

Abstract
Molecular docking is widely applied to computer-aided drug design and has become relatively mature in the recent decades. Application of docking in modeling varies from single lead compound optimization to large-scale virtual screening. The performance of molecular docking is highly dependent on the protein structures selected. It is especially challenging for large-scale target prediction research when multiple structures are available for a single target. Therefore, we have established ProSelection, a docking preferred-protein selection algorithm, in order to generate the proper structure subset(s). By the ProSelection algorithm, protein structures of “weak selectors” are filtered out whereas structures of “strong selectors” are kept. Specifically, the structure which has a good statistical performance of distinguishing active ligands from inactive ligands is defined as a strong selector. In this study, 249 protein structures of 14 autophagy-related targets are investigated. Surflex-dock was used as the docking engine to distinguish active and inactive compounds against these protein structures. Both t test and Mann–Whitney U test were used to distinguish the strong from the weak selectors based on the normality of the docking score distribution. The suggested docking score threshold for active ligands (SDA) was generated for each strong selector structure according to the receiver operating characteristic (ROC) curve. The performance of ProSelection was further validated by predicting the potential off-targets of 43 U.S. Federal Drug Administration approved small molecule antineoplastic drugs. Overall, ProSelection will accelerate the computational work in protein structure selection and could be a useful tool for molecular docking, target prediction, and protein–chemical database establishment research.
Graphical abstract
INTRODUCTION
Molecular docking, which is commonly applied to computer-aided drug design, has become relatively mature in the recent decades. Since the 1980s, pioneering works have been reported to achieve rigid docking of small molecules to protein structures.1 Since then the number of algorithms available to evaluate ligand–protein interaction is large and ever increasing. Docking programs like Autodock,2 Surflex-Dock,3 GOLD,4 Glide,5 FlexX,6 DOCK,7 ICM,8 and FRED9 are now widely used in receptors to small molecule docking. The number of users for relatively new algorithms such as Autodock Vina,2 EADock DSS,10 H-DOCK,11 and ParaDockS12 is still growing. Application of docking in modeling varies from single lead compound optimization to large-scale virtual screening. Typical performance of prediction for widely used programs in docking has a success rate close to 20%.13 This rate may further increase to over 50% when other conditions, like protein conformations from multiple complexes, are optimized.
Computational methods for molecular docking rely heavily on the representation of proteins. However, multiple crystal structures are usually available for a single protein target. For instance, 19 protein targets are randomly selected from the PDB database and listed in the Supplementary Table 1, while 18 of them have more than one crystal structure available. Multiple crystal structures can cause problems when docking an identical ligand that yields different results from these structures. One possible solution to this is incorporating multiple conformations of a single receptor into the docking algorithms.14 Ensemble docking (ED) is one of the primary research focuses within the area of receptor flexibility, in which compounds are docked against multiple conformations of the target receptor.15 This approach mimics the dynamic behavior of the protein by providing a structural degree of freedom, where each ligand may find a matching receptor.16 Combined with ligands’ flexibility, ensemble docking based prediction will achieve a higher success rate, compared to the rigid binding.17
Receptor ensembles for ED can be divided into two categories: (1) ensembles based on experimental structures56–58 and (2) ensembles based on in silico-generated structures. In the experimentally derived ensembles, the area under the ROC curve (AUC) is always employed as the enrichment metric. It reveals that the experimentally derived ensembles only yield better enrichment in some cases when compared to individual members.18 Abagyan’s group even reported an anticooperative behavior of the experimentally derived ensembles, in which the performances of ensembles actually decreased, due to the addition of some ensemble members.19 Encouragingly, the performance of multiple conformations (MRCs) in virtual ligand screening can be further improved by using a proper subset of receptor structures, which is selected from among the originally available conformers.19 Researchers then attempt to define a method to select proper subsets of protein structures for generating the receptor ensembles. Some recommended filtering out receptor subsets that provide only positive docking scores.20 However, this filter protocol is still based on the empirical observation of the correspondence between the runs that yielded positive docking scores for all molecules in the data set and those performing worse than a random selection.
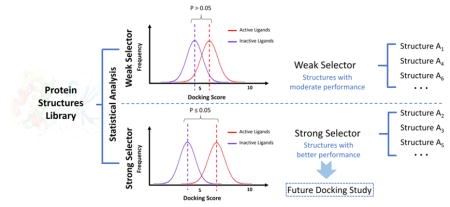
In this work, we introduce the Protein Structure Selection (ProSelection), a new computational and statistical algorithm for picking the proper protein structure subsets according to the user-defined docking protocol. ProSelection is tested using the currently widely used docking algorithm Surflex-dock and is designed for the virtual ligand screening against protein targets. Both t test and Mann–Whitney U test are used as the enrichment metrics in ProSelection, which is different from the previous selection approaches of conformers using AUC.20 This research cannot be considered as traditional virtual compound screening. Instead, our method is more like “structure screening”. That is, our approach is designed to find top structures with better capability for distinguishing compounds. It could be more useful when applying docking approach for target identification. That is, a small molecule will be used to search against thousands of PDB structures. In this situation, if strong selectors are used, we can reduce the prediction errors. ProSelection exhibits a significant power in protein structures selection, making it a promising option for the proper subsets selection for both the experimental structure based receptor ensembles and rigid docking. The research design of this work is presented in the flowchart (Figure 1).
Figure 1.

Method design of ProSelection. The numbers in parentheses and brackets represent the number of protein targets or structures in the corresponding step, respectively.
METHODS AND DATA SETS
Protein and Ligand Test-Set Construction
ProSelection was tested in our in-house Autophagy Knowledgebase (http://www.cbligand.org/autophagy) which was established using a procedure similar to AlzPlatform,21 a chemogenomics knowledgebase for Alzheimer’s disease. In the Autophagy knowledgebase, 102 autophagy-related protein targets were collected from literature and public databases, including PubMed (www.ncbi.nlm.nih.gov/pubmed), PubChem (pubchem.ncbi.nlm.nih.gov/), DrugBank (http://www.drugbank.ca), SciFinder (https://scifinder.cas.org/), UniProt (http://www.uniprot.org/), and ChEMBL (https://www.ebi.ac.uk/chembl/). Protein structures corresponding to the targets in the Autophagy Knowledgebase were downloaded from RCSB Protein Data Bank.22 The number of known active and inactive chemical ligands for each autophagy target was calculated. Only the targets which have more than 40 known ligands (20 active ligands and 20 inactive ligands) were kept in this study as well as their associated protein structures. These targets (14 in total) and their protein structures (249 in total) were then used to generate the protein test-set and structure test-set (Figure 1), respectively.
