≥ 3.
(F) Kinetic model of the exchange cycle. The number in parentheses indicates the koff of 2.9 s−1 calculated from detailed balance relations (see Fig S4C).
(G) Deletion of β hairpin in Cand1 enables formation of a stable complex comprising Cul1, Skp1•Fbxw7, and Cand1. Cand1 or Cand1Δβ (100 nM) was added to 70 nM CFPCul1•Rbx1•Skp1•Fbxw7TAMRA, and formation of SCFFbxw7 was monitored by FRET. + Chase indicates 700 nM Skp1•Skp2.
(H) Deletion of loop regions in Skp1 enables formation of a stable complex comprising Cul1, Skp1•Skp2, and Cand1. Skp1•Skp2 or Skp1ΔΔ•Skp2 (700 nM) was added to 70 nM FlAsHCand1•Cul1AMC•Rbx1 and the persistence of the latter complex was monitored by FRET.
By monitoring the donor Cul1AMC•Rbx1 fluorescence at varying concentrations of FlAsHΔH1Cand1, the kon was determined to be 1.7 × 107 M−1 s−1 (Fig 1B, S1A). This kon together with the koff of 1.2 × 10−5 s−1 we measured previously (Pierce et al., 2013) revealed the KD for Cand1 assembly with Cul1•Rbx1 to be 0.7 pM. We also measured the association of Cand1 with Cul1•Rbx1 that was preassembled with FBP•Skp1, and the kon was 2.0 × 106 M−1 s−1 (Fig 1C, S1B).
Consistent with our previous study (Pierce et al., 2013), the dissociation of the stable Cand1•Cul1•Rbx1 complex was dramatically accelerated by FBP•Skp1 complexes (Fig 1D, 1H, S1F), while Skp1 alone showed a much weaker effect (Fig 1D). Increasing the concentration of FBP•Skp1 led to increased observed rates of Cand1 dissociation from Cul1•Rbx1 (Fig 1E, S1C), and the maximal observed rate, 67 s−1 (Fig 1E), represents the koff of Cand1 from the transient Cand1•Cul1•Skp1•FBP complex. Since FlAsHΔH1Cand1 lacks 14 amino acids from the N-terminus, we tested if this deletion affects the koff of Cand1•Cul1 by comparing the koff of Cand1•Cul1AMC•Rbx1 and ΔH1Cand1•Cul1AMC•Rbx1. In Fig S1D, ΔH1Cand1•Cul1AMC•Rbx1 displayed a two-phase dissociation, with the fast phase koff 21 times higher than for wild type (WT) Cand1. Consistently, a GST pulldown assay suggested the KD increased ~4.5 fold (Fig S1E). These results suggest that truncation of the N-terminal helix of Cand1 modestly destabilized its binding to Cul1•Rbx1, and the koff of 67 s−1 measured in Fig 1E was overestimated by 4.5–21 fold. Since this koff was the only unknown constant in the thermodynamic cycle for Cul1, Cand1 and FBP interactions (Fig 1F), we calculated this constant using the principle of detailed balance (see Fig S4C and Computational method) which yielded a value of 2.9 s−1 (Fig 1F).
Goldenberg et al. (2004) showed that deletion of the β-loop of Cand1 (Cand1Δβ) that is predicted to sterically clash with Skp1 allowed a stable Cand1Δβ •Cul1•Skp1 complex to form. We confirmed this observation using both FRET (Fig 1G) and pulldown assays (Fig S1G). Skp1ΔΔ•Skp2, which lacks a loop in Skp1 predicted to clash with the β-loop of Cand1, failed to disrupt the Cand1•Cul1 complex (Fig 1H, S1F). This was not due to a failure to bind, because stable GSTCand1•Cul1•Skp1ΔΔ could form in vitro (Fig S1G). These results suggest that Cand1, Cul1, and Skp1•FBP form a ternary complex that is exceptionally unstable due to clash of the Skp1 loop with the Cand1 β-loop. As a consequence, the ternary complex rapidly decays to yield either binary complex, each of which is stable (Fig 1F). This mimics the behavior of Ras, Ras-GEF, and guanine nucleotides (Klebe et al., 1995; Goody and Hofmann-Goody, 2002; Guo et al., 2005).
Cand1/2 promote efficient degradation of SCF substrates
To explore the role of Cand1 under physiological conditions, we generated CRISPR knockout human cell lines. Because Cand1 knockdown enhanced the recovery of Cand2 in FLAGCul1 immunoprecipitates (IP) (Fig S2A), we knocked out both Cand1 and Cand2 using pairs of CRISPR nickases to minimize off-target effects (Fig S2B) (Ran et al., 2013; Shen et al., 2014). We generated Cand1 or Cand2 single knockout (KO) cells (Fig S2C), and three independent double knockout (DKO) cells (Fig S2D).
We first looked at TNFα-induced degradation of IκBα by SCFβ-TrCP (Spencer et al., 1999; Kroll et al., 1999), and we found that the t1/2 for IκBα elimination was nearly tripled in the DKO lines compared with the WT (Figs 2A, 2C, S2E). To confirm that the deficiency in IκBα degradation was due to the absence of Cand1, we integrated a single copy of the CAND1 gene into the DKO cell genome through Flp recombinase-mediated insertion. When the Cand1 transgene was induced by tetracycline in DKO cells (Fig 2B), the IκBα degradation defect was fully rescued (Fig 2A, C). Furthermore, IκBα degradation was unaffected in Cand2 KO cells, whereas Cand1 KO had an intermediate effect (Fig S2F), suggesting that Cand1 promotes efficient degradation of IκBα but can be partially substituted by Cand2 upon its deletion. Reduced degradation of IκBα in DKO cells was not due to lack of phosphorylations that trigger IκBα degradation (note the upshift of IκBα in DKO cells at ≥20′ in Fig 2A), nor the lack of β-TrCP (Fig 2B). Also, ubiquitination of phosphorylated IκBα (pIκBα) was greatly reduced in DKO cells relative to WT cells (Fig 2D), suggesting that prolonged IκBα degradation in DKO cells was due to decreased ubiquitination.
Figure 2. Cand1/2 double knockout (DKO) cells display defects in IκBα degradation and SCFβ-TrCP assembly.

(A–C) Cand1/2 DKO cells display defects in IκBα degradation. IκBα levels in indicated cell lines were monitored by western blot (WB) at indicated time points after TNFα treatment. Both phospho-IκBα (pIκBα, upper band) and unmodified IκBα (lower band) were detected by the anti-IκBα antibody. Here and elsewhere in this work, we blotted for GAPDH as a loading control.
(B) WB analysis of Cand1 and β-TrCP in cell lysates from (A). A more intense exposure (dark) of the Cand1 blot and relative levels of Cand1 are also shown.
(C) Quantification of IκBα t1/2 from panel A.
(D) Ubiquitination of pIκBα is significantly reduced in DKO cells. WB analysis (with anti-pIκBα antibody) of the ubiquitination of pIκBα in WT and DKO cells upon TNFα treatment. DKO36 from (A) was used in this experiment and thereafter.
(E–F) TNFα promotes formation of SCFβ-TrCP in WT but not DKO cells. Schematic workflow of the experiment is depicted in (E), and WB analysis of endogenous SCFβ-TrCP in WT and DKO cells before and after 10-min TNFα treatment is shown in (F). Relative levels of SCFβ-TrCP were calculated as the β-TrCP:Cul1 ratio in 3xFLAGCul1 immunoprecipitations (IPs), and all ratios were normalized to that obtained for the IP from WT cells not treated with TNFα. Average fold increase of SCFβ-TrCP induced by TNFα treatment is shown in the graph. Error bars, ± SEM, n = 3, P value = 0.001.
(G–H) Recruitment of Cul1 to pIκBα•β-TrCP is inefficient in DKO cells. Schematic workflow of the experiment is depicted in (G), and WB analysis of the recruitment of β-TrCP and Cul1 to pIκBα following 10-min TNFα treatment is shown in (H). Expression of 3xFLAGIκBα was induced by 100 ng/ml tetracycline for 24 hours. Relative levels of β-TrCP and Cul1 recruited to pIκBα were calculated as the β-TrCP:pIκBα (see also Fig S2I) and Cul1:β-TrCP ratios in the IPs. Average levels of Cul1 recruited to pIκBα•β-TrCP are shown in the graph. Error bars, ± SEM, n = 3, P value = 0.0001.
Cand1/2 is required for rapid assembly of new SCF in response to substrate availability
Because pIκBα is a substrate of SCFβ-TrCP, we measured the level of endogenous SCFβ-TrCP complex before and after TNFα treatment (Fig 2E). We tagged endogenous Cul1 with a 3xFLAG tag using CRISPR, and we lysed the cells in buffer containing recombinant Cul1•Rbx1 ‘sponge’ protein at 100x excess over endogenous 3xFLAGCul1, which blocks the Cand-mediated exchange of FBPs and thus preserves endogenous SCF complexes (Reitsma et al., 2017). In agreement with Reitsma et al. (2017), we found that the level of β-TrCP co-IP’d with endogenous 3xFLAGCul1 was higher in unstimulated DKO cells than in WT cells (Fig 2F). However, after 10-min TNFα treatment, co-IP’d β-TrCP was increased by 80% in the WT cells, but was unchanged in the DKO cells (Fig 2F). These results suggest that when pIκBα substrate appears, new SCFβ-TrCP are quickly assembled in a Cand1/2-dependent manner.
