

Abstract
Human mPGES-1 has emerged as a promising target in exploring a next generation of anti-inflammatory drugs, as selective mPGES-1 inhibitors are expected to discriminatively suppress the production of induced PGE2 without blocking the normal biosynthesis of other prostanoids including homeostatic PGE2. Therefore, this therapeutic approach is believed to reduce the adverse effects associated with the application of traditional non-steroidal anti-inflammatory drugs (tNSAIDs) and selective COX-2 inhibitors (coxibs). Identified from structure-based virtue screening, the compound with (Z)-5-benzylidene-2-iminothiazolidin-4-one scaffold was used as lead in rational design of novel inhibitors. Besides, we further designed, synthesized, and evaluated 5-((1,3-diphenyl-1H-pyrazol-4-yl)methylene)pyrimidine-2,4,6(1H,3H,5H)-triones and structurally related derivatives for their in vitro inhibitory activities. According to in vitro activity assays, a number of these compounds were capable of inhibiting human mPGES-1, with the desirable selectivity for mPGES-1 over COX isozymes.
Keywords: Anti-inflammatory drugs, pyrazole, barbituric acid, mPGES-1 inhibitor
Graphical Abstract
Rational molecular design, followed by synthesis and in vitro activity assays for evaluating both the potency and selectivity, has led to the discovery of a set of novel, potent and selective mPGES-1 inhibitors.
In the eicosanoid pathway, arachidonic acid (AA) is converted to prostaglandin H2 (PGH2) by the action of cyclooxygenases (COX-1 and COX-2). PGH2 serves as a common precursor for various biologically active prostanoids, such as thromboxane A2 (TXA2), PGD2, PGI2, PGF2α, and PGE2, depending on different distal synthases.1, 2 Among these prostanoids, PGE2 is well recognized as an important inflammatory mediator. PGE2 is isomerized from PGH2 catalyzed by three distinct synthases, including microsomal prostaglandin E2 synthase-1 (mPGES-1), mPGES-2, and cytosolic prostaglandin E2 synthase.3 Unlike the other two constitutively expressed enzymes, the expression of mPGES-1, similar to that of COX-2, is highly inducible in response to pro-inflammatory stimuli.4
As two generations of anti-inflammatory drugs, traditional non-steroidal anti-inflammatory drugs (tNSAIDs) and coxibs represent the mainstream for the treatment of inflammation-related symptoms by either non-selectively inhibiting COX isozymes,5 or selectively inhibiting COX-2,6 respectively. However, both categories shut down the biosynthesis of all downstream prostanoids and, so, their application is associated with considerable adverse effects. The tNSAIDs trigger gastrointestinal (GI) ulceration because of the interference with COX-1-derived protective function in GI tract;7 coxibs, as specific COX-2 inhibitors, on the other hand, break the internal balance of vasodilative PGI2 and vasoconstrictive TXA2 and thus result in cardiovascular risk.8 Since PGE2 is the major inducible PG in inflammation, inhibiting mPGES-1 is believed as a promising therapeutic approach in the development of the next generation of anti-inflammatory drugs.9
In the previous study, we reported the discovery of (Z)-5-benzylidene-2-iminothiazolidin-4-one derivative 1 (Figure 1A) as a human mPGES-1 inhibitor with moderate potency (IC50 = 3.5 μM) through structure-based virtual screening.10, 11 Based on the simulated binding mode of 1 with crystal structure (PDB ID: 4BPM,12 Figure 1B) of human mPGES-1, we redesigned the core scaffold and synthesized benzylidenebarbituric acid derivatives (2, with IC50 = 622 nM, as an example, Figure 1A) as mPGES-1 inhibitors. The barbituric acid was introduced in order to maintain and possibly enhance the polar interaction with the enzyme at the active site. Flexible alkoxy side chain was attached to the central benzene ring so as to fit the size of the hydrophobic pocket surrounded by I32, G35, L39, Y130, T131, L135, and A138. As predicted by molecular docking, the barbituric ring interacts with S127 by forming a hydrogen bond (HB) between the barbituric carbonyl group and hydroxyl group of serine, while the central benzene ring with alkoxy substitution occupies the hydrophobic groove where the long hydrocarbon “tail” of PGH2 locates (Figure 1C).
