

Abstract
There are limited biomarkers for substance use disorders (SUDs). Traditional statistical approaches are identifying simple biomarkers in large samples, but clinical use cases are still being established. High-throughput clinical, imaging and “omic” technologies are generating data from SUD studies and may lead to more sophisticated and clinically useful models. However, analytic strategies suited for high dimensional data are not regularly used. We review strategies for identifying biomarkers and biosignatures from high dimensional data types. Focusing on penalized regression and Bayesian approaches, we address how to leverage evidence from existing studies and knowledge-bases, using as an example, nicotine metabolism. We posit that big data and machine learning approaches will considerably advance SUD biomarker discovery. However, translation to clinical practice, will require integrated scientific efforts.
Keywords: Biomarker, Machine Learning, Artificial Intelligence, Substance Use Disorders, Nicotine Metabolism, Genomics
Biosignatures in Substance Use Disorders
Biomarkers for substance use disorders (SUD) are available for drug use based on detection of the substance or its metabolites, e.g., ethyl glucuronide for alcohol [1], tetrahydrocannabinol for marijuana [2], and cotinine for tobacco [3]. They are not, however readily available for the neurobiological modifications that result in the maladaptive behaviors we describe as addiction [4]. Clinical phenotyping has been used to assess the presence and severity of SUDs and comorbid psychiatric disease and to evaluate treatment options. We now have massive data on patients with SUDs (e.g., genomics and other omics (see Glossary), and imaging on the structure and function of the brain). How can we use this data in biomarker/biosignature discovery? The ability to combine omics with each other and with complex neurocognitive or imaging data promises to deliver biosignatures that will reflect the behavioral and biological modifications that occur in addiction. Standard biostatistical analysis that has been so useful in clinical research does not perform well in this high dimensional environment where variables vastly outnumber patients [5]. Studies of SUDs and treatments are beginning to use more comprehensive modeling approaches. In one example, data from diverse concepts (brain, personality, cognition, demographics and genetics) were incorporated into a highly predictive model of current and future alcohol abuse in adolescents [6]. In another recent example, researchers performed an integrative analysis to link genomic variation with expression changes in brains of alcohol dependent individuals [7]. While these studies point to an encouraging trend, there still appears to be a gap between the massive data available and the routine use of computational and statistical tools in biomarker/biosignature discovery in SUDs.
Biosignature discovery provides a way of combining many variables (e.g., genetic effects, voxels in neuroimaging) into meaningful models. Without models of net effects, it is difficult to interpret many small effects, especially given complex correlations in data. High dimensional data is also becoming quite common on large populations, while measures of molecular phenotypes, which may not be cost effective or safely accessed, are less commonly collected. Biobanks of large cohorts with genomic data are becoming available, such as the Millions Veteran’s Program, the All of Us Research Program, UK Biobank, GenomeAsia 100K, and even direct-to-consumer services such as 23andMe and Helix. While there is recognition that additional omics are necessary to understand the influence of genotype on phenotype, the most commonly available data will remain genotypic. Using a biosignature approach, there are opportunities to gain new biological insights and assess multiple predicted phenotypes. This is one of the premises behind transcriptome-wide association studies using genome-wide genotype data [8]. Using studies where both genomic and transcriptomic data are available, the tissue-specific relationship between DNA (genotypes) and RNA (gene expression) can be modeled to generate predictive models [9]. These models are then used in genomic data to predict expression and association to diseases or quantitative traits [10, 11].
The overall strategy of using high dimensional data to profile and predict molecular phenotypes and other outcomes is a new development path for biomarkers and biosignatures for SUDs and their treatments (see Key Figure, Figure 1). Discovery is driven by statistical learning algorithms suited for detection of biosignatures from high dimensional data. The learned biosignatures are then validated and applied in various use-cases (e.g., to identify subgroups at risk of SUDs or as tools to optimally select treatments). We advocate propagating the data from new observations and the predictive performance of the models back into the development cycle to allow for continuous improvement of the biosignatures (Key Figure 1). We see great promise in statistical learning to discover and validate biosignatures, but recognize that the translational path into clinical settings will have some unique challenges. In the next sections, we illustrate the workflow presented for this model (Key Figure, Figure 1) by learning biosignatures of nicotine metabolism in high dimensional genomic data.
Key Figure 1. Biosignature Development Workflow.

Data where the number of variables vastly outnumbers the number of samples (high dimensional data) are becoming commonplace in studies of substance abuse disorders and treatment approaches. We present two approaches (penalized regression and Bayesian learning) for detecting the combination of variables (biosignatures) predictive of SUD phenotypes (e.g., nicotine metabolism). Biosignature detection is followed by validation, then prospective assessment of utility for translation to clinical practice.
Application to Nicotine Metabolism
We use the nicotine metabolism pathway as a motivating example for the utility of statistical learning to discover and use SUD biosignatures from high dimensional data. Nicotine metabolism is strongly influenced by genes; the majority of variance (74%) is due to additive genetic influence [12, 13]. The cytochrome P450 monooxygenase 2A6 (CYP2A6) is the dominant but not exclusive metabolic enzyme in nicotine metabolism [14]. Early work established that the ratio of the first two major metabolites of nicotine (trans-3′hydroxycotinine / cotinine, or the nicotine metabolism ratio; NMR) can serve as a biomarker of nicotine metabolism [15]. The NMR is estimated biochemically [16] or via prediction using CYP2A6 genotypes [17, 18]. Genes coding for numerous additional oxidases (FMO3, AO, CYP2B6, POR, AKR1D1), and the uridine diphosphate glycosyltransferases (UGTs), have been found to be associated with nicotine metabolism through individual candidate gene single nucleotide polymorphisms (SNPs), or, less commonly, gene/protein expression, or protein activity analyses [19, 20, 21, 22, 23, 24, 25, 26, 27]. Moreover, in diverse populations and using blood, saliva and urine biospecimens from smokers, or using labeled nicotine and cotinine in clinical laboratory studies using blood and urine, nicotine metabolism has been reported to vary by ancestry [28, 29, 30, 31], age, gender, body mass index, estrogenic hormones, alcohol and cigarette consumption [32].
