

Heart failure is a multifaceted disease in which mitochondrial dysfunction plays a large contributing role. Mitochondria are essential for maintaining cellular ATP levels sufficient to sustain the beating cardiac myocytes. Their function, however, extends beyond providing myocytes with energy, as they also regulate metabolism, heme synthesis, calcium homeostasis, ROS production, and programmed cell death1. To ensure a network of operational and efficient mitochondria, myocytes use mitochondrial autophagy or mitophagy to eliminate damaged or unwanted mitochondria. The most well-known and studied mitophagy pathway to date is mediated by two proteins, PINK1 and Parkin. The PINK1/Parkin pathway was initially identified as important regulators of mitochondrial integrity and function in muscle cells and dopaminergic neurons in Drosopholia2, 3, but recent studies have additionally confirmed its importance in maintaining cardiac homeostasis and adaptation to stress4–6. Most mechanistic studies on the PINK1/Parkin pathway have been conducted in vitro using harsh mitochondrial toxins to activate mitophagy. However, these conditions do not appropriately mimic conditions in vivo. Thus, physiologic mechanisms of upstream PINK1 and Parkin activation are still under intense investigation. In this issue of Circulation Research, Wang et al. sheds light on the regulation of PINK1/Parkin-mediated mitophagy in the heart7. Specifically, the authors have identified a role for AMPK in activating mitophagy via PINK1 phosphorylation to prevent development of heart failure.
The selective degradation of mitochondria is an essential process for mitochondrial quality control, both under physiologic conditions and in response to pathologic stress. When mitochondria are healthy, the serine/threonine kinase PTEN-inducible kinase 1 (PINK1) is rapidly imported into the mitochondria via the TOM/TIM complex where it is cleaved by the mitochondrial protease PARL (Fig. 1A). Cleaved PINK1 is then retro-translocated into the cytosol and degraded by the proteasome8. However, upon mitochondrial damage, import is terminated and PINK1 accumulates on the outer mitochondrial membrane. PINK1 then recruits the E3 ubiquitin ligase Parkin to the mitochondria to ubiquitinate mitochondrial substrates9. The buildup of ubiquitin flags the mitochondrion for selective elimination by autophagosomes. Thus, PINK1 accumulation acts as the initial mitochondrial stress sensor that activates mitophagy.
Figure 1.
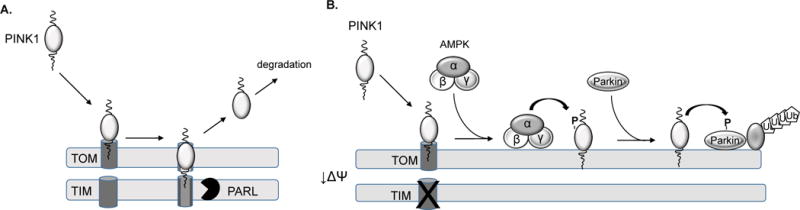
Activation of PINK1 by AMPKα2 Regulation of PINK1. A. Import of PINK1 into healthy mitochondria via the TOM/TIM import complex. PINK1 undergoes proteolytic cleavage by PARL and cleaved PINK1 retro-translocates to the cytosol where it is degraded by the proteasome. B. Upon mitochondrial damage, import of PINK1 is abrogated and it accumulates on the outer mitochondrial membrane which leads to its phosphorylation by AMPKα2.
AMP-activated protein kinase (AMPK) senses changes in cellular energy levels and regulates a number of cellular processes that influence cardiac myocyte survival10. When cellular energy levels are compromised, AMPK reduces energy-consuming pathways while increasing ATP-generating catabolic pathways. In their study, Wang et al. identified an additional function for AMPKα2 in myocytes: activating PINK1/Parkin-mediated mitophagy through PINK1 phosphorylation to protect against the development of heart failure (Fig. 1B).