Figure 1.

Molecular structures of the lead 1, benzylidenebarbiturc acid derivative 2 and the designed scaffold 3 and their binding with human mPGES-1. (A) Ligand structures; (B) binding with the lead 1; (C) binding with 2; (D) binding with 3.
While developing the benzylidenebarbituric acid derivatives, we carefully analyzed the binding mode of these compounds with mPGES-1, and noticed that there is still substantial unoccupied area in the active site, especially the small hydrophobic pocket around the central benzene ring of 2 and above the cofactor glutathione (GSH) of the enzyme. We then decided to introduce pyrazole into the core scaffold not only because of its existence in many bioactive molecules, but also its versatility for multi-functionalization. We kept the barbituric acid “head” because of the importance of the hydrogen bond between carbonyl and hydroxyl group of S127. Substituents on pyrazole-1 and 3 positions were expected to occupy the hydrophobic pockets wrapping the hydrocarbon “tail” of PGH2 and above GSH. Thus, a series of 5-((1,3-diphenyl-1H-pyrazol-4-yl)methylene)pyrimidine-2,4,6 (1H,3H,5H)-trione derivatives and other structurally related compounds were designed and synthesized. The docking study of the simplest scaffold 3 (Figure 1A) with mPGES-1 revealed that 3 binds in a similar region in the active site as 2 in the binding complex (Figure 1D). The strategies of improving the inhibitory efficacy in light of the binding complex include the modification on barbituric acid moiety for maintaining and strengthening a hydrogen bond with S127, and the substitution on 1- and/or 3-phenyl in order to enhance the hydrophobic interaction with the nonpolar groove. As expected, a number of these compounds were active against human mPGES-1 and selective for mPGES-1 over COX isozymes.
The synthesis of this series of compounds followed a straightforward multi-step protocol, as shown in Scheme 1. 4-Alkoxyacetophenone (4b~4g), obtained from the reaction of 4-hydroxyacetophenone and alkyl bromide,13 or acetophenone (4a) was condensed with 4-chlorophenylhydrazine in reflux ethanol containing 5 % glacial acetic acid.14, 15 The ethylidene hydrazine (5a~5g) was formed as precipitate at room temperature and filtered off. The next step was Vilsmeier-Haack-Arnold ring closing formylation,13 by treating 5a~5g with POCl3/DMF. The produced 1H-pyrazole-4-carbaldehyde intermediate (6a~6g) was coupled with barbituric acid or 2-thiobarbituric acid in refluxing EtOH/H2O (4:1, v/v) to afford the final product (7a~7g or 8a~8g).16
Scheme 1.

Reagents and conditions: (a) K2CO3 (2.00 equiv.), DMF, 80 °C; (b) 5 % glacial AcOH in EtOH, reflux; (c) POCl3 (4.00 equiv.), DMF, 0 °C~60 °C; (d) EtOH/H2O (4:1, v/v), reflux.
The length and shape of the aliphatic side chain were investigated in the SAR study. We fixed the substituent at pyrazole-1-position as 4-chlorophenyl and variated the side chain on 3-phenyl. From the in vitro data shown in Table 1, it was observed as compared to that without a side chain (7a and 8a), compounds with linear side chains (7b~7f and 8b~8f) were generally more potent against human mPGES-1, whereas benzyl substitution (7g and 8g), however, did not improve the inhibitory potency. Linear side chains with 4 or 6 carbons yielded compounds with highest potency, whereas longer side chains, such as octyl or decyl, did not show a more potent inhibition. Notably, compounds with 2-thiobarbituric acid “heads” were generally more potent as compared to those with barbituric acid ones. We also changed the substituent in pyrazole-1-position from 4-chlorophenyl to phenyl group. In this case, 4c was used as starting substituted acetophenone. Followed the similar protocol as outlined in Scheme 2, 11 and 12 were prepared. These compounds (11 and 12) were slightly less potent than those with 4-chlorophenyl substituent (7c and 8c, respectively).
