

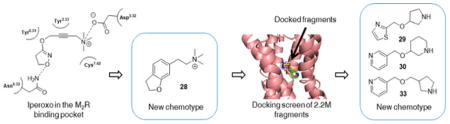
Abstract
Muscarinic receptor agonists are characterized by apparently strict restraints on their tertiary or quaternary amine and their distance to an ester or related center. On the basis of the active state crystal structure of the muscarinic M2 receptor in complex with iperoxo, we explored potential agonists that lacked the highly conserved functionalities of previously known ligands. Using structure-guided pharmacophore design followed by docking, we found two agonists (compounds 3 and 17), out of 19 docked and synthesized compounds, that fit the receptor well and were predicted to form a hydrogen-bond conserved among known agonists. Structural optimization led to compound 28, which was 4-fold more potent than its parent 3. Fortified by the discovery of this new scaffold, we sought a broader range of chemotypes by docking 2.2 million fragments, which revealed another three micromolar agonists unrelated either to 28 or known muscarinics. Even pockets as tightly defined and as deeply studied as that of the muscarinic reveal opportunities for the structure-based design and the discovery of new chemotypes.
Graphical Abstract
INTRODUCTION
With the determination of the atomic resolution structures of ever more G protein-coupled receptors (GPCRs), the question arises of how to exploit them for ligand discovery and design. Although over 30 years of work against soluble proteins have taught a close integration between medicinal chemistry, computation, and structure determination, GPCRs present special challenges. One often wants not only molecules that complement and inhibit a GPCR (inverse agonists), as with enzyme inhibitors, but also agonists that activate the receptors, and the determination of the structures of receptors in their activated states remains rare. Also, most GPCRs have subtypes that recognize identical endogenous agonists but that signal in different organs and that couple to different G proteins, making specificity particularly important and problematic. Finally, structure-based design against GPCRs struggles with the facile determination of cocomplex structures, especially for new ligand series for which affinity is initially weak.1,2
Agonist discovery for the M2 muscarinic receptor illustrates the opportunities and challenges facing GPCRs. On the one hand, there are compelling therapeutic and chemical-probe arguments for new muscarinic agonists, ideally with new scaffolds. The muscarinic (acetylcholine GPCR) receptors are ubiquitous in human organs, regulating functions ranging from heartbeat to smooth muscle contraction to glandular secretion, to cognition.3,4 The receptors are attractive targets for the treatment of conditions like chronic obstructive pulmonary disease, Alzheimer’s disease, and overactive bladder syndrome,4–7 and the use of selective muscarinic ligands has recently been discussed for diseases including cancer, diabetes, cardiovascular disease, pain, and inflammation.3,8–10 Selectivity is challenging, however, owing to the multiple subtypes with related orthosteric sites signaling in often opposed ways in different organs. Among the five major muscarinic receptor subtypes, the M1, M3, and M5 receptors couple to the G protein Gq, activating phospholipase C, while the M2 and M4 subtypes couple to Gi, mediating inhibition of adenylyl cyclase without stimulating PLC, and the differences among the orthosteric sites can be as little as a single amino acid (e.g., the orthosteric sites of the M2 and M3 receptors differ only by a Phe → Leu).11–14 This makes other muscarinic subtypes the major off-target for muscarinic drugs. Meanwhile, for agonists, which are wanted to treat diseases like glaucoma, Alzheimer’s disease, and Sjögren’s syndrome, the design criteria are very tight. Most muscarinic agonists derive from small natural products such as the eponymous muscarine, pilocarpine, and arecoline, and the activated state crystal structure of the M2 receptor15 confirms that the binding site for agonists is highly constrained (Figure 1A).
Figure 1.

(A) The crystal structure of M2 active state in a complex with iperoxo (PDB 4MQS). Residues Asn6.52 and Asp3.32 are represented as sticks and hydrogen bonds as red broken lines. Iperoxo fits tightly in the binding site. (B–D) Docking poses of pilocarpine (B), muscarine (C), and arecoline (D) in the M2 active state structure.
The restricted agonist site in the M2 receptor, and the tight chemotypes of even the natural product agonists (Figure 1B–D), suggested a focused search for new agonist scaffolds. Accordingly, we began with complexed conformation of iperoxo bound to the M2 receptor in its activated state. We initially sought new ligands with an aromatic moiety, substituted with a hydrogen bond acceptor for the interaction with Asn6.52 (Ballesteros–Weinstein numbering system16) and a quaternary amine to ion pair with Asp3.32. This simple strategy succeeded in finding a new scaffold, but to explain the functional effects of the resulting agonists and antagonists, we needed to dock them into the M2 receptor structures. We used the predicted docking poses to exclude compounds unable to interact with Asn6.52, which we expected would prioritize compounds that can activate the receptor; other contact-based filters, such as interactions with the tyrosines that are an important ligand recognition element in the site, did not add to selectivity among the docking hits. As ever, a primary prioritization criterion was docking scores. Almost all predicted agonists received scores in the range of −27 to −41 kcal/mol, with compounds 25 and 28 only slightly outside that range at −23 and −23.5 kcal/mol. Conversely, all compounds predicted not to be agonists received docking scores higher than (worse than) 0 kcal/mol, except for compound 18 that received a docking score of −15.5 kcal/mol. Whereas docking scores are notoriously inaccurate, this range represents a substantial separation. The resulting model allowed us to prioritize the design of still newer analogues, the most promising of which was a dihydrobenzofuran 28 (Figure 2), which only shares a ECFP4-based Tanimoto coefficients (Tc values) of 0.31 from previous M2 ligands and appears to be a new scaffold. With this new chemotype defined, we cast a final, broader net, screening a large library for molecules with similar physical properties but greater chemotype diversity. This led to three more agonists in two distinct scaffolds. The hierarchy of approaches used here, beginning with a pharmacophore from the crystallographic conformation of an agonist bound to the active state of the receptor, followed by detailed structural placement, and ending with a large library screen, though inverted from the more typical discovery-and-optimization flow, may be pragmatic for agonist design against other receptors.
Figure 2.

Structure-based discovery of new muscarinic agonists: project flow. Structure-guided design from the M2R/iperoxo complex (top left) led to 19 candidate ligands chemically distinct from previous agonists. Optimization led to the improved compound 28, a new agonist scaffold (bottom right). A docking screen of a large fragment library (bottom middle) led to three still newer agonists (bottom left).
RESULTS
Structure-Based Design of New Muscarinic Agonists
The manual design of initial set of 19 compounds depended on the use of a benzene unit as a central scaffold. The aromatic ring was functionalized by a hydrogen bond with Asn6.52-accepting group and a quaternary ammonium salt (to form an ion pair with Asp3.32) that was linked to the aromatic system by various spacers consisting of 1–4 carbons (Figure 2, Table 1). To probe distinct orientations of the functional groups to each other, we synthesized different regioisomers. For some compounds, conformationally restricting elements were installed by bridging either the ammonium salt or the hydrogen bond acceptor group with the benzene scaffold. The analogues included conformationally restricted tertiary and quaternary methoxy or hydroxy substituted aminotetralins. Analogous benzofuran derivatives were also prepared. To fine-tune the distance between the aromatic ring and the ammonium ion, we synthesized amino-methyl substituted analogues and tetrahydroisochinolines bearing an endocyclic nitrogen atom. Furthermore, we prepared a set of monocyclic derivatives, where the methoxyphenyl and the quaternary ammonium headgroup are linked by a methylene, ethylene, propylene, cyclopropylene, propenylene, or propynylene chain. The methoxy moiety was added either in the ortho- or meta-position of a benzene ring or incorporated into a fused furan. The compounds were synthesized using solution phase chemical reactions including amide coupling, reductive amination, Henry reaction, nucleophilic displacement, reduction of amides and nitroolefins, methylene transfer, or palladium-catalyzed coupling reactions (Supporting Information). Overall, the 19 molecules had Tc values to previously known muscarinic ligands, annotated in ChEMBL and DrugBank, ranging from 0.19 to 0.47.
Table 1.