Switching Sides: AMPKα isoforms switch during development of heart failure
AMPK is a heterotrimer composed of a catalytic α subunit and two regulatory ß- and γ- subunits. Each subunit has multiple isoforms encoded by distinct genes which allow for the expression of 12 different AMPK complexes. An essential and largely unresolved question is what biological function do distinct AMPK isoforms and complexes serve. In the current study, Wang et al. compared expression of AMPK in normal and failing human hearts and noted that AMPKα2 is the predominant form in the healthy human heart7. In contrast, AMPKα1 is elevated and AMPKα2 reduced in the failing heart. This isoform switch was confirmed using mice subjected to trans-aortic constriction (TAC) as a model of heart failure. In these experiments, the authors performed an extensive time course to distinguish physiologic changes that occur in the heart during early versus late phases of pressure overload. Intriguingly, they observed an increase in AMPKα2 in the early, compensatory phase (3-5 days post TAC) followed by a decrease in the late phase. In contrast, they found a steady increase in AMPKα1 levels up to heart failure development. Consistent with previous reports11, the authors found that both autophagy and mitophagy increase during the early phase of TAC, and subsequently decrease in late-phase. Interestingly, levels of autophagy and mitophagy rose and fell concurrently with levels of AMPKα2. The decline in AMPKα2 also correlates with an accumulation of dysfunctional mitochondria, implicating AMPKα2 as a necessary factor for mitophagy in the heart.
The Mitophagy Connection: AMPKα2 activates PINK1/Parkin-mediated mitophagy
The specific role of AMPKα2 in regulating PINK1/Parkin-mediated mitophagy in vivo was investigated through gain-of-function and loss-of-function experiments7. Overexpression of AMPKα2 in the heart using AAV9-mediated gene delivery led to increased PINK1/Parkin-mediated mitophagy, preserved mitochondrial function and improved cardiac function in the chronic, late phase of TAC compared with control mice. In contrast, AMPKα2-deficiency caused reduced mitophagy and impaired mitochondrial function in hearts and accelerated the progression of TAC-induced heart failure. These findings establish a clear link between AMPKα2, mitophagy, and mitochondrial function in heart failure.
The authors confirm their findings in vitro using cultured myocytes treated with phenylephrine (PE) to mimic the TAC-induced cardiomyocyte hypertrophy7. Although PE is effective at inducing hypertrophy, it only modestly induces mitophagy. Therefore, treatment with the potent mitochondrial uncoupler CCCP was also used to promote mitophagy. In line with the results of the TAC studies, the in vitro data demonstrated that treatment with PE for 24h reduces AMPKα2 levels and PINK1/Parkin-mediated mitophagy, and increases the number of damaged mitochondria. As expected, these effects were diminished with AMPKα2 overexpression. Taken together, these experiments further confirm the link between AMPKa2 and the PINK1/Parkin pathway in myocytes.
The Phosphorylation Game: AMPKα2 phosphorylates PINK1 at Ser495
To determine how AMPKα2 activates PINK1/Parkin-mediated mitophagy, the authors conducted co-immunoprecipitation experiments that confirmed that AMPKα2, but not AMPKα1, interacts with PINK17. To investigate if PINK1 is an AMPKα2 substrate, the authors treated myocytes with PE and ran a phos-tag gels. AMPKα2 overexpression increases phosphorylation-induced mobility shift in PINK1 after PE treatment for 24h. Conversely, si-AMPKα2 depleted the phosphorylated PINK1 in myocytes treated with PE for 6h. They also used software to predict potential phosphorylation sites and LC MS/MS to identify phosphorylated residues in PINK1 in the presence of AMPKα2. Two serine residues were found to be consistently phosphorylated: Ser284 and Ser495. After analyses using site directed mutagenesis, Ser495 emerged as the candidate phosphorylation site. Specifically, the authors found that mutating Ser495 to alanine led to reduced PINK1/Parkin-mediated mitophagy, whereas mutating Ser495 to Asp to mimic phosphorylation enhanced mitophagy. The authors solidify these findings by demonstrating that recombinant AMPKα2β2γ1 interacts with and phosphorylates PINK1 at Ser495 in a cell-free system, and that PINK1 Ser495 is phosphorylated in vivo after TAC surgery with AMPKα2 overexpression, but not in AMPKα2-deficeint hearts. PINK1 activation in response to mitochondrial damage has previously been ascribed to inhibition of its import8; however, the study by Wang et al. suggests that an additional layer of regulation exists in myocytes.
