

Abstract
The selenocysteine (Sec) tRNA[Ser]Sec population consists of two isoforms that differ from each other by a single 2’-O-methylribosyl moiety at position 34 (Um34). These two isoforms, which are encoded in a single gene, Trsp, and modified post-transcriptionally, are involved individually in the synthesis of two subclasses of selenoproteins, designated housekeeping and stress-related selenoproteins. Techniques used in obtaining these isoforms for their characterization include extraction of RNA from mammalian cells and tissues, purifying the tRNA[Ser]Sec population by one or more procedures and finally resolving the two isoforms from each other. Since some of the older techniques for isolating tRNA[Ser]Sec and resolving the isoforms are used in only a few laboratories, these procedures will be discussed briefly and references provided for more detailed information, while the more recently developed procedures are discussed in detail. In addition, a novel technique that was developed in sequencing tRNA[Ser]Sec for identifying their occurrence in other organisms is also presented.
Keywords: Chromatography, Gel electrophoresis, Selenium, Selenocysteine tRNA, Selenocysteine tRNA detection, Selenocysteine tRNA gene modification
1 Introduction
Selenocysteine (Sec) tRNA[Ser]Sec has been described as the central component [1], the key factor [2] and the quintessential constituent in selenoprotein biosynthesis [3]. Indeed, the targeted removal of the gene encoding this tRNA (Trsp) from cells or tissues results in the total loss of selenoprotein expression [4,5]. Sec tRNA[Ser]Sec appears to be among the most essential factors of those that selenoprotein biosynthesis are dependent upon as removal of Trsp completely eliminates expression of this protein class [4,5], whereas deletion of another dependent factor, SECIS-binding protein 2 (Secisbp2), reduces selenoprotein expression substantially, albeit not to the extent as Trsp [6].
Selenoproteins are the only known class of proteins that depend on a single tRNA for their expression. Thus, altering the expression of tRNA[Ser]Sec levels results in modulating the expression of selenoproteins. There are two species of Sec tRNAs that differ from each other by a single methylated base occurring on the 2′-O-hydroxyribosyl moiety at position 34, designated Um34 ([7] and see Fig. 1). The hypermodified base at position 34 is 5-methoxycarbonylmethyluridine and the Um34-methylated form is 5-methoxycarbonylmethyl-2′-O-methyluridine. These two isoforms have only four modified bases, mcm5U, N6-isopentenyladenosine (i6A), pseudouridine (ψ), and 1-methyladenosine (m1A) at positions 34, 37, 55 and 58, respectively, and one nucleoside, mcm5Um [7].
Fig 1.
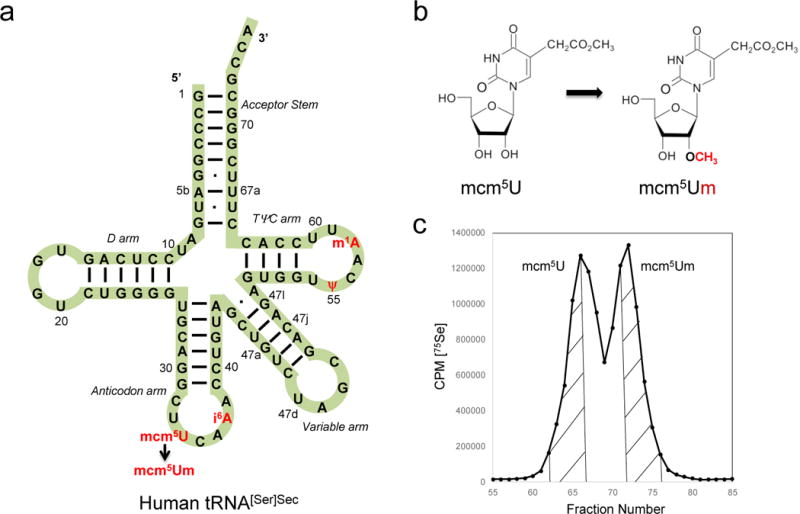
Selenocysteine tRNA[Ser]Sec (a) The primary structure of human tRNA[Ser]Sec is shown in a cloverleaf model. Mammalian tRNA[Ser]Sec is 90 bases in length. The acceptor stem constitutes the paired 5’ and 3’ terminal bases, the D stem and loop constitute the six paired and four unpaired bases of the left portion of the tRNA, the anticodon stem and loop, the six paired and seven unpaired bases of the lower portion of the tRNA, the variable stem and loop, the five paired and 4 unpaired bases, and the TψC stem and loop, the four paired and seven unpaired bases of the right portion of the tRNA. Mammalian tRNA[Ser]Sec contains modifications at positions 34 (mcm5U or mcm5Um), 37 (i6A), 55 (ψ) and 58 (m1A). (b) The two tRNA[Ser]Sec isoforms (containing either mcm5U or mcm5Um at position 34) differ from each other by a single methyl group on the 2’-O-ribosyl moiety. (c) Representative fractionation of [75Se]selenocysteyl-tRNA[Ser]Sec isoforms by RPC5 chromatography. [75Se]selenocysteyl-tRNA[Ser]Sec was extracted from HL-60 cells and run on an RPC5 column as described in Section 3.6. The initial eluting peak from the column is the mcm5U-containing isoform and the later eluting peak is the mcm5Um-containing isoform. The isoforms are pooled as shown by the hatched areas and collected as described in Section 3.6 for further study.