Activities and Structural Complementarity of Compounds 1–19 to the M1R, M2R, and M3R Receptor Subtypes (Ki in μM)
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compound | Ki (μM)a | IP accumulation assayb | docking | |||||||
|
| ||||||||||
| X | n | M1R | M2R | M3R | EC50 [μM]c | Emax [%]d | docking score active state | H-bond to Asn6.52 | predicted agonist? | |
| 1 | 2-OMe | 1 | 26±15 | >50 | >50 | 18±3.3 | 51±15 | −8.13 | N | − |
| 2 | 2-OMe | 2 | 4.9±1.1 | 5.7±2.3 | 4.7±1.2 | 9.1±3.9 | 21±7.8 | −27.11 | N | − |
| 3 | 3-OMe | 2 | 33±6.6 | 14±6.1 | >50 | 12±2.8 | 75±7.5 | −32.89 | Y | + |
| 4 | 3-OMe | 3 | 0.33±0.073 | 0.63±0.17 | 0.62±0.20 | / | <10e | −15.56 | N | − |
| 5 |
|
- | 16±0.88 | 22±2.6 | 21±4.5 | / | / | 3.86 | N | − |
| 6 |
|
- | 20±4.9 | 50±25 | 20±5.9 | / | / | 24.43 | N | − |
| 7 |
|
- | 7.3±1.1 | 15±2.5 | 8.7±4.4 | / | / | 81.61/5.74/9.66/15.82h | N/N/Y/Y | − |
| 8 | 5-OMe | - | 14±3.6 | 26±14 | 24±9.4 | / | / | −18.61 | N | − |
| 9 | 8-OMe | - | 17±8.5 | 6.3±2.3 | 5.8±2.2 | / | / | −10.97 | N | − |
| 10 | 8-OMe | 0 | 1.0±0.19 | 0.93±0.15 | 2.0±0.53 | / | / | 79.83/33.04h | N | − |
| 11 | 8-OH | 0 | 2.9±1.1 | 4.2±2.5 | 16±3.5 | / | / | −4.32/7.83h | Y | − |
| 12 | 8-OMe | 1 | 1.8±0.35 | 1.3±0.18 | 3.9±0.26 | / | / | 28.12/−6.16h | Y | − |
| 13 | 5-OMe | 0 | 2.5±0.35 | 4.4±2.2 | 4.0±0.42 | / | / | 93.73/89.59h | Y | − |
| 14 | 5-OMe | 1 | 17±8.5f | 14±8.9f | 25±2.8f | / | / | 14.86/11.16h | N/Y | − |
| 15 | 8-OMe | 0 | 2.0±1.5f | 1.2±0.88f | 0.83±0.22f | / | / | 74.55/34.28h | Y/N | − |
| 16 | 8-OH | 0 | >50f | >50f | 28±13f | / | / | −6.56/6.86h | Y/Y | − |
| 17 | - | 1 | 0.023±0.0053 | 0.14±0.049 | 0.041±0.011 | 0.22±0.21 | −14±18 | −27.79 | Y | + |
| 18 | - | 2 | 0.063±0.012 | 0.23±0.028 | 0.14±0.053 | 0.027±0.022 | 12±13 | −15.49 | Y | − |
| 19 | - | - | 0.38±0.063 | 0.79±0.17 | 0.69±0.090 | 2.3±1.9 | −8.3±8.0 | 39.28/28.65h | N/N | − |
| iperoxo | 350±50g | 4.9±0.60g | 550±73g | 0.28±0.088g | 125±2.3 | NA | Y | NA | ||
| acetylcholine | 5.8±1.5 | 0.39±0.084 | 4.7±1.3 | 0.056±28 | 92±5.5 | −28.22 | Y | + | ||
| carbachol | 63±12 | 4.1±1.1 | 51±12 | 0.89±0.22 | 100 | −32.23 | Y | + | ||
Ki values ± SEM derived from 3–8 individual competition binding experiments using the radioligand [3H]N-methyl-scopolamine bromide and membranes from HEK cells transiently expressing the human M1R, M2R or M3R.
Second, less sensitive IP accumulation assay with COS cells coexpressing M2R and Gαqi5HA.
EC50 values ± SEM from 3–8 individual experiments each done in triplicate.
Emax values relative to the full effect of carbachol.
Emax at 10 μM (no complete dose–response curve was available).
Ki values ± SD derived from two independent competition binding experiments.
Values are displayed in nM ± SEM.
Racemic mixture. “/” = not determined
Structural Complementarity from Docking
All 19 compounds fit the loose pharmacophore described above, but to guide specific structural complementarity, we wanted a more quantitative metric. In parallel with the synthesis, and blind to biological testing, the 19 compounds were docked into the structures of the active and inactive states of the M2 receptor (PDBs 4MQS15 and 3UON,17 respectively). Docking complexes were scored for electrostatic18,19 and van der Waals complementarity and corrected for ligand desolvation,20,21 and the top scoring configuration of each molecule was retained.
Against the inactive conformation of M2R, all analogues docked favorably, with energy scores ranging from −38.05 to −45.13 kcal/mol, and all posed to interact with Asp3.32 (Supporting Information, Figure S1, Table S1). Conversely, against the active state, only analogues 3 and 17 complemented the more constrained agonist conformation of the orthosteric site, making favorable interactions with Asp3.32 and with Asn6.52 and scoring well, with scores of −32.89 and −27.79, respectively (Figure 3A and Supporting Information, Figure S1; Table 1). All the other compounds in the first set either scored poorly with unfavorable score, typically above 0 kcal/mol (see above), or did not hydrogen bond with Asn6.52. Superposition of the pose of iperoxo in the active state structure of M2R with the docked pose of compound 3 (Figure 3B) shows that the tertiary amine of compound 3, as well as the oxygen forming the hydrogen bond with the Asn6.52, are in the same spatial position as the corresponding moieties of iperoxo.
Figure 3.

(A) Superposition of compound 17 (green) and compound 3 (purple). Residues Asn6.52, Asp3.32, and Ser107 are represented as sticks. Both compounds hydrogen bond (black broken lines) with Asn6.52. (B) Superposition between the iperoxo (silver) pose in the M2R active state structure (PDB 4MQS15) and the docked pose of compound 3 (purple). Both compounds appear to hydrogen bond with Asn6.52, ion pair with Asp3.32, and are enclosed by an aromatic cage composed of Tyr104, Tyr403, and Tyr426.
Binding and Functional Studies at M1, M2, and M3 Muscarinic Receptors
Radioligand binding studies were conducted to evaluate the 19 compounds for their M1R, M2R, and M3R affinity, using [3H]N-methyl-scopolamine bromide and membrane preparations from transiently transfected human embryonic kidney cells (HEK).22,23 To detect agonists, the ability of the compounds to activate the M2 receptor was first investigated using a sensitive IP accumulation assay (HTRF detection, IP-One) in HEK cells transiently expressing the human M2R together with the hybrid G-protein Gαqi5HA.24 Promising compounds were tested in a second, less sensitive but more informative IP accumulation assay in kidney cells from African green monkey (COS) transiently expressing the human M2R and Gαqi5HA;25 it is the results of this second, higher fidelity assay on which we focus here. The affinity and efficacy profiles of the analogues were compared to those of the neurotransmitter acetylcholine, the approved drug carbachol, and the superagonist iperoxo (Table 1 and Supporting Information, Table S2).
Compounds 2, 3 and 5–15 had M1R, M2R, and M3R Ki values in the single- or double-digit micromolar range, and compound 3 had nascent specificity. The conformationally restrained ligands 17–19 had nanomolar Ki values (0.023–0.79 μM). The IP accumulation assay for M2R activation revealed inverse agonism to strong partial agonism, with Emax values ranging from −14% for compound 17 (i.e., inverse agonism) to 75% for compound 3. Antagonist or very weak agonist effects were observed for the bicyclic compounds 8–17. While the monocyclic derivatives 5–7, bearing conformationally restrained moieties, did not substantially stimulate M2R, the methoxyphenyl compounds 1–3 with the flexible alkylene unit had Emax values ranging from 21% to 75% and EC50 values in the 10 μM range. These observations agreed broadly with the docking predictions: compound 3 was the most active, compound 17 was among the tightest binding of the 19 ligands, and 16 compounds were correctly classified as antagonists or inverse agonists (Table 1). Admittedly, the docking prioritization was imperfect: compound 1, which turned out to be a decent agonist, was mispredicted as an antagonist, and compound 17, notwithstanding its affinity, turned out to be an antagonist.