In addition to pharmacologic investigations of nicotine metabolism [14], investigators have studied the influence of nicotine metabolism on smoking cessation retrospectively, using either the biochemical measure of the NMR [33, 34, 35] or genotypes associated with reduced NMR [36, 33, 37]. There has been one prospective analysis of the influence of the NMR on smoking cessation, examining the efficacy of nicotine replacement therapy (NRT) (NRT), varenicline (VAR) and placebo in slow and normal nicotine metabolizers, with the NMR determined from direct biochemical measurement [38]. Note that biomarkers of nicotine metabolism, as studied in the literature, have differed somewhat depending upon genotyping approach, biochemical ratios and cutoff-points selected, as well as clinical or population samples used to establish the biomarker; one common dichotomization stratifies individuals with normal metabolism versus slow metabolism.
In general, retrospective studies of smokers, randomized to NRT or placebo, have shown that individuals with biomarkers of slow metabolism, whether defined by genotype or biochemical ratio, were significantly more likely to remain abstinent than individuals with normal metabolism. In one retrospective analysis [37], individuals with slow nicotine metabolism did not benefit (no reduction in relapse proportions) from active treatment (NRT, bupropion or combined active treatment) compared to placebo treatment, while individuals with fast nicotine metabolism did benefit from active treatment. In the prospective trial stratified by the NMR, individuals with normal nicotine metabolism responded significantly better to active treatment than placebo, and those randomized to varenicline (VAR) responded significantly better than those randomized to NRT [38]. Together, these findings suggest that treatment success can be optimized by assigning treatment to patients by their NMR status, e.g., assigning more active pharmacotherapy to normal metabolizers and less active pharmacotherapy to slow metabolizers. Clinical trials using the NMR to assess nicotine metabolism and provide metabolism-matched pharmacotherapy are in progress [39]. Biosignatures for predicting nicotine metabolism in clinical trials of smoking cessation therapies, and in cohorts being studied for tobacco-related behaviors, diseases and exposures, will be useful to characterize the role of nicotine metabolism in these complex outcomes [40].
The prior knowledge, results, and data described above can be directly used in biosignature development (inputs in Key Figure 1). Statistical learning algorithms (described in the next sections) can be applied to detected biosignatures of nicotine metabolism. These biosignatures (once validated) can be used to predict other outcomes (such as smoking cessation) or to personalize treatments (e.g., bupropion, varenicline, or NRT for a smoker; Key Figure 1).
Biosignature Detection in High Dimensional Data
The data layout for biosignature learning is shown in Figure 2. One or more SUD studies have both high dimensional data (e.g., genomic) and molecular phenotypic data (e.g., metabolites). For simplicity, it was assumed that the clinical factors were binary and that the genotypes were single nucleotide polymorphism (SNPs), coded by the number of copies of the minor allele [41]. Millions of genotyped and imputed SNPs can be available. The relevant variables (the biosignature) and its net effects on a predictable molecular phenotype (denoted Z in Figure 2) are then learned using statistical algorithms. Once the relationships are represented in models, they can be applied to new data for prediction and assessment of the outcomes of interest (denoted Y in Figure 2). For nicotine metabolism, these could be smoking cessation, lung cancer risk, treatment response, etc. Of note, the molecular phenotype does not need to be measured once the model is learned, as it can be predicted from the biosignature, sometimes from summary statistics [10, 11].
Figure 2. Biosignatures of Nicotine Metabolism.
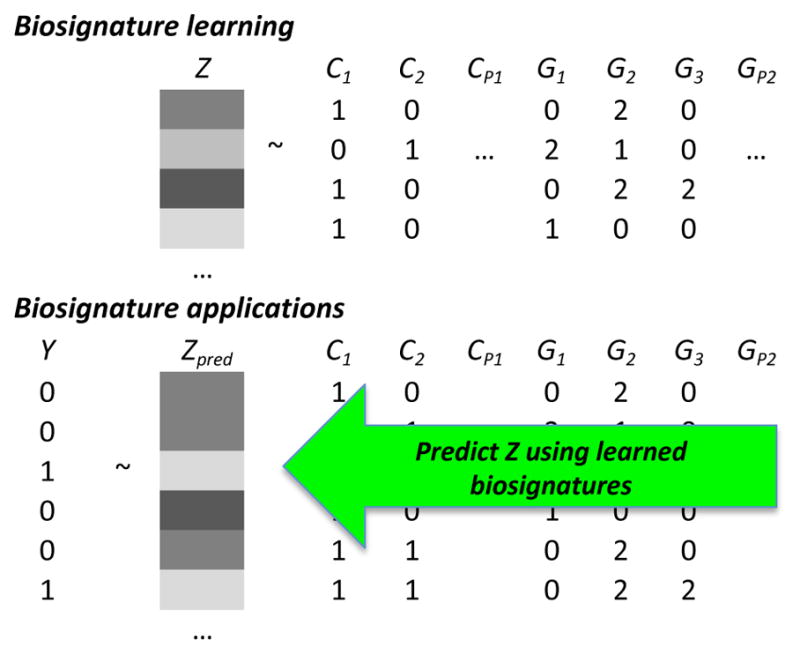
Nicotine metabolism biosignatures are learned from genotypes G and clinical C data in laboratory studies of nicotine metabolism. Nicotine metabolism is then predicted (Zpred) in existing or new observations using the biosignatures and corresponding model weights. The predicted nicotine metabolite ratio can them be associated with clinical outcomes Y, such as smoking cessation (1’s indicate success). Adapted from [10].
