

Abstract
Viral pathogenesis results from changes in host cells due to virus usurpation of the host cell and the innate cellular responses to thwart infection. We measured global changes in protein expression and localization in HIV-1 infected T-cells using subcellular fractionation and the Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH-MS) proteomic platform. Eight biological replicates were performed in two independent experimental series. In silico merging of both experiments identified 287 proteins with altered expression (p< 0.05) between control and infected cells-172 in the cytoplasm, 84 in the membrane, and 31 in nuclei. 170 of the proteins are components of the NIH HIV interaction database. Multiple Reaction Monitoring and traditional immunoblotting validated the altered expression of several factors during infection. Numerous factors were found to affect HIV infection in gain- and loss-of-expression infection assays, including the intermediate filament vimentin which was found to be required for efficient infection.
Keywords: HIV-1, SWATH-MS, proteomics, host response to infection, vimentin
Introduction
Despite the availability of numerous effective therapies that control virus replication, human immunodeficiency virus type 1 (HIV-1) remains incurable and one of the most significant global pathogens. The prospect of life-long treatment for an increasing number of infected individuals makes the emergence of drug resistant viruses a critical concern and highlights the need for the continued development of new antiviral therapies. One under-explored class of inhibitors are compounds that target cellular proteins to disrupt HIV replication. Targeting cellular factors is difficult as the host-virus relationship is complex. As an obligate intracellular parasite, the virus induces cellular changes to establish infection and promote replication. Host cells initiate innate responses to counter the pathogen and signal nearby cells to the presence of the microbe. Consequently, viruses also stimulate additional alterations in the host cell proteome in order to neutralize the innate responses. The cumulative effect of these complex interactions results in pathology and clinical disease manifestations of viral infection. An improved understanding of these multifaceted interactions will increase knowledge of virus replication and accelerate the development of cellular-based antiretroviral therapies.
Many genetic, proteomic, and other “-omics” studies have sought to identify cell factors critical for virus replication and pathogenesis (reviewed in (Bushman et al., 2009; Fellay et al., 2010; Giri et al., 2006) and others). Transcriptome and RNAi studies have provided an important, yet incomplete picture of the HIV interactome as these approaches cannot distinguish alterations in protein level, modification, function, localization, or association in cellular complexes. Proteomic studies examining the host cell response have typically relied on unbiased “shot-gun” approaches. These have included studies of whole cell lysates, individual cellular compartments, or the interactome of individual viral proteins by affinity purification (Donnelly and Ciborowski, 2016; Haverland et al., 2014; Kraft-Terry et al., 2011; Li et al., 2016). A number of HIV-specific studies include analyses of the proteome of infected T cells (Navare et al., 2012; Ringrose et al., 2008; Sheng and Wang, 2009), macrophages (Haverland et al., 2014; Kraft-Terry et al., 2010), intact HIV particles (Bregnard et al., 2013; Chertova et al., 2006; Saphire et al., 2006), purified HIV cores (Fuchigami et al., 2002; Santos et al., 2012), and HIV reverse transcription and preintegration complexes (Raghavendra et al., 2010; Schweitzer et al., 2013). Individual viral protein interactomes have also been studied, including the viral proteins Gag (Le Sage et al., 2015), Nef (Mukerji et al., 2012), Tat (Coiras et al., 2006), and Rev (Naji et al., 2012), as well as a comprehensive study of all HIV-1 proteins (Jager et al., 2011).
Our goal in this study was to map the global intracellular changes induced by HIV-1 infection of Jurkat T cells using a subcellular-focused proteomic approach. Cell lysates were separated into cytosolic, membrane bound, and nuclear fractions by differential lysis. SWATH-based MS was used to quantify protein abundance within the three subcellular compartments of uninfected and HIV-infected T-cells. SWATH is a label-free MS approach in which the acquisition of spectra is divided into sequential segments and all information is collected within each segment (Gillet et al., 2012). This contrasts traditional MS which collects a limited amount of data for a specific analyte within the entire range of spectrum. SWATH requires preconstruction of a spectral library using traditional data-dependent acquisition. The experimental groups are run using a data-independent mode of MS and the spectra are analyzed against the preconstructed library after data acquisition. The collected spectra are quantitative and can be analyzed using traditional statistical methods.
We performed two independent series of biological replicates (Exp1 and Exp2), consisting of five and three repeats of control and infected samples, respectively. The number of candidate proteins with statistically different abundance between infected and control cells ranged from 225 to 578 within individual subcellular fractions. The overlap of proteins between Exp1 and Exp2 ranged between 13.5–25.9% among the three fractions. Therefore, we evaluated the data by combining the spectra from all eight experiments in silico. This approach slightly increased the number of candidate factors. Multiple Reaction Monitoring (MRM) and immunoblotting on a subset of factors validated the change in expression of several factors. Finally, the role of several candidate proteins in HIV infection was investigated by gain- and loss-of-expression studies.
Results
Experimental Infections and SWATH-MS library construction
Jurkat E6-1 T-cells were infected with CXCR4-tropic HIV-1NLX virus (Brown et al., 1999). Infections were performed by spinoculation at a high (>10) MOI to achieve a maximal level of infection (>90%, data not shown). The overall experimental approaches are summarized in Fig. 1. Infections were allowed to proceed for 48 h and then the cells separated into cytosolic, membrane/organelle, and nuclear fractions. SWATH-MS processes spectra after data acquisition, therefore a spectral reference library must first be constructed by DDA. Experimental groups are then run in data-independent mode and all spectra are collected and analyzed post-run against the preconstructed library. This allows for the data to be normalized across replicates and analyzed using traditional statistical methods in silico.
Fig. 1. Experimental approach used for SWATH-MS analysis of HIV-1 infected T-cells.
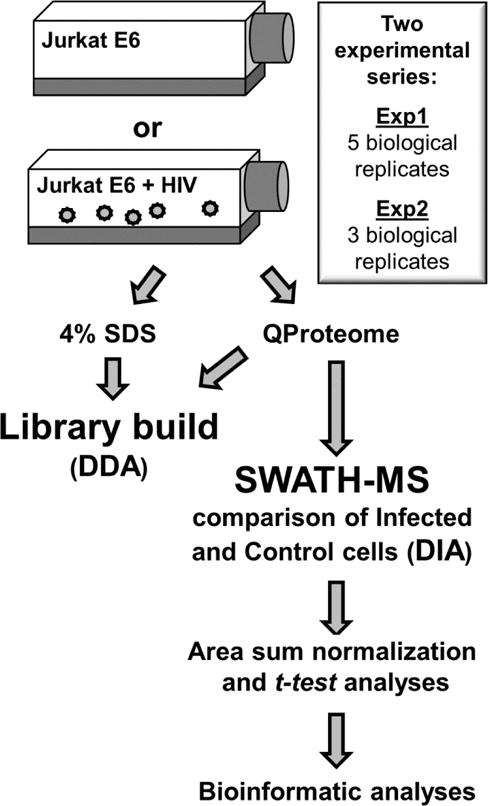
Work-flow representative of one complete biological replicate.
To maximize the size of the SWATH-MS reference library we collected MS data from both whole cell lysates and each subcellular fraction. For each replicate, viral infection was confirmed by immunoblot (Fig 2A), and the isolation and purity of the cell fractionations was assessed by immunoblot of known cellular markers (Fig. 2B), including GAPDH (cytosol), ERp19 (membrane/organelle), and Sumo2/3 (nuclear). A total of three biological replicates were used to construct the reference library, and the final library consisted of 1,515,831 spectra representing 3,992 proteins.
Fig. 2. Example confirmation blots of infections and fractionations.
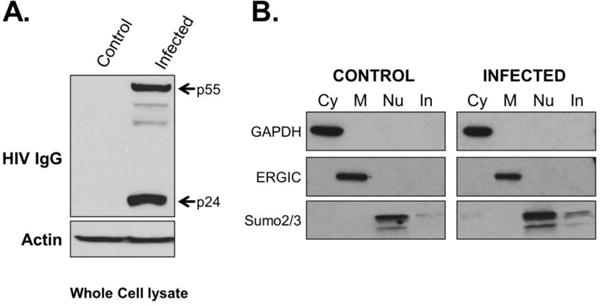
Western blot confirmation of HIV-1 infection in Jurkat cells used for SWATH-MS analysis is shown in (A). Major proteins are indicated, minor bands represent Gag processing intermediates. Control immunoblots against indicated proteins for the integrity of the cell fractionation are shown in (B). Representative blots of fractions from one biological replicate shown. Cy= cytoplasm; M= Membrane; Nu= Nuclear; In= Insoluble/cytoskeletal fraction.
Comparison of the two independent SWATH experiments
Two series of experimental replicates were performed in this study. Exp1 was comprised of 5 biological replicates (infections, subcellular fractionations, and MS runs) of control and infected samples. Exp2 comprised three additional biological replicates. The experimental procedures were identical for both experiments; however, Exp2 was performed with different stocks of Jurkat cells and virus, and an upgraded version of mass spectrometer set to variable SWATH size (see Materials and Methods). Both datasets are summarized in Table 1 and provided as supplemental material. Samples were normalized by Total Area Sums in MarkerView software so the resulting normalized samples had the same area sum calculated using all peaks. Only proteins identified in all biological replicates were included in the analysis. A statistical analysis of average spectral intensities of each protein was used to identify proteins with altered abundance between infected and control cells. In both experimental series, approximately 20% of the proteins in both the cytosolic and membrane fractions were found to be altered in infected versus control cells, representing 497 – 578 proteins. Whereas, only ~10% of nuclear proteins were statistically different between the control and infected cell nuclei (297 and 225, Exp1 and Exp2 respectively; Table 1). Notably, in both experiments the majority of altered proteins exhibited reduced abundance in infected versus control cells.