We further analyzed the assembly of pIκBα•β-TrCP•Cul1 in cells expressing tetracycline-induced 3xFLAGIκBα. We first looked at the interaction of 3xFLAGpIκBα with β-TrCP, by immobilizing 3xFLAGpIκBα, deubiquitinating the immobilized protein with Usp2, and quantifying the ratio β-TrCP:pIκBα (Fig 2G), and we found that the DKO cells had no deficiency in forming the pIκBα•β-TrCP complex (Fig 2H). In a parallel assay, we used the Nedd8 E1 inhibitor MLN4924 (Soucy et al., 2009) to prevent the ubiquitination of pIκBα (Fig S2G–H), and confirmed that pIκBα•β-TrCP formation was not affected in DKO cells (Fig S2I). Finally, we analyzed the association of Cul1 with the pIκBα•β-TrCP complex. Whereas Cul1, especially neddylated Cul1, was efficiently recruited to pIκBα•β-TrCP in WT cells, this recruitment was reduced to 40% in DKO cells (Fig 2H). These results suggest that substrates can bind both free and Cul1-bound FBPs, and the lower degradation rate of IκBα in DKO cells is due to inefficient recruitment of pIκBα•β-TrCP to Cul1, a process that requires the exchange activity of Cand1.
Cand1 stabilizes Cul1•Dcn1
Interestingly, Cul1-bound Dcn1, the E3 of Nedd8, was detected only in WT cells (Fig 3A). Since this could be due to potential difference in Cul1 neddylation status in WT vs. DKO cells, we treated the cells with MLN4924. Again, we could detect Cul1•Dcn1 only in the WT cells (Fig 3A). A prior report established that Cand1, Cul1, and Dcn1 can form a ternary complex (Kim et al., 2008). Unexpectedly, we showed here that Cand1 strongly promotes binding of Dcn1 to Cul1•GSTRbx1 in vitro (Fig 3B). This stabilization could be sustained by the N-terminal half of Cand1 (Fig 3B), which binds the C-terminal domain of Cul1 where Dcn1 also binds. Since Cand1 and Dcn1 formed a complex only in the presence of Cul1•Rbx1 (Fig S2J), this stabilization effect is likely due to a conformational change in the C-terminal domain of Cul1 and/or Rbx1 induced by the binding of Cand1 (Fig S2K). We further quantified that Cand1 increased the GSTDcn1•Cul1 level by 2.9 fold under a specific pulldown condition (Fig 3C). This result together with the known KD of Dcn1•Cul1 (Monda et al., 2013) suggests that Cand1 changed the KD of Dcn1•Cul1 from 1.8 μM to 0.05 μM (Fig 3D), which we validated in additional pulldown assays (Fig S3A).
Figure 3. Cand1 enhances formation of Cul1•Dcn1 complex and subsequent neddylation of Cul1 stabilizes newly formed SCF.

(A) Stable Cul1•Dcn1 complex is dramatically reduced in DKO cells. IP-WB analysis of interactions between endogenous 3xFLAGCul1 and Dcn1 in WT and DKO cells pre-treated with either 0.1% DMSO or 1 μM MLN4924 for 1 hour.
(B) Cand1 stabilizes Cul1•Dcn1 complex in vitro. Pulldown-WB analysis of recombinant Dcn1 (0.2 μM) and Ubc12 (0.2 μM) bound to recombinant Cul1•GSTRbx1 (0.4 μM) in the presence and absence of recombinant Cand1, Cand11-603, or Cand1604-1230 (all 0.4 μM). A more intense exposure (dark) of the Dcn1 blot is also shown.
(C) The Cul1•GSTDcn1 complex is stabilized by Cand1 in vitro. Pulldown (PD) analysis of recombinant Cul1•Rbx1 (1 μM) bound to recombinant GSTDcn1 (0.6 μM) in the presence of 0–3 μM Cand1. Protein samples were fractionated on a SDS-PAGE gel and stained with Coomassie Blue. Normalized levels of Cul1 recovered were calculated as the ratio of Cul1 to GSTDcn1. (See also Fig S3A)
(D) Thermodynamic cycle of Dcn1, Cul1•Rbx1 and Cand1 binding. All numbers are KD values. The KD of 1.8 × 10−6 M for Dcn1 and Cul1•Rbx1 was reported previously (Monda et al., 2013); the KD of 5 × 10−8 M for Dcn1 and Cand1•Cul1•Rbx1 was estimated based on results in Fig 3C and S3A; the KD of 7 × 10−13 M for Cul1•Rbx1 and Cand1 was from Fig 1B; and the KD in parentheses was calculated from detailed balance considerations (see Fig S4C).
(E–F) Cand1-bound Cul1 is neddylated faster than free Cul1 in the presence of FBPs. Schematic workflow of a competitive Cul1 neddylation assay is shown in (E). Free Cul1•Rbx1 and Cand1•Cul1•Rbx1 in which the different Cul1 species are labeled with different fluors (FAM or TAMRA) compete for limiting Dcn1, and neddylation enzymes are provided by DKO lysate. 1x represents 50 nM protein in the final sample mixture. Fluorescence scan of the SDS-PAGE gel containing samples prepared as described in (E) is shown in (F). “Fold increase with Cand1” was calculated as the ratio of percent neddylation of Cand1-bound Cul1 to free Cul1 (see Fig S3B–C for negative controls). A representative result of three replicates is shown.
(G) Neddylation increases the assembly of FBP with Cand1-bound Cul1. Cand1, Dcn1 and Cul1•GSTRbx1 were pre-incubated and then mixed 1:1 (v:v) with Skp1•β-TrCP and Ubc12 or Ubc12 charged to Nedd8 (Ubc12~Nedd8). After 15 min incubation, the protein mixture was incubated with glutathione beads and immobilized proteins were analyzed by WB. (See also Fig S3D.)
Since Cand1 strongly inhibited Cul1 neddylation in vitro and this inhibition was counteracted by FBPs (Pierce et al., 2013), we set up a competitive neddylation assay to test the counter-intuitive prediction that Cand1-bound Cul1 should be a better neddylation substrate than free Cul1 when FBPs are present. Since the koff of Cand1•Cul1•Rbx1 is low, we mixed equimolar free Cul1TAMRA•Rbx1 with Cand1-bound Cul1FAM•Rbx1, or vice versa, incubated the mixture with a limiting amount of Dcn1, then triggered neddylation by adding DKO cell lysate supplemented with recombinant FBPs, and determined the neddylation status of Cul1TAMRA and Cul1FAM individually (Fig 3E). Consistent with the notion that Cand1 stabilized Dcn1•Cul1•Rbx1 and Dcn1 significantly increases the speed of neddylation (Fig S3B) (Monda et al., 2013; Scott et al., 2014), Cand1-bound Cul1 showed up to 50% more neddylation than free Cul1 (Fig 3F), an effect that only occurred when FBPs were present (Fig S3C).
Though Cand1-bound Cul1 cannot be neddylated, formation of Cand1•Cul1•Rbx1•Dcn1 enables immediate neddylation of Cul1 upon removal of Cand1 by FBPs, such that an SCF would be “born” in a neddylated state that is resistant to disassembly by Cand1 and is primed to ubiquitinate substrate. Indeed, when neddylation occurred, more Cand1•Cul1 was dissociated and more SCF was assembled (Fig 3G), and this change occurred over time while neddylation proceeded (Fig S3D). Altogether, these results suggest that Cand1 helps recruit neddylation enzymes to Cul1 such that the formation of an SCF is directly coupled to its activation.
Computational model of the SCF cycle
To understand the impact of SCF assembly and disassembly on the degradation of SCF substrates we developed a deterministic mathematical model based on mass-action equations (Fig 4A and “Computational method” in STAR Methods). To model the SCFβ-TrCP-mediated degradation of IκBα we considered two populations of F-box proteins: one that represents β-TrCP and another one that accounts for all other F-box proteins in the cell. To parametrize our model we used data that we and others have collected on the rate constants of the various processes considered in the model as well as on the cellular concentrations of SCF components and factors that promote the SCF cycle (Pierce et al., 2013; Mosadeghi et al., 2016; Bennett et al., 2010; Reitsma et al., 2017). We specifically incorporated our observations that Cand1 and Dcn1 exhibit positive cooperativity when binding to Cul1•Rbx1 (Fig 3D) and that Dcn1 stabilizes the Cand1•Cul1•Rbx1 complex in the presence of Skp1•FBP by up to 40% (Fig S3E).
Figure 4. Mathematical model of the SCF cycle.

(A) Simplified scheme illustrating the main processes and interactions considered in the mathematical model (see Fig S4 for a detailed reaction scheme). Lines with unidirectional arrows represent irreversible reactions. FB1 stands for Skp1•β-TrCP whereas FB2 represents a pool of auxiliary Skp1•F-box proteins that compete for access to Cul1•Rbx1. Both F-box proteins form SCF ligases with Cul1•Rbx1 that undergo the same cycle of processes including F-box exchange, neddylation, deneddylation, substrate binding and substrate degradation.