Simple models consider only one genetic variant at a time. A genome-wide association scan (GWAS) across the genome represents millions of tests for genetic associations with the trait or outcome [5]. There have been four GWASs using the NMR as the trait to date [13, 42, 43, 44]. These four GWASs used readily available GWAS genotyping arrays, as well as typical statistical pipelines for genotype cleaning and imputation via the 1000 Genomes Project resource (http://www.1000genomes.org). Two GWASs represented meta-analyses of single ancestries [13, 44], one was a multi-ancestry meta-analysis [42], and one was a multi-ancestry mega-analysis [43]. As expected, in all four scans, variants in and near CYP2A6 on human chromosome 19 (the gene encoding the primary nicotine metabolic enzyme) were associated with the NMR at genome-wide significance. In each GWAS, the most significant associations were located proximal, within and distal to CYP2A6, with individual SNPs, significance ranks and span of association being dependent upon study, sample size and ancestry composition. We and others [13, 42, 43] have noted complex patterns of association with the NMR that span into nearby genes, including CYP2B6 (Figure 3) [42]. Given that there are complex patterns of marginal associations and that the number of variables exceed the sample size (known in statistics as P > N), how does one define a biosignature of nicotine metabolism based on genomic data?
Figure 3. Genetic Associations with Nicotine Metabolism in the CYP2A6 Region of Human Chromosome 19.

The variants selected using penalized regression algorithms are overlaid on the marginal genetic association results (−log10 p-values on the y-axis). This shows how penalized regression algorithms can define biosignatures (red boxes) from complex patterns of marginal associations (stars).
The dimensionality problem can be addressed in part by using existing knowledge and results to reduce the model space (Key Figure 1). With available genome-wide data from our GWAS [42], we selected 11 genomic regions implicated in nicotine metabolism for modeling. We identified the relevant regions using a combination of knowledge bases (such as PharmGKB [45]) and recent literature [19, 20, 21, 22, 23, 24, 25, 26, 27]. The second strategy consists of using algorithms to reduce model complexity. The rationale here was that there can be too many variables for a human to decide which should go in a model.
Two classes of statistical learning algorithms can be used for selecting which variables should be in a model (see Box 1) and estimating its joint effects on an outcome [46]. These algorithms explore a trade-off between model complexity, prediction bias, and prediction variance. That is, prediction bias can be reduced by increasing model complexity. Alternatively, one can also trade prediction bias for reduced variance using approaches to reduce complexity. The goal is to find a sweet spot, selecting the right model complexity to minimize prediction error [47, 48].
Box 1. Model Specification.
Generalized linear models can be used as a foundation for biosignature discovery and predicting nicotine metabolism from genomic and clinical data [89]. The conditional mean of Yi, the observed molecular phenotype or outcome of individual i, is related to P explanatory variables through the link function g (·):
where there are P1 clinical factors (e.g, age, sex, BMI), P2 genetic markers, and P3 derived variables, denoted by Cij, Gij, and Zij, respectively, with P = P1 + P2 + P3. The βkj’s are the usual regression coefficients. The derived variables can be interaction terms of other functions that combine sets of other variables. In the analysis described herein, the natural logarithm of NMR is used as the response variable so that g (·) can be taken to be the identity link.
The First Approach: Penalized Regression
Penalized regression approaches add a penalty term to the typical optimization problem [47, 48]. That is, many penalized regression methods estimate the regression coefficients by minimizing a penalized residual sum of squares which is given by:
where λ represents a collection of tuning parameters which implicitly controls the model complexity, β denotes the collection of all the regression coefficients, and P(·, ·) is a penalty function [47, 48]. For example, a penalty function of the form
could be considered, where setting α = 1 results in the least absolute shrinkage and selection operator (LASSO) of [49], α = 0 provides the usual ridge estimator [50], and α ∈ (0, 1) results in the elastic net of [51]. In general, through regularization, the ridge estimator is able to provide better prediction performance by exploiting the so called bias versus variance trade-off, and can be used (unlike standard ordinary least squares) to uniquely estimate the regression coefficients when P > N [50]. Unlike ridge, LASSO provides a parsimonious model through automatic variable selection, though it has been empirically shown that ridge maintains a higher level of prediction accuracy in the face of correlated predictors, when compared to LASSO [49]. The elastic net is a blend of ridge and LASSO, which attempts to gain from their strengths and overcome their individual weaknesses. In particular, the elastic net makes use of a linear combination of the ridge and LASSO penalties and can therefore complete automatic variable selection while maintaining a high degree of prediction accuracy in the face of correlated predictors[51].
There are numerous algorithmic varieties of this general approach, with different properties in terms of handling correlation, sparsity, and picking out features in the data [48]. Moreover, extensive work has shown that none of the penalized regression procedures are universally better in all situations [48, 52]. So it is natural to posit the question “What penalized regression method should I use?”. Our retort, why choose one?