Table 1.
Structural-activity relationship (SAR) on the substitution of central pyrazole ring
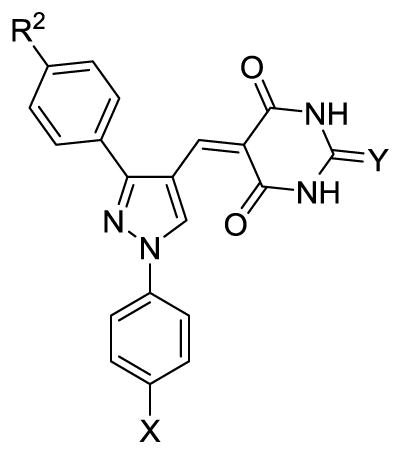
| ||||
|---|---|---|---|---|
|
| ||||
| Comp. | R2 | X | Y | IC50 (nM)a for mPGES-1 |
| 7a | H | Cl | O | 337±85 |
| 7b | C2H5O | Cl | O | 265±96 |
| 7c | nC4H9O | Cl | O | 169±41 |
| 7d | nC6H13O | Cl | O | 285±65 |
| 7e | nC8H17O | Cl | O | 361±51 |
| 7f | nC10H21O | Cl | O | 294±83 |
| 7g | BnO | Cl | O | 598±142 |
| 8a | H | Cl | S | 561±192 |
| 8b | C2H5O | Cl | S | 95±16 |
| 8c | nC4H9O | Cl | S | 56±10 |
| 8d | nC6H13O | Cl | S | 52±15 |
| 8e | nC8H17O | Cl | S | 92±19 |
| 8f | nC10H21O | Cl | S | 93±14 |
| 8g | BnO | Cl | S | 797±160 |
| 11 | nC4H9O | H | O | 212±34 |
| 12 | nC4H9O | H | S | 92±20 |
Data are expressed as means ± SD (standard deviation) of single determinations obtained in triplicate.
Scheme 2.

Reagents and conditions: (a) 5 % glacial AcOH in EtOH, reflux; (b) POCl3 (4.00 equiv.), DMF, 0 °C~60 °C; (c) EtOH/H2O (4:1, v/v), reflux.
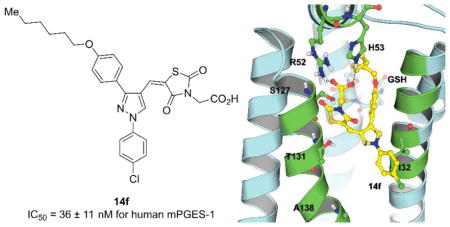
With these (2-thio)barbituric acid derivatives in hand, as depicted in Scheme 3, we broadened the structural diversity with pyrazole core by coupling 1H-pyrazole-4-carbaldehyes (6b and 6d) with various activated methylene compounds such as malononitrile, 2-cyanoacetic acid and 2,4-thiazolidinedione. As shown in Table 2, compounds with malononitirle and 2-cyanoacetamide “heads” (13b, 13c and 14b, 14c) were not very active against human mPGES-1, whereas those with 2-cyanoacetic acid (13a and 14a) showed submicromolar potency. It was noted that compounds 13f and 14f, obtained from the coupling of 6b and 6d with 2,4-thiazolidinedione N-acetic acid, were capable of inhibiting human mPGES-1 with low naonomolar potency (IC50 = 41 ± 5 nM and 36 ± 11 nM, respectively).
