

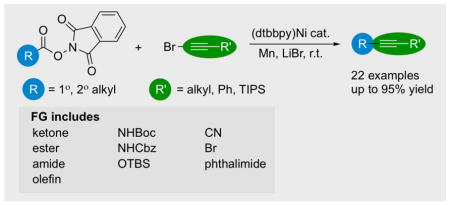
Abstract
A new method for the synthesis of terminal and internal alkynes from the nickel-catalyzed decarboxylative coupling of N-hydroxyphthalimide esters (NHP esters) and bromoalkynes is presented. This reductive cross-electrophile coupling is the first to use a C(sp)-X electrophile, and appears to proceed via an alkynylnickel intermediate. The internal alkyne products are obtained in 41–95% yield without the need for a photocatalyst, light, or strong oxidant. The reaction displays a broad scope of carboxylic acid and alkyne coupling partners and can tolerate an array of functional groups including a carbamate N-H, halogen, nitrile, olefin, ketone, and ester. Mechanistic studies suggest that this process does not involve an alkynylmanganese reagent and involves nickel-mediated bond formation.
Keywords: Cross-electrophile coupling, Homogeneous catalysis, Alkynes, Alkylation, Cross-coupling
Graphical Abstract
A new method for the synthesis of terminal and internal alkynes from the nickel-catalyzed decarboxylative coupling of N-hydroxyphthalimide esters (NHP esters) and bromoalkynes is presented. This reductive cross-electrophile coupling is the first to use a Csp-X electrophile, and appears to proceed via an alkynylnickel intermediate. The internal alkyne products are obtained in 41–95% yield without the need for a photocatalyst, light, or strong oxidant. The reaction displays a broad scope of carboxylic acid and alkyne coupling partners and can tolerate an array of functional groups including a carbamate N-H, halogen, nitrile, olefin, ketone, and ester. Mechanistic studies suggest that this process does not involve an alkynylmanganese reagent and involves nickel-mediated bond formation.
Cross-electrophile coupling[1] has recently been shown to be a general approach to the formation of C-C bonds and the number and type of electrophiles that can be cross-coupled has grown rapidly in the past decade. The formation of C(sp3)-C(sp2) bonds has been the most studied,[2],[3] but the selective formation of C(sp2)-C(sp2)[4] and C(sp3)-C(sp3)[5] bonds has also been demonstrated (Scheme 1). In particular, the coupling of aryl[2] and vinyl halides[3] with alkyl halides has proven to be a useful alternative to other cross-coupling approaches.[6] In contrast, cross-electrophile coupling to form C(sp)-C(spx) bonds has not been demonstrated (Scheme 1), even though bromoalkynes are easily generated from terminal alkynes.[7] [8]
Scheme 1.
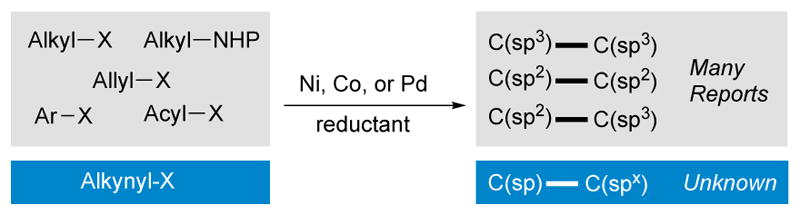
Types of bonds formed by reductive cross-electrophile coupling.
This absence in the literature could be related to a number of potential problems. First, the high reactivity of bromoalkynes could pose a selectivity problem.[9] Second, the low steric bulk of the alkyne could lead to rapid transmetalation and homocoupling to form diynes. Finally, alkynes can act as radical acceptors and strong ligands, potentially leading to catalyst inhibition and side reactions.
We were motivated to explore the reactivity of alkynyl electrophiles in order to develop a new synthesis of alkylated alkynes from carboxylic acids. The most often used solution for this transformation is through the Corey-Fuchs reaction[10] or the Seyferth-Gilbert homologation[11] of aldehydes to terminal alkynes. While this approach has proven useful, it often requires the synthesis of the aldehyde from the more abundant carboxylic acid, strong base to effect the rearrangement, and additional steps if an internal alkyne is desired.