Different penalized regression procedures can be applied to the motivating nicotine metabolism data; for details on these algorithms, their implementation, and their penalty structures see Table 1. In particular, Table 1 provides nine different penalized regression methods, citations that present the relevant background on each procedure, and R-packages that can be utilized to implement each of the methods. As discussed above, these algorithms have different properties and therefore provide diverse insights into the data. We believe that the collection of models (the ensemble) characterizes the biosignature of this molecular phenotype. This is demonstrated in Figure 4, where the rows represent the biosignatures learned by the different algorithms applied to the nicotine metabolism data [53]. The un-shaded portions represent the genetic signature identified by each approach. As expected, a handful of SNPs (out of 3752) were selected in all the models [53]. The largest model included 63 SNPs, but more parsimonious models explained NMR just as well with fewer SNPs (58–62% NMR) [53]. The genetic biosignature found by these methods in human chromosome 19 are overlayed on the marginal results in Figure 3. This highlights additional signals near the CYP2A6 and CYP2B6 genes that may have been missed in a more traditional approach. The collection of variables that is predictive of an individual’s nicotine metabolism is more than those discoverable using standard genetic association scans. To predict nicotine metabolism with new genotypes, one simply applies the learned weights to the SNP biosignatures [53]. The predicted nicotine metabolism can be then applied in additional association or clinical studies.
Table 1.
Penalized Regression Algorithms Applied to the Nicotine Metabolism Dataset. The table includes the R packages implementing it, and the primary research articles describing the algorithms.
| Method | R-Package | Reference |
|---|---|---|
| LASSO | glmnet | Tibshirani (1996) [49] |
| Elastic net | glmnet | Zou and Hastie (2005) [51] |
| Adaptive LASSO | parcor | Zou (2006) [90] |
| SCAD | ncvreg | Fan and Li (2001) [91] |
| MCP | ncvreg | Zhang (2010) [92] |
| SCAD-Ridge | ncvreg | Breheny and Huang (2011) [93] |
| MCP-Ridge | ncvreg | Breheny and Huang (2011) [93] |
| Dantzig Selector | flare | Candes and Tao (2007) [94] |
| lq LASSO | flare | Li et al. (2015) [95] |
Figure 4. An Ensemble of Models to Define Biosignatures.

The rows highlight (unshaded) the sets of SNPs selected by different penalized regression algorithms applied to nicotine metabolism data. Shaded SNPs were not selected as predictors. While there are a core set of SNPs selected by all the approaches, there is diversity in the sets of SNPs selected among the models. We define the biosignature as the entire set of variants selected by any of the algorithms.
The differences in the SNPs selected by the penalized regression algorithms (Figure 4) suggest that there are multiple “good” models and one may want to average over the strengths of a set of models when making predictions [54, 55]. This leads to the second approach where model uncertainty can be quantified [54].
The Second Approach: Bayesian model averaging
In the previous section we discussed the uncertainty with which SNPs belong in a biosignature of nicotine metabolism. Bayesian approaches can account for this uncertainty in the model specification. Here, the posterior probability for a given model is:
Obtaining the denominator would involve exploring all possible biosignatures which may not be computationally feasible. Thus the posterior probability is usually approximated by assessing the relative merit of a subset of models [56].
The likelihood above is actually marginalized over the parameters in the model and again is often approximated.
The priors on the model p(M) and its parameters p(β) can give us the opportunity to formally introduce existing results, knowledge-bases, and assumptions into the modeling [57]; e.g., what variables are biologically important, the direction and magnitude of their importance, and the certainty with which they are involved. In fact, most penalized regression approaches can also be represented by specifying priors on p(β) [48].
More complex relationships among variables can also be learned from the data. Combinations of SNPs or other factors can be condensed into new derived variables Zj. For example, tree structures denoted Λ, can be considered where the output of each node is determined by its input values and a set of edge parameters [58]. One such tree structure is shown in Figure 5, which represents the following system of equations:
Figure 5. Tree-based Structures Can Represent Complex Relationships in Sets of Variables.
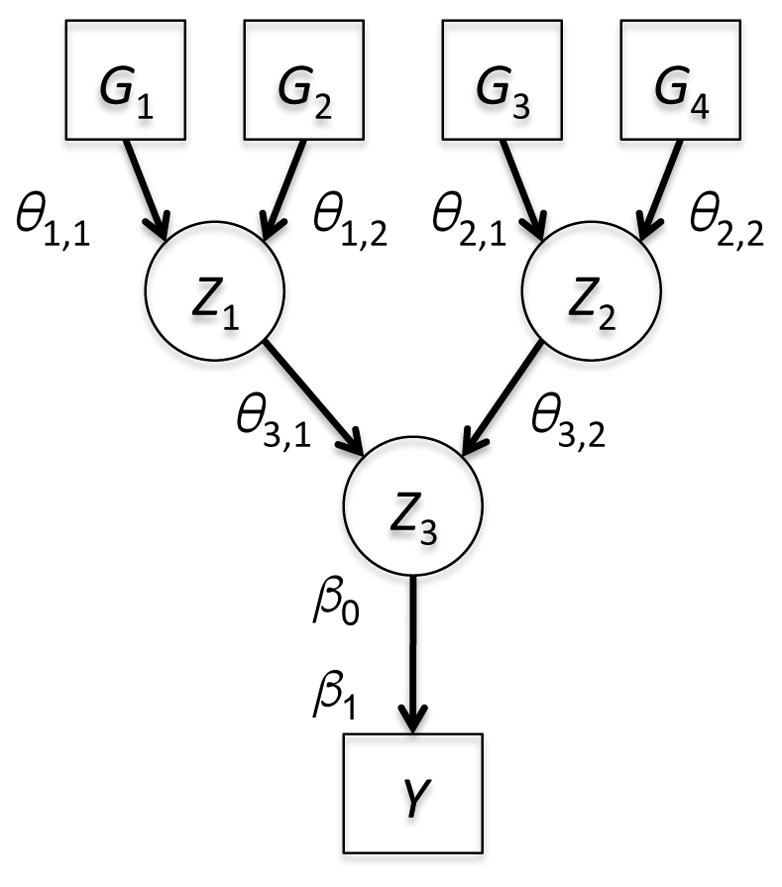
Here each derived variable Z is computed from its inputs (genetic variants, clinical factors, or other derived variables) and a pair of edge parameters θ. The regression coefficient β1 represents the net effect of the entire combination of variables on the outcome of interest Y. These structures were explored using Bayesian algorithms to learn biosignatures of nicotine metabolism.