Scheme 3.
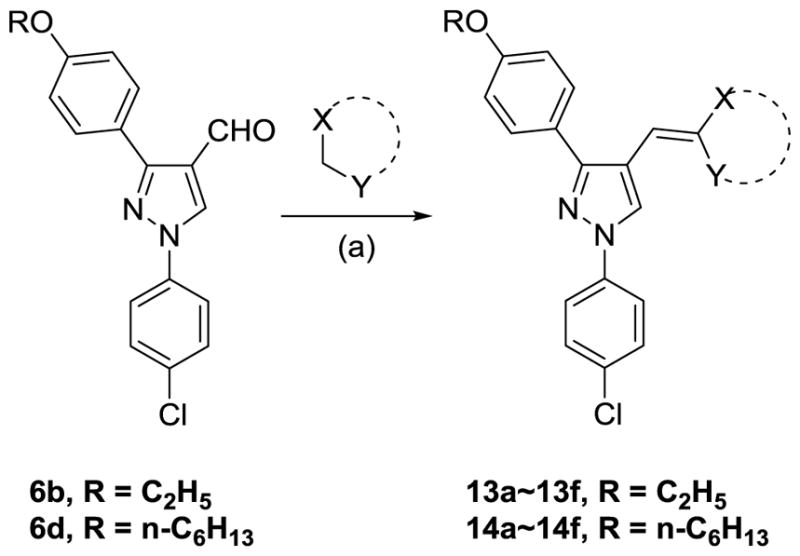
Reagents and conditions: (a) NH4OAc (2.00 equiv.), glacial AcOH, 108 °C.
Table 2.
The SAR on the polar head
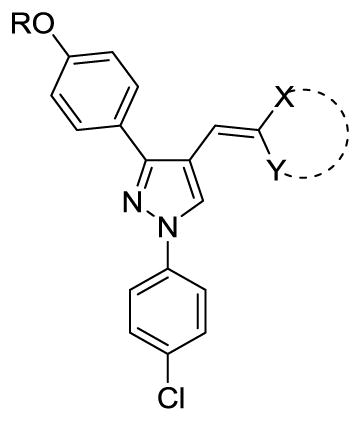
| |||
|---|---|---|---|
|
| |||
| Comp. | R |
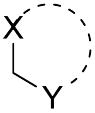
|
IC50 (nM)a for mPGES-1 |
| 13a | C2H5 |
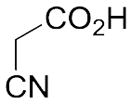
|
283±83 |
| 13b | C2H5 |
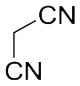
|
n.d.b (51%±10%)c |
| 13c | C2H5 |
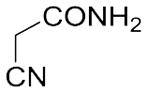
|
n.d. (32%±21%) |
| 13d | C2H5 |
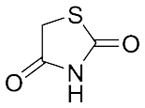
|
1,590±560 |
| 13e | C2H5 |
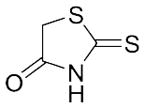
|
1,040±290 |
| 13f | C2H5 |
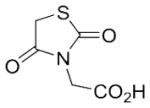
|
41±5 |
| 14a | nC6H13 |
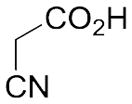
|
83±34 |
| 14b | nC6H13 |
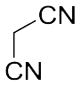
|
n.d. (64%±1.4%) |
| 14c | nC6H13 |
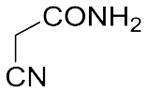
|
n.d. (14%±14%) |
| 14d | nC6H13 |
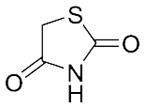
|
1,390±300 |
| 14e | nC6H13 |
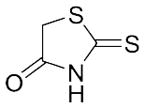
|
1,730±670 |
| 14f | nC6H13 |
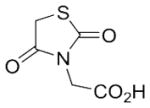
|
36±11 |
Data are expressed as means ± SD (standard deviation) of single determinations obtained in triplicate.
n.d. = not determined.