Table 1.
Summary of proteins with significant change in abundance between infected and control cells.
| Exp1 (5 biological replicates) | Exp2 (3 biological replicates) | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Cytosol | Memb/Org. | Nuclei | Cytosol | Memb/Org. | Nuclei | |
| Total proteins identified: | 2719 | 2719 | 2719 | 2734 | 2350 | 2734 |
| Proteins with significant change in abundance: | 547 (20.1%) | 578 (21.2%) | 297 (10.9%) | 560 (20.5%) | 479 (20.4%) | 225 (8.2%) |
| Increased: | 179 | 257 | 99 | 189 | 120 | 28 |
| Decreased: | 368 | 321 | 198 | 371 | 359 | 197 |
Comparison of the two datasets found a 13.5 – 25.9% overlap of significantly altered proteins between Exp1 and Exp2 (Table 2). Taking into account the direction of change in abundance further reduced the overlap of factors between the two datasets to 10 – 24.5% among the fractions. Manual alignment of the two datasets found 80, 103, and 36 proteins altered in abundance in cytoplasmic, membrane, and nuclear fractions of HIV infected cells, respectively. This represented only 1.47 – 5.28% of the total number of proteins in either Exp1 or Exp2 (Table 2). Consistent with the individual datasets, the majority of these factors were less abundant in infected cells. Of interest, several factors had altered levels in more than one subcellular fraction (Table 3). As expected, among these was HIV-1 Gag and Rev. Five cellular proteins were found to be increased in multiple fractions, including the intermediate filament vimentin (VIM), which was increased in both the cytoplasmic and membrane fractions of cells. Nine cellular proteins were found to be less abundant in multiple fractions of infected cells. This included two members of the mini chromosome maintenance (MCM) family. Expression of MCM5 has been shown to inhibit HIV infection (Santos et al., 2016), suggesting that its reduction in infected cells would benefit virus replication.
Table 2.
Comparison of Exp1, Exp2, and in silico analysis.
| Fraction: | Cytosol | Memb./Org. | Nuclei |
|---|---|---|---|
| Total significantly altered proteins in both experiments (% of Exp. I/II): | 106 (19.4%/18.9%) | 124 (21.4%/25.9%) | 40 (13.5%/17.8%) |
| % total of Exp1 or Exp2 dataset: | 3.90%/3.98% | 4.56 %/5.28% | 1.47%/1.47% |
| Factors with matched direction of expression change in both exps.: | 80 (75.5%) | 103 (83.1%) | 36 (90%) |
| Increased: | 13 | 38 | 7 |
| Decreased: | 67 | 65 | 29 |
| In silico analysis: | 172 | 84 | 31 |
| Increased: | 32 | 28 | 5 |
| Decreased: | 140 | 56 | 26 |
Table 3.
Dual Significant Proteins with Altered Abundance in Multiple Fractions.
| UniProt ID | Gene ID | Name | Subcellular fractions with altered expression1 |
|---|---|---|---|
| Increased in infected cells: | |||
| O14965 | AURKA_HUMAN | Aurora kinase A; AURKA | Cyto./Memb./Nucl. |
| P04591 | GAG_HV1H2 | HIV-1 Gag polyprotein | Memb./Nucl. |
| P04618 | REV_HV1H2 | HIV-1 Rev protein | Cyto./Memb./Nucl. |
| P08670 | VIME_HUMAN | Vimentin; VIM | Cyto./Memb. |
| P52732 | KIF11_HUMAN | Kinesin-like protein KIF11; KIF11 | Cyto./Memb. |
| P53350 | PLK1_HUMAN | Serine/threonine-protein kinase PLK1; PLK1 | Memb./Nucl. |
| Q86U42 | PABP2_HUMAN | Polyadenylate-binding protein 2; PABPN1 | Memb./Nucl.* |
| Decreased in infected cells: | |||
| P26639 | SYTC_HUMAN | Threonine tRNA ligase, cytoplasmic; TARS | Cyto./Memb.* |
| P30154 | 2AAB_HUMAN | Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A beta isoform; PPP2R1B | Cyto./Memb. |
| P33992 | MCM5_HUMAN | DNA replication licensing factor MCM5; MCM5 | Cyto.*/Memb./Nucl. |
| P41250 | SYG_HUMAN | Glycine tRNA ligase; GARS | Cyto./Memb. |
| P49736 | MCM2_HUMAN | DNA replication licensing factor MCM2; MCM2 | Memb./Nucl. |
| P61088 | UBE2N_HUMAN | Ubiquitin-conjugating enzyme E2 N; UBE2N | Cyto./Memb. |
| P61221 | ABCE1_HUMAN | ATP-binding cassette sub-family E member 1; ABCE1 | Cyto./Memb. |
| Q13263 | TIF1B_HUMAN | Transcription intermediary factor 1-beta; TRIM28 | Cyto./Memb./Nucl. |
| Q9BZE4 | NOG1_HUMAN | Nucleolar GTP-binding protein 1; GTPBP4 | Memb./Nucl. |
Subcellular fractions noted with italics and marked with ‘*’ showed inconsistent direction of expression change between Exps. I and II.
Datasets from both experiments were combined in sillico as an alternative approach to collectively analyze all the data. To do this, the SWATH data from both experiments was re-processed computationally with equivalent SWATH size. Similar to the individual experimental series, the majority of altered proteins were less prevalent in infected cells. Statistical testing identified 287 candidate factors (172, 84, and 31 proteins in the cytosolic, membrane, and nuclear fractions, respectively) with altered abundance. A heat map of these factors and their relative change in abundance is provided in Fig. 3. Overall, the merged dataset contained 17 more factors than obtained by manual alignment of Exp1 and Exp2 (Table 2). Notably, the distribution of factors was altered compared to the manual alignment-66 more factors were identified to be altered in the cytosol, but 40 and 9 fewer were identified in the membrane and nuclear fractions, respectively. Next we did a comparison of the dataset to previously published proteomic and siRNA studies of HIV infection (Chertova et al., 2006; DeBoer et al., 2014; Haverland et al., 2014; Konig et al., 2008; Monette et al., 2011; Raghavendra et al., 2010; Zhou et al., 2008). Overall, there were a total of 82 matches among the seven studies analyzed (Table 4). The meta-analysis and specific matches are provided in the supplemental data.
Fig. 3. Heat map plot of proteins with statistically altered expression in indicated subcellular fractions.
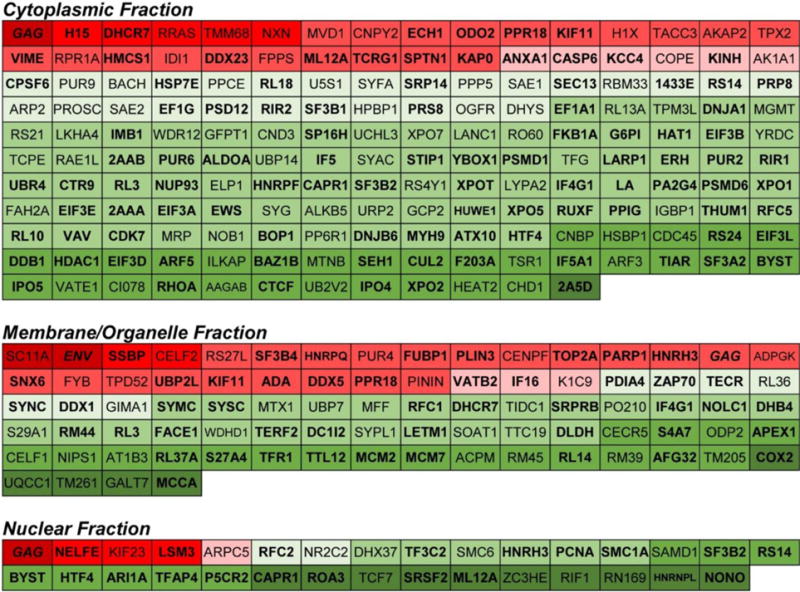
Proteins with higher expression in infected cells are indicated in red and those with lower expression in in green. Shading approximates relative fold-change in expression. Proteins in bold are members of the HIV interaction database. HIV proteins are indicated in italics.
Table 4.
Candidate overlap with previous proteomic and siRNA studies.
| Previous studiesa | |||||||
|---|---|---|---|---|---|---|---|
| Haverland | Chertova | Monette | Raghavendra | DeBoer | Konig | Zhou | |
| Total matchesb | 22 | 9 | 21 | 4 | 8 | 8 | 10 |
| Overlap | 8.0% | 3.3% | 7.6% | 1.5% | 2.9% | 2.9% | 3.6% |
Previous HIV-1 protein interaction and siRNA studies cited in text.
Specific matches can be found in supplementary data.