Active and inactive chemical ligands, which are associated with each of those autophagic targets, are acquired from the ChEMBL database to build the ligand test-set. Specifically, compounds with IC50 values lower than 5 nM against a target are considered as the active ligands for that target. While compounds with IC50 values higher than 50 μM are considered as the inactive ligands, compounds between 5 nM and 50 μM were excluded in this research, because our aim is to find structures with better capability to discriminate well-separated actives and inactives. We use a restrict IC50 threshold to separate structures (5 nM for actives). If a PDB structure cannot even distinguish more distinctive actives/inactives, we will not consider it as a better selector. However, in the validation section, we consider that usually compounds with inhibition IC50 of 10 μM will be categorized as actives in practice, so we use it as a more relaxed threshold. All the cocrystallized inhibitors in the original protein structures are excluded in the ligand test-set. Reasonable molecular weight ranges from 200 to 600 Da.23 Well-defined active/inactives with 1:1 ratio was used in this study. None of the original structure cocrystallized compounds are included in the ligand test set. The searching and generation of compounds test-set are conducted automatically using a Python script. As of November 2016, 14 protein targets in our Autophagy Knowledgebase each have at least 20 active and 20 inactive ligands recorded in the ChEMBL database (Figure 2). These targets and their 249 associated crystal structures are then used to build the protein targets test-set and the structure test-set in this research.
Figure 2.

Ligand information for the 14 targets in Autophagy Knowledgebase. Most of the targets in this protein test-set have more known active ligands than inactive ligands reported in the ChEMBL database. Abbreviations: PDPK1, 3-phosphoinositide-dependent protein kinase 1; B2CL1, Bcl-2-like protein 1; CASP1, Caspase-1; CATB, Cathepsin B; CATD, Cathepsin D; HDAC6, Histone deacetylase 6; INSR, Insulin receptor; SIR1, NAD-dependent protein deacetylase sirtuin-1; SIR2, NAD-dependent protein deacetylase sirtuin-2; PK3CA, Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform; AKT1, RAC-alpha serine/threonine-protein kinase; AKT2, RAC-beta serine/threonine-protein kinase; MTOR, Serine/threonine-protein kinase mTOR; ABL1, Tyrosine-protein kinase ABL1.
Validation Compound Set Establishment
The compound set for testing the performance of “strong selector” structures is generated from the list of U.S. Federal Drug Administration (FDA) approved small molecular antineoplastic drugs since 2005 (www.fda.gov). Although 200–600 was used to select the ligand test set, the range of molecular weight for chemicals within the validation compound set was set as from 200 to 800 Da (4 FDA drugs are between 600 and 800 among all the 43 FDA drugs we used). We do this because the original structure selection is supposed to be more “strict” while the validation can be “loose” (and can include more compounds) since both 600 or 800 are reasonable Mws in the molecular docking. With this criterion, 43 qualified compounds were included in the validation compound set.
Docking Protocol
Docking is conducted for all ligands in the ligand test-set following the same procedure using the Tripos molecular modeling packages SYBYL-X 8.0.24 All molecules for docking are built using standard bond lengths and angles from SYBYL-X 8.0 and are then optimized using the Tripos force field. The flexible docking method, named Surflex-dock, is used with all the parameters setting to default. Before docking, cocrystallized ligands and water molecules are removed from the original PDB structures, and hydrogen atoms are added. Chain A of each structure will be selected for docking by default if no other evidence is available. The active binding pocket of each protein is defined by the residues near the cocrystallized active ligands (most are inhibitors) or is automatically generated by the SYBYL-X utility scripts. Most of the active binding pockets generated for protein kinases in the Autophagy Knowledgebase are pockets which contain the ATP binding domain. This is because most of the cocrystallized ligands we used are ATP competitive inhibitors for these structures. Therefore, most of the ligand predictions in our test of ProSelection are predicting the potential ligands which bind to the ATP domain. The scoring function in Surflex-dock, which contains hydrophobic, polar, repulsive, entropic, and solvation terms, was trained to estimate the dissociation constant (Kd) expressed in log(Kd).25 For large-scale compound screening, it is usually not applicable to check each docking pose of individual compound, even those on the top of the list. Therefore, only the best docking score of a compound against each protein structure is kept for assessing and ranking the potential protein partners or targets.
Selection Metrics
For a protein structure, molecular docking generates higher docking scores against active ligands compared to inactive ligands, statistically. Therefore, the performance of virtual screening based on molecular docking can be quantified using the p-value. This p-value either from the t test of two independent samples26 or the Mann–Whitney U test,27 which generally measures the statistical mean difference between the docking scores in the active ligands set and scores in the inactive ligands set. Choosing of the statistical method was determined by the normality of docking score distribution. Specifically, the t test for two independent samples is used in this work only if the docking scores are normally distributed in both active and inactive ligands set. Otherwise, the Mann–Whitney U test will be used to calculate the p-value instead, due to the required normality assumption of the t test. All statistical tests in this research are one-tailed. A widely used open source python package Scipy (https://www.scipy.org/) is used to conduct the statistical analysis in this research. The detailed selection method is described below:
First, the normality of docking score distributions is determined by the D’Agostino and Pearson’s test,28,29 which combines skew and kurtosis to produce an omnibus test of normality (function name in Python: scipy.stats.m-stats. normaltest). Protein structures which have normal-distributed docking scores for both its active and inactive ligand sets will be selected to run the t test for two independent samples with unequal variance at 95% significance level,30
- Compute the test statistic
- Compute the approximate degrees of freedom d′, where
-
Round d′ down to the nearest integer d″
If t ≤ 0, then p = 2(area to the left of t under a td″ distribution)
If t ≥ 0, then p = 2(area to the right of t under a td″ distribution)
Where d″ is the nearest integer to the approximate degrees of freedom, x̄1 and x̄2 are the averages of docking scores for active ligands and inactive ligands, respectively. Ligand numbers in each ligand set are represented by n1 and n2. Variance of the two input samples is referred as and .