(B–F) Model simulations and predictions. Simulations labeled in orange color were used to estimate unknown parameters. Remaining simulations represent model predictions. Error bars for predictions were obtained from a profile likelihood analysis (Fig S5A). Experimental results are shown as thin bars. To simulate inhibition of Nedd8 conjugation by MLN4924 we set knedd=0. As a result the fraction of Cul1•Rbx1 bound to Cand1 increased while the fraction of Cul1•Rbx1 bound to Skp1•FBP decreased (Reitsma et al. 2017) (B). If Cand proteins are absent (DKO) the latter fraction is predicted to increase to 100% in agreement with observations. The model confirms (C, upper panel) that re-expression of Cand1 (13% of WT level) in a DKO cell line reduces the half-life (t1/2) for IκBα degradation back to WT levels (Fig 2C). The half-life for substrate degradation is predicted to exhibit a U-shaped dependence on the cellular Cand1 concentration with an extended valley where t1/2 ≈20min remains approximately constant (C, lower panel). Dashed lines indicate the Cand1 concentration in WT (black) and DKO cells with Cand1 re-expressed to 13% of WT level (red). When substrate is added the fraction of β-TrCP bound to Cul1 increases ~1.7-fold (D) from its steady state level (46%) as observed in WT cells (Fig 2F). Cul1 overexpression is predicted to reduce the fraction of neddylated Cul1 (E) in agreement with observations. Also, Cul1 overexpression should have no effect on the half-life for IκBα degradation in WT, but should reduce t1/2 in DKO cells back to WT level (F). In contrast, overexpression of β-TrCP is predicted to have no effect on t1/2 in DKO cells (F).
Altogether, our model contains 54 state variables and 35 parameters of which 22 were either directly measured or adapted from previous publications. From the 13 remaining parameters 8 were estimated based on the known value of measured quantities leaving only 5 parameters which had to be estimated from experiments. To fit the model, we chose the following subset of steady state and transient measurements: % of Cul1 bound to Cand1 in WT (Fig 4B), half-life (t1/2) for IκBα degradation in WT and DKO (Fig 4C), % of β-TrCP bound to Cul1 upon substrate addition (Fig 4D) and % of Cul1 conjugated to Nedd8 in WT and DKO (Fig 4E).
Our model correctly anticipates the impact of perturbations to the Nedd8–Cand1 cycle on Cul1 assembly state. For example, 100% of Cul1 is predicted to assemble with Skp1 in DKO cells (Fig 4B) in agreement with observations based on selected reaction monitoring mass spectrometry (SRM-MS; Reitsma et al., 2017). Likewise, inhibition of Nedd8 conjugation by MLN4924 is predicted to increase the fraction of Cul1 bound to Cand1 while that of Cul1 bound to Skp1•FBP is predicted to decline (Fig 4B), which is also consistent with results from SRM-MS (Reitsma et al., 2017; the lower values reported by these authors arise from their estimate that 16% of Cul1 was not bound to either Cand1 or Skp1).
To simulate TNFα-induced degradation of IκBα via SCFβ-TrCP, we first determined the following parameters: (i) the rate of IκBα phosphorylation (by following the shift of the IκBα band in the presence of the ubiquitination inhibitor MLN4924) (Fig S3F), (ii) the cellular concentrations of β-TrCP (64 nM) and IκBα (650 nM) determined by quantitative western blotting (Fig S3G–H), and (iii) the koff of pIκBα•β-TrCP in cell lysate (3.3 × 10−5 s−1; Fig S3I–K) as determined by competitive displacement. This last measurement was quite surprising because prior experiments with a phosphopeptide containing a related degron suggested an off rate in the range of 0.1 sec−1 (Saha and Deshaies, 2008). Using these values for model fitting, we obtained the t1/2 values for IκBα degradation in WT cells (23min) and in DKO cells (46min) which were similar to those determined empirically (Fig 2C). Importantly, the model correctly predicted the surprising observation that re-expression of Cand1 in the DKO cells at 13% of the WT level (as shown for DKO22 in Fig 2B) restored the t1/2 of IκBα degradation to the WT level (Fig 4C). Previous analysis of a mathematical model focusing on the Cand1 cycle suggested that the U-shaped dose-response curve for Cand1 (Fig. 4C, lower panel) results from a trade-off between high SCF ligase activity at low Cand1 concentration and fast F-box exchange at high Cand1 concentration (Straube et al., 2017). One of the predictions of that model is that the presence of substrate would favor the assembly of the corresponding SCF ligase which is precisely what we observed in WT cells (Fig 2F; Reitsma et al. 2017). Interestingly, we obtained the same effect (~1.7-fold increase in the level of β-TrCP bound to Cul1) with our model when simulating the formation of pIκBα using the estimated parameters (Fig 4D). Together, these observations suggest that our model, though it omits much of the complexity that exists in cells (e.g. spatial inhomogeneity and de-novo synthesis of SCF components and accessory proteins), nevertheless captures essential features of the SCF cycle making it effective in predicting both steady-state and dynamic properties of the SCF system in WT cells and in response to genetic and chemical perturbations.
Cellular role of Cand1 revealed by simulations and experimental perturbations
To probe the strength of our model in predicting responses to new perturbations, we simulated the effect of varying the concentration of SCF components on the kinetics of substrate degradation, and compared the results with those obtained empirically. We first increased the Cul1 level in the model, which yielded two predictions: 1) Cul1 overproduction led to a reduction in Cul1 neddylation in both WT and DKO cells (Fig 4E); 2) the t1/2 of IκBα in WT and DKO cells overproducing Cul1 became identical and equaled that in WT cells (Fig 4F). Consistent with these predictions, overproduction of 3xFLAGCul1 did not affect the t1/2 of IκBα in WT cells but restored a normal t1/2 in DKO cells (Fig 5A), and resulted in reduced neddylation of both Cul1 and Cul4a (Fig 5B). Next, we wondered how increasing the β-TrCP level would affect substrate degradation. Consistent with experiments (Fig 5C, S5E) we found that increased levels of β-TrCP had no significant impact on the t1/2 for IκBα in either WT or DKO cells (Fig 4F).
Figure 5. Experimental concordance with mathematical predictions.

(A) 3xFLAGCul1 overexpression rescues the IκBα degradation defect of DKO cells. IκBα levels were monitored by western blot (WB) at indicated time points after TNFα treatment. Overexpression of 3xFLAGCul1 was induced by tetracycline. Average relative t1/2 of IκBα are shown in the graph. Error bars: range of values, n = 2.
(B) 3xFLAGCul1 overexpression impedes cullin neddylation. WB analysis of cullins in cell lysates from (A). Fold increase in total Cul1 levels and percent neddylation of overexpressed 3xFLAGCul1 and endogenous Cul4a are indicated. A representative result of two replicates is shown.
(C) β-TrCP overexpression does not rescue the IκBα degradation defect of DKO cells. IκBα levels were monitored by western blot (WB) at indicated time points after TNFα treatment. Overexpression of β-TrCP was induced by tetracycline. Average relative t1/2 of IκBα are shown in the graph. Error bars: range of values, n = 2. (See Fig S5E for WB of β-TrCP)
(D–E) Overexpression of Cul1 significantly depletes free β-TrCP in the DKO cells. As illustrated in (D), cells with/without tetracycline induced 3xFLAGCul1 were lysed in buffer containing Cul1•GSTRbx1 sponge protein and subjected to GST pulldown, which probes changes in levels of unbound cellular proteins capable of binding to sponge in cell lysate (see Fig S5F for WB images). Average changes in protein levels compared to non-tetracycline induced control are shown in the graph. Overexpression of 3xFLAGCul1 depleted the pool of free β-TrCP in DKO cells by 80%. Error bars: range of values, n = 2.
(F–G) Overproduction of β-TrCP modestly reduces the efficiency of its assembly with Cul1. As illustrated in (F), cells containing endogenous 3xFLAGCul1 were lysed in buffer containing Cul1•GSTRbx1 sponge protein. β-TrCP bound to endogenous 3xFLAGCul1 was probed by anti-FLAG beads, and free cellular β-TrCP capable of binding to sponge in cell lysate was probed by GST beads (see Fig S5G for WB images). Percentage of β-TrCP bound to endogenous 3xFLAGCul1 in WT and DKO cells with or without tetracycline-induced overexpression of β-TrCP is graphed. Error bars: range of values, n = 2.
To understand why the t1/2 of IκBα in DKO cells was rescued by overproducing Cul1 but not β-TrCP, we probed the assembly status of SCFβ-TrCP. Using Cul1•GSTRbx1 “sponge” protein as a bait to capture unbound Skp1•β-TrCP from the cells (Fig 5D), we first found that when Cul1 was overproduced, the amount of free β-TrCP was reduced by 80% in the DKO cells (Fig 5E, S5F), suggesting that almost all β-TrCP was assembled with Cul1, and thus pIκBα was able to access active SCF without relying on the exchange activity of Cand1/2. Second, we quantified the % of β-TrCP bound to endogenous Cul1. Whereas the total amount of SCFβ-TrCP was increased upon β-TrCP overproduction (Fig S5G), the percent of β-TrCP assembled into an SCF complex was not (Fig 5F, G). Thus, despite there being more SCFβ-TrCP, pIκBα was similarly partitioned between pools of SCFβ-TrCP and free β-TrCP. For the ~50% of pIκBα molecules in DKO cells that initially bound free β-TrCP, their degradation required dissociation and re-equilibration with the total pool of β-TrCP, which was slow (Fig S3I–J). This explains why DKO cells could contain an elevated level of SCFβ-TrCP (Fig 2F) but continue to exhibit a reduced rate of IκBα degradation.