Bioinformatic analysis
Additional analyses were performed to identify functional pathways altered in HIV infected cells. Volcano plots from the t-test analyses were used to visualize and identify significant changes between control and HIV-infected samples in the combined dataset for each fraction (Fig. 4, left graphs). The log2 fold change between the control and infected conditions was plotted against the negative log of the p-value. Proteins with p-value ≤0.05 are shown in red, proteins with a fold-change greater than 2 or less than 0.5 are colored blue, and proteins that meet both criteria are colored yellow. Functional analysis of the statistically significant proteins in each subcellular fraction’s combined protein data set was determined using PANTHER (Mi et al., 2017) with the dataset split into upregulated and downregulated proteins (Fig. 4, right panels). Thus, for the cytoplasmic fraction, a list of 140 downregulated protein IDs were entered to obtain functional classifications with terms from Gene Ontology (GO)-slim “biological process.” This input list generated 223 total biological process hits falling under ten GO terms. Next, a list of the cytoplasmic fraction’s upregulated proteins (32) was uploaded resulting in 52 process hits under nine GO terms. This process was repeated for the membrane/organelle and nuclear fractions. Consistent with the overall data, downregulated proteins were more abundant in almost all the process categories. Interestingly, in all three subcellular fractions the largest number of altered factors fell in the “cellular” and “metabolic” processes categories.
Fig. 4. Volcano plots and Gene Ontology of candidate proteins.
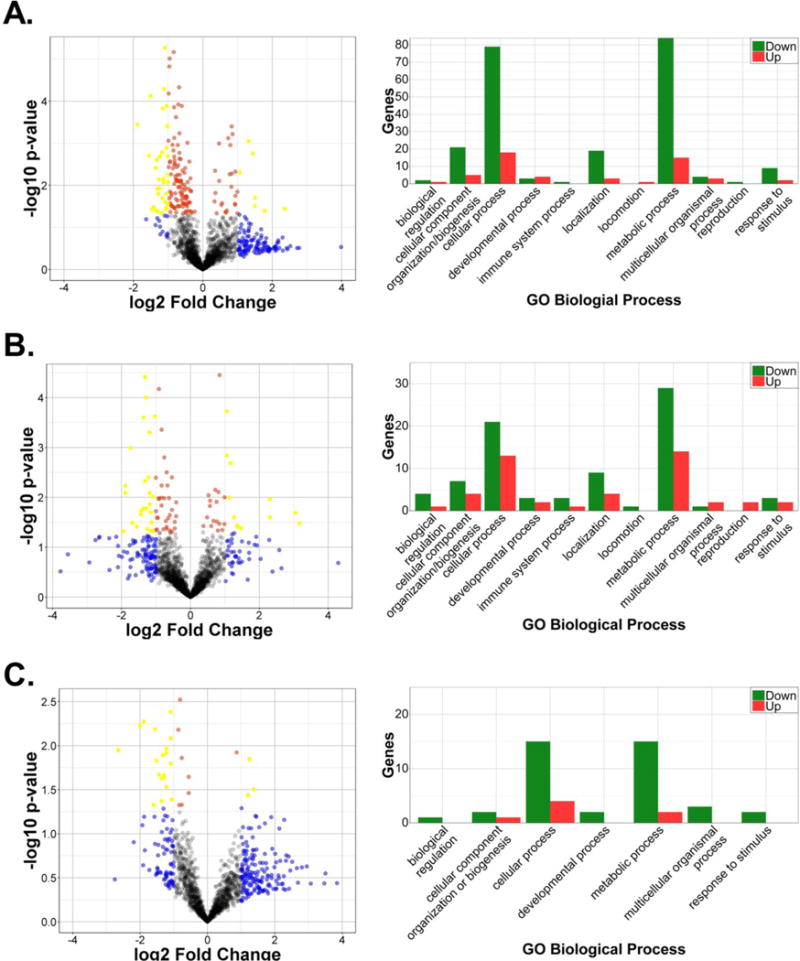
Volcano plots (left panels) and Gene Ontology for Biological Processes (right graphs) for cytoplasmic (a), membrane/organelle (b), and nuclear (c) fractions are shown.
Validation of altered factors
Initially to validate the SWATH-MS data we assessed protein expression via immunoblot of infected and control cells. Candidate proteins were selected randomly, but based on antibody availability. Additional individual infection and fractionation experiments were performed for this purpose. After isolation, equivalent amounts of control and infected cell fractions were separated by SDS-PAGE and the expression of individual candidate factors assessed by western blot. Control blots were performed for each replicate as shown in Fig. 2 to confirm fraction integrity. A number of factors were found to have a similar change in expression profile as predicted by the SWATH-MS analysis, including AURKA, EF1A1, hnRNP L, NONO, PPP2R5D, and PCNA (Fig. 5).
Fig. 5. Alterations in protein expression validated by immunoblot.
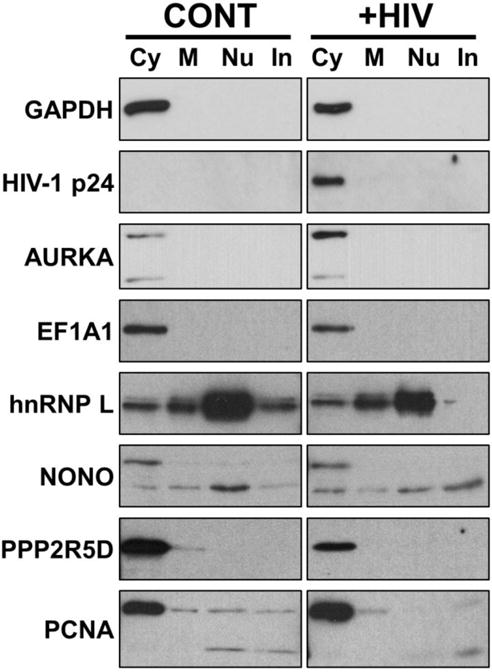
Cell fractionation samples were collected from uninfected (CONT) and HIV-1 infected Jurkat cells at 48 hpi (+HIV). Fractions were normalized by protein concentration, separated by SDS-PAGE, and the indicated factors detected by immunoblot. Cy= cytoplasm; M= Membrane; Nu= Nuclear; In= Insoluble/cytoskeletal fraction.
Several factors were not validated by immunoblot however, including CELF1 and 2, APEX1, and Sec61a. The effectiveness of immunoblot validation is limited by low sensitivity, antibody availability, questionable antibody validation, and inherent error as a result of the multi-step processing of blots. As an alternative method of validation, we performed a different MS-based approach, multiple reaction monitoring (MRM). MRM monitors a limited number of peptide spectra within a sample to provide more accurate quantitative data versus traditional antibody- or MS-based approaches. Two peptides were selected for 100 total proteins identified in the Exp1 dataset. Three infection replicates and subcellular fractionations were performed for these analyses using methodology identical to that used for the experiments for SWATH-MS. HIV-1 Gag was used as a positive control for the MRM. As expected, MRM showed a significant upregulation of Gag in all three of the subcellular fractions of infection cells compared to control cells (Table 5). Twenty-one total peptides were found to be statistically altered in abundance between the infected and control samples by MRM. These represented 18 independent peptides and 17 total proteins (two peptides from cyclophilin B (CypB or PPIB) were present). Next we determined whether or not the MRM data aligned with the SWATH-MS data from the individual experiments. As shown, MRM confirmed the altered expression of 12 of the 17 proteins as predicited by SWATH. Among these was CypB, a factor previously identified to be upregulated in HIV-infected nuclei (DeBoer et al., 2014).
Table 5.