These tree-based derived variables provide a very flexible way of representing interactions [58]. For example, given binary inputs, different edge parameters can represent different operators. If θ1 = θ2 = 0.5, the effects are additive; if θ1 = θ2 = 0 there is an effect only when both variants are present (logical AND); and if θ1 = θ2 = 1 there is an effect if either variant is present (logical OR). The effects of the derived variables represent the net effect of the tree [58].
Under this theme, every pairwise SNP effect on nicotine metabolism can be considered (over 6 million derived variables). The evidence for or against each can be computed using Bayes factors, the ratio of posterior to prior odds. The top associations are shown in Figure 6. Seven genomic regions showed evidence of interactions. The CYP2A6 region is an important hub, with other genetic variants near UGT2B10, UGT1A4/A9, POR, NR1I3, NFE2L2, and HNF4A, contributing to its effects on nicotine metabolism (Figure 6).
Figure 6. Joint SNP Effects on Nicotine Metabolism.
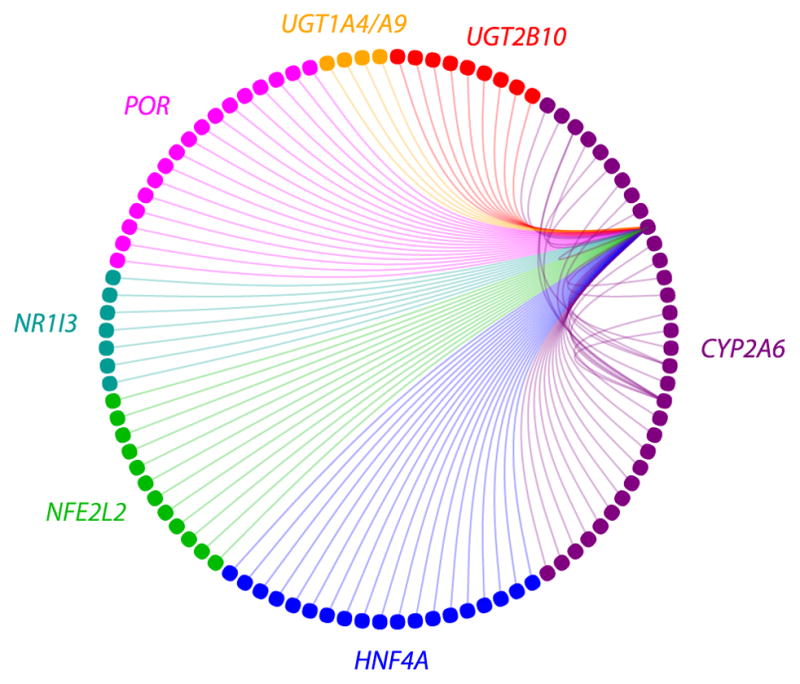
The effects of combination of genetic variant on nicotine metabolism can be explored using Bayesian algorithms [58]. This plot shows that many genetic variants (dots) in different genes (color) can modify the effects of CYP2A6 variants on nicotine metabolism. This presents another way of defining biosignatures from a collection of models for use in prediction or generating new hypotheses.
More complex combinations (starting with one tree and then modifying it in a quest to find better combinations) can be sought. This process can be repeated hundreds of thousands of times. Combinations of variants that had large impacts on nicotine metabolism were found. For example, the best learned model had a posterior probability of 0.90%, explained 43% of nicotine metabolism, and had a rather large effect on natural log NMR (βΛ = −1.35). As in penalized regression, multiple models fit the data well, but the posterior probabilities provide an intuitive way to average models [54, 55]. Trees with the highest posterior probabilities contained a handful of SNPs and had strikingly similar measures of fit [53]. This suggest that one should make nicotine metabolism predictions by averaging over the collection of models. The posterior predictive distribution allowed us to generate predictions of new observations using the entire distribution of explored biosignatures [59].