Structures without normally distributed docking scores against either of the ligand sets will be chosen to run Mann–Whitney U test at 95% significance level31 for which the general calculations after the ranking procedure is shown below:
If R1 ≠ n1(n1 + n2 + 1)/2 and there are no ties, then
If R1 ≠ n1(n1 + n2 + 1)/2 and there are ties, then
If R1 = n1(n1 + n2 + 1)/2, then T = 0.
Where R1 refers to the rank sum in the first sample, ti refers to the number of observations with the same value in the ith tied group, g is the number of tied groups, and n1, n2 represent the number of ligands in the active ligand set and inactive ligand set, respectively. Then, the exact p-value can be computed by
Φ(T) represents the cumulative-distribution function (cdf) for a standard normal distribution and is denoted by Φ(x) = Pr(X ≤ x) where X follows an standard normal distribution (N(0, 1)).
However, multiple structures are usually available for a single protein target in the PDB database. This number can reach over 50 in some cases. Therefore, Bonferroni correction is applied after multiple comparisons in this research because of the relatively large number of comparisons.32 In Bonferroni correction, the significance level α is adjusted to , where m is the number of hypothesis tests.33 The equation used for converting the Mann–Whitney U test or two independent samples t test p-value to Bonferroni corrected p-value is shown below:
Where p refers to the original p-value, p0 is the Bonferroni corrected p-value, and k is the number of PDB structures for a target.
Selection of strong selector structures is based on the p-value after the Bonferroni correction. Specifically, PDB structures with a p-value larger than 0.05 will be considered as weak selectors due to their incapability of distinguishing active ligands from inactive ligands based on the selected docking protocol. In the opposite, structures which can significantly distinguish active ligands from inactive ligands (p < 0.05) will be labeled as a strong selector. Particularly, most of the structures for kinases in this research are labeled based on their ability to distinguish ATP competitive inhibitors. This is because most of the docking pockets we previous defined for kinases in the docking protocol are ATP binding pockets (See the Docking Protocol section). ProSelection is docking protocol sensitive, which further indicates that the weak selector labeled by ProSelection may not be a bad crystal structure if taking other docking pockets (e.g., pocket other than ATP binding pocket) or other research purposes (e.g., predicting a non-ATP-competitive inhibitor) into consideration.
Individual Docking Score Criterion
When conducting the molecular docking on a bunch of protein targets and their associated chemical ligands, how to interpret the docking results may become a major issue. We raised a hypothesis that the distributions of docking scores are different among different protein structures. This assumption indicates that the docking score criterions for active ligands may be specific for each target and protein structure. A suggested docking score threshold for active ligands (SDA) is used in this research as an individual docking score criterion.
SDA is generated based on the receiver operating characteristic (ROC) curves.34 This calculation can be divided into four steps: (1) ROC curve establishment, (2) screening through the ROC curve and measuring the distance from points on the curve to the top-left point on the ROC plot, (3) choosing the point with minimum distance, (4) using the point corresponding docking score as SDA. The ROC curve is established by stepping sequentially through the ranked list of test-set compounds arranged in order of increasing docking scores for each target. Sensitivity and specificity are calculated at each ranking score. Plotting the sensitivity versus (1 – specificity) for all positions in the ranked list will generate the ROC plot. One interesting feature of the ROC curve is the capability of generating the optimized cutoff point to optimize the sensitivity and specificity. In this work, the optimal cutoff point is calculated for each ROC curve using Pathagoras’ theorem, which minimizes the distance between the top-left point in the ROC plot and the selected points on the ROC59 (Supplementary Figure 1). However, in order to keep a relatively low false-positive-rate (FPR) during prediction, we require the FPR3 ≤ 0.0001 for each point to be considered as a “valid point” before using Pathagoras’ theorem. This is similar to requiring at least one correct prediction in the top three predictions when calculating the optimal cutoff points (also means that at least one of the top three predictions is correct). The docking scores associated with these final optimized cutoff points were reported as SDAs. Any ligand that has a docking score higher than its corresponding SDA will be considered as an “active” ligand against its associated crystal structure.
RESULTS
Distributions of Docking Scores
Figure 3 shows the normality of the docking scores distributions for 249 structures in the structure test-set, which belongs to the 14 different autophagic protein targets. For each single protein structure, the distributions of docking scores can be different between active ligands set and inactive ligand set (Figure 4). Generally, 68% of the structures have normally distributed docking scores against their corresponding inactive ligands. Only 47% of the structures have normally distributed docking scores against their corresponding active ligands. These numbers are not surprising because the inactive compounds usually have more “random” chemical structures when compared to active compounds. According to the distributions of docking scores, the Mann–Whitney U test was applied to 167 (67%) crystal structures while the t test was applied to 82 structures (33%) structures in the structure test-set. Using different statistical tests based on the statistical normality in this work is expected to yield a better statistical accuracy during the structure selection.
Figure 3.

Normality test for 249 structures in the structure test-set. The p-value was calculated based on the D’Agostino and Pearson’s test (see section on evaluation metrics). Distributions of docking scores which have a p-value higher than 0.05 can be considered as normal distributions. Each dot represents the p-value of the distribution of docking scores for one protein structure. The normtest_A represents the targets corresponding active ligands while normtest_N represents the corresponding inactive ligands.
Figure 4.

Distributions of docking scores for PDB structure 4JT6. Distribution of docking scores and normal Q–Q plot for (A) active ligands (p < 0.05) and (B) inactive ligands (p = 0.83) against 4JT6, one of the PDB structures for mammalian target of rapamycin (mTOR). The p-value was calculated based on the D’Agostino and Pearson’s test for normality. Here, p-values lower than 0.05 indicates a non-normal distribution of the docking scores. For structure 4JT6, docking scores are normally distributed among negative ligands and non-normally distributed among active ligands.
Structure Selection
Either the t test for two independent samples or the Mann–Whitney U test is used to generate the statistical p-value, which depends on the normality of docking score distribution for each PDB structure. Structures with a p-value lower than 0.05 are categorized as strong selector structures while others are classified as weak selectors for the docking protocol we selected. Percentages of two categories of structures for each target in the test-set are shown in Figure 5.