Proteins validated by Multiple Reaction Monitoring
| Fraction | Uniprot ID and Name | MRM p-value | Fold Change (I/C) | Exp I | Exp II | Confirms SWATH data? |
|---|---|---|---|---|---|---|
| Q1 | P04591 GAG_HV1H2_2 | 0.00002 | 6.66 | + | Yes | |
| CYTO. | P10155 RO60_HUMAN | 0.00874 | 0.53 | + | Yes | |
| P53350 PLK1_HUMAN | 0.01040 | 0.58 | + | No | ||
| Q63HN8 RN213_HUMAN2 | 0.01317 | 0.56 | + | Yes | ||
| O43747 AP1G1_HUMAN2 | 0.01982 | 0.67 | + | Yes | ||
| P23284 PPIB_HUMAN | 0.02807 | 0.74 | + | No | ||
| P41250 SYG_HUMAN2 | 0.03898 | 0.66 | + | + | Yes | |
| P23284 PPIB_HUMAN2 | 0.04214 | 0.70 | + | No | ||
| O00148 DX39A_HUMAN2 | 0.04438 | 0.72 | + | + | Yes | |
| Q2 | P04591 GAG_HV1H2_2 | 0.00006 | 9.08 | + | + | Yes |
| MEMB. | Q14534 ERG1_HUMAN | 0.00259 | 0.57 | + | No | |
| P16402 H13_HUMAN2 | 0.00748 | 1.74 | + | + | Yes | |
| Q96DZ1 ERLEC_HUMAN | 0.01007 | 1.33 | + | No | ||
| Q9NXE4 NSMA3_HUMAN2 | 0.01935 | 0.82 | + | Yes | ||
| P23284 PPIB_HUMAN | 0.02201 | 1.58 | + | Yes | ||
| P56192 SYMC_HUMAN2 | 0.02507 | 0.70 | + | Yes | ||
| O14646 CHD1_HUMAN2 | 0.04714 | 0.74 | + | Yes | ||
| Q3 | P04591 GAG_HV1H2_2 | 0.00342 | 2.18 | + | + | Yes |
| NUCL. | O43390 HNRPR_HUMAN2 | 0.00441 | 0.63 | + | Yes | |
| P39023 RL3_HUMAN | 0.00608 | 1.78 | + | No | ||
| Q07955 SRSF1_HUMAN | 0.03311 | 0.57 | + | No |
Functional analysis of candidate factors
Both independent experiments as well as the merged analysis identified a number of factors altered in expression in one or more subcellular compartments. To evaluate the role of factors in HIV infection, we performed gain- and loss-of-expression infection assays. The gain-of-expression assays included both infection and virus production assays. For the infection assays, 293T cells were pre-transfected with FLAG-tagged expression plasmids 24 h prior to transduction with a VSVg-pseudotyped HIV luciferase marker virus (HIV-Luc). The effect of candidate factors on virus production was assessed by co-transfecting individual factors with HIVNLX molecular clone and measuring reverse transcriptase activity released into cell supernatants at 24 h post-transfection. The results of both overexpression assays are summarized on the left half of Table 6. Overexpression of 12 factors statistically altered HIV transduction and 6 were found to affect virus production; however, the effects were generally modest. Overexpression of DDX39A, RRAS, and TMEM261 showed the greatest potentiation of infection (~1.5 fold). In contrast, increased expression of APEX1 and XRCC1 inhibited infection slightly less than 2-fold. Similarly, none of the selected candidates showed a robust effect on virus production although statistical differences were achieved. The overexpression of two splicing factors, SRPK1 and ASF/SF2, produced the largest effects on virus production among the candidates. Both factors have been previously shown to alter HIV replication. ASF/SF2, a member of the SR family of splicing factors, interacts with HIV-1 Rev and reduces production of unspliced viral RNAs (Jacquenet et al., 2005; Paz et al., 2015; Tange et al., 1996). Consistent with this, overexpression of ASF/SF2 reduced virus production ~3-fold in our experiments. SRPK1, or SR protein kinase 1, modulates SR protein activity through its kinase activity. Interestingly, the main substrate of the second isoform of SRPK1 is ASF/SF2 (Pongoski et al., 2002). In agreement with a previous study (Fukuhara et al., 2006), ectopic expression of SRPK1 increased HIV production from 293T cells. Finally, overexpression of Sec11a, a subunit of the signal peptidase complex also reduced virus production approximately two-fold.
Table 6.
Functional Studies of Candidate Factors.
| Cell Factor | Over-Expression Assays | RNAi Assays | ||
|---|---|---|---|---|
| Infection (% of control)1 |
Virus Production (% of control)2 |
Infection (% of WT control)3 |
Viability (% of control)4 |
|
| ALYREF/THOC4 | 79.4 +/− 4.2(*) | 86.7 +/− 9.7 | 80.2 +/− 11.4 | 83.1 +/− 7.5 |
| APEX1 | 57.4 +/− 2.5(*) | ND | ND | |
| ARHGAP15 | 127.2 +/− 7.2(*) | 125.2 +/− 11.6(*) | 98.9 +/− 3.2 | 94.7 +/− 4.9 |
| ASF/SF2 | 97.6 +/− 12.9 | 30.2 +/− 5.0(*) | 116.8 +/− 5.7(*) | 92.3 +/− 4.9 |
| CNOT10 | 116.9 +/− 7.4 | 110.8 +/− 10.7 | ND | |
| Copine1 | 113.4 +/− 5.2(*) | 103.1 +/− 11.8 | 158.5 +/− 18.9(*) | 104.4 +/− 10.2 |
| DDX39A | 141.7 +/− 11.2(*) | 74.5 +/− 3.2(*) | ND | |
| GSTK1 | 118.0 +/− 8.0 | 103.5 +/− 8.9 | 128.3 +/− 6.4(*) | 63.4 +/− 7.0 (*) |
| LSM5 | 112.0 +/− 11.0 | 100.7 +/− 13.3 | 131.7 +/− 5.5(*) | 68.9 +/− 8.1 (*) |
| MTPN | 122.7 +/− 4.9(*) | 103.8 +/− 8.4 | 89.1 +/− 3.9 | 64.3 +/− 4.5 (*) |
| NCL | 115.2 +/− 10.3 | 112.0 +/− 3.0(*) | 130.5 +/− 9.0(*) | 97.6 +/− 8.7 |
| PPP2R5D | 107.7 +/− 2.0 | ND | ND | |
| RRAS | 137.4 +/− 5.0(*) | ND | ND | |
| SARNP | 102.8 +/− 4.9 | 77.8 +/− 2.8(*) | 86.6 +/− 5.6 | 97.0 +/− 8.8 |
| Sec11a | 83.8 +/− 2.4(*) | ND | ND | |
| SRPK1 | 118.2 +/− 5.6(*) | 141.5 +/− 11.1(*) | 145.2 +/− 8.9(*) | 81.8 +/− 5.4 (*) |
| SSBP1 | 78.6 +/− 2.6(*) | ND | ND | |
| TMEM261 | 168.0 +/− 16.2(*) | 91.3 +/− 4.8 | ND | |
| TXNDC19 | 98.9 +/− 5.6 | 97.5 +/− 4.4 | 126.6 +/− 11.5(*) | 56.6 +/− 6.0 (*) |
| XRCC1 | 58.7 +/− 3.7(*) | ND | ND | |
p<0.05 compared to control group as determined by two-tailed t-test; ND= not done.
Level of infection (+/− SEM) relative to cells mock-transfected with empty vector control.
Level of virus released into supernatants as measured by reverse transcriptase activity and relative to control cells (+/− SEM).
Level of infection (+/− SEM) relative to control cells transfected with pool of scrambled siRNAs.
Viability relative to control cells transfected with pool of scrambled siRNAs and measured by MTT assay (+/− SEM).
To investigate the functional dependency of candidate factors we transiently knocked down expression of candidate factors by siRNA-mediated interference and measured HIV transduction. Seven factors were identified to statistically alter HIV infection when their expression was reduced (Table 6). Surprisingly, in each case knockdown of the protein enhanced transduction. Cell viability was monitored in parallel experiments. Notably, knockdown of four of the seven factors that enhanced infection also significantly reduced cell viability as measured by MTT assay. Among those was COPINE1, a calcium-dependent membrane-binding protein, that may function in membrane trafficking and regulate molecular events at the interface of the cell membrane and cytoplasm (Tomsig et al., 2004). Copine 1 has no published direct HIV-1 interaction, but knockdown by siRNA increases TNFα-stimulated NF-κB transcription which could result in increased HIV-1 transcription (Coiras et al., 2007; Ramsey et al., 2008). Surprisingly, despite the fact that overexpression of SRPK1 mildly enhanced virus production, the knockdown of SRPK1 was also found to enhance infection. This result suggests a multifunctional role of SRPK1 during both the early and late steps of HIV replication.
One of the factors that intrigued us was the intermediate filament protein VIM because it was identified in multiple fractions in both Exp1 and 2, and the merged analysis. Moreover, protein-protein interaction analysis of the cytoplasmic fraction from the merged analysis showed a number of factors from the dataset interact with VIM (Fig. 6A). To confirm the altered expression of VIM during HIV replication, we performed time course infections and monitored VIM expression by immunoblot. The data clearly validated that VIM expression increased in both the cytoplasmic and membrane/organelle fractions during infection (Fig. 6B). Notably, although VIM has been shown to be targeted by HIV protease in vitro (Shoeman et al., 1990), we did not observe any cleavage in infected Jurkat cells.
Fig. 6. VIM distribution is altered in HIV-infected cells.
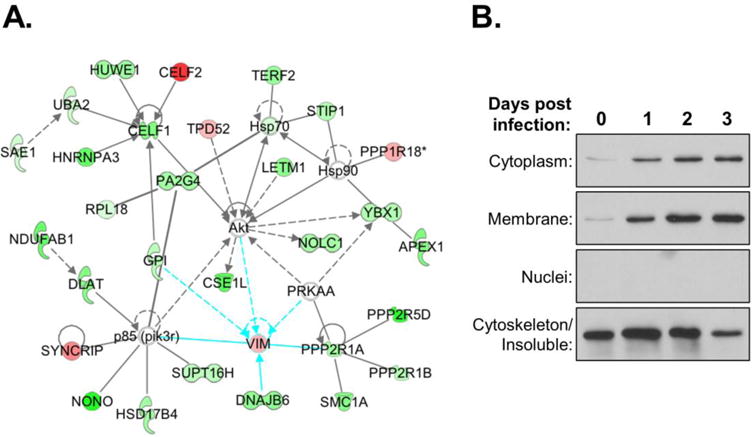
(A) Protein-protein interaction network of VIM with other candidate factors in combined SWATH dataset. (B) Immunoblots of VIM in subcellular fractions of uninfected (day 0) and HIV-1 infected Jurkat cells at dpi shown. Control blots for fractionation were performed as shown in Fig. 2.