We envisioned a more convergent approach, the decarboxylative coupling of an N-hydroxyphthalimide (NHP) ester with an alkynyl bromide (Scheme 2).[12],[13] Strategically, this approach differs from the above strategy because it avoids the aldehyde intermediate. Additionally, the alkyne component can be substituted, allowing for direct synthesis of functionalized internal alkynes. While this exact transformation is unknown, related strategies have been investigated recently. For example, the oxidative decarboxylation of aliphatic acids to form alkyl radicals with subsequent capture by alkynyl electrophiles.[14] Concurrent with these studies, Fu reported on the coupling of terminal alkynes with α-amino NHP esters[15] and Baran reported on the coupling of N-hydroxytetrachlorophthalimide (TCNHPI) esters with alkynylzinc or magnesium reagents.[16] Our proposed approach would avoid the need for organometallic reagents and the more expensive TCNHPI[17] without being limited to α-amino acid derivatives.[15] Although this approach would use alkyne electrophiles, bromoalkynes[7] are more easily prepared than ethynylbenziodoxolones[18] or alkynyl sulfones.[14a–c]
Scheme 2.
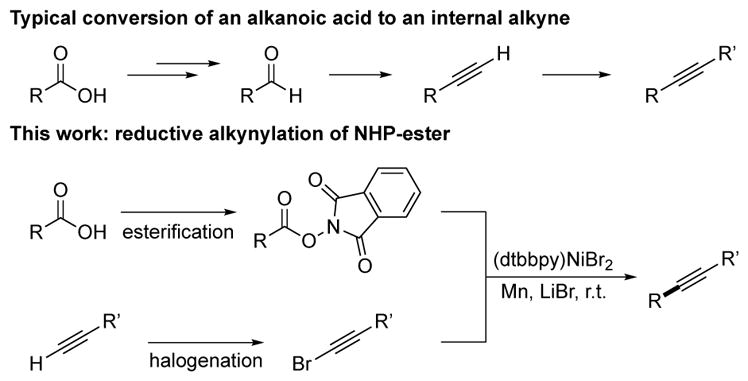
Comparison of alkynylation strategies.
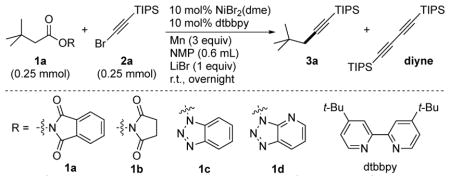
Initial results using conditions we reported for the coupling of iodoarenes to NHP esters[13a] resulted in a low yield of the cross-product (Table 1, entry 1, 3a) and a large amount of the homodimer (diyne). Two changes solved this selectivity problem (entries 1–3): changing the reductant from Zn to Mn (from 11 to 43% yield) and adding in LiBr as an additive (43% to 78% yield).[4c, 19] Examination of several other salts showed that both LiCl and LiBr improve the yield of cross-product at the expense of diyne formation. Stoichiometric amounts of LiBr were required to observe a beneficial effect (entries 8 and 9). Small improvements in yield were obtained when using an excess of either coupling partner (entries 10 and 11).
Table 1.
Optimization of the decarboxylative alkynylation.
| |||
|---|---|---|---|
|
| |||
| Entry | Change from Conditions in Scheme | Yield of 3a[a] | Yield of diyne[a] |
| 1 | Zn instead of Mn, omit LiBr | 11 | 40 |
| 2 | omit LiBr | 43 | 23 |
| 3 | none | 78 | 11 |
| 4 | LiCl instead of LiBr | 67 | 9 |
| 5 | KF instead of LiBr | 42 | 21 |
| 6 | KBr instead of LiBr | 43 | 16 |
| 7 | NaI instead of LiBr | 46 | 19 |
| 8 | LiBr (2 equiv) | 78 | 14 |
| 9 | LiBr (0.5 equiv) | 53 | 13 |
| 10 | 1.5 equiv of 1a | 89 (86) | 8 |
| 11 | 1.5 equiv 2a | 84 | 18 |
| 12 | omit dtbbpy | n.d. | 6 |
| 13 | omit NiBr2(dme) | n.d. | n.d |
| 14 | 1b–d instead of 1a | n.d. | >40 |
Corrected GC yield using n-dodecane as the internal standard; isolated yield in parentheses.