The two tRNA[Ser]Sec isoforms are involved in the expression of separate subclasses of selenoproteins, housekeeping and stress-related selenoproteins [8,9], which provides an additional handle to control the expression of these two subclasses of proteins and regulating the expression of the two tRNA[Ser]Sec isoforms by specific base modifications in the Trsp gene transcript [8,10]. This chapter describes the isolation of tRNA[Ser]Sec from mammalian cells and tissues, resolution of the two isoforms, identification and sequencing of tRNA[Ser]Sec to further elucidate the roles of these two unique tRNAs in selenoprotein synthesis and regulation. Traditional techniques used in isolating tRNA[Ser]Sec, such as isolating total tRNA by diethylaminoethyl (DEAE)-cellulose chromatography [11], resolving the two tRNA[Ser]Sec isoforms by RPC5 chromatography [12,13], and aminoacylation of tRNA[Ser]Sec [14] are described herein, since they are highly significant in the characterization of the tRNA[Ser]Sec isoforms. However, the details regarding techniques such as enriching tRNA[Ser]Sec by benzoylated DEAE (BD)-cellulose chromatography [15–17], sequencing the isoforms by the [32P]-labeling techniques of Silberklang et al. [18] as modified [7] and identification of the modified bases and nucleoside by [32P]-labeling [18], can be found in the given references.
The present chapter emphasizes the more recently developed procedures. One of these protocols involves the isolation, partial purification and sequencing of tRNA[Ser]Sec from lower eukaryotic cells by a novel procedure [19]. This procedure was previously used to determine the primary sequences of tRNA[Ser]Sec from Chlamydomonas reinhardtii [19,20], Dictyostelium discoideum and Tetrahymena thermophile [21].
2 Materials
All solutions were prepared with molecular biology grade H2O and all reagents were of the highest grade commercially available. Routine reagents with no listed supplier should be purchased from your supplier of choice. Buffers are stored at room temperature unless otherwise noted. Purchased reagents are stored as recommended by the supplier. Freshly grown cells or freshly harvested tissues were either frozen immediately by placing into liquid nitrogen and storing at −80°C until ready for use or by immediately beginning RNA extraction followed by total tRNA and subsequent tRNA[Ser]Sec purification. For large scale tRNA preparations, calf liver was obtained from freshly euthanized animals, and the liver (~10 kg) maintained on ice until the isolation procedure.
2.1 Extraction and Isolation of Total RNA from Cells and Tissues
Extraction buffer: 0.14 M sodium acetate, pH 4.5.
H2O-saturated phenol, pH 4.5.
Chloroform (CHCl3).
DE52 preswollen microgranular DEAE cellulose (GE Healthcare Life Sciences, Pittsburgh, PA).
Disposable chromatography columns (e.g., 20 ml, 1.5 × 12 cm columns from Bio-Rad, Hercules, CA).
DE52 column buffer A: 0.1 M Tris, pH 7.4, 0.1 M NaCl.
DE52 column buffer E: 0.1 M Tris, pH 7.4, 1.0 M NaCl.
Pure ethanol (EtOH).
2.2 RPC5 Chromatography
Reverse phase chromatography 5 (RPC5) column [13] (see Section 3.3 for preparation).
RPC5 starting buffer (+ Mg2+): 10 mM NaOAc, pH 4.5, 0.45 M NaCl, 10 mM MgOAc2, 1 mM EDTA.
RPC5 starting buffer (− Mg2+): 10 mM NaOAc, pH 4.5, 0.5 M NaCl, 1 mM EDTA.
RPC5 buffer A: 10 mM NaOAc, pH 4.5, 0.525 M NaCl, 10 mM MgOAc2, 1 mM EDTA.
RPC5 buffer B: 10 mM NaOAc, pH 4.5, 0.675 M NaCl, 10 mM MgOAc2, 1 mM EDTA.
RPC5 buffer C: 10 mM NaOAc, pH 4.5, 1.5 M NaCl, 10 mM MgOAc2, 1 mM EDTA.
RPC5 buffer D: 10 mM NaOAc, pH 4.5, 0.6 M NaCl, 1 mM EDTA.
RPC5 buffer E: 10 mM NaOAc, pH 4.5, 0.825 M NaCl, 1 mM EDTA.
0.45 μm HA nitrocellulose filters (Millipore, Billerica, MA).
Column fraction collector.
Ecoscint A scintillation cocktail (National Diagnostics, Atlanta, GA) and scintillation vials.
2.3 Northern Blotting and Aminoacylation
Isotopes: [3H]serine (specific activity ~20 Ci/mmol; Moravek Biochemicals, Brea, CA); γ-[32P]-ATP (specific activity ~3000 Ci/mmol; Perkin Elmer, Waltham, MA).
Positively charged nylon blotting membrane.
15% TBE/urea gels (handcast or precast).
Quikhyb hybridization solution (Stratagene, San Diego, CA).
Trizol® reagent (Thermo Fisher Scientific, Waltham, MA).
T4 polynucleotide kinase.
TAM (10×): 0.5 M Tris-HCl, pH 7.4, 0.06 M ATP, 0.2 M MgC12.
19 unlabeled amino acid mix (minus serine): 10 mM final concentration of each amino acid.
Purified seryl-tRNA synthetase (MyBioSource, Inc., San Diego, CA)
Trichloroacetic acid (TCA).
Wash buffer 1: 2X SSC, 0.1% SDS.
Wash buffer 2: 0.1X SSC, 0.1% SDS.
1X TBE buffer: 89 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 7.6.
Betafluor scintillation cocktail (National Diagnostics, Atlanta, GA) and scintillation vials.
2.4 Extraction and Isolation of [75Se]selenocysteyl-tRNA[Ser]Sec from Cells
[75Se]selenious acid (specific activity ~1000 Ci/mmol) in HNO3 was obtained from the University of Missouri Research Reactor Center (MURR), Columbia, MO.
Phosphate buffered saline (PBS): 9 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4, 137 mM NaCl, 2.7 mM KCl.
Sodium selenite (Sigma-Aldrich, St. Louis, MO).
Cycloheximide.
Extraction buffer B: 5 mM NaOAc, pH 4.5, 225 mM NaCl, 5 mM MgOAc2, 1 mM EDTA, 0.2% SDS, 6 mM 2-mercaptoethanol.
H2O-saturated phenol, pH 4.5.
CHCl3.