Cand1/2 buffer changes in the expression level of F-box proteins
A major conundrum that emerged from the interplay between model predictions and experiments was the finding that Cul1 overexpression rescued the degradation defect of DKO cells. This begs the question of why such a complex system of FBP exchange exists when similar substrate degradation rates could be achieved by simply increasing the level of Cul1? Remarkably, a hint to resolving this conundrum came from the matrix of normalized response coefficients (Fig S5B) that we computed to quantify the impact of selected model parameters on the experimentally accessible quantities of interest such as the fraction of neddylated Cul1 or the t1/2 of substrate. Strikingly, the total FBP concentration was one of the two most sensitive parameters which were predicted to have a strong effect on the substrate t1/2 in DKO cells, but not in WT cells. Consistent with this prediction, expression of HAFbxo6 in DKO cells through lentiviral infection further increased the t1/2 of IκBα by ~40% (Fig 6A, S6A), and decreased the level of SCFβ-TrCP (Fig S6C), without altering levels of β-TrCP and Skp1 (Fig S6B). Similarly, cyclin E and p27, which are substrates of SCFFbxw7 and SCFSkp2, respectively, were also stabilized in DKO cells with overproduced Fbxo6 (DKOFbxo6; Fig 6B).
Figure 6. Overexpression of single F-box proteins suppresses cell proliferation in DKO cells by sequestering Cul1.

(A) Overexpression of Fbxo6 increases the t1/2 of IκBα only in DKO cells (see Fig S6A–B for WB images). Fbxo6 was overexpressed by transduction with a recombinant lentivirus expressing HAFbxo6. The assay was performed four days after the viral transduction. Average fold increase of IκBα t1/2 by Fbxo6 overexpression in WT and DKO cells are graphed. Error bar: ± SD, n = 3, P value < 0.01.
(B) Overexpression of Fbxo6 in DKO cells reduces degradation of SCF substrates. All samples were analyzed on the same gel and blot, but one lane between WT and DKO samples on the blot image was eliminated and indicated as a space.
(C) Overexpressed Fbxo6 sequesters Cul1 in DKO cells. Cells were infected with recombinant lentiviruses carrying the HAFbox6 gene five days before HAFbxo6 was IP’d from WT3xFLAG-Cul1 and DKO3xFLAG-Cul1 cells in the presence of recombinant Cul1•GSTRbx1 (+ sponge). Equal percent volumes of Input (In), immunoprecipitation eluent (IP), and flow-through (FT) were analyzed by WB. Long (L) and short (S) exposures of endogenous 3xFLAGCul1 are shown. Quantifications of percent Cul1 in the HAFbxo6 IPs are graphed. Error bars: ± SD, n = 3, P value < 0.01.
(D) Fbxo6 overexpression reduces proliferation of DKO cells in a specific, FBP-dependent manner. Cells were treated with recombinant lentiviruses carrying different FBP constructs as indicated. Three days after lentiviral infection, cells were equally seeded and counted every 24 hrs for 4 days. Average cell doubling time is graphed. Error bars: ± SD, n = 3, P value < 0.01. Note that Fbxl16 bound at least as much Skp1 as Fbxo6 but did not bind Cul1 (compare Fig S6G with panel D), and that re-introduction of Cand1 rescued the DKO cells.
(E) Overexpression of Cul1 partially rescues toxicity of overproduced Fbxo6 in DKO cells. Cul1 overexpression was induced by tetracycline. Cell doubling was measured as in (D). Error bars: ± SD, n = 3.
(F) Overexpression of HASkp2 or HASkp2ΔLRR slows cellular proliferation in DKO cells. Cells were infected by lentiviruses and cell doubling was measured as in (D). Error bars: ± SD, n = 3, P value < 0.01.
(G) Overexpression of HASkp2ΔLRR increased the level of apoptosis marker in DKO cells. A representative result of two replicates is shown.
When we immunoprecipitated (IP) HAFbxo6 in the presence of the Cul1•GSTRbx1 “sponge”, the amount of endogenous Cul1 co-IP’d with HAFbxo6 was dramatically increased in the DKOFbxo6 cells, and therefore, the amount of Cul1 available for the remaining FBP pool was reduced by > 50% (Fig 6C). More strikingly, cell proliferation of DKOFbxo6 cells slowed dramatically and the cells displayed abnormal morphology and inviability (Fig 6D, S6D–E), which were not observed in WTFbxo6 cells (Fig 6D, S6D), in DKOFbxo6 cells that re-expressed Cand1 (Fig 6C–D), when the F-box motif of overexpressed Fbxo6 was deleted (Fbxo6ΔFbox) or when Fbxl16 that forms Skp1•Fbxl16 incapable of binding to Cul1 (Honarpour et al., 2014) was overexpressed (Fig 6D, S6F–G). Consistently, the increased doubling time in DKOFbxo6 cells was partially rescued by overexpression of Cul1 (Fig 6E), suggesting that the defects in DKOFbxo6 cells were at least partially due to the sequestration of Cul1 by HAFbxo6. Moreover, the increased cell doubling time and the depletion of the free Cul1 pool were also observed in DKO cells overexpressing Skp2, or Skp2 with its substrate binding region deleted (Skp2ΔLRR) (Fig 6F, S6H–J), suggesting the observed defects in FBP-overexpressing DKO cells were not due to altered stability of substrate proteins. In agreement with the dramatic reduction in cell proliferation, DKOSkp2ΔLRR cells exhibited increased levels of cleaved PARP, a marker for apoptosis (Fig 6G).
DISCUSSION
In this study, we expand upon our prior work (Pierce et al., 2013) to devise a kinetic model for the Cand1-mediated cycle of SCF assembly and disassembly. Consistent with the demonstrated substrate receptor exchange factor (SREF) activity of Cand1, we show that the degradation of SCF substrates is inefficient in Cand1/2 DKO human cells. We propose that this defect arises because substrates bind equivalently to FBPs regardless of whether they are assembled with Cul1 or not. In WT cells, this is immaterial, because for at least some substrates (e.g. pIκBα), a Skp1•β-TrCP•pIκBα complex gains access to Cul1 within a few minutes through dynamic assembly/disassembly of SCFs (Fig 7; see below), which is much faster than the dissociation of pIκBα from β-TrCP (Fig S3I–J). But this is not the case in DKO cells, where the pIκBα bound to free β-TrCP can access Cul1 only through successive rounds of dissociation and re-equilibration with the entire pool of β-TrCP molecules, which is expected to be very slow. An implication of this is that in WT cells the degradation rate of a substrate should not be related to the percent assembly of its FBP because of the rapid flux of SCF assembly/disassembly, whereas in DKO cells, which have a static complement of SCFs, there should be a direct correlation between these parameters. Indeed, there is remarkable agreement between the fraction of pIκBα•β-TrCP not associated with Cul1 in DKO cells, and the fold defect in degradation of IκBα, suggesting that inefficient access of substrate to Cul1 is the major deficiency of DKO cells. Consistent with this argument, slower degradation of IκBα in DKO cells was rescued by overexpression of Cul1, which drove assembly of almost all β-TrCP into an SCF, rendering it independent of Cand1/2.
Figure 7. Rapid cycling of Cul1 in human cells.

(A) Cycling of Cul1 summarized from biophysical, cellular and computational studies. Association rates are computed based on kon and steady state cellular concentrations of unbound proteins, and the cycle time for Cul1 is computed using effective rates for the reversible binding steps (see also Fig S5D). The reversal of the de-neddylation reaction by Dcn1 (dashed lines) is discouraged in WT cells due to preferential association of Dcn1 with Cand1•Cul1, but is expected to occur more frequently in DKO cells. The substrate of the SCF complex can bind the FBP either in its free or assembled state. Substrate binding stabilizes the SCF complex by preventing CSN from binding. The 55 s cycle time for Cul1 represents the average time it takes a Cul1 molecule to be deneddylated and exchanged into a different SCF if it is not bound by substrate.
(B) Deneddylation of Cul1 is fast in human cells. HEK293 cells were treated with 3 μM MLN4924 to inhibit the Nedd8 E1 and were maintained at 37°C fo r the indicated time before being directly lysed on culture plates. Average t1/2 for deneddylation is shown. Error bars, ± SD, n = 3.
(C) Neddylation of Cul1 is fast in human cells. Assay condition was similar to (B) but 3 μM CSN5i-3 was used to inhibit CSN (Schlierf et al., 2016). Average t1/2 for neddylation is shown. Error bars, ± SD, n = 3.
Prior work had suggested that Cand1 is not important for regulation of SCF in human cells (Bennett et al., 2010). Given that low (13%) re-expression of Cand1 fully rescued the deficiency of IκBα degradation in DKO cells, and that Cand2 partially compensated for the Cand1 KO, our data suggest that RNAi underestimates the significance of Cand-mediated exchange in cells.