Control experiments showed that both nickel and ligand were essential for this transformation (entries 12 and 13). A redox active ester appears to be required because 1b was not consumed under these reaction conditions (entry 14). Other redox-active esters (HOBt or HOAt esters 1c and 1d) were also not consumed under these conditions (entry 14).
Under the optimized conditions, various alkyl NHP esters and bromoalkynes were cross-coupled successfully in 41–95% yield (Table 2). Functional group compatibility is promising, and the reaction tolerates ketones (3b and 3c), esters (3d) and the N-H of secondary carbamates (3e). The high reactivity of the NHP esters compared to other alkyl electrophiles was demonstrated in product 3f, where an alkyl bromide was tolerated, albeit in moderate yield. Another advantage of the NHP esters is that readily available amino acids can be easily converted into useful alkynyl amino acids (3g) and alkynyl amines (3k and 3m). Propargylglycine, obtainable from 3g by routine deprotection, has been made on large scale to support pharmaceutical synthesis.[20] Our route, from aspartic acid, is an attractive alternative to the chiral auxiliary approach that was recently reported.[21] Linoleic acid was coupled to form 3i in high yield without isomerization of the Z-olefins. Although these reactions were set up in a glovebox for convenience, the chemistry could be run on preparative scale (5 mmol NHP ester) on the benchtop without the need for strict exclusion of moisture and air. Subsequent removal of the TIPS protecting group with nBu4NF (1 M in THF) gave the terminal alkyne in 84% yield over the two steps.
Table 2.
Substrate scope of NHP-ester and bromoalkyne.

|
This reaction was run on 3.3 mmol scale and the crude product was deprotected with TBAF before isolation. Yield is over two steps.
Reaction run at 50 °C.
Reaction run at lower concentration (1.8 mL of NMP).
α-Branched redox-active esters were suitable substrates for the alkynylation reaction (3j-n), but reactions with dibranched substrates did not form product. The value of using an aliphatic acid as a source for the alkyl moiety was demonstrated with these compounds, as the corresponding alkyl halides are not commercially available or fail to couple.[22] For example, 3-halotetrahydrofurans, 2-halopyrrolidines, and 2-amido alkyl halides are not easily accessible starting materials, whereas the corresponding aliphatic acids are relatively affordable and can be successfully transformed into the alkynylation products in moderate to good yields (3j, 3k and 3m).
Next, the scope of the bromoalkynes was tested in our reductive coupling method. Both the alkyl substituted and the phenyl substituted bromoalkynes were coupled with 3,3-dimethylbutanoic acid NHP ester in good yields (3o and 3p). Several functional groups can be appended onto the alkyl chain of the bromoalkynes including malonate (3q), OTBS (3r), cyanide (3s), phthalimide (3t), and alkene (3u), without preventing product formation.
Not all substrates tested coupled in high yield under these conditions. An NHP ester with a free carboxylic acid and a bromoalkyne with a free hydroxyl group resulted in low yields. Finally, NHP esters that would generate an allylic or benzylic radical failed to give alkynylation products.
Three potential mechanisms were proposed: 1) in-situ formation of an alkynyl manganese reagent and subsequent nickel-catalyzed cross-coupling,[16] 2) nickel-catalyzed radical generation[13b, 23],[13a] and outer-sphere addition of the free radical to the bromoalkyne;[14a–c] 3) sequential reaction of two electrophiles with the nickel catalyst and bond-formation at nickel.[24] At this time, we can rule out the first two mechanistic possibilities (vide infra).
Given the high reactivity of alkynyl bromides and the positive effect of LiBr on the reaction outcome, an additive known to increase the rate of insertion of Mn or Zn,[25] we suspected an alkynylmanganese reagent might be an intermediate. However, when silylated bromoalkyne 2a was subjected to standard conditions without adding nickel or ligand (Scheme 3a), the bromoalkyne was not consumed and only a trace of hydrodebrominated alkyne was observed. This shows that direct insertion of Mn into the C-Br bond is slow and suggests that Mn serves to reduce a nickel intermediate. Although we are unsure of the role of LiBr in minimizing formation of diyne, our own work and that of Osakada and Yamamoto[26] suggests that diyne arises from dialkynylnickel(II) intermediates. Added LiBr could slow ligand exchange between alkynylnickel(II) intermediates, inhibiting diyne formation.