DE52 column equilibration buffer: 5 mM NaOAc, pH 4.5, 112.5 mM NaCl, 5 mM MgOAc2, 1 mM EDTA.
DE52 elution buffer: 10 mM NaOAc, pH 4.5, 1.5 M NaCl, 10 mM MgOAc2, 1 mM EDTA.
Pure 200 proof EtOH.
2.5 Acid-urea gel electrophoresis
Molecular biology grade urea.
1 M NaOAc, pH 4.5.
40% acrylamide stock solution: acrylamide:bis-acrylamide (29:1).
Ammonium persulfate: 10% solution in molecular biology grade H2O.
Tetramethylethylenediamine (TEMED).
Acid gel running buffer: 100 mM NaOAc, pH 4.5.
2X acid-urea sample buffer: 8 M Urea, 100 mM NaOAc, pH 4.5.
Xylene cyanol and bromophenol blue dyes.
Gel fixative solution: 25% ethanol, 10% acetic acid.
2.6 Affinity Purification of tRNA[Ser]Sec
Purified total tRNA.
Streptavidin-conjugated magnetic M-280 Dynabeads (Thermo Fisher Scientific, Waltham, MA).
Buffer A: 10 mM Tris-HCl, pH 7.5, 2 M NaCl, 2 mM EDTA.
6X Saline Sodium Citrate (SSC) buffer: 90 mM sodium citrate, pH 7.0, 0.9 M NaCl.
RQ1 RNase-Free DNase (Promega, Madison, WI).
2.7 tRNA Sequencing
Escherichia coli (E. coli) poly(A) polymerase (New England Biolabs, Ipswich, MA).
RNase A (10 μg/μl) and RNase H (2 U/μl).
T4 polynucleotide kinase (New England Biolabs, Ipswich, MA).
Adenosine 5′-triphosphate (ATP) disodium salt hydrate.
Terminal Deoxynucleotidyl Transferase (TdT) and ddATP (Thermo Fisher Scientific, Waltham, MA).
T4 RNA ligase (New England Biolabs, Ipswich, MA).
DH5α competent E. coli cells (Thermo Fisher Scientific, Waltham, MA).
Luria broth (LB) and LB agar plates.
Taq DNA polymerase.
pCR2.1-TOPO vector (Thermo Fisher Scientific, Waltham, MA).
QIAprep Spin Miniprep Kit and QIAquick spin columns (Qiagen, Valencia, CA).
3 Methods
Carry out all procedures at room temperature unless otherwise specified.
3.1 Extraction of Total RNA from Cells and Tissues
Combine equal parts of H2O-saturated phenol and RNA extraction buffer in a 50 ml polypropylene tube. Typically, ~20 ml of extraction buffer mix is used per gram of frozen tissue or cells (see Note1).
To frozen cells or tissues, add extraction buffer mix and immediately homogenize for 3-4 min using an Omni Macro Homogenizer with a 50 mL stainless steel chamber for tissues weighing less than 2 g (see Note2).
Centrifuge homogenized sample at 4,000 × g for 10 min.
Transfer the upper aqueous layer to a clean polypropylene tube.
Extract upper layer with an equal volume of CHCl3. Vortex vigorously for 1-2 min.
Centrifuge at 4,000 × g for 10 min.
Take top layer and transfer to clean polypropylene tube and load sample onto a prepared DE52 column (see Section 3.2).
3.2 Isolation of Total tRNA from Cells and Tissues
DE52 is prepared by adding DE52 to a glass beaker or flask. Gently add 4-5 volumes of molecular biology grade H2O, let DE52 settle, pour off H2O, and repeat wash 5-6 times to remove fine particles (see Note3).
Gently pipette DE52 into empty chromatography column (see Note4).
Equilibrate column with several column volumes of column buffer A.
Add the supernatant obtained following CHCl3 extraction to the column.
Wash ~5-6 times with 25 ml of buffer A or until absorbance (A260) of eluate is virtually undetectable.
Elute tRNA from column with ~12-15 ml of column buffer E.
Add MgCl2 to the eluate to a final concentration of 10 mM.
Precipitate tRNA with 3 volumes of EtOH. Leave at -20°C for at least 3 hours.
Collect precipitated tRNA on Millipore 0.45 μm HA filter. Alternatively, precipitate in 3 volumes of EtOH and collect precipitant by centrifugation at 20,000 × g for 30 min at 4°C.
Wash collected tRNA 2-3 times with 75% EtOH.
Gently dry, but do not over-dry the filter or pellet.
Elute tRNA from filter with molecular biology grade H2O by placing filter in microcentrifuge tube, adding 250 μl of H2O, vortexing and centrifuging.
Transfer eluted tRNA to a clean tube. Repeat elution with another 250 μl of H2O. (Typically use a final volume of 500 μl for tRNA from 1 g of tissue or cells).
Deacylate purified Sec-tRNA[Ser]Sec in 1.0 M Tris, pH 8.0, at 37°C for 1 h. Deacylated tRNA[Ser]Sec is precipitated with 3 volumes of EtOH, collected by centrifugation at 20,000 × g for 30 min, washed with 75% EtOH, dried and dissolved in nuclease-free H2O (see Note5).
3.3 Purification of tRNA[Ser]Sec Isoforms by RPC5 Column Chromatography and Northern Blot Analysis
RPC5 columns can be prepared using the technique outlined by Kelmers and Heatherly [13]. Briefly, 100 g of polychlorotrifluoroethylene (Plaskon CTFE) powder is coated in 4 ml of Adogen 464 (methyltrialkyl (C8-C10) ammonium chloride; Sigma-Aldrich, St. Louis, MO) that has been dissolved in 200 ml of chloroform. The solution is blended in a fume hood and ground vigorously with a mortar and pestle to break down the Plaskon powder to ~10 μ size particles and until the chloroform is evaporated. The coated Plaskon powder is then suspended in RPC5 starting buffer (+ Mg2+) and used to pack a 30 cm × 1.5 cm glass chromatographic column (see Note6).