Next we utilized CRISPR technology to create VIM(−) 293T cells. Guide RNAs were designed targeting exon 1 of VIM and four clonal cell lines were isolated that lacked detectable VIM expression (Fig. 7A). The susceptibility of each cell line to HIV infection was assessed using a VSVg-pseudotyped HIV-Luc marker virus. Three of the four cell lines showed reduced susceptibility to HIV compared to the parental 293T cells (Fig. 7B, dark bars), suggesting that VIM is important, but may not be required for HIV infection. Given that complete sequencing was not performed on the cell lines, we cannot rule out that off-target effects of the CRISPR treatment may have occurred in the F6 cell line and compensate for the deficiency in VIM. To test if the altered susceptibility was HIV-specific, we investigated the ability of the cells to support MLV transduction (Fig. 7B, light bars). Surprisingly, all the cell lines, including F6, showed reduced susceptibility to MLV compared to the control cell lines. This data suggests that VIM expresson is critical for retroviral transduction. Next we assessed if reconstituting VIM expression would rescue HIV infection of the VIM(−) cells. To do this, the cells were pretransfected with a VIM expression plasmid one day prior to infection with HIV-Luc. Unexpectedly, ectopic expression of VIM did not noticeably alter the level of HIV transduction in any of the cell lines (Fig. 7C). We suspect that the presence of C-terminal myc and FLAG epitope tags in the expression vector may have interfered with VIM function in these experiments. Further studies will be necessary to discern specifically how the absence of VIM in these cell clones alters retroviral infection.
Fig. 7. Role of VIM in HIV infection.
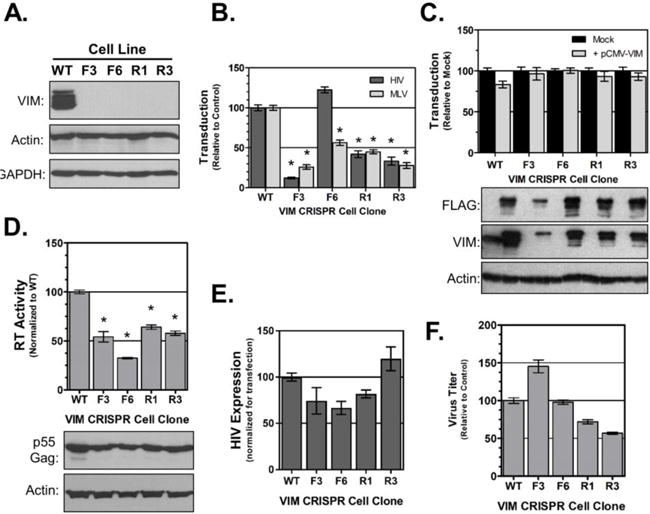
(A) Immunoblot analysis demonstrating knockout of VIM in 293T cell clones. (B) HIV- and MLV-Luc transduction of VIM knockout cell lines. (C) Exogenous supplementation of VIM does not rescue HIV transduction. Cell lines were pre-transfected with empty vector control (Mock; black columns) or pCMV-VIM (grey columns) prior to transduction with HIV-Luc. (D) Virus release from VIM(−) cells. Each cell line was transfected with pNLX molecular clone for 24 h. Release assessed by exogenous RT assay of supernatants. Transfections were confirmed by immunoblot of cell lysates as shown in bottom panels. (E) HIV gene expression in VIM-null cells. Indicated cell lines were co-transfected with pNLX-luc and pCMV-lacZ. HIV expression was normalized to beta-galactoside activity. (F) Titer of viruses produced from VIM(−) cells. Supernatants collected from experiments shown in (d) were normalized for RT activity and titered on TZM-bl indicator cells in triplicate. In all graphs data is normalized to WT control group, error bars denote SEM, and (*) indicates p<0.01 compared to WT control group as determined by two-tailed t-test.
Next we tested if the loss of VIM would affect virus production and infectivity. Each cell line was transfected with pNLX molecular clone for 24 h and both supernatants and cell lysates collected. Virus production was measured by exogenous RT assay and the cell lysates used to confirm transfection of the molecular clone. As shown in Figure 7D, all four VIM(−) cell lines showed an approximately two-fold reduction in virus production compared to the WT cells, suggesting that the absence of VIM reduced the efficiency of virus release. To test if the loss of virus release was caused by altered transcription, we performed gene expression studies. The pNLX-luc plasmid was co-transfected with pCMV-lacZ in each cell line and HIV expression normalized to beta-galactoside expression (Fig 7E). HIV expression was mildly reduced in three of the four cell lines, suggesting that reduced transcription may account in part for the reduced infection and virus production in those cell lines. However, HIV expression was slightly elevated in the R3 cell line. Finally, the supernatants from these cells were then normalized for RT content and their titer determined on TZM-bl indicator cells to see if the absence of VIM would alter virus infectivity. As shown in Figure 7F, the results were mixed. Virus from the F3 cells was slightly more infectious than WT, but virus from both the R1 and R3 cells was slight less infectious. However the combined differences were not statistically different, suggesting that VIM expression in producer cells is not required for virus infectivity.
Discussion
In this study, we investigated the alterations in the subcellular proteome of HIV-infected T-cells. The data encompassed eight independent infections across two experimental series. Hundreds of candidate factors were identified in each fraction with an ~20% overlap between the two experiments. This lower than expected reproducibility between experiments has been observed by us in the past, and we attribute it to the dynamic nature of viral infections. In contrary to uniform exposure of all cells to an agent (e.g. drug, stimulant etc.), the rate of viral infection varies and consists of multiple rounds of infections resulting from the constant production of virus particles. Viral infection is also associated with strong stimulation of factors such as cytokines and chemokines and may have profound influence on the state of intracellular proteome at any given time. To address this, we merged the datasets in silico. This did not substantially alter the number of factors identified with significant alterations of expression in infected versus control cells, but resulted in a different distribution of factors identified among the three subcellular fractions. Overall, this demonstrates the significance of biological variability and suggests that there may be an even greater challenge when comparing proteomic experiments from different laboratories.
Thus, our results support shifting the application of proteomics from global, unbiased profiling studies in which cataloging and comparing proteins in relevant samples is the major goal, to more focused and “surgical” proteomic studies in which we measure specific functional changes in a biological system with defined manipulations (Chen et al., 2012; Vuckovic et al., 2013). Yet, biological variation will still complicate matters. We attempted to focus our screen by individually analyzing subcellular compartments of cells. The idea being to detect proteins with altered localization during infection, which may identify critical pathways of activation or repression within the cell. Several factors were identified with altered levels in more than one fraction, suggesting changes in localization. This included the MCM complex which has been shown to inhibit HIV infection (Santos et al., 2016), and CypB, which we recently demonstrated potentiates HIV transduction (DeBoer et al., 2016). Further studies with other candidate proteins may identify additional factors that alter HIV infection.
The total number of factors precluded complete validation of all candidates, but the altered expression of several proteins was confirmed by immunoblot or MRM. Host factors with altered expression may represent proteins manipulated by the virus to support productive replication, or proteins altered because of HIV-induced pathology. This may complicate functional studies as not all the factors altered in infected cells may play direct roles in virus replication. Preliminary functional screens identified several cellular proteins that affected HIV infection when overexpressed or knocked down by RNA interference. These factors add to the growing list of candidate factors that may play a role in HIV replication and pathogenesis.
Functional characterization of candidate factors still remains the greatest bottleneck of large-scale “−omics” studies. We screened a number of candidate factors through gain- and loss-of-expression studies. Several factors were found to alter HIV infection when their expression was increased or decreased, but no one factor was found to be essential for infection. These data add to the growing knowledgebase of cellular factors with roles in HIV infection. As we were unable to quantitatively measure T-cell pathology, we have yet to identify the role of candidate factors in terms of pathogenesis.
Among the candidate factors we focused our attention on VIM since it displayed such a distinctive phenotype in the immunoblot validations. Interestingly, VIM is found to be increased in oral epithelial cells of HIV-infected patients (Yohannes et al., 2011). VIM is cleaved in vitro by the HIV-1 protease (Shoeman et al., 1990), and it has been reported that knockdown of VIM by shRNA reduced HIV infection (Fernandez-Ortega et al., 2016). Our data also suggest that VIM is important for HIV infection. Three of four VIM(−) cell lines showed reduced susceptibility to HIV transduction and all were resistant to MLV transduction. Moreover, all four cell lines had a reduced capacity to produce HIV. In three of the cell lines this may be partially due to reduced HIV expression. Off-target effects of CRISPR-Cas9 treatment are suspected to occur at a higher than expected level (Haeussler et al., 2016), and the F3 cell line may have a compensatory alteration that maintains susceptibility to HIV infection. Additional genome-wide sequence analysis will likely be necessary to discover what changes the F3 cell line possesses to maintain HIV susceptibility. However, identifying those adaptations may discover a factor or pathway important for HIV infection.