Scheme 3.
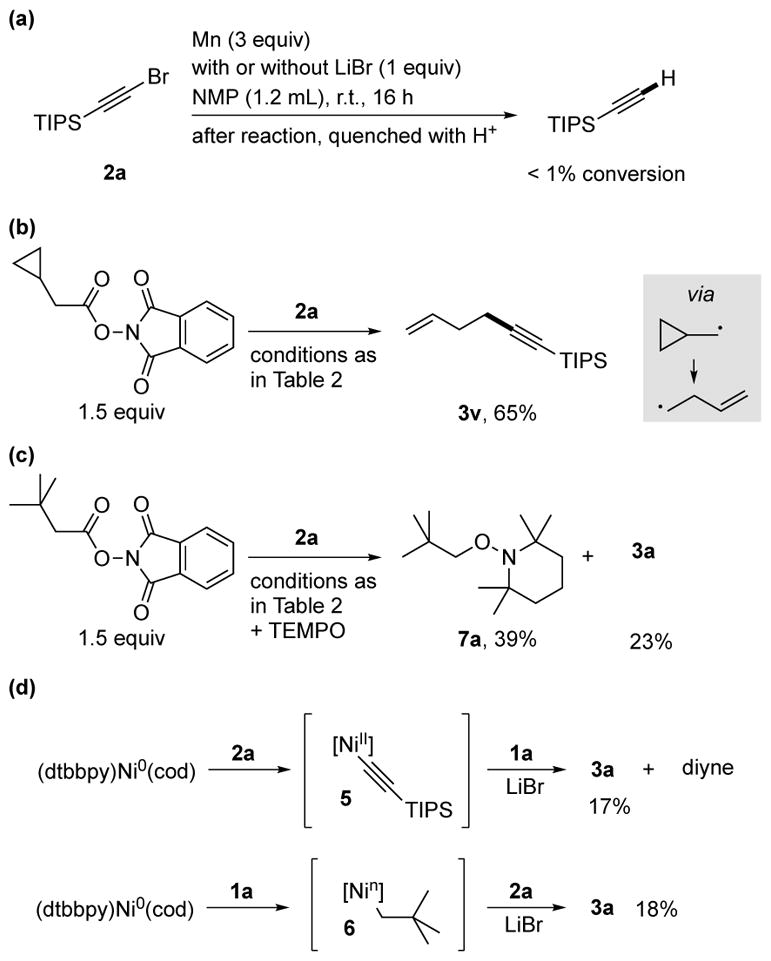
Mechanistic studies: (a) reaction of bromoalkyne with Mn, (b) cyclopropylmethyl radical rearrangement, (c) TEMPO radical trap, and (d) reactivity of nickel towards the two electrophiles.
Two experiments suggested the presence of a free alkyl radical in the reaction. The NHP ester of cyclopropylacetic acid (1v) reacted to form the rearranged product exclusively (Scheme 3b).[24], [27–28] In addition, a standard reaction run in the presence of TEMPO formed both the expected cross product and alkylated TEMPO (Scheme 3c).[27a] This product required nickel, confirming that nickel is required for radical generation.
While these results are consistent with mechanism two, the coupling of a free-radical with the bromoalkyne to form product is unlikely for three reasons. First, it has been reported that this process is not high yielding.[14a, 14c, 18] Second, our studies have shown that the ligand on nickel is essential to bond formation but not consumption of the NHP ester (Table 1, entry 12).[13a] Third, the major challenge during reaction optimization was too-rapid consumption of the bromoalkyne and subsequent diyne formation, a process that requires an alkynylnickel intermediate (Table 1 and Scheme 3d).