Total tRNA (5-8 mg maximum) following the DE52 column is dissolved in RPC5 starting buffer (+ Mg2+) and loaded onto an RPC5 column that has been equilibrated in RPC5 starting buffer (+ Mg2+) (see Note7).
A 160 ml linear gradient (80 ml of RPC5 buffer A as the starting buffer and 80 ml of RPC5 buffer B as the terminal buffer) is run at a flow rate of 2 ml/min and 2 ml fractions collected in a fraction collector.
Wash column with 60 ml RPC5 buffer C and collect eluted fractions (see Note8).
The absorbance at A260 in the eluted fractions is measured and tRNA[Ser]Sec is detected by dot blot hybridization.
Perform dot blot analysis by spotting 5 μl of each column fraction on a Hybond-N+ membrane followed by UV cross-linking of membrane.
Hybridize membrane using Quikhyb or similar hybridization solution for 4 h at 58°C in a rotating hybridization oven with a [32P]-end labeled oligo probe complementary to the 3′-end of tRNA[Ser]Sec (5′-CGCCCGAAAGGTGGAATTGA-3′) prepared using T4 polynucleotide kinase and [γ-32P]-ATP according to manufacturer’s instructions.
Following hybridization, wash membrane 3 times with 2X SSC, 0.1% SDS, wash twice with 0.1X SSC, 0.1% SDS, expose to a PhosphorImager screen and quantitate spots using ImageQuant (GE Healthcare Life Sciences, Pittsburgh, PA) or similar software.
The two eluting peaks represent mcm5U- (initial eluting peak) and mcm5Um- (later eluting peak) containing isoforms of tRNA[Ser]Sec. Pool column fractions containing mcm5U or mcm5Um-containing isoforms, precipitate the tRNA with 3 volumes of EtOH.
Collect the pooled samples either on Millipore 0.45 μm HA nitrocellulose filters or by centrifugation at 20,000 × g for 30 min at 4°C.
Dissolve tRNA samples containing either mcm5U or mcm5Um isoforms (or both if purified isoform is not needed) in RPC5 starting buffer (- Mg2+) and individually load onto the RPC5 column. A 160 ml linear gradient (80 ml of RPC5 buffer D as the initial buffer and 80 ml of RPC5 buffer E) is run at a flow rate of 2 ml/min and 2 ml fractions collected, as described above.
The absorbance at A260 in the eluted fractions is measured and plotted, and tRNA[Ser]Sec is detected by dot blot hybridization as above.
Samples are pooled and collected as given above in steps 8 and 9 (see Note9).
tRNA[Ser]Sec isoforms can be further purified by running on a 15% TBE/urea gel in 1X TBE buffer according to standard techniques (see Note10) or by binding to immobilized oligonucleotides complementary to tRNA[Ser]Sec (see Section 3.8), if more highly purified tRNA[Ser]Sec is needed for further examination (e.g., modified base analysis).
3.4 Aminoacylation of tRNA
Add in the following order to a final volume of 25 μl: 10X TAM (2.5 μl); H2O (to 25μl final volume); 19 unlabeled amino acid mix (2.5 μl); [3H]serine (2.5 μl); tRNA (0-5 μl); purified SARS (2.0 μl or ~0.5 μg).
tRNA is typically added in 0, 0.5, 1, 2, 3 and 5 μl volumes to generate a curve and determine linearity (see Note11).
Incubate at 37°C for 15 min.
Stop reaction by adding 0.5 mL of cold H2O and immediately precipitate tRNA with ~5 ml of 10% TCA.
Collect each reaction on a Millipore 0.45 μm HA nitrocellulose filter.
Dry filter, place in liquid scintillation vial, add scintillation cocktail and count in liquid scintillation counter.
3.5 Quantification of tRNA[Ser]Sec from Cells or Tissues using Total RNA
Isolate total RNA from tissue or cells using Trizol® according to manufacturer’s instructions.
Load total RNA (1-2 μg) on 15% TBE/urea gels and electrophorese in 1X TBE buffer according to manufacturer’s instructions.
Stain gel with ethidium bromide in 1X TBE buffer, photograph, and transfer to Hybond-N+ membrane in 0.5X TBE buffer at 25V for 60 min.
UV-crosslink the membrane.
Hybridize membrane using Quikhyb solution for 3-4 hours at 58°C in rotating hybridization oven using a [32P]-end labeled oligo probe complementary to the 3’-end of tRNA[Ser]Sec (5′-CGCCCGAAAGGTGGAATTGA-3′) prepared using T4 polynucleotide kinase and [γ-32P]-ATP according to manufacturer’s instructions.
Following hybridization, wash membrane 3 times with wash buffer 1, wash twice with wash buffer 2. Wrap membrane and expose to PhosphorImager. After exposure, strip membrane by gentle rocking with ~100 ml wash buffer 2 that has been heated to 95-100°C until radioactivity is no longer detected on the membrane (~5-10 min).
Remove the blot from the solution, shake off excess liquid and wrap the membrane with plastic wrap or reuse immediately. Do not let the membrane dry.
Re-hybridize stripped membrane with [32P]5’-end labeled tRNASer1 (5’-CGTAGTCGGCAGGATTCGAA-3’) oligo probe as a loading control.
Expose to a PhosphorImager and quantitate spots using ImageQuant (GE Healthcare Life Sciences, Pittsburgh, PA) or similar software.
3.6 Isolation of [75Se]-Labeled Selenocysteyl-tRNA[Ser]Sec
Grow several flasks of HL-60 cells (~5 g wet weight) in RPMI-1640 media in log phase and in the presence of 300 nM sodium selenite (see Note12).
Collect cells by centrifugation at 300 × g for 5 min at 4°C, wash with PBS and re-suspend in 75 ml of RPMI-1640 media containing 1% fetal bovine serum without sodium selenite.
Add 5 mCi of neutralized 75Se and gently shake the cells for 3 h at 37°C.