Materials and methods
Cells, Virus Production, and Infections
Jurkat E6-1 cells ((Weiss et al., 1984); NIH AIDS Reagent Program, Germantown, MD) were cultured in RPMI 1640 media supplemented with 10% Fetalclone III (Hyclone, Logan, UT USA), 8 mM L-glutamine, 100 U/mL penicillin, and 100 U/ml streptomycin. 293T and TZM-bl cells (NIH AIDS Reagent Program) were cultured in DMEM media with the same supplementation. All cells were cultured in humidified incubators at 37°C and 5% CO2. HIV-1 NLX virus stocks were produced by transient transfection of 293T cells with pNLX molecular clone using PolyJet as described by the manufacturer (SignaGen, Gaithesburg, VA). NLX-Luc+VSVg virus was produced by transfection of the viral molecular clone (pNLX-luc) DNA and pMD2.G vesicular stomatitis virus glycoprotein G (VSVg) expression vector (Addgene Plasmid Repository, Cambridge, MA). Viral supernatants were collected over 48 hours, clarified by centrifugation at 4000 × g for 5 minutes and stored at −80° C.
For the SWATH-MS analyses, 1× 107 Jurkat cells were infected using the spinoculation technique as described previously (O’Doherty et al., 2000; Schweitzer et al., 2013). Uninfected Jurkat cells were processed in parallel with no virus as control cells. At 48 hpi the cells were washed with phosphate-buffered saline (PBS) and fractionated into subcellular compartments using the Qproteome Cell Compartment Kit as described by the manufacturer (Qiagen, Valencia CA). For construction of the reference library, an additional three biological replicates (infected and control) of 1× 107 Jurkat cells were lysed in 4% (w/v) SDS, 0.1M dithiothreitol, 0.1M Tris-HCl, and 100 units/mL Benzonase Nuclease, pH 7.6 (Life Technologies, Grand Island, NY). The protein concentration of all samples was determined by BCA assay supplemented with 50 mM ionic detergent compatible reagent (Thermo Fisher Scientific, Waltham, MA). For immunoblot experiments, fractions were normalized to each other by the addition of PBS.
VIM-null 293T cells were constructed using CRISPR-Cas9 gene targeting essentially as described previously (Bauer et al., 2015). Two guide RNA sequences (5′-tcctaccgcaggatgttcgg-3′ and 5-gccgaacatcctgcggtagg-3′) targeting exon 1 of VIM were synthesized (Integrated DNA Technologies, Coralville IA USA) and cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9 (plasmid # 42230, Addgene Plasmid Repository, Cambridge, MA; (Cong et al., 2013)), and verified by DNA sequencing. 293T cells were transfected with individual VIM-CRISPR plasmids using PolyJet reagent; 48 h post-transfection the cells were seeded at 1 cell/well into 96-well plates and single cell colonies expanded. Cell clones were screened for VIM expression by immunoblot using anti-VIM antibody (V9; Santa Cruz Biotechnology, Santa Cruz, CA).
Generation of Library of Spectra – data dependent acquisition
Jurkat T cell reference library of spectra was generated using DDA method and nano-LC Sciex 5600 TripleTOF mass spectrometer as described previously (Haverland et al., 2014). All samples were processed using filter-assisted sample preparation (Wisniewski et al., 2009) and 25 μg of 4% SDS lysate and 20 μg (each) of QProteome fraction 1–3 were used. For this purpose, samples were fractionated by isoelectric point using OFFGEL electrophoresis using pH 3–10 OFFGEL strips (Agilent, Santa Clara, CA), cleaned using an Oasis mixed cation exchange cartridge following manufacturers protocols (Waters, Milford, MA) and 2 μg of protein prepared for mass spectrometry using C18 Zip-Tips® (Millipore, Billerica, MA). Samples were digested with trypsin and cleaned using C18 spin columns following manufacturer protocols (Thermo Fisher Scientific). The reference library was generated by running samples in the traditional DDA mode with a 250-ms survey scan and the top 50 ions selected for subsequent MS/MS. Ion selection criteria included a charge state from +2 to +5, intensity of greater than 100 counts/s, mass tolerance of 50 mDa, and were not found on a dynamic exclusion list. Ions that were fragmented and analyzed by MS/MS were excluded from further analysis for 15 seconds.
SWATH-MS – data independent acquistion
For SWATH-MS, as for DDA acquisition as described above, all samples were processed using filter-assisted sample preparation (Wisniewski et al., 2009) and 20 μg of each subcellular fraction was used. DIA for SWATH-MS was performed as described previously (Haverland et al., 2014). Peptide samples were re-suspended in 0.1% formic acid in HPLC grade water and were analyzed by reverse-phase high-pressure liquid chromatography electrospray ionization tandem mass spectrometry (RP-HPLC-ESI-MS/MS) using an Eksigent NanoLC-Ultra 1D plus (Exsigent, Dublin, CA) and nanoFlex cHiPLC system coupled to either a 5600 Triple-TOF mass spectrometer for the first set of biological replicates or a 6600 Triple-TOF mass spectrometer for the second set of biological replicates (AB Sciex; Concord, Canada). In either case, the nanospray needle voltage was set to 2400V. Samples were loaded using a stepwise flow rate of 10 μL/min for 8.5 min and 2 μL/min for 1 min using 0.1% (v/v) formic acid in HPLC water (solvent A). Peptides were eluted from the analytical column using a 5–35% linear gradient of solvent B (95% (v/v) acetonitrile with 0.1% (v/v) formic acid over the course of 180 minutes with a flow rate of 0.3 μL/min. Experimental samples were processed using cyclic DIA of mass spectra using either 25-Da swaths (Exp1) or variable swaths (Exp2) as described in Liu et al (Liu et al., 2013). Briefly, a 50-ms survey scan (MS1) was performed and all precursors within a given swath were fragmented and analyzed (MS2). Each cycle was composed of 34 25-Da swaths which covered 400Da to 1200 Da. Total Cycle time was 3.314 s using an accumulation time of 96 ms per 25-Da swath.
Protein Identification and data analysis
The DDA spectral data was searched using ProteinPilot software v5.0 (AB Sciex). Complete spectral data is available upon request. Mass tolerance for precursor ions and transitions are automatically set up by algorithm for specific instrument used, in this case 5600 or 6600 TripleTof. Protein identification was through the UniProt Swiss-Prot database (November 2015 release) containing both human and HIV-1 proteins with FDR less than 5%. The DIA data was subject to spectral alignment and targeted data extraction by PeakView v1.2 (AB Sciex) software using the Jurkat reference spectral library. Identification of proteins used an extraction window of 20 min and the following parameters: 8 peptides, 5 transitions, exclude shared peptides, extracted ion chromatogram (XIC) width set at 50 ppm, and peptide confidence of >99%. Reversed sequences and laboratory contaminants were removed from the data set. For both experiments (Exp1 and Exp2), the protein lists (peak areas) were exported into text files using PeakView software and then imported into MarkerView software. Samples in each data set were normalized in MarkerView using Total Area Sums. This approach was used to normalize each sample so that the resulting normalized samples have the same area sum calculated using all peaks. For each sample the total response (Σarea) was calculated where the sum was over all peaks. The scale factor for a given sample was the average total response for all samples divided by the total response for the given sample. This was useful as all major peaks for all the samples are expected to be in common with similar intensity. Next, HIV and control groups were compared for differences in MarkerView by t-Test analyses. Protein IDs with p-value ≤0.05 were considered to have significantly different expression between infected and control samples. Functional analysis of the statistically significant proteins in each subcellular fraction’s combined protein data set was done using PANTHER v. 11.1 (Mi et al., 2017). Bar charts were generated using the ggplot2 R package, R version 3.3.2.
Multiple Reaction Monitoring
MRM was performed employing established methodology as described previously using a LC - Sciex QTrap 6500, QQQ/ion trap mass spectrometer (Ebhardt et al., 2015; Sherwood et al., 2009; Shi et al., 2016; Timm et al., 2015). A reverse phase C18 column and water:acetonitrile gradient was used for peptides fractionation. For each precursor ion representing peptide of interest, two transitions were used based on data from the library of spectra generated from previous data dependent acquisition experiments. (Haverland et al., 2014). MRM-MS was performed in triplicate on each sample. Triplicate infections and cell fractionations were prepared as done for the SWATH-MS experiments. Statistically significant differences between infected and control samples were determined by t-test analysis of area counts.
Viral assays
For all assays, 293T cells were seeded in triplicate wells of a 6-well plate at 2×105 cells/well. For overexpression assays, each well was transfected with 1 μg plasmid using PolyJet reagent. For siRNA knockdown assays, 2 pmol siRNA was transfected using PepMute reagent according to manufacturer’s directions (Signagen). The next day, wells were inoculated with NLX-Luc+VSVg virus and incubated for 48 h at 37°C. Cells were lysed with M-PER solution (Thermo Fisher Scientific) and clarified by centrifugation. One-glo luciferase reagent (Promega, Madison, WI) was used to determine Luciferase activity. Total protein concentration was determined by BCA protein assay and luciferase activity normalized to the total protein. Data shown represents at least three independent experiments for each factor. MTT assays were performed on siRNA transfected cells using the CellTiter 96 non-radioactive cell proliferation assay per the manufacturer’s protocol (Promega). For virus production assays, cells were transfected with 0.5 μg of candidate factor expression vector and 0.5 μg pNLX. At 24 h post-transfection, supernatants and cells were collected. Supernatants were clarified by centrifugation (12,000 × g) and stored at −20°C until assayed for exogenous reverse transcriptase (RT) activity. Cells were washed with PBS, lysed with M-PER solution and immunoblotted with anti-HIV Ig (NIH AIDS Reagent Program) to confirm transfection efficiency (data not shown). HIV expression assays were performed in 24-well plates in quadruplicate using 5×104 cells/well. 250 ng each of pNLX-luc and pCMV-lacZ (Life Technologies, Carlsbad, CA) were co-transfected per well. After 24 h, the cells were lysed with M-PER solution and assayed for luciferase and beta-galactosidase activity (Thermo Fisher Scientific). Luciferase levels in each sample were normalized to beta-galactosidase levels. Data is reported relative to wild-type 293T cell line. Statistical analyses for all experiments were performed using GraphPad Prism 5 software.