Going Forward: Insights into PINK1 Regulation, Mitophagy, and Heart Failure
This study has several major strengths. Because heart failure development is very complex process, it is especially crucial to study both the early and late phases in pressure overload induced heart failure. Additionally, the authors very convincingly demonstrate that PINK1 is phosphorylated by AMPKα2 in a cell-free system, in vitro, and in vivo. However, limitations must also be considered when analyzing the findings of this study. The main limitation of this study is the lack of a physiologically relevant in vitro model. The authors use PE to simulate heart failure and treat the cells for 6h and 24h to mimic the early and late phases, respectively. While PE is able to faithfully activate cardiomyocyte hypertrophy, it has not been reported to replicate many other signaling cascades involved in the development of heart failure including inflammation, fibrosis, and mitophagy. The absence of robust mitophagy activation prompts the authors to use CCCP in addition to PE. CCCP, however, is notoriously un-physiologic and completely depolarizes the entire pool of mitochondria. Whereas during the development of early and late phase heart failure in mice, the time frame is set at days and weeks and involves the activation of a myriad of signaling cascades, these un-physiologic in vitro conditions use a much shorter time period and stimulate only a couple of these pathways.
Nevertheless, the findings in this study raise many thought-provoking questions. For instance, how AMPKα2-mediated phosphorylation affects the kinase activity of PINK1 needs to be investigated in further detail. The enhanced Parkin-mediated mitophagy could be due to either enhanced PINK1 kinase activity or increased total PINK1 levels. Another important unanswered question is under what conditions AMPKα2 regulates mitophagy. Is it specific for pressure overload or does it play a role in mitophagy activated by a myocardial infarction or ischemia/reperfusion injury? These are just some of the questions that arise from the current study.
Overall, the findings presented by Wang et al. demonstrate that AMPKα2 is an important regulator of PINK1/Parkin-mediated mitophagy in the heart. Also, the suppression of mitophagy during the chronic phase of TAC is, in part, due to a decrease in AMPKα2. The data also reveal distinct functions for AMPKα1 and AMPKα2 catalytic subunits in the heart, which highlights the importance of developing isoform-specific drug targets for the treatment of cardiovascular disease.
Acknowledgments
Sources of Funding
ÅB Gustafsson is supported by an AHA Established Investigator Award, and by NIH R21AG052280, R01HL132300, R01HL138560 and P01HL085577. S Shires is supported by an AHA Pre-doctoral fellowship.
Footnotes
Disclosures
None.
References
- 1.Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 3.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 4.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW., 2nd Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siddall HK, Yellon DM, Ong SB, Mukherjee UA, Burke N, Hall AR, Angelova PR, Ludtmann MH, Deas E, Davidson SM, Mocanu MM, Hausenloy DJ. Loss of PINK1 increases the heart's vulnerability to ischemia-reperfusion injury. PLoS One. 2013;8:e62400. doi: 10.1371/journal.pone.0062400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, Zou MH, Chen C, Wang DW. AMPKalpha2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ Res. 2017 doi: 10.1161/CIRCRESAHA.117.312317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekine S, Youle RJ. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC biology. 2018;16:2. doi: 10.1186/s12915-017-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salt IP, Hardie DG. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway With Key Roles in the Cardiovascular System. Circ Res. 2017;120:1825–1841. doi: 10.1161/CIRCRESAHA.117.309633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation. 2016;133:1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]