Cand1, Nedd8 and CSN cooperatively regulate the rapid cycling of Cul1
Using the kinetic parameters for the Cul1 assembly/disassembly cycle, coupled with quantitative measurements of SCF protein and substrate levels in cells reported here and elsewhere (Bennett et al., 2010; Reitsma et al., 2017), we developed a mathematical model that allowed us to study the dynamics of SCF assembly and SCF substrate degradation. Our model accurately predicts the effect of Cand1/2 DKO on IκBα degradation and recapitulates the general features of the steady-state architecture of the SCF network and how it changes in DKO cells and upon inhibition of neddylation. Strikingly, the model reveals that with no bound substrate, Cul1 progresses through an entire exchange cycle with an average time of 87 s (Fig 7A, S5D). In agreement with this prediction, chemical ablation of neddylation or deneddylation resulted in conversion of Cul1 to fully deneddylated or neddylated species, respectively, with a t1/2 of 54–90 s (Fig 7B–C). Given the molecular ratio of Skp1:Cul1 (Reitsma et al., 2017), an F-box protein should cycle through an SCF complex every ~4 minutes. The rapid pace of this cycle allows a cell to remodel its network of SCFs far faster than could be achieved by regulated transcription or translation.
The key gate in our model is substrate occupancy. If an SCF is born with no substrate, it can immediately enter the exchange cycle. If it contains bound substrate, it persists until the substrate is degraded. Then, CSN binds and removes Nedd8, and the SCF is either disrupted by Cand1 or re-neddylated by Dcn1. Because Cand1 binds Cul1 faster than Dcn1 and Dcn1 prefers Cand1•Cul1 to Cul1, we predict that re-neddylation is discouraged and the SCF proceeds to the exchange state. Removal of Skp1•FBP from Cul1 by Cand1 establishes a substantial reservoir of Cand1•Cul1•Dcn1 (up to 42% of the total Cul1; Reitsma et al., 2017) that is primed to form new, active SCFs by drawing from the pool of Skp1•FBPs.
From a broader perspective, the mechanism that drives the SCF cycle resembles that of a Brownian ratchet which converts random (undirected) motion into directed motion through input of energy (Peskin et al., 1993). In the SCF cycle the “Brownian motion” is provided by the Cand1-mediated exchange of FBPs. Input of energy, which enforces directionality, is provided by neddylation, which prevents re-binding of Cand1 to a newly-formed, neddylated SCF.
The Adaptive Exchange Hypothesis
The degradation defect of cells lacking the Cand SREFs was compensated by overexpression of Cul1, such that the vast majority of FBPs were assembled with Cul1 and thus there was no need for an exchange mechanism to link substrates to Cul1. This begs the question, why does the exchange mechanism exist? Both the mathematical model and experimental observations converged on a simple explanation: Cand-mediated exchange renders the SCF system tolerant of large changes in the expression of individual FBPs. Notably, bioinformatics analyses revealed that such large changes occur routinely during development (Fig S7). This could explain why the multicellular organism Arabidopsis is dramatically perturbed by disruption of Cand function (Cheng et al., 2004; Chuang et al., 2004; Feng et al., 2004), whereas cultured cells and single-celled yeasts are not (Liu et al., 2009; Zemla et al., 2013). If it were not for the Cand system, cells would require counting mechanisms to ensure that the total level of Cul1 was adequate to saturate all available Skp1•FBP modules. Having a system in which all SCF enzymes were always assembled and active could create other problems, especially considering the millisecond rate at which SCF enzymes can initiate and extend ubiquitin chains (Pierce et al., 2009; Scott et al., 2016). The exchange mechanism, by creating a delay between substrate binding to an FBP and its assembly into an active SCF, may have the side benefit of increasing the specificity of the system by setting a ceiling on the maximal koff value of a substrate.
At its heart, the assembly/disassembly cycle is controlled by the koff of substrate from an active SCF complex. The molecular logic of the SCF cycle resembles the control of microtubule networks by dynamic instability (Kirschner and Mitchison, 1986). In that case, individual microtubules sample the cytoplasm through constant, randomly-directed growth and shrinkage. Formation of favorable contacts retards the koff of tubulin subunits, thereby stabilizing the microtubule and driving morphogenesis of a mitotic spindle. In the case of SCFs, the same Darwinian process of variation-selection occurs, but instead of enabling morphogenesis of a specific structure, it controls the repertoire of assembled SCFs such that the cell is biased to accumulate those that are needed at a given time. We refer to this mechanism as ‘adaptive exchange’. It is adaptive both in a functional, biochemical sense at the cellular and organismal levels as described here, as well as in an evolutionary sense. This exchange mechanism could enable expansion/contraction of FBP gene repertoires without the maladaptive effects that would occur in a system dependent on stoichiometric Cul1. Duplicated FBP genes could then diverge and acquire new functions beneficial to the organism. This could be the basis for massive variations in FBP gene number found in different organisms (Xu et al., 2009; Srinivasan et al., 2013).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Raymond J. Deshaies (deshaies@caltech.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Flp-In T-REx 293 cells (RRID:CVCL_U427) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% tetracycline-free fetal bovine serum (Clontech) and penicillin at 37 °C. The generation of stable cell lines is described below in method details.
METHOD DETAILS
Constructs
ΔH1Cand1 was generated by replacing the first 14 codons of full length Cand1 cDNA with DNA sequence encoding CCPGCCGSG. The resulting construct was inserted into the Xma1/Not1 sites of pGEX-4T for expression in E. coli. Cand11-603 and Cand1604-1230 were designed by truncating the full length Cand1 cDNA at the indicated codons and inserting the PCR products into the Xma1/Not1 sites of pGEX-4T for expression in E. coli. Constructs for CRISPR nickase- mediated gene knockout in mammalian cells were designed as described (Ran et al., 2013). Briefly, oligos containing sgRNA sequences were annealed and inserted into the Bbs1 site of pX335 (Addgene). The homologous recombination template was generated by first cloning the 300-bp homologous recombination regions using genomic DNA purified from WT 293 cells as the PCR templates, then inserting the antibiotic resistance gene plus a terminator between the two homologous recombination DNA fragments using overlapping PCR (Heckman & Pease, 2007), and finally inserting the resulting PCR products into the EcoR1/Xho1 sites of pGEX-4T. For generation of stable cells lines using the Flp-In system, cDNAs encoding Cand1HA, 3xFLAGCul1, 3xFLAGIκBα and 2xStrepIIβ-TrCP were individually inserted into pcDNA5/FRT/TO vector (Thermo Fisher Scientific). For expression of exogenous genes in Figure 6, the lentiviral backbone pCDH-EF1-MCS-IRES-NEO (System Biosciences) was used to direct the expression of HAFbxo6, HAFbxo6ΔF-box(Δ10-57), HASkp2, HASkp2ΔLRR(1-234), HAFbxl16, and MycCand1.
Protein Expression and Purification
Cul1•Rbx1, Skp1•Skp2, and Skp1ΔΔ•β-TrCP139-569 were purified as described (Saha and Deshaies, 2008). Cand1, Skp1ΔΔ•Skp2, Skp1•Fbxw7TAMRA, and Skp1•β-TrCP139-569 were purified as described (Pierce et al., 2013). Cul1•GSTRbx1 was purified as described for Cul1•Rbx1, omitting the thrombin digestion step. Cul1Sortase-Tag•Rbx1 was expressed by cotransforming BL21 E. coli with RDB 2080 and RDB 2557 and inducing overnight at 16°C; it was then purified on glutathione resin followed by digestion with thrombin and chromatography on HiTrap SP cation exchange column (GE Healthcare), and was exchanged to buffer containing 50 mM Tris (pH 7.6), 150 mM NaCl, 10 mM CaCl2 using PD-10 columns (GE Healthcare). Cul1Sortase-Tag•Rbx1 was incubated with 60 μM Sortase and 250 μM GGGGAMC, GGGGKTAMRA, or GGGGKFAM peptides (New England Peptide) at room temperature for 24 hr. Cul1AMC•Rbx1, Cul1TAMRA•Rbx1, and Cul1FAM•Rbx1 were further purified by S200 size exclusion chromatography. ΔH1Cand1 was expressed in Rosetta E. coli with IPTG induction overnight at 16°C, and was purified on glutathione resin followed by digestion with thrombin and chromatography on HiTrap Q cation exchange column (GE Healthcare) and S200 size exclusion column. Then 50 μl of 40 μM ΔH1Cand1 was incubated with 1 μl Lumio Green (FlAsH) dye (Thermo Fisher Scientific) in buffer containing 20 mM Tris (pH7.5), 100 mM NaCl, 2 mM TCEP (Thermo Fisher Scientific), 1 mM EDTA, and 5% glycerol at room temperature for at least two hours to generate FlAsHΔH1Cand1. Cand11-603, Cand1604-1230, Skp1, and Dcn1 were expressed and purified similarly to the preparation of Cand1. Ubc12 was expressed in Sf9 cells from a recombinant baculovirus and was prepared as previously described (Scott et al., 2014).