Structure-Guided Optimization
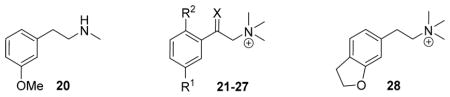
We sought to optimize for activity by designing a second set of ligands. Two approaches were used: (1) docking of a library of analogues and (2) structure-based design from the docking pose. All nine of the resulting analogues preserve the ethylene linker between the aromatic moiety and the ammonium headgroup of compound 3 that seem important to superpose with iperoxo and well-complement the activated conformation of the M2R.
In the first approach, a library of 54 analogues was generated and docked against the active state structure. Out of these, four compounds were predicted to be agonists from their ability to hydrogen bond with Asn6.52 and their favorable docking scores. These include the secondary amine 20, the hydroxy analogue 21, the secondary alcohol 23, and compound 22 (Table 2 and Supporting Information, Table S1 and Figure S1). On synthesis and testing, compound 22 displayed an improved Ki of 1 μM while retaining specificity over the M1 and M3 receptors and substantial agonist activity (Table 2 and Supporting Information, Table S2). Of the other three compounds, the Ki for 20 also improved to micromolar but it lost agonism and specificity, 21 retained activity but was less active than the lead agonist 3, and 23 lost both affinity and most of its activity. These results, which represent docking failures, are consistent with the idea that while the method can select for fit, both optimization and selection for activation remain challenging it.
Table 2.
Screening of the Second Set Compounds 20–28 (Receptor Binding Affinities to M1, M2, and M3 (Ki in μM), M2 Receptor Activation and Docking Data)
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | X | Ki [μM]a | IP accumulation assayb | docking | |||||
|
|
|
|
|||||||||
| M1R | M2R | M3R | EC50 [μM]c | Emax [%]d | docking score active state | H-bond to Asn6.52 | predicted agonist? | ||||
| 20 | 0.74 ± 0.13 | 1.0 ± 0.25 | 0.78 ± 0.10 | 8.2 ± 5.5 | −12 ± 1.5 | −40.99 | Y | + | |||
| 21 | OH | H | H,H | 20 ± 11 | 14 ± 4.7 | 13 ± 4.8 | 10 ± 4.1 | 44 ± 10 | −36.29 | Y | + |
| 22 | OMe | F | H,H | 7.1 ± 2.5 | 0.98 ± 0.61 | 14 ± 5.9 | 23 ± 10 | 44 ± 4.7 | −29.73 | Y | + |
| 23 | OMe | H | H,OH | 13 ± 5.5 | 29 ± 8.7 | 12 ± 4.2 | <10e | −32.23/−30.97g | Y/N | +/− | |
| 24 | OMe | H | O | 0.17 ± 0.014f | 2.4 ± 0.42f | 0.21 ± 0.028f | <10e | −34.56 | Y | + | |
| 25 | OEt | H | H,H | 42 ± 14 | 27 ± 9.9 | 43 ± 10 | 40e | −23.00 | Y | + | |
| 26 | Cl | H | H,H | 4.5 ± 1.1 | 4.8 ± 1.4 | 5.8 ± 1.8 | <10e | −39.07 | N | − | |
| 27 | OCF3 | H | H,H | 7.8 ± 1.6 | 9.2 ± 2.1 | 11 ± 6.6 | <10e | −28.6 | Y | + | |
| 28 | 6.4 ± 0.96 | 13 ± 3.3 | 6.3 ± 1.7 | 21 ± 7.6 | 62 ± 6.6 | −23.47 | Y | + | |||
Ki values ± SEM derived from three to eight individual competition binding experiments using the radioligand [3H]N-methyl-scopolamine bromide.
Second, less sensitive IP accumulation assay with COS cells coexpressing M2R and Gαqi5HA.
EC50 values ± SEM from three individual experiments each done in triplicate.
Emax values ± SEM relative to the full effect of carbachol.
Maximum effect at 100 μM; no complete dose–response curve could be determined.
Ki values ± SD derived from 2 individual competition binding experiments.
Racemic mixture.
In the second approach, which also began with the docking pose, we manually designed and then synthesized analogues of compound 3 by replacing the meta-methoxy substituent by an ethoxy, chloro, or trifluoromethoxy group (25–27). Additionally, the ketone 24 and the dihydrobenzofuran analogue of compound 17 (compound 28) were prepared. The conformationally restricted ligand 28 was expected to be a closer surrogate of the lead 3 than the previous unsaturated analogues 17–19 because the electronic properties of the sp3 oxygen with two lone pairs are more isosteric than the respective sp2 atom of the benzofuran system. On testing, compound 24 had improved affinity and 25 retained substantial efficacy, but only compound 28, the conformationally restricted analogue, retained both decent affinity and agonist efficacy for the M2 receptor in the higher fidelity IP accumulation assay (Table 2 and Supporting Information, Table S2). Overall, the docking prioritized compounds 3 and its phenyl-fluorinated analogue 22, along with the conformationally restricted 28, emerged as the most active of the new analogues.
With these results in hand, we decided to more thoroughly investigate 3 and 28 in whole cell assays using Chinese Hamster Ovary (CHO) cells stably expressing the muscarinic receptor subtypes M1, M2, or M3 (Table 3, Figure 4). Unlike the assays reported in Tables 1 and 2, these cells recapitulate the native Gi/0 coupling of the M2 receptor and the Gq/11 coupling of the M1 and M3 receptors. Whereas the binding affinity and selectivity for compound 3 remained little changed, the apparent affinity of compound 28 improved in the CHO cells to 2 μM, while its selectivity over M1 and M3 receptors improved to 4- and 10-fold, respectively (Table 3 and Figure 4A–C). Looking at [35S]GTPγS binding assay, a classic functional assay for Gi/o protein-coupled receptors, 28 was a full agonist with an EC50 of 3.3 μM and a 100% Emax compared to acetylcholine (Figure 4F); compound 3 was also a full agonist in this assay with an EC50 only slightly higher at 8 μM. These values were largely confirmed by a cAMP accumulation assay (Figure 4G). In IP accumulation assays (IP-One assay), which flows from Gq/11 stimulation through the M1 and M3 receptors, both 3 and 28 showed only weak agonist behavior, suggesting specificity for the M2R, with potencies consistent with their binding affinities (Figure 4D,E), while no IP accumulation was measured via M2, as expected given its native Gi/o coupling. In a more downstream functional assay, looking at the level of ligand-mediated ERK1/2 phosphorylation, both 3 and 28 behaved as full agonists (Emax = 100%) with potencies of 0.83 and 0.23 μM for M2R (Figure 4J). Against the M1 receptor, only 3 had reliable agonist activity, displaying partial agonist profile, while against M3 neither compound showed substantial activation, suggesting that amplification of signal corresponds with amplification of the new agonists’ subtype selectivity (Figure 4I–K). Finally, we determined the ability of 28 to stimulate β-arrestin recruitment via M2R activation. Compared to acetylcholine, 28 was a less potent but more efficacious agonist and showed 20-fold bias toward arrestin versus acetylcholine as a reference in the cAMP assay (Table 3, Figure 4H).
Table 3.