Figure 5.
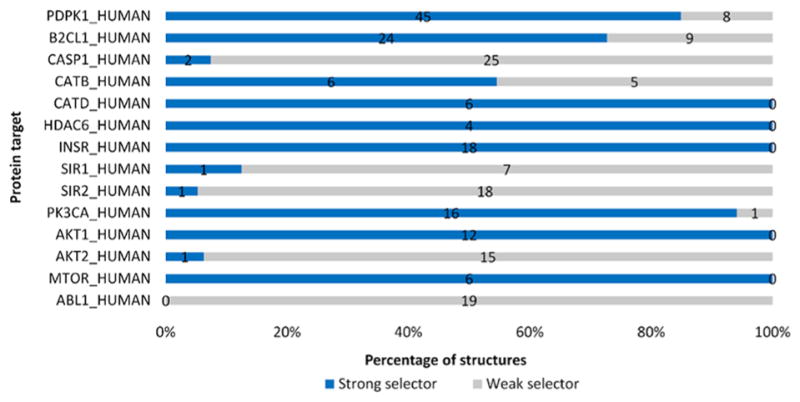
Percentage of the strong selector structures among 14 targets in the protein test-set. Crystal structures which have a good performance in distinguishing active ligands from inactive ligands are labeled as strong selectors. The number on the bar represents the quantity of strong or weak selector structures for each target. In total, 249 PDB structures in the structure test-set are examined while 142 structures are strong and 107 structures are weak. Among all the 14 targets in this test, 13 targets have at least one strong selector structure according to ProSelection. Abbreviations: PDPK1, 3-phosphoinositide-dependent protein kinase 1; B2CL1, Bcl-2-like protein 1; CASP1, Caspase-1; CATB, Cathepsin B; CATD, Cathepsin D; HDAC6, Histone deacetylase 6; INSR, Insulin receptor; SIR1, NAD-dependent protein deacetylase sirtuin-1; SIR2, NAD-dependent protein deacetylase sirtuin-2; PK3CA, Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform; AKT1, RAC-alpha serine/threonine-protein kinase; AKT2, RAC-beta serine/threonine-protein kinase; MTOR, Serine/threonine-protein kinase mTOR; ABL1, Tyrosine-protein kinase ABL1.
This result suggests that for the molecular docking protocol we chose (Sybyl 8.0 Surflex-dock with default parameters used), a significant portion of PDB structures are weak selectors (Figure 5). These weak selector structures may result in unseparated docking scores against active ligands and inactive ligands during docking process. If no modifications are made either for these structures or the docking protocol, high score decoys can be expected, which can be misleading for further research approaches. However, these results of the structure selection, specifically strong and weak selector structures, are docking protocol sensitive. Either the docking parameter settings, pocket definitions, active and inactive ligand choices, or the docking mode selection can contribute to the results, independent of the protein structure itself. If other binding pockets, such as the allosteric binding pockets, are used, different results of selection can be expected. Therefore, the exact results of structural selection in this work can only be referred when a similar docking protocol is conducted. For a closer look at the weak selector structures, several examples are used below to show why these structures are weak selectors in our docking protocol.
PDB Structures 3NAY and 3NAX
3NAY and 3NAX are X-ray structures of 3-phosphoinositide dependent protein kinase-1 (PDPK1) binding to compounds 2 and 7 (Supplementary Figure 3), which are PDPK1 inhibitors used by Jannik’s group.35 In our structure selection, most of the known active ligands for this target are ATP competitive inhibitors. The typical active ATP binding site is used as the docking pocket. As a result, 3NAY is a strong selector structure (p = 3.9 × 10−3) while 3NAX is labeled as a weak selector (p = 0.12). Compound 2 in 3NAY binds to the active DFG-in conformation of PDPK1 ATP binding site, like most typical ATP competitive inhibitors (type I). Compound 7 in 3NAX binds in the ATP pocket and induces substantial conformational changes, causing the DFG loop to exist in an “out” conformation (DFG-out) that typifies inactive form kinase inhibitors (type II).35 Compound 7 also disrupts the αC helix which is a unique feature to this compound among inactive form kinase inhibitors.35 The conformational change in the αC helix results in catalytic residue Glu-130 being displaced from the active site. It can be concluded that compound 7 binds to the inactive kinase conformation of PDPK1 and induces conformational changes. Therefore, compared to the structure of active ATP binding site, the PDPK1 protein structure cocrystallized with compound 7 is an inactive kinase conformation with some additional conformational changes. For verification, we use another known PDPK1 ATP competitive inhibitor, CHEMBL3640476, to dock both the structure 3NAY and 3NAX. In 3NAY, CHEMBL3640476 forms H-bonds to Ala162, Glu166, and Thr222 within the ATP-site of PDPK1 (Figure 6A). Glu130 is also in the active binding site. However, only one H-bond to Glu166 is formed and Glu130 is out of the active site in the 3NAX (Figure 6B). This indicates that the inactive kinase conformation, like 3NAX, is unsurprisingly being a weak selector structure for the typical ATP competitive inhibitors (type I) binding and prediction. Similar results can also be found for other pairs of kinase structures like 4L2Y and 4L2336 (Supplementary Figure 4), 1MRY/1MRV,37,38 and 2X39 (Supplementary Figure 5).
Figure 6.

Compound CHEMBL3640476 bound to the PDPK1 ATP binding site. Structures of the compound CHEMBL3640476 binds to the PDPK1 ATP binding site in the kinase domain. (A) CHEMBL3640476 binds to the PDPK1 crystal structure 3NAY (docking score 8.49). (B) CHEMBL3640476 binds to the PDPK1 crystal structure 3NAX (docking score 5.35). (C) Original structure of 3NAX (PDPK1 cocrystallized with compound 7). Blue sticks represent the ATP competitive inhibitor of PDPK1, named CHEMBL3640476. Yellow sticks represent the important residues of PDPK1 ATP binding domain which are also labeled. H-bonds to the important residues are displayed as dashed red lines. In 3NAY, CHEMBL3640476 has H-bonds to Ala162, Glu166, and Thr222 within the ATP-site of PDPK1. Glu130 is also in the active binding site. However, only one H-bond to Glu166 is formed while Glu130 is out of the active site in the 3NAX.