FRET Assay
Fluorimeter scans were performed on a Fluoromax-4 Spectrofluorometer (Jobin Yvon) in a buffer containing 30 mM Tris (pH 7.6), 100 mM NaCl, 0.5 mM DTT, and 1 mg/ml Ovalbumin (Sigma). Mixtures containing Cul1AMC•Rbx1 were excited at 350 nm and the emissions were scanned from 400 nm to 600 nm. Mixtures containing CFPCul1•Rbx1 were excited at 430 nm and the emissions were scanned from 450 nm to 650 nm. Stopped flow reactions were performed on a Kintek stopped flow machine in the same buffer as the fluorimeter scans.
Antibodies
The following primary antibodies were used in Western Blot analyses: anti-Cand1 (Bethyl Laboratories # A302-901, Santa Cruz Biotechnology # 10672), anti-Cand2 (Bethyl Laboratories # A304-046A), anti-IκBα (Abcam # ab32518), anti-β-TrCP (Cell Signaling # 4394S), anti-GAPDH (Millipore # MAB374), anti-Cul1 (Thermo Fisher Scientific # 32-2400), anti-phospho-IκBα (Cell Signaling Technology # 9246S), anti-FLAG (Sigma # F1804), anti-Dcn1 (Novus Biologicals # H00054165-A01), anti-Ubc12 (Rockland # 600-401-865), anti-Irp2 (Santa Cruz Biotechnology # 33682), anti-Fbxl5 (Neoclone # N0036), anti-Cul4a (Cell Signaling Technology # 2699S), anti-Rbx1 (Bethyl Laboratories # A303-462A), anti-Skp1 (Thermo Fisher Scientific # MA5-15928), anti-HA-HRP (Sigma # 12013819001), anti-cleaved PARP (Asp214) (Cell Signaling # 5625), anti-Fbxo6 (Abcam # 103635), anti-Cyclin E (Santa Cruz Biotechnology # 247), anti-Skp2 (D3G5) (Cell Signaling # 2652S), anti-p27 (Abcam # 32034). Alexa Fluor 680 conjugated anti-mouse IgG (Thermo Fisher Scientific # A10038), Alexa Fluor 790 conjugated anti-rabbit IgG (Thermo Fisher Scientific # A11374), and Alexa Fluor 680 conjugated anti-rabbit IgG (Abcam # ab175772) secondary antibodies were used to detect the primary antibodies on an Odyssey Imager (LI-COR Biosciences). HRP-conjugated anti-rabbit IgG (Sigma), HRP-conjugated anti-mouse IgG (Sigma), and anti-mouse IgG HRP Mouse TrueBlot Ultra (Rockland # 18-8817-33) were used to detect the primary antibodies using chemiluminescence.
Generation of Stable Cell Lines
To construct Cand1/2 DKO cells, the CAND2 gene was first knocked out in Flp-In T-REx 293 cells to generate Cand2 single KO cells, followed by disruption of the CAND1 gene. To knock out the CAND2 gene with CRISPR-Cas9 nickases, WT cells in a 12-well plate were cotransfected with three plasmids: 0.2 μg pX335 containing “GTGGAAGGCGGCGGTGCTCA” guide RNA, 0.2 μg pX335 containing “GAAGATGACGTCCAGCGACA” guide RNA, and 0.2 μg pGEX-4T containing neomycin resistance gene plus a SV40 terminator which was placed between two 300-bp DNA sequences that are identical to CAND2 genomic DNA regions flanking the first exon. Twenty-four hours after cotransfection, cells were transferred to 15-cm plates and cultured with medium containing 800 μg/ml G418 (Thermo Fisher Scientific). After two weeks, neomycin-resistant colonies were isolated and screened for the loss of Cand2 protein by immunoblot with an antibody recognizing the C-terminus of Cand2. Colonies that showed loss of Cand2 were further confirmed for complete knock out of Cand2 by sequencing PCR products of the genomic region surrounding the first exon of Cand2. Forward primer “AGCTGGCACCTACGGGAATAACAAGGA” and reverse primer “ACACACACGAGGGAGGAGAG” were used for PCR. The sequencing results also revealed independent KO cell colonies. A similar approach was used to knock out the Cand1 gene with CRISPR-Cas9 nickases. WT or Cand2 single KO cells were cotransfected with three plasmids: 0.2 μg pX335 containing “GCAAATTGGAAATGTGGTACG” guide RNA, 0.2 μg pX335 containing “GCATCCAGCGACAAGGACTTT” guide RNA, and 0.2 μg pGEX-4T containing puromycin resistance gene plus a BGH terminator placed between two 300-bp DNA sequences that are identical to Cand1 genomic DNA regions flanking the first exon. Cells were then selected for resistance to 1 μg/ml puromycin, loss of Cand1 protein in immunoblot analysis, and disruption of WT Cand1 Exon 1 in the genomic DNA. Forward primer “TGTCTGGCTCCCCGTAGAGGCCCTTCT” and reverse primer “CCTATTCGCTTGCCATCCT” were used for PCR.
Site-specific fusion of sequences encoding the 3xFLAG tag to the 5’ end of the coding region of endogenous CUL1 alleles is described elsewhere (Reitsma et al., 2017).
Targeted integrations of the coding sequences of Cand1HA, 3xFLAGCul1, 3xFLAGIκBα, or 2xStrepIIβ-TrCP into the Flip-In T-REx 293 cells were carried out as described in the manual (Thermo Fisher Scientific). pcDNA5/FRT/TO vector containing the gene insert and pOG44 vector containing Flp recombinase were cotransfected into cells using Lipofectamine 3000 (Invitrogen). Cells were then selected for resistance to 100 μg/ml hygromycin and confirmed for tetracycline induced expression of integrated genes.
Lentiviral Infection
Lentiviral constructs were co-transfected with packaging (psPAX2) and enveloping (pMD.2G) plasmids (System Biosciences) into 293FT cells using Fugene HD (Promega). Virus-containing supernatants were harvested at 48 and 72 hrs after transfection. Relative viral titer was determined using Lenti-X GoStix (Clontech). HEK293 cells were infected at a multiplicity of infection of 2.0 at 24 hrs and 48 hrs after seeding, to ensure 100% cell infection rate which was verified by immunofluorescence.
IκBα Degradation Assay
Cells (0.6 million) of desired genotypes were seeded on 6-well plates and allowed to grow overnight with or without 100 ng/ml tetracycline in the medium. Cells were then incubated in DMEM medium containing no serum for 6 hours, and 100 μg/ml cycloheximide was added to the medium 10 minutes before the start of the assay. Cells were taken out of the cell culture incubator five minutes before the addition of 25 ng/ml TNFα (Sigma-Aldrich), and were kept at room temperature for the duration of the degradation assay. At different time points after the addition of TNFα, cells were washed with PBS and lysed by adding 2x SDS sample buffer to the plate. Cell lysates were collected in tubes and sonicated before fractionation by SDS-PAGE for Western Blot analyses. IκBα signals (both phosphorylated and unmodified IκBα) measured on an Odyssey Imager (LI-COR Biosciences) were normalized to GAPDH signals in the same sample and were fit to a single exponential in Prism to calculate half-lives.
In vivo IκBα Ubiquitination Assay
Similar to IκBα degradation assay, except that 1 μM bortezomib was added to the medium 30 minutes before TNFα treatment. Ubiquitinated phospho-IκBα was detected by anti-phospho-IκBα primary antibody.
Co-Immunoprecipitation Assay
To IP the endogenous 3xFLAGCul1 and probe the formation of SCFβ-TrCP, cells were resuspended in buffer containing 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 1x protease inhibitors (Roche), 50 μM quinoline-8-thiol (8TQ, Sigma-Aldrich)(Li et al., 2017), 0.6% IGEPAL CA-630, and recombinant Cul1•Rbx1 protein at ~100x of the endogenous Cul1 level (1.5x cell pellet volume of 35 μM Cul1•Rbx1 solution)(Reitsma et al., 2017). Cells were lysed by sonication, and the supernatant of the cell lysate after 10-min centrifugation at 15,000 g was incubated with anti-FLAG beads (Sigma-Aldrich) for 30 minutes at 4 °C. The beads were then washed by the lysis buffer three times and eluted by 2x SDS-PAGE sample buffer. To IP 3xFLAGIκBα, a similar method was used except that no recombinant Cul1•Rbx1 protein was added in the lysis buffer. To IP the endogenous 3xFLAGCul1 and probe the formation of the Cul1•Dcn1 complex, cells were lysed in buffer containing 50 mM HEPES (pH 7.5), 5 mM Mg(OAc)2, 70 mM KOAc, 50 μM 8TQ, and protease inhibitors. Supernatant of the cell lysate after centrifugation was incubated with anti-FLAG beads (BioLegend) for 30 minutes at 4 °C, and the b eads were washed in micro Bio-Spin columns (Bio-Rad) by the lysis buffer three times and eluted by 2x SDS-PAGE sample buffer. To determine percent β-TrCP bound to Cul1, cells were lysed by brief sonication after being mixed with Pierce IP lysis buffer (Thermo Fisher) containing 1x protease inhibitor (Roche), 50 μM 8TQ, and recombinant Cul1•GSTRbx1 protein at ~100x of the endogenous Cul1 level (1.5x cell pellet volume of 35 μM Cul1•GSTRbx1 solution), followed by sequential incubations with anti-FLAG beads and glutathione sepharose 4B beads (Reitsma et al., 2017). The precipitated proteins in each pull-down were eluted by 30 μl 2x SDS-PAGE sample buffer prior to WB analysis. To determine percent Cul1 bound to overexpressed HA-tagged F-box protein, cells were lysed by brief sonication after being mixed with Pierce IP lysis buffer (Thermo Fisher) containing 1x protease inhibitor (Roche) and recombinant Cul1•GSTRbx1 ‘sponge’ protein. Then 100 μl cell lysate (input) was withdrawn and incubated with anti-HA EZVIEW Red affinity beads (Sigma). After separating and collecting the flowthrough, the beads were eluted in 100 μl 2x SDS-PAGE sample buffer (IP), and 10 μl each of input, IP and flowthrough samples were fractionated by SDS-PAGE and analyzed by WB.