Finally, we briefly studied the reactivity of two possible nickel intermediates to shed light on the mechanism (Scheme 3d). The nickel(0) complex (dtbbpy)Ni0(cod) reacted rapidly with both electrophiles at rt, in each case forming an as-yet unidentified organonickel intermediate (5 and 6).[29] Both of these intermediates, either after isolation and washing to remove excess electrophile or without isolation, react with the complementary electrophile to form cross-product.[22e],[30] While these studies demonstrate that an inner-sphere, nickel-mediated mechanism is possible, further studies will be required before a complete catalytic cycle can be proposed.[24]
In conclusion, the reductive alkynylation to form a C(sp)-C(sp3) bond has been achieved, opening a new class of reactions to cross-electrophile coupling and demonstrating that even highly reactive electrophiles can be coupled selectively. The resulting method provides a convenient and direct synthesis of a wide variety of internal alkynes from convenient precursors and useful alternative to methods that proceed via olefination or use alkyl halides.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge funding from the NIH NIGMS (R01 GM097243 to DJW) and the NSF (NSF DGE-1419118 to AMO). DJW is a Camille Dreyfus Teacher-Scholar. Additional funding from Novartis, Pfizer, and Boehringer Ingelheim is also gratefully acknowledged. Matthew Goldfogel, Amanda Spiewak, and Keywan Johnson (Univ. of Rochester) are acknowledged for assistance with substrate synthesis. Theresa Iannuzzi is thanked for assistance with EPR spectroscopy experiments.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Knappke CEI, Grupe S, Gärtner D, Corpet M, Gosmini C, Jacobi von Wangelin A. Chem – Eur J. 2014;20:6828–6842. doi: 10.1002/chem.201402302. [DOI] [PubMed] [Google Scholar]; b) Weix DJ. Acc Chem Res. 2015;48:1767–1775. doi: 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gu J, Wang X, Xue W, Gong H. Org Chem Front. 2015;2:1411–1421. [Google Scholar]; d) Moragas T, Correa A, Martin R. Chem – Eur J. 2014;20:8242–8258. doi: 10.1002/chem.201402509. [DOI] [PubMed] [Google Scholar]
- 2.a) Krasovskiy A, Duplais C, Lipshutz BH. J Am Chem Soc. 2009;131:15592–15593. doi: 10.1021/ja906803t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Amatore M, Gosmini C. Chem – Eur J. 2010;16:5848–5852. doi: 10.1002/chem.201000178. [DOI] [PubMed] [Google Scholar]; c) Everson DA, Shrestha R, Weix DJ. J Am Chem Soc. 2010;132:920–921. doi: 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]; d) Hansen EC, Pedro DJ, Wotal AC, Gower NJ, Nelson JD, Caron S, Weix DJ. Nat Chem. 2016;8:1126–1130. doi: 10.1038/nchem.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wang X, Wang S, Xue W, Gong H. J Am Chem Soc. 2015;137:11562–11565. doi: 10.1021/jacs.5b06255. [DOI] [PubMed] [Google Scholar]; f) Li X, Feng Z, Jiang ZX, Zhang X. Org Lett. 2015;17:5570–5573. doi: 10.1021/acs.orglett.5b02716. [DOI] [PubMed] [Google Scholar]
- 3.a) Johnson KA, Biswas S, Weix DJ. Chem – Eur J. 2016;22:7399–7402. doi: 10.1002/chem.201601320. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Krasovskaya V, Krasovskiy A, Bhattacharjya A, Lipshutz BH. Chem Commun. 2011;47:5717–5719. doi: 10.1039/c1cc11087j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Krasovskiy A, Duplais C, Lipshutz BH. Org Lett. 2010;12:4742–4744. doi: 10.1021/ol101885t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Qiu C, Yao K, Zhang X, Gong H. Org Biomol Chem. 2016;14:11332–11335. doi: 10.1039/c6ob02269c. [DOI] [PubMed] [Google Scholar]