Signaling Selectivity of New Muscarinic Agonists 3 and 28 vs Acetylcholine
| test system | receptor subtype | test compounds | |||
|---|---|---|---|---|---|
|
| |||||
| acetylcholine | 3 | 28 | |||
| whole cell bindinga | pKi [M]/Ki [μM] | M1R | 4.64 ± 0.04/23 | 4.28 ± 0.09/52 | 5.17 ± 0.03/6.8 |
| pKi [M]/Ki [μM] | M2R | 6.42 ± 0.06/0.38 | 5.04 ± 0.08/9.1 | 5.69 ± 0.07/2.0 | |
| pKi [M]/Ki [μM] | M3R | 5.17 ± 0.07/6.8 | 4.09 ± 0.15/81 | 5.11 ± 0.10/7.8 | |
| IP accumulationb | pEC50 [M]/EC50 [μM] | M1R | 6.21 ± 0.11/0.62 | 5.04 ± 0.17/9.1 | 5.32 ± 0.33/4.8 |
| Emax [% ± SEM]c | 100 | 48 ± 5 | 8 ± 2 | ||
| pEC50 [M]/EC50 [μM] | M2R | ||||
| pEC50 [M]/EC50 [μM] | M3R | 6.62 ± 0.16/0.24 | 4.98 ± 0.07/10 | 5.43 ± 0.23/3.7 | |
| Emax [% ± SEM]c | 100 | 43 ± 2 | 17 ± 2 | ||
| GTPγS bindingd | pEC50 [M]/EC50 [μM] | M1R | |||
| pEC50 [M]/EC50 [μM] | M2R | 6.29 ± 0.07/0.51 | 5.11 ± 0.06/7.8 | 5.48 ± 0.07/3.3 | |
| Emax [% ± SEM]c | 100 | 100 | 100 | ||
| pEC50 [M]/EC50 [μM] | M3R | ||||
| inhibition of cAMP accumulatione | pEC50 [M]/EC50 [μM] | M1R | |||
| pEC50 [M]/EC50 [μM] | M2R | 7.31 ± 0.19/0.049 | 5.20 ± 0.18/6.3 | 4.96 ± 0.13/11 | |
| Emax [%]c | 100 | 108 ± 9 | 102 ± 16 | ||
| pEC50 [M]/EC50 [μM] | M3R | ||||
| β-arrestin recruitmentf | pEC50 [M]/EC50 [μM] | M1R | |||
| pEC50 [M]/EC50 [μM] | M2R | 5.72 ± 0.09/1.9 | 4.85 ± 0.05/14 | 4.76 ± 0.11/17 | |
| Emax [%]c | 100 | 133 ± 7 | 118 ± 9 | ||
| pEC50 [M]/EC50 [μM] | M3R | ||||
| ERK1/2 phosphorylationg | pEC50 [M]/EC50 [μM] | M1R | 7.33 ± 0.15/0.047 | 6.27 ± 0.23/0.54 | nah |
| Emax [%]c | 100 | 53 ± 6 | |||
| pEC50 [M]/EC50 [μM] | M2R | 7.90 ± 0.08/0.013 | 6.08 ± 0.14/0.83 | 6.62 ± 0.11/0.24 | |
| Emax [%]c | 100 | 92 ± 8 | ~100 | ||
| pEC50 [M]/EC50 [μM] | M3R | 7.64 ± 0.08/0.023 | nah | nah | |
| Emax [%]c | 100 | ||||
Affinity to M1R, M2R, or M3R from whole cell receptor binding performed with CHO cells stably expressing a given receptor subtype and with the antagonist radioligand [3H]N-methyl scopolamine; pKi ± SEM values are the means of 4–6 individual experiments each in duplicate.
Gq-protein mediated functional activity was measured with the same cells using the IP-One assay (Cisbio); pEC50 ± SEM values are the means of 3–4 individual experiments each in duplicate.
Maximum efficacy vs acetylcholine.
Gi/o-Protein activation was measured using [35S]GTPγS binding with membranes from the same cells stably expressing M2R; pEC50 ± SEM values are mean values from three individual experiments each in duplicate.
Inhibition of cAMP accumulation was done with HEK cells stably expressing the Epac cAMP sensor and M2R; pEC50 ± SEM were means of 4–5 individual experiments each in duplicate.
β-Arrestin recruitment assay was performed with HEK cells stably expressing the β-arrestin-TEVprotease and transiently transfected with M2-TEV-tTA; pEC50 ± SEM values are the means of 4–5 individual experiments, each in triplicate.
ERK1/2 phosphorylation was measured by AlphScreen with CHO cells stably expressing M1R, M2R, or M3R; pEC50 ± SEM values are the means of 3–4 individual experiments, each in duplicate.
na: could not be estimated.
Figure 4.

Detailed investigation of the new muscarinic agonists 3 and 28. (A–C) Binding behavior at whole cells expressing the muscarinic receptor subtypes M1 (A), M2 (B), and M3 (C) in comparison to the natural ligand acetylcholine. (D–G) M2 selective signaling of 28 indicated by a weak activation of M1 and M3 stimulated IP accumulation (D and E, respectively) and full agonist effect in M2 mediated GTPγS binding (F) and inhibition of cAMP accumulation (G). (H) M2 mediated β-arrestin recruitment displays full agonist effect for 28. (I–K) Downstream signaling shows M2 selective agonist properties for 28 as determined in a ERK1/2 phosphorylation assay for M1 (I), M2 (J), and M3 (K).
These results suggest that compounds 3 and 28 are micromolar to submicromolar full agonists of the M2 receptor in native signaling, with 28 having moderate arrestin bias versus acetylcholine. The aryl methoxy groups on the “left-hand side” of each molecule represent a new chemotype for the muscarinic receptors, while the distal quaternary nitrogen is well-established. To investigate the replacement of this cationic group and find still newer scaffolds, we turned to large library docking.
Prospective Fragments Library Docking Screen Selection of 10 Compounds
Heartened by the discovery of the new agonists, we sought still more novel agonists from a structure-based screen of a larger chemical library. We screened the “clean fragments” subset of the open access ZINC26,27 (http://zinc15.docking.org), then just over 2.2 million commercially available compounds, with xlogP ≤ 3.5, molecular weight ≤250 Da, and rotatable bonds ≤5, with DOCK3.6.28 Each library molecule was screened in an average of 337.5 orientations in the orthosteric site, and in each orientation an average of 32.6 conformations was sampled. Overall, over 24 billion molecular complexes were evaluated (in a lead-like screen, by comparison, we might evaluate 50-fold more complexes, as the increased molecular size demands more sampling). Configurations were ranked by their electrostatic (using a point charge model of the Poisson–Boltzmann equation, as implemented in QNIFFT)29,30 and van der Waals complementarity (using the AMBER potential31) to the M2 active state, corrected for context-dependent ligand desolvation28 (using GB/SA electrostatics implemented in AMSOL20,21), and the top scoring configuration of each molecule was retained. The screen took 37.6 total core hours or less than an hour of elapsed time on our lab cluster.
The result of the calculation was a ranked list of fragments, from most to least complementarity to the M2 orthosteric active state pocket. The top ranked 1000 (best 0.05%) fragments were inspected for those that interacted with both Asp3.32 and Asn6.52. Ten were selected for testing by radioligand displacement and IP accumulation, again using the sensitive (IP-One) and the more informative IP accumulation assay ([3H]inositol based) (Table 4, Supporting Information, Table S3). Three of the 10 fragments had micromolar EC50 values in the more stringent IP accumulation assay of between 9.9 and 29 μM, and M2 receptor Emax values ranging from 60% to 74% (Table 4, Figure 5). Most of the other fragments had midmicromolar affinities for the M2R, and several even had substantial agonism in the IP screening assay, but this activity was not retained in the more stringent functional assay (Supporting Information, Table S3). By design, the three new agonists have little similarity to known muscarinic ligands, with ECFP4-based Tc values ranging from 0.20 to 0.34 to annotated ligands in ChEMBL and DrugBank (Table 4). While all three retain the ubiquitous cation of aminergic agonists, the conserved ester/amide of muscarinic agonists has been replaced with either a thiazole (29) or a pyridine (30 and 31), which has little precedence; in the docked configurations, these heterocycles interact with the same Asn6.52 with which the carbonyl system of classic agonists interact. Consistent with the degree of these changes, further alkylation of the aminergic group, which ordinarily would increase activity, for the new agonists diminished it substantially (Table 4 and Supporting Information, Table S3). Structurally, these new agonists represent an even greater departure from known agonist scaffolds than even compounds 3, 22, and 28.