PDB Structures 2YXJ and 4HNJ
2YXJ and 4HNJ are structures of the Bcl-2-like protein 1 (also named Bcl-xL). Bcl-xL is a potent nonkinase protein inhibitor of cell death and caspases activation. Structure 2YXJ is the Bcl-xL in complex with ABT-737 (Supplementary Figure 3), a known Bcl-xL inhibitor which belongs to the class of BH3 mimics.60 While 4HNJ is the crystallographic structure of BCL-xL domain-swapped dimer in complex with PUMA (p53-upregulated modulator of apoptosis), which induced partial unfolding of two α-helix within Bcl-xL BH3 binding pocket.39 In our research, 2YXJ is a strong selector (p = 0.029) and 4HNJ is a weak selector (p = 0.49). This categorization is validated by the docking research where the known BH3 mimics inhibitor of Bcl-xL (CHEMBL2312484) forms two H-bonds to Phe150 in the BH3 binding pocket (Figure 7A). However, no H-bond is formed in the BH3 binding pocket in the 4HNJ (Figure 7B). Therefore, 4HNJ is unsurprisingly a weak selector for BH3 mimics because of its structural change in the BH3 binding domain induced by PUMA.
Figure 7.

Structures of the compound CHEMBL2312484 bind to the BH3 binding domain within the Bcl-xL. (A) CHEMBL2312484 binds to the Bcl-xL crystal structure 2YXJ (docking score 5.42). (B) CHEMBL2312484 binds to the Bcl-xL crystal structure 4HNJ (docking score 3.35). Blue sticks represent the inhibitor compound CHEMBL2312484. Yellow sticks represent the important residues in the BH3 binding domain which are also labeled. H-bonds to the important residues are displayed as dashed red lines. In 2YXJ, CHEMBL2312484 has two H-bonds to Phe105. In 4HNJ, however, no H-bond is formed between the compound and the important residues. One residue, Tyr195, is even “out” of the BH3 binding domain due to the protein structural change induced by PUMA in 4HNJ.
Individual Docking Score Criterion
Before validating the target-prediction using the structures selected by ProSelection, we raise the hypothesis that each protein structure of a protein target may have its unique distribution of docking scores for even the same set of ligands. This individualism is due to the existence of diverse protein/compound structures and docking protocols in real life. For instance, a docking score of 6.0 may be high enough for those compounds against one structure to be considered as active ligands. However, the same score 6.0 may not be sufficient for another structure to determine its active ligands. This phenomenon further indicates that if an individual docking score criterion can be established for each structure, a better understanding of docking score in the molecular docking can be expected to distinguish active ligands from inactive ligands.
In this work, the SDA is used as a score criterion for individual protein structures base on the ROC curve with both a restriction of FPR and an optimized sensitivity versus specificity (see more details in the Methods and Data Sets section). Supplementary Figure 2 presents the ROC curves for 4 of the strong selector structures in our structure test-set. SDA is then calculated by finding the optimal cutoff points in the ROC curves for each crystal structure. Particularly, SDA is a new concept raised in this work which is designed for a better understanding of the docking scores in the molecular docking research.
The SDAs for all the 142 strong selector structures in the structure test-set distribute from 4.0 to 10.0 (Supplementary Table 2 and Figure 8). It presents a non-normal distribution with a mean value of 7.37 and standard deviation of 1.265. Although most of the SDAs fall in the range of 6 to 8, few reach an even higher score of 10.0 which was previously considered as “very high” in molecular docking. On the other hand, 4PPI, one of the PDB structures of the Bcl-2 family protein Bcl-xL, has a low SDA of 3.76. These results are particularly inspiring for active ligands prediction in the future because previously compounds with a docking score lower than 4.0 will tend to be recognized as “inactive ligands” no matter which target the score is against in the Sybyl-X.
Figure 8.

Suggested docking score threshold for active ligands (SDA) of 142 strong selector structures.
Validation of the Target Prediction for Selected Targets
Target identification of small molecules is essential for unraveling the underlying mechanisms of their bioactivities or side effects. In cancer therapeutics, autophagy is considered as one of the most critical drug resistance mechanisms, due to its attempt at removing damaged cellular components in the drug administrated cancer cells.40 As a validation procedure, we used our established ProSelection algorithm to predict the potential autophagy related off-targets for 43 FDA-approved small molecule antineoplastic agents. Only the strong selector structures are used. Previously, 13 targets in our protein test-set have at least one strong selector structure among all the 14 targets we explored according to the ProSelection (human ABL1 target has no strong selector). ChEMBL is then used as the source for searching the experimental bioactivity data (IC50) for the drug binding of these targets. Two autophagy related targets in the protein test-set, insulin receptor (INSR) and histone deacetylase 6 (HDAC6), are selected for validation due to their relatively sufficient data in the ChEMBL database.
INSR is commonly considered as a drug target for antihyperglycemia and the treatment of diabetes. However, it also regulates the phagophore formation, which is one of the early stages in the macroautophagy. Enhanced insulin signaling will inhibit the autophagy in the initiation process by inducing the downstream PI3K/AKT/mTOR pathway.41 A molecular docking study for 43 FDA-approved antineoplastic drugs against insulin receptor was conducted (Figure 9A). In this docking process, only the 18 strong selector structures of insulin receptor were used. Among these 43 drugs, six agents have IC50 data for binding in the ChEMBL database (Table 1). Their associated docking scores were summarized in Figure 9B.
Figure 9.

Molecular docking results for FDA-approved drugs against insulin receptor. (A) Docking scores for 43 FDA-approved antineoplastic drugs. (B) Docking scores for 6 FDA-approved drugs with experimental IC50 data of inhibition in ChEMBL. All docking scores are shown in folds compared to the corresponding SDA for each structure. Sybyl software generates negative scores for some compounds during the docking process. Scores lower than 0 are manipulated to 0 for figure generating. The folds are negative because the original docking scores are below zero. Sybyl 8.0 software sometimes generates negative scores for some compounds during the docking process. But, the number of such compounds is small enough and can be ignored.
Table 1.