- 4.a) Gomes P, Gosmini C, Périchon J. Tetrahedron. 2003;59:2999–3002. [Google Scholar]; b) Amatore M, Gosmini C. Angew Chem Int Ed. 2008;47:2089–2092. doi: 10.1002/anie.200704402. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;120:2119–2122. [Google Scholar]; c) Ackerman LKG, Lovell MM, Weix DJ. Nature. 2015;524:454–457. doi: 10.1038/nature14676. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu J, Ren Q, Zhang X, Gong H. Angew Chem Int Ed. 2016;55:15544–15548. doi: 10.1002/anie.201607959. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2016;128:15773–15777. [Google Scholar]
- 5.a) Qian X, Auffrant A, Felouat A, Gosmini C. Angew Chem Int Ed. 2011;50:10402–10405. doi: 10.1002/anie.201104390. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2011;123:10586–10589. [Google Scholar]; b) Yu X, Yang T, Wang S, Xu H, Gong H. Org Lett. 2011;13:2138–2141. doi: 10.1021/ol200617f. [DOI] [PubMed] [Google Scholar]; c) Anka-Lufford LL, Prinsell MR, Weix DJ. J Org Chem. 2012;77:9989–10000. doi: 10.1021/jo302086g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Xu H, Zhao C, Qian Q, Deng W, Gong H. Chem Sci. 2013;4:4022–4029. [Google Scholar]
- 6.a) Molander GA, Traister KM, O’Neill BT. J Org Chem. 2014;79:5771–5780. doi: 10.1021/jo500905m. [DOI] [PubMed] [Google Scholar]; b) Molander GA, Traister KM, O’Neill BT. J Org Chem. 2015;80:2907–2911. doi: 10.1021/acs.joc.5b00135. [DOI] [PubMed] [Google Scholar]; c) Fegheh-Hassanpour Y, Arif T, Sintim HO, Al Mamari HH, Hodgson DM. Org Lett. 2017 doi: 10.1021/acs.orglett.7b01513. [DOI] [PubMed] [Google Scholar]
- 7.a) Hofmeister H, Annen K, Laurent H, Wiechert R. Angew Chem Int Ed. 1984;23:727–729. [Google Scholar]; Angew Chem. 1984;96:720–722. [Google Scholar]; b) Brandsma L, Verkruijsse HD. Synthesis. 1990:984–985. [Google Scholar]
- 8.a) Brand JP, Waser J. Chem Soc Rev. 2012;41:4165–4179. doi: 10.1039/c2cs35034c. [DOI] [PubMed] [Google Scholar]; b) Wu W, Jiang H. Acc Chem Res. 2014;47:2483–2504. doi: 10.1021/ar5001499. [DOI] [PubMed] [Google Scholar]
- 9.a) Thaler T, Guo LN, Mayer P, Knochel P. Angew Chem Int Ed. 2011;50:2174–2177. doi: 10.1002/anie.201006879. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2011;123:2222–2225. [Google Scholar]; b) Corpet M, Bai XZ, Gosmini C. Adv Synth Catal. 2014;356:2937–2942. [Google Scholar]
- 10.a) Desai NB, McKelvie N, Ramirez F. J Am Chem Soc. 1962;84:1745–1747. [Google Scholar]; b) Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769–3772. [Google Scholar]; c) Habrant D, Rauhala V, Koskinen AMP. Chem Soc Rev. 2010;39:2007–2017. doi: 10.1039/b915418c. [DOI] [PubMed] [Google Scholar]
- 11.a) Seyferth D, Marmor RS, Hilbert P. J Org Chem. 1971;36:1379–1386. [Google Scholar]; b) Ohira S. Synth Commun. 1989;19:561–564. [Google Scholar]; c) Müller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996:521–522. [Google Scholar]
- 12.a) Okada K, Okamoto K, Oda M. J Am Chem Soc. 1988;110:8736–8738. [Google Scholar]; b) Okada K, Okamoto K, Oda M. J Chem Soc, Chem Commun. 1989:1636–1637. [Google Scholar]; c) Okada K, Okamoto K, Morita N, Okubo K, Oda M. J Am Chem Soc. 1991;113:9401–9402. [Google Scholar]; d) Schnermann MJ, Overman LE. Angew Chem Int Ed. 2012;51:9576–9580. doi: 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2012;124:9714–9718. [Google Scholar]; e) Pratsch G, Lackner GL, Overman LE. J Org Chem. 2015;80:6025–6036. doi: 10.1021/acs.joc.5b00795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Huihui KMM, Caputo JA, Melchor Z, Olivares AM, Spiewak AM, Johnson KA, DiBenedetto TA, Kim S, Ackerman LKG, Weix DJ. J Am Chem Soc. 2016;138:5016–5019. doi: 10.1021/jacs.6b01533. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Toriyama F, Cornella J, Wimmer L, Chen TG, Dixon DD, Creech G, Baran PS. J Am Chem Soc. 2016;138:11132–11135. doi: 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Le Vaillant F, Courant T, Waser J. Angew Chem Int Ed. 2015;54:11200–11204. doi: 10.1002/anie.201505111. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:11352–11356. [Google Scholar]; b) Yang J, Zhang J, Qi L, Hu C, Chen Y. Chem Commun. 2015;51:5275–5278. doi: 10.1039/c4cc06344a. [DOI] [PubMed] [Google Scholar]; c) Zhou Q-Q, Guo W, Ding W, Wu X, Chen X, Lu L-Q, Xiao W-J. Angew Chem Int Ed. 2015;54:11196–11199. doi: 10.1002/anie.201504559. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:11348–11351. [Google Scholar]; d) Yang C, Yang JD, Li YH, Li X, Cheng JP. J Org Chem. 2016;81:12357–12363. doi: 10.1021/acs.joc.6b02385. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Zhang P, Jiang M, Yang H, Fu H. Org Lett. 2017;19:1016–1019. doi: 10.1021/acs.orglett.6b03888. [DOI] [PubMed] [Google Scholar]
- 16.Smith JM, Qin T, Merchant RR, Edwards JT, Malins LR, Liu Z, Che G, Shen Z, Shaw SA, Eastgate MD, Baran PS. Angew Chem Int Ed. 2017 doi: 10.1002/anie.201705107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2017 doi: 10.1002/ange.201705107. [DOI] [Google Scholar]
- 17.Currently, TCNHPI is two orders of magnitude more expensive than NHP. Sigma-Aldrich prices: $42/mole for NHP vs. $2560/mole for TCNHPI.
- 18.Liu X, Wang Z, Cheng X, Li C. J Am Chem Soc. 2012;134:14330–14333. doi: 10.1021/ja306638s. [DOI] [PubMed] [Google Scholar]
- 19.a) Zhao C, Jia X, Wang X, Gong H. J Am Chem Soc. 2014;136:17645–17651. doi: 10.1021/ja510653n. [DOI] [PubMed] [Google Scholar]; b) Suzuki N, Hofstra JL, Poremba KE, Reisman SE. Org Lett. 2017;19:2150–2153. doi: 10.1021/acs.orglett.7b00793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun G, Wei M, Luo Z, Liu Y, Chen Z, Wang Z. Org Process Res Dev. 2016;20:2074–2079. [Google Scholar]
- 21.a) Belokon YN, Bulychev AG, Vitt SV, Struchkov YT, Batsanov AS, Timofeeva TV, Tsyryapkin VA, Ryzhov MG, Lysova LA. J Am Chem Soc. 1985;107:4252–4259. [Google Scholar]; b) Belokon YN, Bakhmutov VI, Chernoglazova NI, Kochetkov KA, Vitt SV, Garbalinskaya NS, Belikov VM. J Chem Soc, Perkin Trans 1. 1988:305–312. [Google Scholar]; c) Collet S, Bauchat P, Danion-Bougot R, Danion D. Tetrahedron: Asymmetry. 1998;9:2121–2131. [Google Scholar]; d) Houck D, Luis Aceña J, Soloshonok VA. Helv Chim Acta. 2012;95:2672–2679. [Google Scholar]
- 22.a) Eckhardt M, Fu GC. J Am Chem Soc. 2003;125:13642–13643. doi: 10.1021/ja038177r. [DOI] [PubMed] [Google Scholar]; b) Altenhoff G, Würtz S, Glorius F. Tetrahedron Lett. 2006;47:2925–2928. [Google Scholar]; c) Vechorkin O, Barmaz D, Proust V, Hu X. J Am Chem Soc. 2009;131:12078–12079. doi: 10.1021/ja906040t. [DOI] [PubMed] [Google Scholar]; d) Yi J, Lu X, Sun YY, Xiao B, Liu L. Angew Chem Int Ed. 2013;52:12409–12413. doi: 10.1002/anie.201307069. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:12635–12639. [Google Scholar]; e) Pérez García PM, Ren P, Scopelliti R, Hu X. ACS Catal. 2015;5:1164–1171. [Google Scholar]