Table 4.
Experimentally Active Molecules from Docking and Their Synthesized Analogues
| compound | Ki [μM]a | IP accumulation assayb | docking | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| • | Rank | Structure | M1 | M2 | M3 | EC50 [μM]c | Emax [%]d | Tc to closest muscarinic ligand | ZINC IDs of closest muscarinic ligande |
| 29 | 324 |
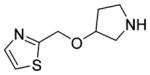
|
4.2±0.65 | 6.0±2.5 | 4.1±1.6 | 9.9±1.7 | 74±4.6 | 0.20 | C13739835 |
| 30 | 383 |
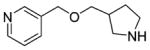
|
18±9.7 | 11±2.0 | 36±6.7 | 29±13 | 74±14 | 0.34 | C34802190 |
| 33 | 449 |
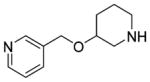
|
10±2.9 | 17±4.9 | 10±2.8 | 13±4.8 | 60±6.5 | 0.33 | C27984351 |
| 29a | NA |
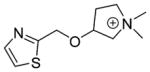
|
6.1±1.8 | 10±1.1 | 12±1.4 | 10±4.3 | 28±1.2 | 0.28 | C00000346 |
| 30a | NA |
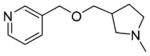
|
38±16 | 39±4.4 | 75±6.4 | >100 | 38±4.8f | 0.33 | C34802190 |
| 33a | NA |
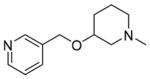
|
20±3.2 | 46±2.0 | 42±2.2 | 50±7.4 | 33±10 | 0.31 | C27984351 |
Ki values ± SEM derived from 3–8 individual competition binding experiments using the radioligand [3H]N-methyl-scopolamine bromide.
Second, less sensitive IP accumulation assay with COS cells coexpressing M2R and Gαqi5HA.
EC50 values ± SEM from 4–6 individual experiments each done in triplicate.
Emax values ± SEM relative to the full effect of carbachol.
2D structures are presented in Supporting Information, Table S4.
Maximum effect at 100 μM; no complete dose–response curve could be determined.
Figure 5.

Docking poses of compounds 29 (A), 30 (B), and 33 (C) in the M2R active state structure (PDB 4MQS). Residues Asn6.52, Asp3.32, Ser107, and Tyr403 are represented as sticks. Hydrogen bonds are represented in black.
Like many primary neurotransmitters, acetylcholine activates receptors from more than one protein family; such cross-family polypharmacology provides an uniquely chemical organization for signaling.32,33 Besides the five muscarinic GPCR subtypes, acetylcholine also activates ligand-gated ion channels as primary targets. These nicotinic acetylcholine receptors (nAChRs) are widely expressed throughout the central and peripheral nervous system and at the neuromuscular junction. Functional nAChRs are a heterogenic group of pentameric ion channels composed of various subunits. Whereas the new docking compounds were chosen for their novelty against known muscarinic ligands, we initially did not consider their similarity to nicotinic ion channel ligands. Unexpectedly, certainly not by design but during the review of this article, we discovered that compounds 3, 28, 29, 30, 33, 29a, 30a, and 33a had meaningful similarities to known nicotinic ligands, with Tc values ranging from 0.33 to 0.62 (Supporting Information, Table S5). Whereas none were identical to known nicotinic ligands, these similarities are high enough to suggest the new M2 muscarinic agonists might also activate the nicotinic receptor, a cross pharmacology that remains relatively rare, though not completely unknown, outside of acetylcholine itself and its close cogeners.
Accordingly, the affinity for the (−)-nicotine binding site at α 4β2 nAChR was determined for representative compounds 28, 30, and 33, against the potent radioligand [3H]cytisine. Although all three compounds display nearly identical Ki values for M2 mAChR (11–17 μM), their affinity for the α4β2 nAChR differs in more than 3 orders of magnitude, resulting in distinct selectivity profiles. While 28 shows equipotent affinities to the muscarinic and nicotinic acetylcholine receptor (Ki = 9.7 μM for α4β2 nAChR), the alkyloxymethylpyridines 30 and 33 have pronounced selectivity toward nAChR (Ki values 200 nM for 30 and an extraordinary 1.6 nM for 33, respectively). Indeed, the affinity of 33 for nAChR resembles the 1.5 nM affinity of nicotine itself. Notably, compounds 30 and 33 share structural similarity with selective α4β2 nAChR ligands of the 3-pyridylether family such as pozanicline (ABT-089, 2-methyl-3-(2-(S)-pyrrolidinylmethoxy)pyridine). Pozanicline, epibatidine, and other high affinity α4β2 nAChR (partial-) agonists have been under clinical investigation as cognitive enhancers, including for attention deficit/hyperactivity disorder and Alzheimer’s disease, and as analgesics and anxiolytics. Moreover, the α4β2 nAChR partial agonist varenicline is used to effectively enhance smoking cessation. Whereas the functional activity of compounds 30 and 33 is out of scope for this study, such properties may merit future study, as to their role as joint activators of the acetyl cholinergic circuit.
DISCUSSION AND CONCLUSION
Two key observations emerge from this study. First, whereas classical muscarinic agonists reflect a highly constrained pharmacophore, structural complementarity to an activated receptor reveals novel agonists topologically unrelated to those previously known. The modeling that discovered the new agonists suggests that they are recognized via the same interactions made by the classic agonists but using different agonist functional groups. This suggests that there might be many more agonist recognition motifs readily accessible, as neither our pharmacophore-like design nor our library screen pretend to comprehensiveness. Several of these new molecules, like the more optimized 28, have intriguing signaling properties, including a 20-fold bias toward arrestin signaling vs the endogenous acetylcholine and a signaling specificity for the M2 vs the M1 or M3 receptor subtypes. Second, the new agonists flowed both from a large library docking screen and also from by-hand modeling, the traditional domain of the medicinal chemist. This study supports an alloy between the designing chemist and facile quantitative techniques by which their inspiration can be rapidly checked.
Certain caveats bear mentioning. None of the new agonists have strong activities against muscarinic receptors: none are even at probe levels of activity or specificity, far less than what one would expect from a therapeutic lead, and while novel chemotypes can lead to new biological effects,34–36 the evidence for such here, even with the nascent selectivity and signaling bias of 28, remains at an early stage. While both the by-hand design and the unbiased molecular docking screen both support the possibility of discovering new agonists for the muscarinic receptors, the docking hits stumbled into chemotypes with high activities against the nicotinic ion channel, against which muscarinic GPCR activity would ordinarily be optimized against (the coactivity against both the ionotropic and metabotropic acetyl choline receptors may itself merit further study).
Still, this study supports a structure-based effort to discover new chemotypes, even in a field as well-ploughed as the muscarinic. We have only undertaken an early reconnaissance into the design of or screens for such agonists; we suspect many more are readily accessible, and a screen of a larger, more elaborated lead-like or drug-like molecules might reveal a broader array of more potent and more selective molecules, as would optimization of the early agonists discovered here. An advantage of these new chemotypes is that by engaging the receptor with new functionalities they can stabilize activated ensembles in manners unexplored by the precedented agonists, engaging effectors in new ways; biased signaling is one example of that. More concretely, they provide templates for the optimization of pharmacokinetic properties that have long been exploited among muscarinic ligands, such as blood–brain penetration, typically defined by tertiary vs quaternary ammoniums. The new agonists, with their new responses to well-established optimization moves, provide new points of departure for medicinal chemistry and probe development programs.