Usp2 on-bead treatment
After the immunoprecipitation of 3xFLAGIκBα, anti-FLAG beads were washed and incubated with 0.5 μM Usp2 catalytic domain (Boston Biochem) in 30 μl buffer containing 50 mM HEPES (pH 8.0), 0.01% IGEPAL CA-630, and 3 mM DTT at 37 °C for 2.5 hours. The treatment was stopped and proteins were eluted by adding 10 μl of 4x SDS-PAGE sample buffer.
Quantification of Protein Concentration by WB
A few million WT cells were collected and resuspended in 150 μl PBS buffer containing full length recombinant GSTIκBα (Novus Biologicals) and GSTβ-TrCP (Novus Biologicals) and lysed by mixing with 150 μl 4x SDS sample buffer. The total cell volume per sample was estimated by multiplying the total cell number and 2 pl/cell (BioNumbers.hms.harvard.edu). Recombinant GSTIκBα and GSTβ-TrCP were added to a level corresponding to 200 nM and 26 nM of cellular concentration, respectively, as internal standards. Both endogenous and recombinant IκBα and β-TrCP were detected by immunoblot analysis to reveal the ratio of endogenous vs. internal standard protein for quantification. The concentration of added recombinant protein standard was determined by a serial dilution experiment prior to the quantification experiment to avoid over- or under-loading, and it was also verified that all quantified signals were within the linear range of detection.
In vitro Neddylation Assay
Neddylation reactions were conducted at room temperature in buffer containing 30 mM Tris (pH 7.5), 5 mM MgCl2, and 2 mM ATP, with Dcn1 and NAE purified from E. coli and Ubc12 purified from Sf9 insect cells. Concentrations of Dcn1, NAE, and Ubc12 used in each experiment are specified in figures and figure legends. Nedd8 (Boston Biochem) was first thioesterified onto Ubc12 by NAE in an individual tube, and the Ubc12~Nedd8 was then mixed with Cul1 to start the neddylation reaction. When cell lysate was used for neddylation reaction, DKO cells pooled from a 10-cm culture plate were lysed in 200 μl buffer containing 30 mM Tris (pH 7.5), 5 mM MgCl2, 2 mM ATP, protease inhibitors, and 50 μM 8TQ. After mixing the neddylation enzymes with Cul1, samples were incubated at room temperature for desired time periods, and the reaction was stopped by adding 4x SDS-PAGE sample buffer.
Dissociation of pIκBα•β-TrCP in Cell Lysate
DKO cells expressing 3xFLAGIκBα were treated with 25 ng/ml TNFα for 15 min, and were collected and lysed in buffer containing 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 0.6% IGEPAL CA-630, protease inhibitors, and phosphatase inhibitors (Thermo Fisher Scientific). An aliquot of cell lysate (100 μl) was incubated with anti-FLAG beads for 30 min to get 0-hr sample. Another 100 μl aliquot was kept at room temperature for 9.5 hr and then incubated with anti-FLAG beads for 30 min to get the 10-hr control sample. For the rest of the lysate, recombinant Skp1ΔΔ•β-TrCP139-569 (0.8x cell pellet volume of 12 μM Skp1•β-TrCP139-569 solution, ~100x of endogenous β-TrCP level) was added as a chase, and the lysate was incubated at room temperature. Thirty min prior to each time point, 100 μl was drawn from the lysate-chase mixture and incubated with anti-FLAG beads for 30 min. All the proteins bound by the beads were eluted by 2x SDS-PAGE sample buffer and analyzed by SDS-PAGE followed by Western Blot.
Analysis of FBP Expression Levels
RNA-seq data for mouse development across multiple tissues was obtained from ENCODE (Mouse ENCODE Project; BioProject accession number PRJNA66167). Specifically, data for 66 tissues across embryonic and birth (day 0) timepoints generated by Barbara Wold’s lab was used, along with data for ES cells from an E14 mouse embryo generated by Michael Snyder’s lab. Processed data was downloaded in an automated manner and the average of Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values for two replicates was retained for further analyses. Accession numbers or processed data for the 134 ENCODE datasets available upon request. For analyses, only transcripts expressed in ES cells (25130 transcripts) were considered. FBPs were defined as those previously described (Jin et al., 2004).
QUANTIFICATION AND STATISTICAL ANALYSIS
Western Blots with fluorescent signals were scanned on an Odyssey Imager (LI-COR Biosciences), and when necessary, multiple scans with different detection sensitivity levels were taken to avoid oversaturation. Images were exported as tif files, and protein band intensities were quantified by ImageJ (NIH) or Image Studio Lite (LI-COR Biosciences). Western Blots with chemiluminescence were detected by BioMax MR Film (Carestream) with varied exposure time lengths, and films with appropriate exposure strength were scanned and quantified using ImageJ (NIH). Protein gels stained by Coomassie blue were either imaged by Gel Doc™ XR+ Gel Documentation System (Bio-Rad) or scanned after drying between cellophane sheets, and the protein band intensities were quantified by ImageJ (NIH). Kinetic analyses were performed by regressions in Prism. Fluorescence signals detected by the Typhoon scanner were quantified by ImageQuant (GE Healthcare). Statistical parameters are reported in the Figures and Figure Legends. Data are judged to be statistically significant when p < 0.05 by two-tailed Student’s t test. Statistical analysis was performed in GraphPad QuickCalcs.
Supplementary Material
Acknowledgments
We thank Brenda Schulman, Ning Zheng, William den Besten for gifts of reagents, Novartis for providing the CSN5i-3 compound, Shu-Ou Shan for sharing instruments, Lea Goentoro and Noah Olsman for insightful discussion on the mathematical model, Robert J. Flassig for discussions on parameter estimation and all the members of the Deshaies lab for helpful discussions. XL is a fellow of The Jane Coffin Childs Memorial Fund for Medical Research (JCCMF), and this investigation has been aided by a grant from JCCMF. JMR is supported by F32 grant GM112308 from NIH. JLM is supported by Life Sciences Research Foundation. RJD was an investigator of the Howard Hughes Medical Institute (HHMI), and this work was supported in part by HHMI and NIH GM065997 to RJD.
Conceptualization, X.L., R.S., R.J.D.; Methodology and Investigation, X.L., J.M.R, Y.Z., J.L.M.; Methodology and Software, R.S.; Writing, X.L, R.S., R.J.D; Supervision, X.L, R.S., R.J.D; Funding Acquisition, X.L., J.M.R., R.J.D..