- 23.a) Cornella J, Edwards JT, Qin T, Kawamura S, Wang J, Pan CM, Gianatassio R, Schmidt M, Eastgate MD, Baran PS. J Am Chem Soc. 2016;138:2174–2177. doi: 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS. Science. 2016;353:801–805. doi: 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang J, Qin T, Chen TG, Wimmer L, Edwards JT, Cornella J, Vokits B, Shaw SA, Baran PS. Angew Chem Int Ed. 2016;55:9676–9679. doi: 10.1002/anie.201605463. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2016;128:9828–9831. [Google Scholar]; d) Edwards JT, Merchant RR, McClymont KS, Knouse KW, Qin T, Malins LR, Vokits B, Shaw SA, Bao D-H, Wei F-L, Zhou T, Eastgate MD, Baran PS. Nature. 2017 doi: 10.1038/nature22307. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Qin T, Malins LR, Edwards JT, Merchant RR, Novak AJE, Zhong JZ, Mills RB, Yan M, Yuan C, Eastgate MD, Baran PS. Angew Chem Int Ed. 2017;56:260–265. doi: 10.1002/anie.201609662. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2017;129:3367–3371. [Google Scholar]; f) Sandfort F, O’Neill MJ, Cornella J, Wimmer L, Baran PS. Angew Chem Int Ed. 2017;56:3319–3323. doi: 10.1002/anie.201612314. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2017;129:3367–3371. [Google Scholar]
- 24.Biswas S, Weix DJ. J Am Chem Soc. 2013;135:16192–16197. doi: 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Feng C, Cunningham DW, Easter QT, Blum SA. J Am Chem Soc. 2016;138:11156–11159. doi: 10.1021/jacs.6b08465. [DOI] [PubMed] [Google Scholar]; b) Krasovskiy A, Malakhov V, Gavryushin A, Knochel P. Angew Chem Int Ed. 2006;45:6040–6044. doi: 10.1002/anie.200601450. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:6186–6190. [Google Scholar]; c) Piller FM, Appukkuttan P, Gavryushin A, Helm M, Knochel P. Angew Chem Int Ed. 2008;47:6802–6806. doi: 10.1002/anie.200801968. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;120:6907–6911. [Google Scholar]; d) Sase S, Jaric M, Metzger A, Malakhov V, Knochel P. J Org Chem. 2008;73:7380–7382. doi: 10.1021/jo801063c. [DOI] [PubMed] [Google Scholar]; e) Sämann C, Schade MA, Yamada S, Knochel P. Angew Chem Int Ed. 2013;52:9495–9499. doi: 10.1002/anie.201302058. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:9673–9677. [Google Scholar]; f) Fillon H, Gosmini C, Périchon J. J Am Chem Soc. 2003;125:3867–3870. doi: 10.1021/ja0289494. [DOI] [PubMed] [Google Scholar]
- 26.a) Osakada K, Yamamoto T. Coord Chem Rev. 2000;198:379–399. [Google Scholar]; b) Yamamoto T, Wakabayashi S, Osakada K. J Organomet Chem. 1992;428:223–237. [Google Scholar]
- 27.a) Kinney RJ, Jones WD, Bergman RG. J Am Chem Soc. 1978;100:7902–7915. [Google Scholar]; b) Ash CE, Hurd PW, Darensbourg MY, Newcomb M. J Am Chem Soc. 1987;109:3313–3317. [Google Scholar]
- 28.Note that cyclopropylnickel intermediates are also known to rearrange via a presumed non-radical process. Pinke PA, Stauffer RD, Miller RG. J Am Chem Soc. 1974;96:4229–4234.Masarwa A, Marek I. Chem--Eur J. 2010;16:9712–9721. doi: 10.1002/chem.201001246.Nakamura I, Yamamoto Y. Adv Synth Catal. 2002;344:111–129.
- 29.For further details on these experiments and preliminary characterization of the intermediates, see the Supporting Information. Although the organonickel intermediates are paramagnetic, EPR data suggests a integer-spin complex, such as tetrahedral or octahedral nickel(II), and not a ½-integer spin complex.
- 30.Similar to this situation, both alkylnickel(II) and arylnickel(II) intermediates are known to react with the complementary electrophile to form alkylated arenes. See ref. 24 and references cited therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.