EXPERIMENTAL METHODS
Docking against Active and Inactive State of M2R
We used DOCK3.628 to dock molecules against the M2R active state crystal structure bound to the agonist iperoxo (PDB 4MQS15) and to the M2R inactive state bound to QNB (PDB 3UON17). The same program was used in a docking screen of the “fragments-now” subset of the ZINC database (http://zinc15.docking.org). Partial charges from the united-atom AMBER force field were used for all receptor atoms except for Asn6.52, for which the magnitude of the local partial atomic charges were increased to accentuate electrostatic interactions with this particular residue (the net charge of the residue remained neutral), a technique we have widely used previously.36,37 Forty-five matching spheres were used. The number of ligand orientations sampled is determined by the values of the bin size, bin size overlap, and distance tolerance, set at 0.3, 0.1, and 1.2 Å, respectively, for both the matching spheres and the docked molecules. The ligand conformations sampled were precalculated using Openeye’s Omega program38 (Openeye Software, Santa Fe NM). Ligand charges and initial solvation energies were calculated using AMSOL (http://comp.chem.umn.edu/amsol/).20,21
Ballesteros–Weinstein (BW) Numbering
Receptor residues are referred to by their three-letter code, followed by their Ballesteros–Weinstein (BW) number. In this method, TM residues are identified by a superscript numbering system, in which the residue corresponding to the family A GPCRs most conserved residue in a given TM is assigned the index X.50, where X is the TM number and the remaining residues are numbered relative to this position.16
Tanimoto Coefficient (Tc) Calculation
We extracted a data set of 2422 ligands from CHEMBL2039–41 and DrugBank.42 Using the GenerateMD program (version 5.10.3) in the Chemaxon package, we calculated the EFCP4 fingerprints which were used to calculate the Tc43 between our hits and all of the 2422 ligands in Table 4.
Membrane-Based Radioligand Binding Experiments
Affinities of the test compounds toward the human M1, M2, and M3 receptors were determined using homogenates of membranes as described previously.22,23,44 In brief, HEK293T cells were transiently transfected with the cDNA of the appropriate receptor (purchased from cDNA Resource Center, Bloomsberg, PA) using a solution of linear polyethylenimine in PBS.45 Receptor binding experiments were performed in 96-well plates using homogenates of the corresponding receptor together with the radioligand [3H]N-methyl-scopolamine bromide (specific activity of 70 Ci/mmol, PerkinElmer, Rodgau, Germany) at a final concentration of 0.20–0.30 nM for M1R and M2R and 0.10–0.20 nM for M3R at a receptor density (Bmax) of 1500 ± 260, 1400 ± 140, and 2200 ± 530 fmol/mg, a protein concentration of 3–6, 5–10, and 2–10 μg/test tube, and a Kd value of 0.18 ± 0.052, 0.20 ± 0.018, and 0.086 ± 0.005 nM, for M1R, M2R, and M3R, respectively. Unspecific binding was determined in the presence of 10 μM atropine. Protein concentration was established by the method of Lowry using bovine serum albumin as standard.46 Resulting competition curves were analyzed by nonlinear regression using the algorithms for one-site competition of PRISM 6.0 (GraphPad, San Diego, CA).
Whole Cell Radioligand Binding Assays
Radioligand binding experiments were performed on CHO-FlpIn whole cells stably expressing the human M1, M2, and M3. receptor constructs of choice. After plating 20000 cells in complete DMEM into 96-well ISOPLATE TC plates (all amounts are per well), cells were allowed to grow overnight at 37 °C. The next day, cells were washed with phosphate-buffered saline (100 mL) and resuspended in binding buffer (10 mM HEPES, 100 mM NaCl, 10 mM MgCl2, pH 7.4). Assay mixtures, in a total volume of 100 μL with a 1/10 dilution of drug, were incubated at room temperature (22 °C) for 6 h. Assays were terminated by buffer removal followed by rapid washing, twice, with ice-cold 0.9% NaCl (100 μL). Plates were allowed to dry inverted for 30 min; OptiPhase Supermix scintillation cocktail (100 μL) was added, plates were sealed (TopSeal), and radioactivity was measured in a MicroBeta2 LumiJET microplate counter. Saturation binding experiments were performed in the absence or presence of atropine (10 μM) with 0.003–3 nM [3H]NMS. Inhibition binding experiments were performed with ~0.2 nM [3H]NMS (the approximate K) in the presence of various concentrations of analogues.
IP Accumulation Assay
For validation of the screening data derived with the IP-One assay (see Supporting Information), the most promising compounds were tested on M2R activation with an IP accumulation assay as described previously.15 In brief, COS-7 cells were transiently cotransfected with M2R and Gαqi53HA (Gαq protein with the last five amino acids at the C-terminus replaced by the corresponding sequence of Gαi, a gift from The J. David Gladstone Institutes, San Francisco, CA, for which we are grateful) by applying the TransIT-2020 Mirus transfection reagent (MoBiTec, Goettingen, Germany). Eighteen h before, the test cells were incubated with myo-[3H]inositol (specific activity = 20.1 Ci/mmol, PerkinElmer, Rodgau, Germany). Test compounds (six different concentrations for each compound, total range from 0.1 pM up to 300 μM) were incubated for 2 h at 37 °C in triplicate, and resulting radioactivity was measured by scintillation counting. Activation curves were normalized to the maximum effect of carbachol (100%) and buffer (0%) and analyzed using the algorithms for nonlinear regression in PRISM 6.0. For all compounds, 3–8 individual dose–response curves were measured, and the corresponding EC50 and Emax values of each mean curve were calculated and summarized to get the average EC50 and Emax values ± SEM.
IP-One Accumulation Assays
The IP-One assay kit (Cisbio, France) was used for the direct quantitative measurement of myoinositol 1-phosphate (IP1) in FlpIn CHO cells stably expressing hM1 and hM3 mAChRs. This is a competitive immunoassay that measures the homogeneous time-resolved fluorescence signal transferred between a cryptate-labeled IP1-specific monoclonal antibody and d2-labeled IP1. The fluorescence signal measured is inversely proportional to the concentration of native IP1. Briefly, cells were seeded into 96-well plates at 20000 cells per well and allowed to grow overnight at 37 °C, 5% CO2. The following day, cells were washed once with PBS then incubated with stimulation buffer (HEPES 10 mM, CaCl2 1 mM, MgCl2 0.5 mM, KCl 4.2 mM, NaCl 146 mM, glucose 5.5 mM, LiCl 50 mM, pH 7.4) for 60 min at 37 °C, 5% CO2. Following this incubation, ligands were added at 10× their final concentrations (ACh or test compounds) and incubated for a further 40 min prior to terminating the ligand-mediated stimulation by removing the buffer and adding 25 μL of lysis buffer. Finally, 14 μL of lysate was transferred into 384-well Optiplate, followed by the addition of 3 μL of IP1-d2, then 3 μL of Ab-Cryp, and incubated for 60 min at room temperature. Time resolved fluorescence resonance energy transfer (HTRF) was determined using the Envision plate reader (PerkinElmer).
[35S]GTPγS Binding Assay
Membrane homogenates (15 μg) were equilibrated in a 500 μL total volume of assay buffer containing 10 mM guanosine 5′-diphosphate and a range of concentrations of ACh or test compounds (1 nM tp 100 mM) at 30 °C for 60 min. After this time, 50 μL of [35S]GTPγS (1 nM) was added and incubation continued for 30 min at 30 °C. Termination of the reaction and determination of radioactivity were performed by rapid filtration through Whatman GF/B filters using a Brandell cell harvester (Gaithersburg, MD). Filters were washed three times with 3 mL aliquots of ice-cold 0.9% NaCl buffer and dried before the addition of 4 mL of scintillation mixture (Ultima Gold, PerkinElmer Life Sciences). Vials were then left to stand until the filters became uniformly translucent before radioactivity was determined in dpm using scintillation counting.
cAMP Accumulation Assay
HEK293 cells stably expressing the Epac cAMP sensor,47 obtained as a gift from Jesper Mathiesen, were stably transfected with M2AChR-tetO (a gift from Brian Kobilka). HEK-Epac-M2tetO cells were grown to confluency and then treated with 2 μg/mL doxycycline and 1 mM sodium butyrate for 40 h to induce expression of the M2AChR-tetO. Cells were harvested with lifting buffer (0.68 mM EDTA, 150 mM NaCl, 20 mM HEPES, pH 7.4), centrifuged, resuspended in HBSS-HEPES (Hank’s Balanced Salt Solution plus 20 mM HEPES, pH 7.4), and pipetted into a 96-well plate (black with clear bottom). After 20 min in the dark at 37 °C, the basal CFP/YFP ratio of the Epac-cAMP FRET sensor was measured at 436 exc and 480/535 ems for 2 min using a SpectraMax M5. Then forskolin (2 μM final), IBMX (1 mM final), and test compound (0–100 μM final) in HBSS-HEPES, pH 7.4, were added and the CFP/YFP ratio area under the curve was measured for 10 min at 37 °C. Basal values were subtracted, and data was analyzed using GraphPad Prism 6.