The value in the parenthesis refers to the % inhibition of the compound at a concentration of 10 μM against the mPGES-1 (IC50 values were determined only for the compounds that showed ≥70% inhibition).
To further understand the SAR of these synthesized compounds, we selected compounds 7c, 7e and 14f and used the AutoDock Vina program17 as the molecular docking tool to investigate their binding modes with human mPGES-1. 14f was chosen for further docking study because it is the most active one with the scaffold (possessing a carboxylic acid polar “head”) for the mPGES-1 inhibitors in Table 2. 7c and 7e were chosen from compounds in Table 1 with another scaffold (barbituric acid derivatives). Of the barbituric acid derivatives in Table 1, we are mainly interested in the compounds with Y = O, because the compounds with Y = S are less soluble in water. Within all of the barbituric acid derivatives with Y = O in Table 1, 7c is the most potent one for human mPGES-1. 7e was also chosen for comparison with 7c because they are different only in the length of the side chain (R2).
The predicted binding modes are shown in Figure 2. The substituted phenyl group on pyrazole-1-position occupies the hydrophobic groove where the long hydrocarbon side chain of PGH2 locates. The other substituted phenyl group on pyrazole-3-position fits into the small hydrophobic pocket above GSH. Since this pocket is small, a bulky group on pyrazole-3-position is not favorable as reflected in the inhibitory data that 7e (with longer octyl side chain) is an inferior inhibitor as compared with 7c (with a shorter butyl side chain). The substituents on pyrazole-4-position attaches on the surface of the protein. When the barbituric acid is present, the carbonyl group on the barbituric ring forms a hydrogen bond with the hydroxyl group of S127. . While replacing the barbituric acid with 2,4-thiazolidinedione N-acetic acid, however, the carboxyl group forms hydrogen bonds with the NH groups of R52 and H53. So, 14f is a potent inhibitor against human mPGES-1.
Figure 2.
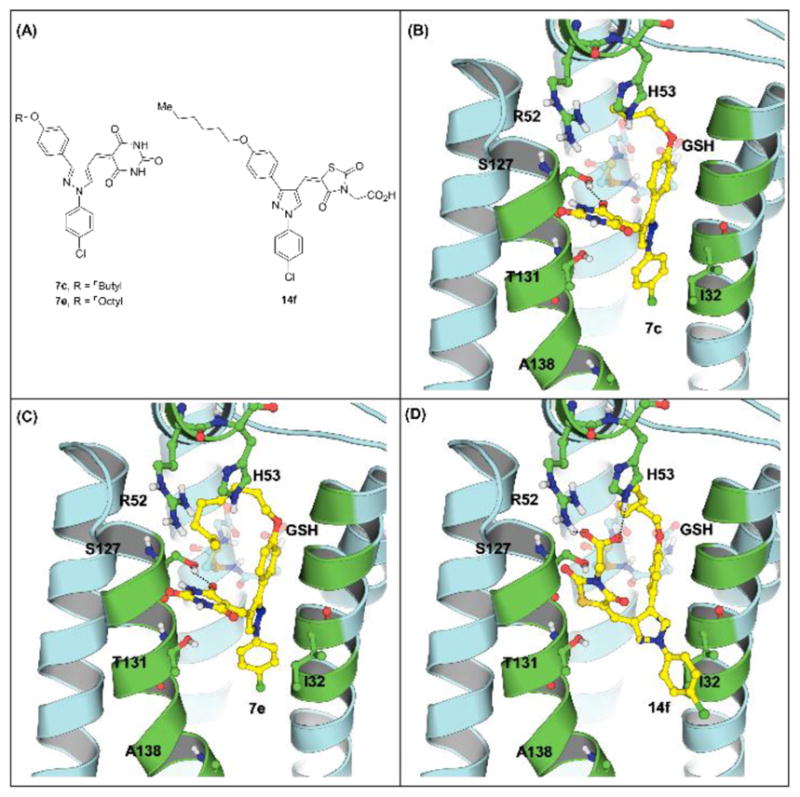
Molecular structures of 7c, 7e and 14f and their binding with human mPGES-1. (A) Compound structures; (B) binding with the lead 7c; (C) binding with 7e; (D) binding with 14f.