Add cycloheximide (100 μM final concentration) and incubate cells for an additional 45 min.
Collect cells by centrifugation at 300 × g for 5 min at 4°C, wash with ice cold PBS, and store at -80°C until ready to use.
To frozen cell pellet, add H2O-saturated phenol and extraction buffer B and immediately vortex vigorously for 3-4 min.
Centrifuge homogenized sample at 4,000 × g for 10 min.
Transfer the upper aqueous layer to a clean polypropylene tube.
Extract upper layer with an equal volume of CHCl3. Vortex vigorously for 1-2 min.
Centrifuge at 4,000 × g for 10 min.
Take top layer and transfer to clean polypropylene tube and load sample onto a prepared DE52 column (see Section 3.2) equilibrated with several column volumes of DE52 column equilibration buffer.
Add the supernatant obtained following CHCl3 extraction to the column.
Wash 5-6 times with 25 ml of DE52 column equilibration buffer or until absorbance (A260) of eluate is virtually undetectable.
Elute tRNA from column with ~12-15 ml of DE52 elution buffer.
Dilute eluate to a final concentration of 0.45 M NaCl with ice-cold H2O containing 0.02 M 2-mercaptoethanol, and load onto an RPC5 column that has been equilibrated with RPC5 starting buffer (+ Mg2+).
Purification of [75Se]-labeled Sec-tRNA[Ser]Sec by RPC5 chromatography is carried out using a 160 ml linear gradient (80 ml of RPC5 buffer A as the starting buffer and 80 ml of RPC5 buffer B as the terminal buffer), run at a flow rate of 2 ml/min and 2 ml fractions collected.
Wash column with 60 ml RPC5 buffer C and collect eluted fractions.
Analyze all fractions for 75Se-containing tRNA using a gamma counter (see Fig. 1C for a representative RPC5 profile).
Pool the mcm5U and mcm5Um-containing isoforms, precipitate with 2.5 volumes of EtOH, store at -20°C for 2-3 h and collect the resulting precipitate by centrifugation at 20,000 × g for 30 min at 4°C.
Wash pellet in 75% EtOH, gently dry and dissolve in ~100 μl of H2O. Measure A260 and cpm/μl and store the [75Se]selenocysteyl-tRNA[Ser]Sec at -80°C after gently blowing nitrogen gas over the sample before sealing the tube.
3.7 Analysis of [75Se]-Labeled Selenocysteyl-tRNA[Ser]Sec by Acid-Urea Gel Electrophoresis
The following method is adapted from [22].
Prepare 7% acid-urea gel mix by dissolving 48 g of urea into a solution containing 35 ml of H2O, 10 ml of 1 M NaOAc, pH 4.5 and 17.5 ml of a 40% acrylamide stock solution. Protected from light, this solution is stable for ~2 weeks.
Polymerize gel by adding 1/10 volume of 10% ammonium persulfate and 1/100 volume of TEMED. Pour 7.5 × 10 cm mini-gel with 0.75 mm or 0.5 mm comb and spacers. Allow polymerization to occur for at least 1 h.
Set up the gel with acid gel running buffer at 4˚C. Rinse wells thoroughly with running buffer using a 10 ml syringe and 21-gauge needle. Pre-run the gel at 12 V/cm (~100 V) for at least 30 minutes.
Concentrate purified [75Se]selenocysteyl-tRNA[Ser]Sec to at least 500 cpm per microliter and add 2 μl of tRNA to 2 μl of 2X acid-urea sample buffer and 0.05% (w/v) each of xylene cyanol and bromophenol blue dyes).
After the pre-run, thoroughly rinse the wells again and load the entire 8 μl sample. Run gel at 100 V constant voltage for about 1 hour until the bromophenol blue reaches the bottom of the gel.
Disassemble the rig and transfer the gel to gel fixative solution and incubate with gentle rocking for 30 min.
Dry the gel and expose to a PhosphorImager screen overnight. The [75Se]selenocysteyl-tRNA[Ser]Sec will appear as a single band migrating approximately two-thirds of the way down the gel, migrating slightly slower than a control [14C]phenylalanyl-tRNAPhe [23].
3.8 Isolation of tRNA[Ser]Sec from Cells and Tissues by Affinity Capture
The following method is adapted from [24].
Total tRNA is purified from tissues or cells as described in Sections 3.1 and 3.2.
Immobilize 3′-biotinylated DNA oligonucleotide (5’-CGCCCGAAAGGTGGAATTGAACCACTCTGTCGCTA-biotin-3’) corresponding to the 3’-end of tRNA[Ser]Sec on streptavidin-conjugated magnetic M-280 Dynabeads as detailed below.
For nucleic acid purification, beads must be made RNase-free according to the manufacturer’s instructions.
Wash desired amount of beads with an equal volume of buffer A using a magnet to pellet the beads.
Suspend beads in buffer A to a final concentration of 5 μg/μl.
Add 2 μM 3′-biotinylated oligonucleotide in H2O to an equal volume of Dynabeads in buffer A and incubate at room temperature for 30 min with gentle mixing.
Wash the oligonucleotide-coated Dynabeads 4-5 times using a magnet in 0.5X buffer A.
Equilibrate in 6X Saline Sodium Citrate (SSC) buffer.
Incubate the oligonucleotide-coated Dynabeads and total tRNA suspended in 6X SSC (binding capacity using single-stranded biotinylated oligonucleotides is ~200 pmol/mg of Dynabeads) for 5 min at 75°C.
Pool oligonucleotide-coated Dynabeads and tRNA and incubate for 5 min at 75°C.
Bind tRNA by incubating at room temperature for 1-2 h with gentle mixing (see Note 13).
Wash the beads 3 times with 3X SSC, twice with 1X SSC, and 3-4 times with 0.1X SSC or until the absorbance (A260) of the wash buffer is undetectable.
Elute tRNA from beads with 0.1X SSC at 65°C for 5 min. Perform elution 3 times in succession and pool eluate.