Exogenous RT Activity
The procedure used was a modification of one previously described (Quan et al., 1996). Standard reaction mixtures contained 50 mM Tris (pH 7.9), 75 mM KCl, 2 mM DTT, 0.1875 mM ATP, 5 mM MgCl2, 0.05% Nonidet P-40, 2 μM dTTP, and 2 μCi [32P]-α-TTP, and 10μL supernatant in a final volume of 40 μL. Reactions were run in triplicate and incubated for 4 hours at 37°C. 10μL RT products were added to as individual spots on Whatman filter paper and allowed to dry completely. The paper was washed three times with 2x saline-sodium citrate (SSC) buffer and allowed to dry. After drying, 32P activity was visualized and quantitatively analyzed using a storage phosphor plate and scanned on a Typhoon 9410 scanner (GE Healthcare Bio-Sciences Corp. Piscataway, NJ). The data presented is normalized to the activity produced from cells co-transfected with empty vector control and pNLX.
Immunoblotting
Protein were normalized to equal concentrations with PBS and mixed 1:1 with 2x SDS-PAGE loading buffer and boiled for 5 min. Samples (20 μg/lane) separated by SDS-PAGE using Tris-glycine gels, and transferred to PVDF. Proteins were detected by immunoblot using the following primary antibodies: anti-HIV-1 p24 (Toohey et al., 1995; Wehrly and Chesebro, 1997), anti-ERGIC (H-245), Actin (C-4), Sumo2/3 (N-18), GAPDH (6C5), EEF1A1 (CBP-KK1), hnRNP-L (4D11), NONO (H-85), PCNA (PC10), and the HRP conjugated anti-rabbit, anti-mouse, and anti-human IgG secondary antibodies were all obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-AURKA was obtained from Cell Signaling Technology (Danvers, MA). Anti-PPP2R5D was obtained from Abcam (Cambridge, MA). HRP conjugated anti-Flag (M2) primary antibody was from Sigma-Aldrich (St Louis, MO). All antibodies were diluted in blot wash buffer (TBS with 0.4% tween-20). HRP secondary antibodies were detected using West Pico chemiluminescent staining (Thermo Fisher Scientific) and exposure to radiographic film. Images were scanned to computer, adjusted for brightness and contrast if necessary, and cropped for size.
Supplementary Material
Highlights.
SWATH-MS was used to identify changes in protein expression during HIV-1 infection.
Eight biological replicates in two experimental series were analyzed in silico.
287 proteins, including 117 novel factors, were identified with altered expression.
Candidate factors were validated by immunoblot or multiple reaction monitoring.
Many candidate factors altered HIV infection in gain- and loss-of-expression studies.
Acknowledgments
This study was funded by Public Health Service grants AI080348 (M.B.), DA030962 (P.C.), DA0443258 (P.C.), and MH062261 (P.C.) from the National Institutes of Health. Jason DeBoer was funded by the US Army’s Medical Service Corp Long Term Health Education Program. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Jurkat Clone E6-1 from Dr. Arthur Weiss; HIV-IG from NABI and NHLBI; anti-HIV-1 p24 Monoclonal (183-H12-5C) from Dr. Bruce Chesebro and Kathy Wehrly; and TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information
Four supplementary data files are associated with this manuscript. Three files contain a summary of the MS data of the cytosolic, membrane, and nuclear subcellular fractions for each experimental series as well as the in silico merged data. The fourth file contains the knowledge- based comparison of the merged dataset with previous proteomic and siRNA studies.
References
- Bauer DE, Canver MC, Orkin SH. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. J Vis Exp. 2015:e52118. doi: 10.3791/52118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregnard C, Zamborlini A, Leduc M, Chafey P, Camoin L, Saib A, Benichou S, Danos O, Basmaciogullari S. Comparative proteomic analysis of HIV-1 particles reveals a role for Ezrin and EHD4 in the Nef-dependent increase of virus infectivity. J Virol. 2013;87:3729–3740. doi: 10.1128/JVI.02477-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HE, Chen H, Engelman A. Structure-based mutagenesis of the human immunodeficiency virus type 1 DNA attachment site: effects on integration and cDNA synthesis. J Virol. 1999;73:9011–9020. doi: 10.1128/jvi.73.11.9011-9020.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman FD, Malani N, Fernandes J, D’Orso I, Cagney G, Diamond TL, Zhou H, Hazuda DJ, Espeseth AS, König R, Bandyopadhyay S, Ideker T, Goff SP, Krogan NJ, Frankel AD, Young JAT, Chanda SK. Host Cell Factors in HIV Replication: Meta-Analysis of Genome-Wide Studies. PLoS Pathog. 2009;5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Na CH, Peng J. Quantitative proteomics to decipher ubiquitin signaling. Amino Acids. 2012;43:1049–1060. doi: 10.1007/s00726-012-1286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chertova E, Chertov O, Coren LV, Roser JD, Trubey CM, Bess JW, Jr, Sowder RC, 2nd, Barsov E, Hood BL, Fisher RJ, Nagashima K, Conrads TP, Veenstra TD, Lifson JD, Ott DE. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol. 2006;80:9039–9052. doi: 10.1128/JVI.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coiras M, Camafeita E, Ureña T, López JA, Caballero F, Fernández B, López-Huertas MR, Pérez-Olmeda M, Alcamí J. Modifications in the human T cell proteome induced by intracellular HIV-1 Tat protein expression. Proteomics. 2006;6:S63–S73. doi: 10.1002/pmic.200500437. [DOI] [PubMed] [Google Scholar]
- Coiras M, Lopez-Huertas MR, Rullas J, Mittelbrunn M, Alcami J. Basal shuttle of NF-kappaB/I kappaB alpha in resting T lymphocytes regulates HIV-1 LTR dependent expression. Retrovirology. 2007;4:56. doi: 10.1186/1742-4690-4-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoer J, Jagadish T, Haverland NA, Madson CJ, Ciborowski P, Belshan M. Alterations in the nuclear proteome of HIV-1 infected T-cells. Virology. 2014;468–470:409–420. doi: 10.1016/j.virol.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoer J, Madson CJ, Belshan M. Cyclophilin B enhances HIV-1 infection. Virology. 2016;489:282–291. doi: 10.1016/j.virol.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly MR, Ciborowski P. Proteomics, biomarkers, and HIV-1: A current perspective. Proteomics Clin Appl. 2016;10:110–125. doi: 10.1002/prca.201500002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebhardt HA, Root A, Sander C, Aebersold R. Applications of targeted proteomics in systems biology and translational medicine. Proteomics. 2015;15:3193–3208. doi: 10.1002/pmic.201500004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellay J, Shianna KV, Telenti A, Goldstein DB. Host genetics and HIV-1: the final phase? PLoS Pathog. 2010;6:e1001033. doi: 10.1371/journal.ppat.1001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Ortega C, Ramirez A, Casillas D, Paneque T, Ubieta R, Dubed M, Navea L, Castellanos-Serra L, Duarte C, Falcon V, Reyes O, Garay H, Silva E, Noa E, Ramos Y, Besada V, Betancourt L. Identification of Vimentin as a Potential Therapeutic Target against HIV Infection. Viruses. 2016;8 doi: 10.3390/v8060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchigami T, Misumi S, Takamune N, Takahashi I, Takama M, Shoji S. Acid-labile formylation of amino terminal proline of human immunodeficiency virus type 1 p24(gag) was found by proteomics using two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry. Biochem Biophys Res Commun. 2002;293:1107–1113. doi: 10.1016/S0006-291X(02)00329-7. [DOI] [PubMed] [Google Scholar]
- Fukuhara T, Hosoya T, Shimizu S, Sumi K, Oshiro T, Yoshinaka Y, Suzuki M, Yamamoto N, Herzenberg LA, Hagiwara M. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11329–11333. doi: 10.1073/pnas.0604616103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri MS, Nebozhyn M, Showe L, Montaner LJ. Microarray data on gene modulation by HIV-1 in immune cells: 2000–2006. J Leukoc Biol. 2006;80:1031–1043. doi: 10.1189/jlb.0306157. [DOI] [PubMed] [Google Scholar]
- Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, Joly JS, Concordet JP. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverland NA, Fox HS, Ciborowski P. Quantitative proteomics by SWATH-MS reveals altered expression of nucleic acid binding and regulatory proteins in HIV-1-infected macrophages. J Proteome Res. 2014;13:2109–2119. doi: 10.1021/pr4012602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquenet S, Decimo D, Muriaux D, Darlix JL. Dual effect of the SR proteins ASF/SF2, SC35 and 9G8 on HIV-1 RNA splicing and virion production. Retrovirology. 2005;2:33. doi: 10.1186/1742-4690-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D’Orso I, Fernandes J, Fahey M, Mahon C, O’Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ. Global landscape of HIV-human protein complexes. Nature. 2011;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft-Terry S, Gerena Y, Wojna V, Plaud-Valentin M, Rodriguez Y, Ciborowski P, Mayo R, Skolasky R, Gendelman HE, Melendez LM. Proteomic analyses of monocytes obtained from Hispanic women with HIV-associated dementia show depressed antioxidants. Proteomics Clin Appl. 2010;4:706–714. doi: 10.1002/prca.201000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft-Terry SD, Engebretsen IL, Bastola DK, Fox HS, Ciborowski P, Gendelman HE. Pulsed stable isotope labeling of amino acids in cell culture uncovers the dynamic interactions between HIV-1 and the monocyte-derived macrophage. J Proteome Res. 2011;10:2852–2862. doi: 10.1021/pr200124j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Sage V, Cinti A, Valiente-Echeverria F, Mouland AJ. Proteomic analysis of HIV-1 Gag interacting partners using proximity-dependent biotinylation. Virol J. 2015;12:138. doi: 10.1186/s12985-015-0365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Frederick KM, Haverland NA, Ciborowski P, Belshan M. Investigation of the HIV-1 matrix interactome during virus replication. Proteomics Clin Appl. 2016;10:156–163. doi: 10.1002/prca.201400189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Huttenhain R, Surinova S, Gillet LC, Mouritsen J, Brunner R, Navarro P, Aebersold R. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. PROTEOMICS. 2013;13:1247–1256. doi: 10.1002/pmic.201200417. [DOI] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monette A, Pante N, Mouland AJ. HIV-1 remodels the nuclear pore complex. The Journal of cell biology. 2011;193:619–631. doi: 10.1083/jcb.201008064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukerji J, Olivieri KC, Misra V, Agopian KA, Gabuzda D. Proteomic analysis of HIV-1 Nef cellular binding partners reveals a role for exocyst complex proteins in mediating enhancement of intercellular nanotube formation. Retrovirology. 2012;9:33. doi: 10.1186/1742-4690-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naji S, Ambrus G, Cimermancic P, Reyes JR, Johnson JR, Filbrandt R, Huber MD, Vesely P, Krogan NJ, Yates JR, 3rd, Saphire AC, Gerace L. Host cell interactome of HIV-1 Rev includes RNA helicases involved in multiple facets of virus production. Molecular & cellular proteomics: MCP. 2012;11 doi: 10.1074/mcp.M111.015313. M111 015313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navare AT, Sova P, Purdy DE, Weiss JM, Wolf-Yadlin A, Korth MJ, Chang ST, Proll SC, Jahan TA, Krasnoselsky AL, Palermo RE, Katze MG. Quantitative proteomic analysis of HIV-1 infected CD4+ T cells reveals an early host response in important biological pathways: Protein synthesis, cell proliferation, and T-cell activation. Virology. 2012;429:37–46. doi: 10.1016/j.virol.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol. 2000;74:10074–10080. doi: 10.1128/jvi.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz S, Lu ML, Takata H, Trautmann L, Caputi M. SRSF1 RNA Recognition Motifs Are Strong Inhibitors of HIV-1 Replication. J Virol. 2015;89:6275–6286. doi: 10.1128/JVI.00693-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongoski J, Asai K, Cochrane A. Positive and negative modulation of human immunodeficiency virus type 1 Rev function by cis and trans regulators of viral RNA splicing. J Virol. 2002;76:5108–5120. doi: 10.1128/JVI.76.10.5108-5120.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan Y, Gu Z, Li X, Li Z, Morrow CD, Wainberg MA. Endogenous reverse transcription assays reveal high-level resistance to the triphosphate of (−)2′-dideoxy-3′- thiacytidine by mutated M184V human immunodeficiency virus type 1. J Virol. 1996;70:5642–5645. doi: 10.1128/jvi.70.8.5642-5645.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra NK, Shkriabai N, Graham R, Hess S, Kvaratskhelia M, Wu L. Identification of host proteins associated with HIV-1 preintegration complexes isolated from infected CD4+ cells. Retrovirology. 2010;7:66. doi: 10.1186/1742-4690-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey CS, Yeung F, Stoddard PB, Li D, Creutz CE, Mayo MW. Copine-I represses NF-kappaB transcription by endoproteolysis of p65. Oncogene. 2008;27:3516–3526. doi: 10.1038/sj.onc.1211030. [DOI] [PubMed] [Google Scholar]
- Ringrose JH, Jeeninga RE, Berkhout B, Speijer D. Proteomic studies reveal coordinated changes in T-cell expression patterns upon infection with human immunodeficiency virus type 1. J Virol. 2008;82:4320–4330. doi: 10.1128/JVI.01819-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos S, Obukhov Y, Nekhai S, Bukrinsky M, Iordanskiy S. Virus-producing cells determine the host protein profiles of HIV-1 virion cores. Retrovirology. 2012;9:65. doi: 10.1186/1742-4690-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos S, Obukhov Y, Nekhai S, Pushkarsky T, Brichacek B, Bukrinsky M, Iordanskiy S. Cellular minichromosome maintenance complex component 5 (MCM5) is incorporated into HIV-1 virions and modulates viral replication in the newly infected cells. Virology. 2016;497:11–22. doi: 10.1016/j.virol.2016.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphire AC, Gallay PA, Bark SJ. Proteomic analysis of human immunodeficiency virus using liquid chromatography/tandem mass spectrometry effectively distinguishes specific incorporated host proteins. J Proteome Res. 2006;5:530–538. doi: 10.1021/pr050276b. [DOI] [PubMed] [Google Scholar]
- Schweitzer CJ, Jagadish T, Haverland N, Ciborowski P, Belshan M. Proteomic analysis of early HIV-1 nucleoprotein complexes. Journal of Proteome Research. 2013;12:559–572. doi: 10.1021/pr300869h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng WY, Wang TC. Proteomic analysis of the differential protein expression reveals nuclear GAPDH in activated T lymphocytes. PLoS One. 2009;4:e6322. doi: 10.1371/journal.pone.0006322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood CA, Eastham A, Lee LW, Risler J, Mirzaei H, Falkner JA, Martin DB. Rapid optimization of MRM-MS instrument parameters by subtle alteration of precursor and product m/z targets. J Proteome Res. 2009;8:3746–3751. doi: 10.1021/pr801122b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Song E, Nie S, Rodland KD, Liu T, Qian WJ, Smith RD. Advances in targeted proteomics and applications to biomedical research. Proteomics. 2016;16:2160–2182. doi: 10.1002/pmic.201500449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoeman RL, Honer B, Stoller TJ, Kesselmeier C, Miedel MC, Traub P, Graves MC. Human immunodeficiency virus type 1 protease cleaves the intermediate filament proteins vimentin, desmin, and glial fibrillary acidic protein. Proc Natl Acad Sci U S A. 1990;87:6336–6340. doi: 10.1073/pnas.87.16.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange TO, Jensen TH, Kjems J. In vitro interaction between human immunodeficiency virus type 1 Rev protein and splicing factor ASF/SF2-associated protein, p32. J Biol Chem. 1996;271:10066–10072. doi: 10.1074/jbc.271.17.10066. [DOI] [PubMed] [Google Scholar]
- Timm T, Lenz C, Merkel D, Sadiffo C, Grabitzki J, Klein J, Lochnit G. Detection and site localization of phosphorylcholine-modified peptides by NanoLC-ESI-MS/MS using precursor ion scanning and multiple reaction monitoring experiments. J Am Soc Mass Spectrom. 2015;26:460–471. doi: 10.1007/s13361-014-1036-3. [DOI] [PubMed] [Google Scholar]
- Tomsig JL, Sohma H, Creutz CE. Calcium-dependent regulation of tumour necrosis factor-alpha receptor signalling by copine. Biochem J. 2004;378:1089–1094. doi: 10.1042/BJ20031654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toohey K, Wehrly K, Nishio J, Perryman S, Chesebro B. Human Immunodeficiency Virus Envelope V1 and V2 Regions Influence Replication Efficiency in Macrophages by Affecting Virus Spread. Virology. 1995;213:70–79. doi: 10.1006/viro.1995.1547. [DOI] [PubMed] [Google Scholar]
- Vuckovic D, Dagley LF, Purcell AW, Emili A. Membrane proteomics by high performance liquid chromatography-tandem mass spectrometry: Analytical approaches and challenges. Proteomics. 2013;13:404–423. doi: 10.1002/pmic.201200340. [DOI] [PubMed] [Google Scholar]
- Wehrly K, Chesebro B. p24 Antigen Capture Assay for Quantification of Human Immunodeficiency Virus Using Readily Available Inexpensive Reagents. Methods. 1997;12:288–293. doi: 10.1006/meth.1997.0481. [DOI] [PubMed] [Google Scholar]
- Weiss A, Wiskocil RL, Stobo JD. The role of T3 surface molecules in the activation of human T cells: a two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. Journal of immunology. 1984;133:123–128. [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Yohannes E, Ghosh SK, Jiang B, McCormick TS, Weinberg A, Hill E, Faddoul F, Chance MR. Proteomic Signatures of Human Oral Epithelial Cells in HIV-Infected Subjects. PLOS ONE. 2011;6:e27816. doi: 10.1371/journal.pone.0027816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.