Incorporating these Approaches into Research and Clinical Translation
Statistical learning algorithms can help identify biosignatures of SUD outcomes. Once learned, these models can provide biological insights on their own as well as be applied to existing or new data to generate predictions. While the focus of the nicotine metabolism application in this example relied on genomics, the approach can be applied to other data commonly available or becoming available in studies of substance use disorders, such as metabolomic [60, 61], personality assessment [62, 63], neuroimaging [64, 65], and mobile health applications [66]. The first strategy described involves learning using penalized regression algorithms. These algorithms select variables while simultaneously estimating their effects. As demonstrated in the nicotine metabolism application, they extract different features from the data. We advocate using the entire ensemble to characterize SUD biosignatures because each algorithm can extract unique features in the data. The second approach involves learning a distribution of models and leveraging that distribution in prediction [54]. These Bayesian algorithms allow us to consider more complex relationships among variables [58]. In the nicotine metabolism example, variants near CYP2A6 and other parts of the pathway jointly influenced nicotine metabolism (Figure 6). This suggests that learning algorithms can identify combinations of genetic variances that explain molecular phenotypes that may be missed using traditional analyses. The next steps involve: validating the biosignatures in other datasets with both genomic data and nicotine metabolites, and then applying the learned biosignatures and weights in other datasets, to observe how the biosignatures influence smoking related outcomes (e.g., smoking-related behaviors, cessation, disease and comorbidities).
Many statistical learning approaches have been around for some time, but are just now being applied to SUDs. Computing used to be a major bottleneck in applying these algorithms. But with new acceleration computing and software stacks, algorithms are being retooled to handle much more complex and large datasets. As in all industries, data is now in abundance. There are more data on individuals with SUDs available now than ever before [67]. Several groups have begun to explore applying these algorithms to learn predictors of response to treatment for SUDs [68, 69, 70], but this is just the beginning of a new wave of discovery. There has been a recent increase of deep learning algorithms being applied to health applications: automatic detection of new tumors from imaging data [71], discovery of new drug targets [72], and precision treatments in cancer patients (http://candle.cels.anl.gov/). The availability of data, computation, and algorithms have profound implications for the future of biosignature and biomarker development in SUD screening, diagnosis, and treatment.
Retrospective biomarker/biosignature discovery has strengths (available data) and weaknesses (older, possibly less relevant trials, biospecimen availability) or biases (lack of biospecimens, older molecular datasets). Similarly, hypothesis-driven prospective biomarker discovery and validation has strengths (ability to define variables/study domains, state of the art biospecimen and biomarker data collection) and weaknesses (candidate hypotheses may miss predictive biomarker variables/domains). The choice of a retrospective versus a prospective approach may depend upon currently available resources or the ability to leverage existing public datasets. A prospective discovery and validation design offers the theoretical ability to include all domains or selected (hypothesis-driven) domains; the former is limited by practical considerations and the latter may unfortunately restrict variable discovery. The necessity of validation for replication, and clinical utility analysis to fulfill regulatory and reimbursement requirements, means that designing discovery and validation studies will involve both retrospective and prospective designs. Excluding logical incompatibilities, practical and contingent limitations are more likely to limit or slow biosignature development and translation to practice than the choice of study design or whether either type of design is hypothesis-driven.
Guidelines for biomarker development and translation to treatment of omics-based tests has been a hot topic for almost a decade [73, 74]. Omics-based tests are defined as an assay composed of or derived from many molecular measurements and interpreted by a fully specified computational model to produce a clinically actionable result [75]. There are currently no regulatory guidelines on versioning of biosignatures whose specifications may adapt and improve with more data (see Key Figure 1). Current guidelines require that biomarker tests for any application be fixed before moving into a clinical trial for assessment of their utility [76, 74]. Current recommendations regarding evaluating evidence on a biomarker prior to final utility testing are focused on the limitations derived from development from retrospective analyses, the complexity of the bioassay, and the nature of the mathematical model [77, 78]. Many challenges remain in the translation of biosignatures to clinical care; the guidelines, roadmap, and regulatory ecosystem will need to be recalibrated as models and predictions become more dynamic.
Some progress has been made in developing dynamic systems that collect and analyze data from its own processes in order to improve outcomes. Recommendations for development of a rapid learning system for biomarkers encompassing policy, data infrastructure and patient care are part of an evolutionary process of the biomedical enterprise [74]. This represents extensions of older ideas of a learning [79], continuously improving [80], and, a genomics-enabled learning [81] health care system. Adding biosignature discovery and translation to these ideas implies much greater efforts to align patient care, and provider and health care system practice than introduction of a single biomarker with a single context of use. Rising to the challenges of biosignature translation have perhaps been most thoroughly addressed in oncology [82, 83]. However, the FDA has provided guidance on: the multiple domain challenges [84, 85]; the general pathway for biomarker qualification [86]; and, biomarkers for specific SUDs [87, 88].
Concluding Remarks
Here, new strategies for biosignature development have been described that acknowledge the complexities of disease and data and touch on the ongoing challenges of translation to clinical care. In both biosignature development and in translation to clinical care, complex challenges require comprehensive, integrated solutions. We encourage addiction researchers to share data, organize themselves to enable secondary data analyses, and consider applying these and other learning algorithms to their data to generate new biological insights and prediction models. We realize there are some remaining big questions on how the strategy presented fits into existing biomarker development, clinical translation, and regulation paradigms (see Outstanding Questions and Box 2). There is a delicate balance between encouraging standardization and enabling a learning healthcare system that requires scientific and regulatory leadership to advance biosignatures into clinical care.
Outstanding Questions Box.
What tools can be developed to help researchers accelerate SUD biomarker/biosignature innovation? Learning algorithms need high quality data to grow models. Current challenges include culture (data sharing), data merging, and patient consent and regulatory issues.
How will predictive biosignatures or learning algorithms be clinically validated? Traditional biomarker development emphasizes a discovery to clinical validation path, followed by evaluation of a fixed model for clinical utility and use. How will this be adapted for versioned biosignatures in a continuous improvement system? What best practices should there be to encourage innovation, yet maintain patient safety?