Treat eluate with RNase-Free DNase to remove any bound oligonucleotide.
Concentrate eluted tRNA by adding MgCl2 (10 mM final concentration) and precipitate with 3 volumes of EtOH.
Centrifuge at 20,000 × g for 15 min at 4°C, wash with 75% EtOH and centrifuge at 14,000 × g for 10 min at 4°C. Dry pellet and dissolve in molecular biology grade H2O or buffer of choice needed for further analysis of tRNA[Ser]Sec, such as modified base analysis by mass spectrophotometry.
3.9 Identification and Sequencing of tRNA[Ser]Sec from Any Organism
This method can be used to identify and sequence tRNA[Ser]Sec from organisms whose tRNA[Ser]Sec has not been characterized [19–21] and can also be used to verify mutations in tRNA[Ser]Sec, such as those that have been shown to occur in human tRNA[Ser]Sec [25]. See Figure 2 for a schematic illustration of this procedure.
Fig 2.
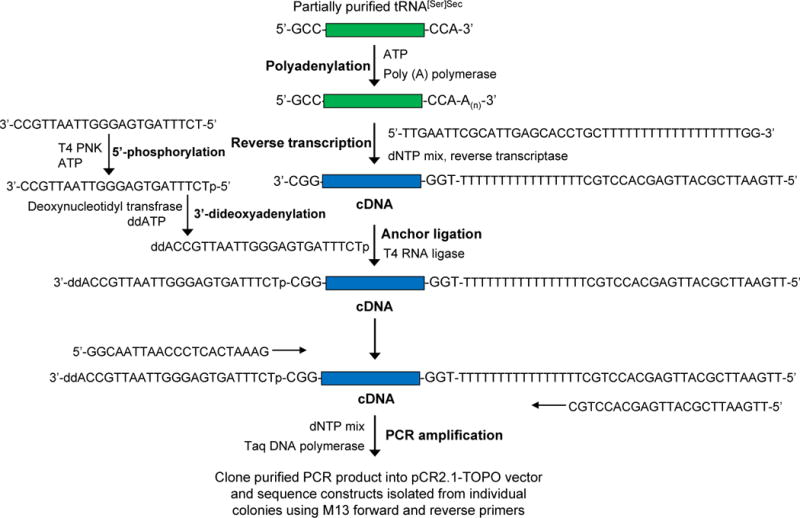
Schematic diagram demonstrating the procedure for sequencing uncharacterized tRNA[Ser]Sec from any organism as described in the text.
To isolate tRNA[Ser]Sec for sequencing from organisms whose tRNA[Ser]Sec has not been characterized, grow ~5 g of cells and isolate total tRNA as described in Sections 3.1 and 3.2.
Label ~0.5 g cells with 0.5 mCi of 75Se, isolate tRNA as described in Section 3.6 and combine 75Se-labeled tRNA with the unlabeled tRNA obtained from Step 1.
Chromatograph the pooled tRNAs by RPC5 column chromatography, and collect [75Se]selenocysteyl-tRNA[Ser]Sec-containing fractions as described in Section 3.6.
Polyadenylate the 3’-end of the partially purified tRNA[Ser]Sec using E. coli poly(A) polymerase according to manufacturer’s instructions.
Denature the polyadenylated tRNAs at 65°C for 10 min along with primer 5′-TTGAATTCGCATTGAGCACCTGCTTTTTTTTTTTTTTTTTTGG-3′ (100 nM final concentration) and a mixture of dNTPs (0.5 mM final concentration/each), cool on ice, briefly centrifuge, and then transcribe with reverse transcriptase according to the manufacturer’s protocol.
Add RNase A (50 μg/ml final concentration) and RNase H (1 U/nmol tRNA) and incubate at 37°C for 20 min to remove any remaining tRNA.
Purify cDNA on QIAquick spin columns to separate fragments greater than 100 bp in length from the other components in the reaction mix.
Phosphorylate the anchor oligonucleotide, 3′-CCGTTAATTGGGAGTGATTTCT-5′, on its 5′-end using T4 polynucleotide kinase and ATP, and then block its 3′-end by incubating with ddATP and terminal deoxynucleotidyl transferase according to the manufacturer’s protocols (see Note14).
Ligate the phosphorylated, blocked anchor-oligonucleotide, 3′-ddACCGTTAATTGGGAGTGATTTCTp-5′, to the 3′-terminus of cDNA using T4 RNA ligase according to the manufacturer’s instructions.
Purify the resulting cDNA-anchor-oligonucleotide product by loading on a QIAquick spin column to remove protein and unreacted primers.
Amplify the purified cDNA-anchor-oligonucleotide by PCR using Taq DNA polymerase according to manufacturer’s instructions with the forward primer 5′-TTGAATTCGCATTGAGCACCTGC-3’ and reverse primer 5′-GGCAATTAACC CTCACTAAAG-3′ under the following conditions: 4 min at 94°C; 5 cycles of 94°C for 30 sec, 40°C for 1 min, 55°C for 1 min; 30 cycles of 94°C for 30 sec, 50°C for 1 min, 70°C for 1 min; 10 min at 72°C.
Purify the PCR products (100-200 bp) on 1% agarose gels.
Clone PCR products into the pCR2.1-TOPO vector using the TOPO TA cloning kit according to the manufacturer’s instructions.
Transform competent DH5α E. coli cells with the construct by standard techniques and plate onto LB agar.
Isolate individual colonies and purify plasmids from each colony using a plasmid miniprep kit according to the manufacturer’s instructions.
Sequence the plasmid DNA constructs encoding the cloned tRNAs from both ends using M13 forward (5´-GTAAAACGACGGCCAG-3´) and reverse (5´-CAGGAAACAGCTATGAC-3´) primers by standard techniques.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, NCI, Center for Cancer Research to DLH, NIH grants CA080946, GM061603 and GM065204 to VNG, DFG Priority Program 1784 to US (Schw914/5-1) to US, Towson University’s Jess and Mildred Fisher College of Science and Mathematics to PAT, who is a Jess and Mildred Fisher Endowed Chair of Biological Sciences, and NIH grants GM077073 and HD083616 to PRC.