Experimental Bioactivities of Six Drugs against Human Insulin Receptor
One of the commonly used IC50 threshold for active compounds for inhibition is 10 μM.42 Specifically, in this research, one compound can be considered as active once it has an IC50 lower than 10 μM (10000 nM). Osimertinib is an active compound for INSR in the experiment (Table 1), and it shows a docking score higher than SDA for 8 of total 18 INSR structures in the molecular docking after ProSelection. A similar result was also observed for the active drugs Crizotinib, Ceritinib, and Sunitinib. However, for inactive compounds like Lapatinib, scores against some structures are unexpectedly higher than SDA (Figure 9B). This incorrect prediction is acceptable since molecular docking has some extent of “randomness”43 and most of the inactive compounds in our test have a lower quantity of high scores ( all available structures) compared to active compounds ( all available structures). On the functional level, drugs that inhibit the INSR will further inhibit the downstream signaling of PI3K/AKT/mTOR pathway. An enhancement of autophagy function can be observed as the final result of this signaling, which is consistent to the autophagy-related drug resistant nature of many antineoplastic agents.40
HDAC6 is another target which is closely related to autophagy. Autophagy acts as a compensatory degradation system when the ubiquitin–proteasome system (UPS) is impaired. Histone deacetylase 6 (HDAC6), a microtubule-associated deacetylase that interacts with polyubiquitinated proteins, is an essential mechanistic link in this compensatory interaction.49 Although HDAC6 is not required for autophagy activation, it rather controls the fusion of autophagosomes to lysosomes during the autophagy process.50 Molecular docking results of 43 FDA-approved antineoplastic drugs against HDAC6 were presented in Figure 10A. Again, only the strong selector structures are used. Belinostat, Panobinostat, and Bendamustine are known to bind HDAC6 with IC50 of 15, 11, and 6 nM respectively (Table 2). Belinostat and Panobinostat are successfully predicted by strong selector structures, while Bendamustine is accidently predicted as inactive (Figure 10B). For known HDAC6 inactive drugs Lapatinib and Romidepsin, none of the strong selector structures show a docking score higher than SDA (Figure 10B). These results indicate that the protein structures selected by ProSelection work accurately during active ligands prediction.
Figure 10.

Molecular docking results for FDA-approved drugs against histone deacetylase 6. (A) Docking scores for 43 FDA-approved antineoplastic drugs. (B) Docking scores for 5 FDA-approved drugs with experimental inhibition data in ChEMBL. All docking scores are shown in folds compared to the corresponding SDA for each structure. Scores lower than 0 are manipulated to 0 for figure generating. Sybyl 8.0 software sometimes generates negative scores for some compounds during the docking process. But the number of such compounds is small enough and can be ignored.
Table 2.
Experimental Bioactivities for Five Antineoplastic Drugs against Human Histone Deacetylase 6
CONCLUSION
ProSelection, a computational statistical algorithm designed for generating the proper protein structure subset, is developed for docking-based target identification research. The virtual screening performances of different PDB structures were measured by using targets associated active and inactive ligands with Sybyl-X default Surflex-dock docking protocol. This study examined 249 crystal protein structures from 14 autophagy related targets. Either the t test or the Mann–Whitney U test was used to distinguish strong selector structures from weak selector structures based on the normality of the docking score distributions for each protein structure. The receiver operating characteristic (ROC) curve was made and suggested docking scores threshold for active ligands (SDA) were generated for each strong selector structure according to the ROC curve. The performance of the target prediction using only strong selector structures was validated by recently FDA approved small molecule antineoplastic drugs for predicting their potential off-targets among autophagy-related proteins.
Protein selection is getting more and more important in the docking-based virtual screening. The structures come from the crystallization environments, which may yield different outcomes even when using the same compound for docking. One approach to incorporate multiple protein structures into the molecular docking is the using of an ensemble consisting of multiple conformations of the same protein.17 This method can be categorized as the “ensemble docking” (ED) strategy, which achieves the protein “flexibility” compared to the rigid docking. In rigid docking, protein structure is not able to change its conformation during the docking process. By using ensemble docking, the docking performance can be increased.54,55 Although ED strategy can be relatively fast compared to the sequentially docking during large-scale database screening and even more accurate, it could still be “overfitting” if some weak selector structures are taken into consideration during ED. It is common that different crystal structures are generated based on various research purpose. Thus, the strong and weak selector protein structures may be distinct when facing diverse docking protocols and research aims. Therefore, we developed the ProSelection, an algorithm designed for protein structure selection based on the known ligands and docking protocols. After using the personalized ligands and protocols to run ProSelection, users can put the structures selected by ProSelection directly into their rigid docking or act as a new source for their further ensemble docking approaches.
It is worth considering the weaknesses of this current study to explore the directions for further research. ProSelection depends heavily on the abundance of known active and inactive ligands. As shown in Figure 2, the number of active ligands is usually much larger than inactive ligands in the public databases. This ratio can even approach 50 for some targets. One solution to rebalance the number of ligands is replacing the inactive ligands with random ligands. This is expected to slightly reduce the performance of the structure selection because of the existence of active ligands in a random compound-set. However, this modification is still acceptable because a strong selector structure is also expected to distinguish active ligands from random ligands. More importantly, the number of random compounds is not limited. More protein targets will be available for ProSelection if the active-ligands set and the random-ligands set are used. Specifically, every target, with the size of active ligand test-set larger than 20, is sufficient for ProSelection if a random-ligands set is used. However, for targets which do not have enough active ligands, statistical approaches like ProSelection cannot be used. Therefore, machine learning algorithms can be an alternative solution to predict the proper protein structure subset based on the similar logic of designing ProSelection. Using of the DUD data set as source is another consideration, however, it will become a new study design and only a limited number of the targets included in the DUD data set. This means the proteins can only be from those DUD-targets. This is not the original purpose of this study.
A further consideration in this work is the collaboration of ProSelection with ensemble docking. Because of the backstage usage of popular docking tools like Sybyl-x, statistical methods like ProSelection are still categorized as “rigid” docking but with flexible ligands. If ensemble docking (which has taken receptor flexibility into consideration) can be integrated into ProSelection, a much better performance is expected because of the consideration of the flexibility in both the protein target and the ligand.