Footnotes
DECLARATION OF INTERESTS
Raymond J. Deshaies is an employee and shareholder of Amgen.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143:951–965. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosu DR, Feng H, Min K, Kim Y, Wallenfang MR, Kipreos ET. C. elegans CAND-1 regulates cullin neddylation, cell proliferation and morphogenesis in specific tissues. Developmental biology. 2010;346:113–126. doi: 10.1016/j.ydbio.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Xiong S, Li J, Li X, Liu Y, Zou C, Mallampalli RK. The ubiquitin E3 ligase SCF-FBXO24 recognizes deacetylated nucleoside diphosphate kinase A to enhance its degradation. Molecular and cellular biology. 2015;35:1001–1013. doi: 10.1128/MCB.01185-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Dai X, Zhao Y. AtCAND1, a HEAT-repeat protein that participates in auxin signaling in Arabidopsis. Plant Physiol. 2004;135:1020–1026. doi: 10.1104/pp.104.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HW, Zhang W, Gray WM. Arabidopsis ETA2, an apparent ortholog of the human cullin-interacting protein CAND1, is required for auxin responses mediated by the SCF(TIR1) ubiquitin ligase. The Plant cell. 2004;16:1883–1897. doi: 10.1105/tpc.021923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland WW. Partition analysis and the concept of net rate constants as tools in enzyme kinetics. Biochemistry. 1975;14:3220–3224. doi: 10.1021/bi00685a029. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annual review of biochemistry. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Duda MD, Scott DC, Calabrese MF, Zimmerman ES, Zheng N, Schulman BA. Structural regulation of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol. 2011;21:257–264. doi: 10.1016/j.sbi.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberley ED, Mosadeghi R, Deshaies RJ. Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-F-box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism. The Journal of biological chemistry. 2012;287:29679–29689. doi: 10.1074/jbc.M112.352484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nature reviews Molecular cell biology. 2015;16:30–44. doi: 10.1038/nrm3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enchev RI, Scott DC, da Fonseca PC, Schreiber A, Monda JK, Schulman BA, Peter M, Morris EP. Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep. 2012;2:616–627. doi: 10.1016/j.celrep.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Shen Y, Sullivan JA, Rubio V, Xiong Y, Sun TP, Deng XW. Arabidopsis CAND1, an unmodified CUL1-interacting protein, is involved in multiple developmental pathways controlled by ubiquitin/proteasome-mediated protein Degradation. The Plant cell. 2004;16:1870–1882. doi: 10.1105/tpc.021949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg SJ, Cascio TC, Shumway SD, Garbutt KC, Liu J, Xiong Y, Zheng N. Structure of the Cand1-Cul1-Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell. 2004;119:517–528. doi: 10.1016/j.cell.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Goody RS, Hofmann-Goody W. Exchange factors, effectors, GAPs and motor proteins: common thermodynamic and kinetic principles for different functions. Eur Biophys J. 2002;31:268–274. doi: 10.1007/s00249-002-0225-3. [DOI] [PubMed] [Google Scholar]
- Guo Z, Ahmadian MR, Goody RS. Guanine nucleotide exchange factors operate by a simple allosteric competitive mechanism. Biochemistry. 2005;44:15423–15429. doi: 10.1021/bi0518601. [DOI] [PubMed] [Google Scholar]
- Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nature protocols. 2007;2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- Honarpour N, Rose CM, Brumbaugh J, Anderson J, Graham RL, Sweredoski MJ, Hess S, Coon JJ, Deshaies RJ. F-box protein FBXL16 binds PP2A-B55α and regulates differentiation of embryonic stem cells along the FLK1+ lineage. Mol Cell Proteomics. 2014;13:780–791. doi: 10.1074/mcp.M113.031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang GY, Zhang XP, Wang L, Lin XY, Yu JH, Wang EH, Zhang Y. FBXO25 promotes cell proliferation, invasion, and migration of NSCLC. Tumour Biol. 2016;37:14311–14319. doi: 10.1007/s13277-016-5298-1. [DOI] [PubMed] [Google Scholar]
- Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes & development. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamran M, Long ZJ, Xu D, Lv SS, Liu B, Wang CL, Xu J, Lam EW, Liu Q. Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis. 2017;6:e298. doi: 10.1038/oncsis.2016.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama K, Noguchi K, Sugimoto Y. FBXO15 regulates P-glycoprotein/ABCB1 expression through the ubiquitin--proteasome pathway in cancer cells. Cancer Sci. 2013;104:694–702. doi: 10.1111/cas.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keuss MJ, Thomas Y, McArthur R, Wood NT, Knebel A, Kurz T. Characterization of the mammalian family of DCN-type NEDD8 E3 ligases. J Cell Sci. 2016;129:1441–1454. doi: 10.1242/jcs.181784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AY, Bommelje CC, Lee BE, Yonekawa Y, Choi L, Morris LG, Huang G, Kaufman A, Ryan RJ, Hao B, et al. SCCRO (DCUN1D1) is an essential component of the E3 complex for neddylation. The Journal of biological chemistry. 2008;283:33211–33220. doi: 10.1074/jbc.M804440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner M, Mitchison T. Beyond self-assembly: from microtubules to morphogenesis. Cell. 1986;45:329–342. doi: 10.1016/0092-8674(86)90318-1. [DOI] [PubMed] [Google Scholar]
- Klebe C, Prinz H, Wittinghofer A, Goody RS. The kinetic mechanism of Ran—nucleotide exchange catalyzed by RCC1. Biochemistry. 1995;34:12543–12552. doi: 10.1021/bi00039a008. [DOI] [PubMed] [Google Scholar]
- Kroll M, Margottin F, Kohl A, Renard P, Durand H, Concordet JP, Bachelerie F, Arenzana-Seisdedos F, Benarous R. Inducible degradation of IkappaBalpha by the proteasome requires interaction with the F-box protein h-betaTrCP. The Journal of biological chemistry. 1999;274:7941–7945. doi: 10.1074/jbc.274.12.7941. [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Kurz T, Chou YC, Willems AR, Meyer-Schaller N, Hecht ML, Tyers M, Peter M, Sicheri F. Dcn1 functions as a scaffold-type E3 ligase for cullin neddylation. Molecular cell. 2008;29:23–35. doi: 10.1016/j.molcel.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Li J, Yakushi T, Parlati F, Mackinnon AL, Perez C, Ma Y, Carter KP, Colayco S, Magnuson G, Brown B, et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat Chem Biol. 2017;13:486–493. doi: 10.1038/nchembio.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Furukawa M, Matsumoto T, Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Molecular cell. 2002;10:1511–1518. doi: 10.1016/s1097-2765(02)00783-9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Mimura S, Kishi T, Kamura T. A longevity protein, Lag2, interacts with SCF complex and regulates SCF function. The EMBO journal. 2009;28:3366–3377. doi: 10.1038/emboj.2009.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SC, Hannink M. CAND1-mediated substrate adaptor recycling is required for efficient repression of Nrf2 by Keap1. Molecular and cellular biology. 2006;26:1235–1244. doi: 10.1128/MCB.26.4.1235-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Lee BH, King RW, Finley D, Kirschner MW. Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science. 2015;348:1250834. doi: 10.1126/science.1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–1061. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monda JK, Scott DC, Miller DJ, Lydeard J, King D, Harper JW, Bennett EJ, Schulman BA. Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure. 2013;21:42–53. doi: 10.1016/j.str.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosadeghi R, Reichermeier KM, Winkler M, Schreiber A, Reitsma JM, Zhang Y, Stengel F, Cao J, Kim M, Sweredoski MJ, et al. Structural and kinetic analysis of the COP9-Signalosome activation and the cullin-RING ubiquitin ligase deneddylation cycle. eLife. 2016:5. doi: 10.7554/eLife.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskin CS, Odell GM, Oster GF. Cellular motions and thermal fluctuations: the Brownian ratchet. Biophys J. 1993;65:316–324. doi: 10.1016/S0006-3495(93)81035-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce NW, Lee JE, Liu X, Sweredoski MJ, Graham RL, Larimore EA, Rome M, Zheng N, Clurman BE, Hess S, et al. Cand1 Promotes Assembly of New SCF Complexes through Dynamic Exchange of F Box Proteins. Cell. 2013;153:206–215. doi: 10.1016/j.cell.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raue A, Kreutz C, Maiwald T, Bachmann J, Schilling M, Klingmüller U, Timmer J. Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics. 2009;25:1923–1929. doi: 10.1093/bioinformatics/btp358. [DOI] [PubMed] [Google Scholar]
- Reitsma JM, Liu X, Reichermeier KM, Moradian A, Sweredoski MJ, Hess S, Deshaies RJ. Composition and regulation of the cellular repertoire of SCF ubiquitin ligases. Cell. 2017;171:1326–1339. doi: 10.1016/j.cell.2017.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Molecular cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, Grishin NV, Bruick RK. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science. 2009;326:722–726. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MW, McQuary PR, Wee S, Hofmann K, Wolf DA. F-box-directed CRL complex assembly and regulation by the CSN and CAND1. Molecular cell. 2009;35:586–597. doi: 10.1016/j.molcel.2009.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman BA, Carrano AC, Jeffrey PD, Bowen Z, Kinnucan ERE, Finnin MS, Elledge SJ, Harper JW, Pagano M, Pavietich NP. Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature. 2000;408:381–386. doi: 10.1038/35042620. [DOI] [PubMed] [Google Scholar]
- Scott DC, Sviderskiy VO, Monda JK, Lydeard JR, Cho SE, Harper JW, Schulman BA. Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell. 2014;157:1671–1684. doi: 10.1016/j.cell.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nature methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Spencer E, Jiang J, Chen ZJ. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes & development. 1999;13:284–294. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan J, Dillman AR, Macchietto MG, Heikkinen L, Lakso M, Fracchia KM, Antoshechkin I, Mortazavi A, Wong G, Sternberg PW. The draft genome and transcriptome of Panagrellus redivivus are shaped by the harsh demands of a free-living lifestyle. Genetics. 2013;193:1279–1295. doi: 10.1534/genetics.112.148809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straube R, Shah M, Flockerzi D, Wolf DA. Trade-off and flexibility in the dynamic regulation of the cullin-RING ubiquitin ligase repertoire. PLoS Comput Biol. 2017;13:e1005869. doi: 10.1371/journal.pcbi.1005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhu W, Nhan T, Toth JI, Petroski MD, Wolf DA. CAND1 controls in vivo dynamics of the cullin 1-RING ubiquitin ligase repertoire. Nat Commun. 2013;4:1642. doi: 10.1038/ncomms2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Ma H, Nei M, Kong H. Evolution of F-box genes in plants: different modes of sequence divergence and their relationships with functional diversification. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:835–840. doi: 10.1073/pnas.0812043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemla A, Thomas Y, Kedziora S, Knebel A, Wood NT, Rabut G, Kurz T. CSN- and CAND1-dependent remodelling of the budding yeast SCF complex. Nat Commun. 2013;4:1641. doi: 10.1038/ncomms2628. [DOI] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. Structure of the Cul1-Rbx1-Skp1-F-boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.