β-Arrestin Recruitment Assay
HEK293 cells stably expressing tTA dependent luciferase and β-arrestin-TEVprotease were transiently transfected with M2-TEV-tTA (cells and DNA construct were a gift from Bryan Roth) for measurement of M2AChR stimulated of β-arrestin recruitment, basically as described at https://pdspdb.unc.edu/pdspWeb/content/PDSP%20Protocols%20II%202013-03-28.pdf. The day after transfection, cells were lifted, resuspended in DMEM with 1% FBS, and plated into a poly-D-lysine coated 384-well clear-bottom plate at 15000 cells/well. After at least 6 h, 0–100 μM test compounds in HBSS-HEPES (Hank’s Balanced Salt Solution plus 20 mM HEPES, pH 7.4) were added to the cells. The following day, media was replaced with diluted Bright-Glo reagent (Promega, Madison, WI), and after 20 min in the dark, luminescence was measured using a SpectraMax M5. Data was analyzed using GraphPad Prism 6.
ERK1/2 Phosphorylation Assays
These assays were performed using the AlphaScreen-based SureFire kit as described in detail previously.48 All data were expressed as a percentage of ERK1/2 phosphorylation mediated by 100 μM of ACh.
Nicotinic Acetylcholine Receptor (nAChR, α4β2-type) Binding Assays
Binding affinities for the three new muscarinic agonists were determined by Eurofins Panlabs, Inc.49 Briefly, membranes from human recombinant SH-SY5Y cells with a nAChR expression level of 2000 fmol/mg protein were incubated with the radioligand [3H]cytisine (KD 0.30 nM) at a concentration of 0.60 nM together with the test compounds (0.1 nM to 300 μM) in binding buffer (50 mM Tris-HCl, pH 7.4) for 120 min at 4 °C. Nonspecific binding was determined in the presence of 10 μM (−)-nicotine. Concentration–response curves were analyzed using MathIQTM (ID Business Solutions Ltd., UK) to obtain IC50 values by nonlinear, least-squares regression analysis. IC50 values were subsequently converted to Ki values using the Cheng and Prusoff equation.50
Compound Synthesis and Purity
Compound synthesis and purity is described in the Supporting Information. The purity of all compounds tested was ≥95% and was confirmed by reverse phase HPLC by applying different elution systems and detecting the UV absorption at two different wavelengths (220 and 254 nm).
Supplementary Material
Acknowledgments
This work was supported by the U.S. National of Health grants GM106990 and R35GM122481 and by the NHMRC of Australia program grant APP1055134, project grant APP1082318 and by BaCaTeC. C.V. is an Australian Research Council Future Fellow, and A.C. is Senior Principal Research Fellow of the NHMRC.
ABBREVIATIONS USED
- Asn
asparagine
- Asp
aspartatic acid
- BW
Ballesteros–Weinstein
- CFP
cyan fluorescent protein
- CHO
Chinese hamster ovary
- COS
kidney cells from african green monkey
- DMEM
Dulbecco’s Modified Eagle’s Medium
- ERK
extracellular-signal regulated kinases
- FBS
fetal bovine serum
- FRET
fluorescence resonance energy transfer
- Gαx
G protein α subunit subtype x
- GPCR
G protein-coupled receptor
- GTPγS
guanosine 5′-O-(thiotriphosphate)
- HBSS
Hank’s Balanced Salt Solution
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HTRF
homogeneous time-resolved fluorescence
- IBMX
3-isobutyl-1-methylxanthine
- IP
inositol phosphate
- Leu
leucine
- Mx
muscarinic Mx receptor
- NMS
N-methylscopolamine
- nAChRs
nicotinic acetylcholine receptors
- Phe
phenylalanine
- PLC
phospholipase C
- SD
standard deviation
- SEM
standard error of mean
- Ser
serine
- Tc
Tanimoto coefficient
- TM
transmembrane
- Tyr
tyrosine
- YFP
yellow fluorescent protein
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.7b01113.
Docking data, figures of docking poses, functional assay data and experimental procedures, and analytical data of synthesized compounds (PDF)
Molecular formula strings of the target compounds (CSV)
References
- 1.Shoichet BK, Kobilka BK. Structure-Based Drug Screening for G-Protein-Coupled Receptors. Trends Pharmacol Sci. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jazayeri A, Dias JM, Marshall FH. From G Protein-Coupled Receptor Structure Resolution to Rational Drug Design. J Biol Chem. 2015;290:19489–19495. doi: 10.1074/jbc.R115.668251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matera C, Tata AM. Pharmacological Approaches to Targeting Muscarinic Acetylcholine Receptors. Recent Pat CNS Drug Discovery. 2014;9:85–100. doi: 10.2174/1574889809666141120131238. [DOI] [PubMed] [Google Scholar]
- 4.Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of Muscarinic Acetylcholine Receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- 5.Karakiulakis G, Roth M. Muscarinic Receptors and Their Antagonists in Copd: Anti-Inflammatory and Antiremodeling Effects. Mediators Inflammation. 2012;2012:409580. doi: 10.1155/2012/409580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dale PR, Cernecka H, Schmidt M, Dowling MR, Charlton SJ, Pieper MP, Michel MC. The Pharmacological Rationale for Combining Muscarinic Receptor Antagonists and Beta-Adrenoceptor Agonists in the Treatment of Airway and Bladder Disease. Curr Opin Pharmacol. 2014;16:31–42. doi: 10.1016/j.coph.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levey AI. Muscarinic Acetylcholine Receptor Expression in Memory Circuits: Implications for Treatment of Alzheimer Disease. Proc Natl Acad Sci U S A. 1996;93:13541–13546. doi: 10.1073/pnas.93.24.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broadley KJ, Kelly DR. Muscarinic Receptor Agonists and Antagonists. Recent Res Dev Org Chem. 2002;6:747–792. [Google Scholar]
- 9.Ruiz de Azua I, Gautam D, Guettier JM, Wess J. Novel Insights into the Function of Beta-Cell M3Muscarinic Acetylcholine Receptors: Therapeutic Implications. Trends Endocrinol Metab. 2011;22:74–80. doi: 10.1016/j.tem.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pieper MP. The Non-Neuronal Cholinergic System as Novel Drug Target in the Airways. Life Sci. 2012;91:1113–1118. doi: 10.1016/j.lfs.2012.08.030. [DOI] [PubMed] [Google Scholar]
- 11.Nathanson NM. Molecular Properties of the Muscarinic Acetylcholine Receptor. Annu Rev Neurosci. 1987;10:195–236. doi: 10.1146/annurev.ne.10.030187.001211. [DOI] [PubMed] [Google Scholar]
- 12.Caulfield MP. Muscarinic Receptors–Characterization, Coupling and Function. Pharmacol Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- 13.Peralta EG, Ashkenazi A, Winslow JW, Ramachandran J, Capon DJ. Differential Regulation of Pi Hydrolysis and Adenylyl Cyclase by Muscarinic Receptor Subtypes. Nature. 1988;334:434–437. doi: 10.1038/334434a0. [DOI] [PubMed] [Google Scholar]
- 14.Richards MH. Pharmacology and Second Messenger Interactions of Cloned Muscarinic Receptors. Biochem Pharmacol. 1991;42:1645–1653. doi: 10.1016/0006-2952(91)90498-t. [DOI] [PubMed] [Google Scholar]
- 15.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Huebner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Activation and Allosteric Modulation of a Muscarinic Acetylcholine Receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballesteros JA, Weinstein H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure–Function Relations in G Protein-Coupled Receptors. In: Stuart CS, editor. Methods in Neurosciences. Vol. 25. Academic Press; New York: 1995. pp. 366–428. [Google Scholar]