For the in vitro evaluation of these compounds, we first conducted the single-concentration screening at 10 μM against human mPGES-1. Compounds that showed significant inhibition (≥70%) were tested further for their IC50 values against human mPGES-1. The protocol for the protein preparation and in vitro activity assays were the same as described previously.10, 11, 18, 19 Further, the inhibitory activity against COX isozymes was also evaluated for some of the more promising compounds (with IC50 < 100 nM against human mPGES-1). As shown in Table 3, at a concentration as high as 100 μM, compounds 8b~8f, 12, 13f, 14a and 14f inhibited COX-1/2 for less than 20 %. So, these compounds are highly selective for the mPGES-1 over COX-1/2.
Table 3.
Inhibition against COX-1/2
| Compound | % Inhibition of COX-1/2 at 100 μMa |
|---|---|
| 8b | 1.0±4.5 |
| 8c | 4.4±8.6 |
| 8d | 0.7±5.4 |
| 8e | 0±0.2 |
| 8f | 11±16 |
| 12 | 16±4.1 |
| 13f | 6.8±6.4 |
| 14a | 19±0.1 |
| 14f | 1.2±29 |
The % inhibition of the compound at a concentration of 100 μM against the COX-1/2 (mixed COX-1 and COX-2). The enzyme mixture contained equal amounts of COX-1 and COX-2 in terms of their enzyme activities. In this way, when a compound can significantly inhibit either COX-1 or COX-2, it will show the significant inhibitory effects against the mixed COX-1 and COX-2.
In summary, in light of the binding complex of both the lead and the benzylidenebarbituric acid scaffold with human mPGES-1, we designed and synthesized a novel series of 5-((1,3-diphenyl-1H-pyrazol-4-yl)methylene)-pyrimidine-2,4,6 (1H,3H,5H)-triones and other structurally related derivatives.20 These compounds were evaluated in vitro for their inhibitory potency against human mPGES-1 and selectivity over COX-1/2, leading to discovery of various potent and selective inhibitors of human mPGES-1. The most potent one is 14f (IC50 = ~36 nM against human mPGES-1) without significant inhibition against COX-1/2.
Acknowledgments
This work was supported in part by the funding of the Molecular Modeling and Biopharmaceutical Center at the University of Kentucky College of Pharmacy, the National Science Foundation (NSF grant CHE-1111761), and the National Institutes of Health via the National Center for Advancing Translational Sciences (UL1TR001998) grant. The authors also acknowledge the Computer Center at University of Kentucky for supercomputing time on a Dell Supercomputer Cluster consisting of 388 nodes or 4,816 processors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES AND NOTES
- 1.Norberg JK, Sells E, Chang HH, Alla SR, Zhang S, Meuillet EJ. Pharm Pat Anal. 2013;2:265. doi: 10.4155/ppa.12.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garavito RM, DeWitt DL. Biochim Biophys Acta. 1999;1441:278. doi: 10.1016/s1388-1981(99)00147-x. [DOI] [PubMed] [Google Scholar]
- 3.Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandins Other Lipid Mediat. 2002;68–69:383. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- 4.Funk CD. Science. 2001;294:1871. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 5.Samuelsson B, Morgenstern R, Jakobsson PJ. Pharmacol Rev. 2007;59:207. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 6.Hawkey CJ. Best Pract Res Clin Gastroenterol. 2001;15:801. doi: 10.1053/bega.2001.0236. [DOI] [PubMed] [Google Scholar]