What are the challenges of deploying these models into health care systems? Predictive biosignatures should be applicable across demographics and environments by qualification across diverse populations. Enabling comparative effectiveness evaluation will require data sharing across regions and practice environments.
Box 2. Clinician’s Corner.
Biomarkers of SUDs are now used clinically to detect substance use and relapse from abstinence. Emerging biomarkers will enable stratification of diagnosed SUD patients for greatest therapy efficacy.
Future SUD biosignatures will aggregate multiple predictors through omic analysis and statistical learning and dramatically expand diagnostic and predictive utility.
Translation of SUD biosignatures into clinical care requires support for clinical biospecimen testing and clinical counseling.
Improving the effectiveness of SUD biosignatures will be facilitated by integration of SUD biosignature assessment and counseling into clinical care - as smoking status assessment has become routine in multiple medical specialities.
Highlights Box.
Substance use disorders are complex diseases with significant heritibility and psychosocial susceptibility factors that develop with neurobiological and neurocognitive adaptations after chronic exposure to drugs of abuse. Individual effects of genes and psychosocial factors are modest in most cases. Recent large sample collections have began to identify single factors with confidence.
New molecular, imaging and environmental collection technologies have vastly increased the depth of data per individual. New computation and data management technologies no longer limit complex statistical modeling routines.
Machine learning algorithms are beginning to be applied to SUD data. New or streamlined artificial intelligence algorithms are disrupting all industries. These have direct applications to biomarker/biosignature development.
Acknowledgments
We thank the participants of the PKPD of Nicotine, PKTWIN and SMO-FAM studies, and Drs. Benowitz, Hops and Swan, and we acknowledge NIH/NIDA grant R43DA041211 for funding support, and Amazon Web Services and NVIDIA for computing support. We also acknowledge the work of the entire BioRealm team: https://biorealm.ai/team.
Glossary
- Bayes factors
A statistical measure that quantifies the evidence for a hypothesis relative to an alternative hypothesis.
- Bayesian
A method of statistical inference in which Bayes’ theorem is used to update the probability for a hypothesis as more evidence or information becomes available.
- bupropion
A FDA approved non-nicotine smoking cessation pharmacotherapy, most commonly prescribed in an extended-release formulation.
- edge
An element that connects two nodes in a graph, sometimes defining relationships or assigning weights.
- genome-wide association scan
An epidemiological study designed to evaluate the statistical association among genotypes and traits or diseases of interest.
- high dimensional data
Data characterized by a large number of dimensions. The refinement of “large” within this context is still a topic that is debatable. Many define “large” relative to the size of the available data; i.e., the data is high dimensional when the number of dimensions (P) exceeds the number of observations (N).
- learned weights
The estimated model parameters, such as regression coefficients.
- measures of fit
Statistical techniques designed to assess how well a model fits a data set.
- model complexity
Typically refers to the size of the model; i.e., the number of free parameters.
- nicotine replacement therapy (NRT)
Smoking cessation therapy providing the patient with nicotine; there are five FDA-approved modes of administration of NRTs, i.e., gum, patch, lozenge, spray, and inhaler.
- node
A point at which lines or pathways intersect or branch.
- omics
The exhaustive and systematic study of a molecular analytes (DNA, RNA, protein, or metabolites, and modifications to the same) from one or multiple (“meta”) species, sometimes in relation to a disease or trait, e.g., substance use behavior.
- operators
A mapping that takes as inputs elements of a space and returns other elements of the same space.
- Penalized regression
Regression method that make use of a penalty structure to regularize regression coefficients, also known as regularized and shrinkage regression.
- posterior predictive distribution
The distribution of possible unobserved values conditional on the observed values.
- posterior probability
The conditional probability of an event after observed/known evidence is taken into account.
- prediction bias
The bias associated with a prediction, with bias referring to the tendency of a measurement process to over- or under-estimate the value of a population parameter.
- prediction variance
The variability associated with a prediction, with variance being a measure of dispersion/deviation from a mean.
- priors
Probability models which are meant to reflect a modeler’s a priori knowledge of the parameters in the data model before some evidence is taken into account.
- single nucleotide polymorphism
A variation in a single nucleotide that occurs at a specific position in the genome.
- statistical learning algorithms
Algorithms which implement different statistical learning techniques. That is, algorithms which complete function estimation from a given collection of data.
- varenicline
A smoking cessation aid that is a partial agonist of α4β2 nicotinic acetylcholine receptors.
- versioning
The variables selected in a model and its estimated weights may change as more data becomes available. Careful tracking of each iteration of the data, algorithms, models, predictions, and validations allows biomarkers and biosignatures to continually improve.