- 17.Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T. Structure of the Human M2Muscarinic Acetylcholine Receptor Bound to an Antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallagher K, Sharp K. Electrostatic Contributions to Heat Capacity Changes of DNA-Ligand Binding. Biophys J. 1998;75:769–776. doi: 10.1016/S0006-3495(98)77566-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharp KA. Polyelectrolyte Electrostatics: Salt Dependence, Entropic, and Enthalpic Contributions to Free Energy in the Nonlinear Poisson–Boltzmann Model. Biopolymers. 1995;36:227–243. [Google Scholar]
- 20.Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG. Model for Aqueous Solvation Based on Class Iv Atomic Charges and First Solvation Shell Effects. J Phys Chem. 1996;100:16385–16398. [Google Scholar]
- 21.Li J, Zhu T, Cramer CJ, Truhlar DG. New Class Iv Charge Model for Extracting Accurate Partial Charges from Wave Functions. J Phys Chem A. 1998;102(10):1820–1831. [Google Scholar]
- 22.Huebner H, Schellhorn T, Gienger M, Schaab C, Kaindl J, Leeb L, Clark T, Moeller D, Gmeiner P. Structure-Guided Development of Heterodimer-Selective Gpcr Ligands. Nat Commun. 2016;7:12298. doi: 10.1038/ncomms12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huebner H, Haubmann C, Utz W, Gmeiner P. Conjugated Enynes as Nonaromatic Catechol Bioisosteres: Synthesis, Binding Experiments, and Computational Studies of Novel Dopamine Receptor Agonists Recognizing Preferentially the D(3) Subtype. J Med Chem. 2000;43:756–762. doi: 10.1021/jm991098z. [DOI] [PubMed] [Google Scholar]
- 24.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Substitution of Three Amino Acids Switches Receptor Specificity of Gq Alpha to That of Gi Alpha. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 25.Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Muscarinic Acetylcholine Receptors: Novel Opportunities for Drug Development. Nat Rev Drug Discovery. 2014;13:549–560. doi: 10.1038/nrd4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sterling T, Irwin JJ. Zinc 15 - Ligand Discovery for Everyone. J Chem Inf Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irwin JJ, Shoichet BK. Zinc–a Free Database of Commercially Available Compounds for Virtual Screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mysinger MM, Shoichet BK. Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J Chem Inf Model. 2010;50:1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 29.Shoichet BK, Kuntz ID. Matching Chemistry and Shape in Molecular Docking. Protein Eng, Des Sel. 1993;6:723–732. doi: 10.1093/protein/6.7.723. [DOI] [PubMed] [Google Scholar]
- 30.Gilson MK, Honig BH. Calculation of Electrostatic Potentials in an Enzyme Active Site. Nature. 1987;330:84–86. doi: 10.1038/330084a0. [DOI] [PubMed] [Google Scholar]
- 31.Meng EC, Shoichet BK, Kuntz ID. Automated Docking with Grid-Based Energy Evaluation. J Comput Chem. 1992;13:505–524. [Google Scholar]
- 32.Lin H, Sassano MF, Roth BL, Shoichet BK. A Pharmacological Organization of G Protein-Coupled Receptors. Nat Methods. 2013;10:140–146. doi: 10.1038/nmeth.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Meara MJ, Ballouz S, Shoichet BK, Gillis J. Ligand Similarity Complements Sequence, Physical Interaction, and Co-Expression for Gene Function Prediction. PLoS One. 2016;11:e0160098. doi: 10.1371/journal.pone.0160098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Huebner H, Huang XP, Sassano MF, Giguere PM, Loeber S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK. Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang XP, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, Saddoris KA, Nikolova VD, Farrell MS, Wang S, Mangano TJ, Deshpande DA, Jiang A, Penn RB, Jin J, Koller BH, Kenakin T, Shoichet BK, Roth BL. Allosteric Ligands for the Pharmacologically Dark Receptors Gpr68 and Gpr65. Nature. 2015;527:477–483. doi: 10.1038/nature15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powers RA, Morandi F, Shoichet BK. Structure-Based Discovery of a Novel, Noncovalent Inhibitor of Ampc Beta-Lactamase. Structure. 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 37.Carlsson J, Yoo L, Gao ZG, Irwin JJ, Shoichet BK, Jacobson KA. Structure-Based Discovery of A2a Adenosine Receptor Ligands. J Med Chem. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. Current Protocols in Molecular Biology. John Wiley & Sons; Hoboken, NJ: 2006. High-Throughput Real-Time Quantitative Reverse Transcription PCR. doi:15.8.110.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- 39.Davies M, Nowotka M, Papadatos G, Dedman N, Gaulton A, Atkinson F, Bellis L, Overington JP. Chembl Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015;43:W612–620. doi: 10.1093/nar/gkv352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, Mutowo P, Atkinson F, Bellis LJ, Cibrian-Uhalte E, Davies M, Dedman N, Karlsson A, Magarinos MP, Overington JP, Papadatos G, Smit I, Leach AR. The Chembl Database in 2017. Nucleic Acids Res. 2017;45:D945–D954. doi: 10.1093/nar/gkw1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. Chembl: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012;40:D1100–1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, Tang A, Gabriel G, Ly C, Adamjee S, Dame ZT, Han B, Zhou Y, Wishart DS. Drugbank 4.0: Shedding New Light on Drug Metabolism. Nucleic Acids Res. 2014;42:D1091–1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rogers DJ, Tanimoto TT. A Computer Program for Classifying Plants. Science. 1960;132:1115–1118. doi: 10.1126/science.132.3434.1115. [DOI] [PubMed] [Google Scholar]
- 44.Tschammer N, Elsner J, Goetz A, Ehrlich K, Schuster S, Ruberg M, Kuehhorn J, Thompson D, Whistler J, Huebner H, Gmeiner P. Highly Potent 5-Aminotetrahydropyrazolopyridines: Enantioselective Dopamine D3 Receptor Binding, Functional Selectivity, and Analysis of Receptor-Ligand Interactions. J Med Chem. 2011;54:2477–2491. doi: 10.1021/jm101639t. [DOI] [PubMed] [Google Scholar]
- 45.Moeller D, Banerjee A, Uzuneser TC, Skultety M, Huth T, Plouffe B, Huebner H, Alzheimer C, Friedland K, Mueller CP, Bouvier M, Gmeiner P. Discovery of G Protein-Biased Dopaminergics with a Pyrazolo[1:5-a]Pyridine Substructure. J Med Chem. 2017;60:2908–2929. doi: 10.1021/acs.jmedchem.6b01857. [DOI] [PubMed] [Google Scholar]
- 46.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein Measurement with the Folin Phenol Reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 47.Mathiesen JM, Vedel L, Brauner-Osborne H. Camp Biosensors Applied in Molecular Pharmacological Studies of G Protein-Coupled Receptors. Methods Enzymol. 2013;522:191–207. doi: 10.1016/B978-0-12-407865-9.00011-X. [DOI] [PubMed] [Google Scholar]
- 48.Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM, Christopoulos A. A Novel Mechanism of G Protein-Coupled Receptor Functional Selectivity. Muscarinic Partial Agonist Mcn-a-343 as a Bitopic Orthosteric/Allosteric Ligand. J Biol Chem. 2008;283:29312–29321. doi: 10.1074/jbc.M803801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gopalakrishnan M, Monteggia LM, Anderson DJ, Molinari EJ, Piattoni-Kaplan M, Donnelly-Roberts D, Arneric SP, Sullivan JP. Stable Expression, Pharmacologic Properties and Regulation of the Human Neuronal Nicotinic Acetylcholine Alpha 4 Beta 2 Receptor. J Pharmacol Exp Ther. 1996;276:289–297. [PubMed] [Google Scholar]
- 50.Cheng YC, Prusoff WH. Relationship between the Inhibition Constant (Ki) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.