Footnotes
Procedure can be scaled up for isolating RNA from large amounts of tissues or cells and will require larger amounts of materials and equipment.
It is very important to keep the tissues/cells frozen during RNA extraction by adding the phenol/buffer mix directly onto the frozen sample and begin homogenizing IMMEDIATELY.
DE52 can be stored in molecular biology grade H2O at 4°C with a few drops of CHCl3.
Typically, use 2 ml of bed volume for 1 g of tissue/cells used.
Transfer RNA can be stored at -20°C, but should be stored at -80°C, if storing for an extended period of time. Never store tRNA samples in liquid nitrogen as the extremely low temperature will shear off the CCA 3’-terminus.
RPC5 chromatography [13] is the only procedure described thus far for separating the two Sec tRNA isoforms. This procedure is currently actively being used in the laboratory of Dr. Byeong J. Lee. Dr. Lee can be contacted regarding any needs for RPC5 chromatography.
The amount of starting material will depend upon the investigator’s objective. For example, if the objective is to a) resolve small amounts of labeled tRNA[Ser]Sec isoforms, apply >100,000 cpm of [3H]seryl-tRNA; b) resolve double-labeled samples, apply ~250,000 cpm and ~25,000 cpm of [3H]- and [14C]seryl-tRNA, respectively; and c) resolve larger amounts of unlabeled tRNA[Ser]Sec, add up to 5-8 mg of sample.
It is extremely important to regenerate the RPC5 column with 1.5 M NaCl in the wash buffer to remove any remaining tRNA[Ser]Sec from the column. Washes at concentrations of 1.0 M NaCl [13] will leave residual, late eluting tRNA[Ser]Sec, which may affect ensuing runs; especially when carrying out multiple runs, wherein the first run was carried out with labeled material and the following run also involved material labeled with the same isotope yielding an eluted, contaminated product with false results.
It is recommended to precipitate the more highly purified mcm5U- and mcm5Um- containing samples in 10 mM MgCl2 with 3 volumes of EtOH and collect by high speed centrifugation as substantial losses of highly purified samples can occur by collection on filters.
Although gel electrophoresis was employed numerous times attempting to separate the two isoforms without success, this procedure would still seem to be a fruitful area to explore as a potential technique to achieve their separation.
The aminoacylation reaction can be scaled up using total tRNA at limiting conditions to produce large amounts of [3H]seryl-tRNA[Ser]Sec for analysis by RPC5 chromatography.
HL-60 cells contain a relatively high level of tRNA[Ser]Sec compared to the total seryl-tRNA population [26], whereas other mammalian cell types contain relatively smaller amounts, but many other cell types and media may be used and can be adapted to this procedure.
This method can also be used to prepare a tRNA[Ser]Sec-depleted tRNA population for use in in vitro translation systems or other assays requiring such a tRNA[Ser]Sec-deficient population. It may take 3-4 passes over the column to completely remove tRNA[Ser]Sec from the total tRNA population depending on the amount of starting material and beads used (e.g., 200 μg total tRNA and 2 ml of coated beads yields a tRNA population depleted of tRNA[Ser]Sec).
To achieve the complete sequence of tRNA, the 3′-end of the cDNA is extended by ligation using an anchor-oligonucleotide primer.
References
- 1.Hatfield DL, Choi IS, Ohama T, Jung JE, Diamond AM. Selenocysteine tRNA(Ser)sec isoacceptors as central components in selenoprotein biosynthesis in eukaryotes. In: Burk RF, editor. Selenium in Biology and Human Health. Springer-Verlag; New York, NY: 1994. pp. 25–44. [Google Scholar]
- 2.Baron C, Sturchler C, Wu XQ, Gross HJ, Krol A, Böck A. Eukaryotic selenocysteine inserting tRNA species support selenoprotein synthesis in Escherichia coli. Nucleic Acids Res. 1994;22(12):2228–2233. doi: 10.1093/nar/22.12.2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlson BA, Lee BJ, Tsuji PA, Tobe R, Park JM, Schweizer U, Gladyshev VN, Hatfield DL. Selenocysteine tRNA[Ser]Sec: From nonsense suppressor tRNA to the quintessential constituent in selenoprotein biosynthesis. In: Hatfield DL, Schweizer U, Tsuji PA, Gladyshev VN, editors. Selenium: Its molecular biology and role in human health. 4th. Springer Science+Business Media, LLC; New York, NY: 2016. (In Press) [Google Scholar]
- 4.Carlson BA, Novoselov SV, Kumaraswamy E, Lee BJ, Anver MR, Gladyshev VN, Hatfield DL. Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J Biol Chem. 2004;279(9):8011–8017. doi: 10.1074/jbc.M310470200. [DOI] [PubMed] [Google Scholar]
- 5.Kumaraswamy E, Carlson BA, Morgan F, Miyoshi K, Robinson GW, Su D, Wang S, Southon E, Tessarollo L, Lee BJ, Gladyshev VN, Hennighausen L, Hatfield DL. Selective removal of the selenocysteine tRNA [Ser]Sec gene (Trsp) in mouse mammary epithelium. Mol Cell Biol. 2003;23(5):1477–1488. doi: 10.1128/MCB.23.5.1477-1488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seeher S, Carlson BA, Miniard AC, Wirth EK, Mahdi Y, Hatfield DL, Driscoll DM, Schweizer U. Impaired selenoprotein expression in brain triggers striatal neuronal loss leading to co-ordination defects in mice. Biochem J. 2014;462(1):67–75. doi: 10.1042/BJ20140423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diamond AM, Choi IS, Crain PF, Hashizume T, Pomerantz SC, Cruz R, Steer CJ, Hill KE, Burk RF, McCloskey JA, Hatfield DL. Dietary selenium affects methylation of the wobble nucleoside in the anticodon of selenocysteine tRNA([Ser]Sec) J Biol Chem. 1993;268(19):14215–14223. [PubMed] [Google Scholar]