Supplementary Material
Acknowledgments
The project is supported by funding to Xie laboratory from the NIH NIDA (P30 DA035778A1) and DOD (W81XWH-16-1-0490).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00277.
SDA of 142 strong selector protein structures, SDA generation, ROC examples of strong selector structures, structures of the chemical inhibitors, and the docking comparisons between strong and weak selectors for known inhibitors (PDF)
References
- 1.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–88. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 2.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 4.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 5.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 6.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 7.Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput-Aided Mol Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 8.Brylinski M. Nonlinear Scoring Functions for Similarity-Based Ligand Docking and Binding Affinity Prediction. J Chem Inf Model. 2013;53:3097. doi: 10.1021/ci400510e. [DOI] [PubMed] [Google Scholar]
- 9.McGann M. FRED and HYBRID docking performance on standardized datasets. J Comput-Aided Mol Des. 2012;26:897–906. doi: 10.1007/s10822-012-9584-8. [DOI] [PubMed] [Google Scholar]
- 10.Grosdidier A, Zoete V, Michielin O. Fast docking using the CHARMM force field with EADock DSS. J Comput Chem. 2011;32:2149–2159. doi: 10.1002/jcc.21797. [DOI] [PubMed] [Google Scholar]
- 11.Luo W, Pei J, Zhu Y. A fast protein-ligand docking algorithm based on hydrogen bond matching and surface shape complementarity. J Mol Model. 2010;16:903–913. doi: 10.1007/s00894-009-0598-7. [DOI] [PubMed] [Google Scholar]
- 12.Meier R, Pippel M, Brandt F, Sippl W, Baldauf C. ParaDockS: a framework for molecular docking with population-based metaheuristics. J Chem Inf Model. 2010;50:879–889. doi: 10.1021/ci900467x. [DOI] [PubMed] [Google Scholar]
- 13.Jain AN. Effects of protein conformation in docking: improved pose prediction through protein pocket adaptation. J Comput-Aided Mol Des. 2009;23:355–374. doi: 10.1007/s10822-009-9266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lill MA. Efficient incorporation of protein flexibility and dynamics into molecular docking simulations. Biochemistry. 2011;50:6157–6169. doi: 10.1021/bi2004558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuriev E, Ramsland PA. Latest developments in molecular docking: 2010–2011 in review. J Mol Recognit. 2013;26:215–239. doi: 10.1002/jmr.2266. [DOI] [PubMed] [Google Scholar]
- 17.Huang SY, Zou X. Ensemble docking of multiple protein structures: considering protein structural variations in molecular docking. Proteins: Struct, Funct Genet. 2007;66:399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- 18.Craig IR, Essex JW, Spiegel K. Ensemble docking into multiple crystallographically derived protein structures: an evaluation based on the statistical analysis of enrichments. J Chem Inf Model. 2010;50:511–524. doi: 10.1021/ci900407c. [DOI] [PubMed] [Google Scholar]
- 19.Rueda M, Bottegoni G, Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J Chem Inf Model. 2010;50:186–193. doi: 10.1021/ci9003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bottegoni G, Rocchia W, Rueda M, Abagyan R, Cavalli A. Systematic exploitation of multiple receptor conformations for virtual ligand screening. PLoS One. 2011;6:e18845. doi: 10.1371/journal.pone.0018845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H, Wang L, Lv M, Pei R, Li P, Pei Z, Wang Y, Su W, Xie XQ. AlzPlatform: an Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J Chem Inf Model. 2014;54:1050–1060. doi: 10.1021/ci500004h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berman HM, Battistuz T, Bhat T, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, et al. The protein data bank. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 23.Balius TE, Mukherjee S, Rizzo RC. Implementation and evaluation of a docking-rescoring method using molecular footprint comparisons. J Comput Chem. 2011;32:2273–2289. doi: 10.1002/jcc.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geldenhuys WJ, Nakamura H. 3D-QSAR and docking studies on transforming growth factor (TGF)-β receptor 1 antagonists. Bioorg Med Chem Lett. 2010;20:1918–1923. doi: 10.1016/j.bmcl.2010.01.140. [DOI] [PubMed] [Google Scholar]
- 25.Gu SX, Li ZM, Ma XD, Yang SQ, He QQ, Chen FE, De Clercq E, Balzarini J, Pannecouque C. Chiral resolution, absolute configuration assignment and biological activity of racemic diarylpyrimidine CH (OH)-DAPY as potent nonnucleoside HIV-1 reverse transcriptase inhibitors. Eur J Med Chem. 2012;53:229–234. doi: 10.1016/j.ejmech.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Paternoster R, Brame R, Mazerolle P, Piquero A. Using the correct statistical test for the equality of regression coefficients. Criminology. 1998;36:859–866. [Google Scholar]
- 27.Ruxton GD. The unequal variance t-test is an underused alternative to Student’s t-test and the Mann–Whitney U test. Behavioral Ecology. 2006;17:688–690. [Google Scholar]
- 28.d’Agostino RB. An omnibus test of normality for moderate and large size samples. Biometrika. 1971;58:341–348. [Google Scholar]
- 29.D’Agostino R, Pearson ES. Tests for departure from normality. Empirical results for the distributions of b2 and √ b1. Biometrika. 1973;60:613–622. [Google Scholar]
- 30.Rosner B. Fundamentals of Biostatistics. 7. Cengage Learning; Boston, MA: 2010. Two-Sample t Test for Independent Samples with Unequal Variance; pp. 287–293. [Google Scholar]
- 31.Rosner B. Fundamentals of Biostatistics. 7. Cengage Learning; Boston, MA: 2010. The Wilcoxon Rank-Sum Test; pp. 339–344. [Google Scholar]
- 32.Curtin F, Schulz P. Multiple correlations and Bonferroni’s correction. Biol Psychiatry. 1998;44:775–777. doi: 10.1016/s0006-3223(98)00043-2. [DOI] [PubMed] [Google Scholar]
- 33.Shaffer JP. Multiple hypothesis testing. Annu Rev Psychol. 1995;46:561–584. [Google Scholar]
- 34.Fawcett T. An introduction to ROC analysis. Pattern recognition letters. 2006;27:861–874. [Google Scholar]