- 7.FitzGerald GA, Patrono C. N Engl J Med. 2001;345:433. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- 8.Buttgereit F, Burmester GR, Simon LS. Am J Med. 2001;110(Suppl 3A):13S. doi: 10.1016/s0002-9343(00)00728-2. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. J Clin Invest. 2006;116:1391. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamza A, Zhao X, Tong M, Tai HH, Zhan CG. Bioorg Med Chem. 2011;19:6077. doi: 10.1016/j.bmc.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Z, Yuan Y, Zhou S, Ding K, Zheng F, Zhan CG. Bioorg Med Chem Lett. 2017;27:3739. doi: 10.1016/j.bmcl.2017.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D, Howe N, Dukkipati A, Shah ST, Bax BD, Edge C, Bridges A, Hardwicke P, Singh OM, Giblin G, Pautsch A, Pfau R, Schnapp G, Wang M, Olieric V, Caffrey M. Cryst Growth Des. 2014;14:2034. doi: 10.1021/cg500157x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamasaki K, Hishiki R, Kato E, Kawabata J. ACS Med Chem Lett. 2011;2:17. doi: 10.1021/ml100171x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ragab FA, Abdel Gawad NM, Georgey HH, Said MF. Eur J Med Chem. 2013;63:645. doi: 10.1016/j.ejmech.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Harikrishna N, Isloor AM, Ananda K, Obaid A, Fun HK. RSC Advances. 2015;5:43648. [Google Scholar]
- 16.Baron R, Huang CH, Bassani DM, Onopriyenko A, Zayats M, Willner I. Angew Chem Int Ed Engl. 2005;44:4010. doi: 10.1002/anie.200463055. [DOI] [PubMed] [Google Scholar]
- 17.Trott O, Olson AJ. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamza A, Tong M, AbdulHameed MD, Liu J, Goren AC, Tai HH, Zhan CG. J Phys Chem B. 2010;114:5605. doi: 10.1021/jp100668y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang XQ, Yan WL, Gao DQ, Tong M, Tai HH, Zhan CG. Bioorg Med Chem. 2006;14:3553. doi: 10.1016/j.bmc.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Analytical data and yields for the most potent compounds: (13f) 1H NMR (400 MHz, DMSO) δ 13.47 (s, 1H), 8.76 (s, 1H), 8.05 (d, J = 8.7 Hz, 2H), 7.68 (s, 1H), 7.57 (dd, J = 26.7, 8.6 Hz, 4H), 7.09 (d, J = 8.6 Hz, 2H), 4.35 (s, 2H), 4.10 (q, J = 6.9 Hz, 2H), 1.36 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 168.00, 166.61, 164.74, 159.29, 153.83, 137.53, 131.57, 130.03, 129.48, 128.35, 124.12, 123.13, 120.93, 119.57, 115.31, 114.84, 63.24, 42.37, 14.63. HRMS (ESI+) m/z calcd for C23H19ClN3O5S (MH)+: 484.0728, found: 484.0728. Yield: 74 %. (14f) 1H NMR (400 MHz, DMSO) δ 8.78 (s, 1H), 8.06 (d, J = 8.8 Hz, 2H), 7.69 (s, 1H), 7.63 (s, 1H), 7.61 – 7.47 (m, 3H), 7.09 (d, J = 8.7 Hz, 2H), 4.36 (s, 2H), 4.03 (t, J = 6.5 Hz, 2H), 1.82 – 1.64 (m, 2H), 1.48 – 1.28 (m, 6H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 168.01, 166.62, 164.75, 159.47, 153.86, 137.55, 131.59, 130.04, 129.50, 128.40, 124.15, 123.13, 120.97, 119.59, 115.32, 114.90, 67.64, 42.34, 31.03, 28.63, 25.21, 22.11, 13.95. HRMS (ESI+) m/z calcd for C27H27ClN3O5S (MH)+: 540.1354, found: 540.1352. Yield: 71 %.