- 8.Carlson BA, Xu XM, Gladyshev VN, Hatfield DL. Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine tRNA. J Biol Chem. 2005;280(7):5542–5548. doi: 10.1074/jbc.M411725200. [DOI] [PubMed] [Google Scholar]
- 9.Hatfield DL, Carlson BA, Xu XM, Mix H, Gladyshev VN. Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog Nucleic Acid Res Mol Biol. 2006;81:97–142. doi: 10.1016/S0079-6603(06)81003-2. [DOI] [PubMed] [Google Scholar]
- 10.Carlson BA, Moustafa ME, Sengupta A, Schweizer U, Shrimali R, Rao M, Zhong N, Wang S, Feigenbaum L, Lee BJ, Gladyshev VN, Hatfield DL. Selective restoration of the selenoprotein population in a mouse hepatocyte selenoproteinless background with different mutant selenocysteine tRNAs lacking Um34. J Biol Chem. 2007;282(45):32591–32602. doi: 10.1074/jbc.M707036200. [DOI] [PubMed] [Google Scholar]
- 11.Roe BA. Studies on human tRNA. I. The rapid, large scale isolation and partial fractionation of placenta and liver tRNA. Nucleic Acids Res. 1975;2(1):21–42. doi: 10.1093/nar/2.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatfield D, Diamond A, Dudock B. Opal suppressor serine tRNAs from bovine liver form phosphoseryl-tRNA. Proc Natl Acad Sci U S A. 1982;79(20):6215–6219. doi: 10.1073/pnas.79.20.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelmers AD, Heatherly DE. Columns for rapid chromatographic separation of small amounts of tracer-labeled transfer ribonucleic acids. Anal Biochem. 1971;44(2):486–495. doi: 10.1016/0003-2697(71)90236-3. [DOI] [PubMed] [Google Scholar]
- 14.Hatfield D, Matthews CR, Rice M. Aminoacyl-transfer RNA populations in mammalian cells chromatographic profiles and patterns of codon recognition. Biochim Biophys Acta. 1979;564(3):414–423. doi: 10.1016/0005-2787(79)90032-7. [DOI] [PubMed] [Google Scholar]
- 15.Carlson BA, Hatfield DL. Transfer RNAs that insert selenocysteine. Methods Enzymol. 2002;347:24–39. doi: 10.1016/s0076-6879(02)47005-x. [DOI] [PubMed] [Google Scholar]
- 16.Diamond A, Dudock B, Hatfield D. Structure and properties of a bovine liver UGA suppressor serine tRNA with a tryptophan anticodon. Cell. 1981;25(2):497–506. doi: 10.1016/0092-8674(81)90068-4. [DOI] [PubMed] [Google Scholar]
- 17.Sharp SJ, Stewart TS. The characterization of phosphoseryl tRNA from lactating bovine mammary gland. Nucleic Acids Res. 1977;4(7):2123–2136. doi: 10.1093/nar/4.7.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silberklang M, Gillum AM, RajBhandary UL. Use of in vitro 32P labeling in the sequence analysis of nonradioactive tRNAs. Methods Enzymol. 1979;59:58–109. doi: 10.1016/0076-6879(79)59072-7. [DOI] [PubMed] [Google Scholar]
- 19.Rao M, Carlson BA, Novoselov SV, Weeks DP, Gladyshev VN, Hatfield DL. Chlamydomonas reinhardtii selenocysteine tRNA[Ser]Sec. RNA. 2003;9(8):923–930. doi: 10.1261/rna.5510503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novoselov SV, Rao M, Onoshko NV, Zhi H, Kryukov GV, Xiang Y, Weeks DP, Hatfield DL, Gladyshev VN. Selenoproteins and selenocysteine insertion system in the model plant cell system, Chlamydomonas reinhardtii. EMBO J. 2002;21(14):3681–3693. doi: 10.1093/emboj/cdf372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shrimali RK, Lobanov AV, Xu XM, Rao M, Carlson BA, Mahadeo DC, Parent CA, Gladyshev VN, Hatfield DL. Selenocysteine tRNA identification in the model organisms Dictyostelium discoideum and Tetrahymena thermophila. Biochem Biophys Res Commun. 2005;329(1):147–151. doi: 10.1016/j.bbrc.2005.01.120. [DOI] [PubMed] [Google Scholar]
- 22.Köhrer C, Rajbhandary UL. The many applications of acid urea polyacrylamide gel electrophoresis to studies of tRNAs and aminoacyl-tRNA synthetases. Methods. 2008;44(2):129–138. doi: 10.1016/j.ymeth.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merrick WC. Assays for eukaryotic protein synthesis. Methods Enzymol. 1979;60:108–123. doi: 10.1016/s0076-6879(79)60011-3. [DOI] [PubMed] [Google Scholar]
- 24.Songe-Møller L, van den Born E, Leihne V, Vagbo CB, Kristoffersen T, Krokan HE, Kirpekar F, Falnes PO, Klungland A. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol Cell Biol. 2010;30(7):1814–1827. doi: 10.1128/MCB.01602-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoenmakers E, Carlson B, Agostini M, Moran C, Rajanayagam O, Bochukova E, Tobe R, Peat R, Gevers E, Muntoni F, Guicheney P, Schoenmakers N, Farooqi S, Lyons G, Hatfield D, Chatterjee K. Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J Clin Invest. 2016;126(3):992–996. doi: 10.1172/JCI84747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatfield D, Lee BJ, Hampton L, Diamond AM. Selenium induces changes in the selenocysteine tRNA[Ser]Sec population in mammalian cells. Nucleic Acids Res. 1991;19(4):939–943. doi: 10.1093/nar/19.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]