- 35.Nagashima K, Shumway SD, Sathyanarayanan S, Chen AH, Dolinski B, Xu Y, Keilhack H, Nguyen T, Wiznerowicz M, Li L, et al. Genetic and pharmacological inhibition of PDK1 in cancer cells characterization of a selective allosteric kinase inhibitor. J Biol Chem. 2011;286:6433–6448. doi: 10.1074/jbc.M110.156463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Zhang X, Chen Y, Lu S, Peng Y, Wang X, Guo C, Zhou A, Zhang J, Luo Y, et al. Crystal structures of PI3Kα complexed with PI103 and its derivatives: new directions for inhibitors design. ACS Med Chem Lett. 2014;5:138–142. doi: 10.1021/ml400378e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McHardy T, Caldwell JJ, Cheung KM, Hunter LJ, Taylor K, Rowlands M, Ruddle R, Henley A, de Haven Brandon A, Valenti M, et al. Discovery of 4-amino-1-(7 H-pyrrolo [2, 3-d] pyrimidin-4-yl) piperidine-4-carboxamides as selective, orally active inhibitors of protein kinase B (Akt) J Med Chem. 2010;53:2239–2249. doi: 10.1021/jm901788j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang X, Begley M, Morgenstern KA, Gu Y, Rose P, Zhao H, Zhu X. Crystal structure of an inactive Akt2 kinase domain. Structure. 2003;11:21–30. doi: 10.1016/s0969-2126(02)00937-1. [DOI] [PubMed] [Google Scholar]
- 39.Follis AV, Chipuk JE, Fisher JC, Yun MK, Grace CR, Nourse A, Baran K, Ou L, Min L, White SW, et al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat Chem Biol. 2013;9:163–168. doi: 10.1038/nchembio.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu H-Y, Lin L-T, Siegelin MD, Fimognari C, Kumar NB, Dou QP. Seminars in cancer biology. Vol. 35. Elsevier; 2015. Broad targeting of resistance to apoptosis in cancer; pp. S78–S103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hildmann C, Wegener D, Riester D, Hempel R, Schober A, Merana J, Giurato L, Guccione S, Nielsen TK, Ficner R, Schwienhorst A. Substrate and inhibitor specificity of class 1 and class 2 histone deacetylases. J Biotechnol. 2006;124:258–270. doi: 10.1016/j.jbiotec.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 43.Erickson JA, Jalaie M, Robertson DH, Lewis RA, Vieth M. Lessons in molecular recognition: the effects of ligand and protein flexibility on molecular docking accuracy. J Med Chem. 2004;47:45–55. doi: 10.1021/jm030209y. [DOI] [PubMed] [Google Scholar]
- 44.Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, Jiang T, Kim S, Li N, Warmuth M, et al. Synthesis, structure–activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-Chloro-N 2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl) phenyl)-N 4-(2-(isopropylsulfonyl) phenyl) pyrimidine-2, 4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 2013;56:5675–5690. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- 45.Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung PP, Pairish M, Jia L, Meng J, Funk L, Botrous I, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J Med Chem. 2011;54:6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 46.Mahboobi S, Sellmer A, Winkler M, Eichhorn E, Pongratz H, Ciossek T, Baer T, Maier T, Beckers T. Novel chimeric histone deacetylase inhibitors: a series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and histone deacetylase activity. J Med Chem. 2010;53:8546–8555. doi: 10.1021/jm100665z. [DOI] [PubMed] [Google Scholar]
- 47.Finlay MRV, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, Bradbury RH, Brown SJ, Butterworth S, Campbell A. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations That Spares the Wild Type Form of the Receptor. J Med Chem. 2014;57:8249. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 48.Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:860–864. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 50.Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones P, Altamura S, De Francesco R, Gallinari P, Lahm A, Neddermann P, Rowley M, Serafini S, Steinkühler C. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg Med Chem Lett. 2008;18:1814–1819. doi: 10.1016/j.bmcl.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 52.Fredenhagen A, Kittelmann M, Oberer L, Kuhn A, Kühnöl J, Délémonté T, Aichholz R, Wang P, Atadja P, Shultz MD. Biocatalytic synthesis and structure elucidation of cyclized metabolites of the deacetylase inhibitor panobinostat (LBH589) Drug Metab Dispos. 2012;40:1041–1050. doi: 10.1124/dmd.111.043620. [DOI] [PubMed] [Google Scholar]
- 53.Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Med Chem. 2003;46:5097–5116. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 54.Evangelista W, Weir RL, Ellingson SR, Harris JB, Kapoor K, Smith JC, Baudry J. Ensemble-based docking: From hit discovery to metabolism and toxicity predictions. Bioorg Med Chem. 2016;24:4928–4935. doi: 10.1016/j.bmc.2016.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ellingson SR, Miao Y, Baudry J, Smith JC. Multiconformer ensemble docking to difficult protein targets. J Phys Chem B. 2015;119:1026–1034. doi: 10.1021/jp506511p. [DOI] [PubMed] [Google Scholar]
- 56.Shen M, Tian S, Pan P, Sun H, Li D, Li Y, Zhou H, Li C, Lee SM-Y, Hou T. Discovery of novel ROCK1 inhibitors via integrated virtual screening strategy and bioassays. Sci Rep. 2015 doi: 10.1038/srep16749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tian S, Sun H, Li Y, Pan P, Li D, Hou T. Development and evaluation of an integrated virtual screening strategy by combining molecular docking and pharmacophore searching based on multiple protein structures. J Chem Inf Model. 2013;53:2743–2756. doi: 10.1021/ci400382r. [DOI] [PubMed] [Google Scholar]
- 58.Tian S, Sun H, Pan P, Li D, Zhen X, Li Y, Hou T. Assessing an ensemble docking-based virtual screening strategy for kinase targets by considering protein flexibility. J Chem Inf Model. 2014;54:2664–2679. doi: 10.1021/ci500414b. [DOI] [PubMed] [Google Scholar]
- 59.Cleophas TJ, Droogendijk J, van Ouwerkerk BM. Validating diagnostic tests, correct and incorrect methods, new developments. Curr Clin Pharmacol. 2008;3:70–76. doi: 10.2174/157488408784293697. [DOI] [PubMed] [Google Scholar]
- 60.Van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.