- voxels
Much like a pixel in a 2-D image, a voxel is a tiny cube that contains information and is used to build a 3-D image.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boscolo-Berto R, Viel G, Montisci M, Terranova C, Favretto D, Ferrara SD. Ethyl glucuronide concentration in hair for detecting heavy drinking and/or abstinence: a meta-analysis. Int J Legal Med. 2013;127(3):611–619. doi: 10.1007/s00414-012-0809-0. [DOI] [PubMed] [Google Scholar]
- 2.Cone EJ, Bigelow GE, Herrmann ES, Mitchell JM, LoDico C, Flegel R, Vandrey R. Nonsmoker exposure to secondhand cannabis smoke. III. oral fluid and blood drug concentrations and corresponding subjective effects. J Anal Toxicol. 2015;39(7):497. doi: 10.1093/jat/bkv070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benowitz NL, Jain S, Dempsey DA, Nardone N, Helen GS, Jacob P., 3rd Urine cotinine screening detects nearly ubiquitous tobacco smoke exposure in urban adolescents. Nicotine Tob Res. 2017;19(9):1048. doi: 10.1093/ntr/ntw390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volkow ND, Koob G, Baler R. Biomarkers in substance use disorders. ACS Chem Neurosci. 2015;6(4):522. doi: 10.1021/acschemneuro.5b00067. [DOI] [PubMed] [Google Scholar]
- 5.Yi H, Breheny P, Imam N, Liu Y, Hoeschele I. Penalized multimarker vs. single-marker regression methods for genome-wide association studies of quantitative traits. Genetics. 2015;199(1):205. doi: 10.1534/genetics.114.167817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whelan R, Watts R, Orr CA, Althoff RR, Artiges E, Banaschewski T, Barker GJ, Bokde ALW, Büchel C, Carvalho FM, Conrod PJ, Flor H, Fauth-Bühler M, Frouin V, Gallinat J, Gan G, Gowland P, Heinz A, Ittermann B, Lawrence C, Mann K, Martinot J-L, Nees F, Ortiz N, Paillère-Martinot M-L, Paus T, Pausova Z, Rietschel M, Robbins TW, Smolka MN, Ströhle A, Schumann G, Garavan H IMAGEN Consortium. Neuropsychosocial profiles of current and future adolescent alcohol misusers. Nature. 2014;512(7513):185. doi: 10.1038/nature13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mamdani M, Williamson V, McMichael GO, Blevins T, Aliev F, Adkins A, Hack L, Bigdeli T, van der Vaart AD, Web BT, Bacanu S-A, Kalsi G, Kendler KS, Miles MF, Dick D, Riley BP, Dumur C, Vladimirov VI Consortium COGA. Integrating mRNA and miRNA weighted gene Co-Expression networks with eQTLs in the nucleus accumbens of subjects with alcohol dependence. PLoS One. 2015;10(9):e0137671. doi: 10.1371/journal.pone.0137671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, Nicolae DL, Cox NJ, Im HK. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J, Fleming J, Siminoff L, Traino H, Mosavel M, Barker L, Jewell S, Rohrer D, Maxim D, Filkins D, Harbach P, Cortadillo E, Berghuis B, Turner L, Hudson E, Feenstra K, Sobin L, Robb J, Branton P, Korzeniewski G, Shive C, Tabor D, Qi L, Groch K, Nampally S, Buia S, Zimmerman A, Smith A, Burges R, Robinson K, Valentino K, Bradbury D, Cosentino M, Diaz-Mayoral N, Kennedy M, Engel T, Williams P, Erickson K, Ardlie K, Winckler W, Getz G, DeLuca D, MacArthur D, Kellis M, Thomson A, Young T, Gelfand E, Donovan M, Meng Y, Grant G, Mash D, Marcus Y, Basile M, Liu J, Zhu J, Tu Z, Cox NJ, Nicolae DL, Gamazon ER, Im HK, Konkashbaev A, Pritchard J, Stevens M, Flutre T, Wen X, Dermitzakis ET, Lappalainen T, Guigo R, Monlong J, Sammeth M, Koller D, Battle A, Mostafavi S, McCarthy M, Rivas M, Maller J, Rusyn I, Nobel A, Wright F, Shabalin A, Feolo M, Sharopova N, Sturcke A, Paschal J, Anderson JM, Wilder EL, Derr LK, Green ED, Struewing JP, Temple G, Volpi S, Boyer JT, Thomson EJ, Guyer MS, Ng C, Abdallah A, Colantuoni D, Insel TR, Koester SE, Roger Little A, Bender PK, Lehner T, Yao Y, Compton CC, Vaught JB, Sawyer S, Lockhart NC, Demchok J, Moore HF. The Genotype-Tissue expression (GTEx) project. Nat Genet. 2013;45(6):580. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Brenda WJ, Jansen R, de Geus EJC, Boomsma DI, Wright FA, Sullivan PF, Nikkola E, Alvarez M, Civelek M, Lusis AJ, Lehtimäki T, Raitoharju E, Kähönen M, Seppälä I, Raitakari OT, Kuusisto J, Laakso M, Price AL, Pajukanta P, Pasaniuc B. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mancuso N, Shi H, Goddard P, Kichaev G, Gusev A, Pasaniuc B. Integrating gene expression with summary association statistics to identify genes associated with 30 complex traits. Am J Hum Genet. 2017;100(3):473. doi: 10.1016/j.ajhg.2017.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swan GE, Lessov-Schlaggar CN, Bergen AW, He Y, Tyndale RF, Benowitz NL. Genetic and environmental influences on the ratio of 3′hydroxycotinine to cotinine in plasma and urine. Pharmacogenet Genomics. 2009;19(5):388. doi: 10.1097/FPC.0b013e32832a404f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loukola A, Buchwald J, Gupta R, Palviainen T, Hällfors J, Tikkanen E, Korhonen T, Ollikainen M, Sarin A-P, Ripatti S, Lehtimäki T, Raitakari O, Salomaa V, Rose RJ, Tyndale RF, Kaprio J. A Genome-Wide association study of a biomarker of nicotine metabolism. PLoS Genet. 2015;11(9):e1005498. doi: 10.1371/journal.pgen.1005498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benowitz NL, Hukkanen J, Jacob P., 3rd Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 2009;(192):29–60. doi: 10.1007/978-3-540-69248-5\_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dempsey D, Tutka P, Jacob P, 3rd, Allen F, Schoedel K, Tyndale RF, Benowitz NL. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther. 2004;76(1):64. doi: 10.1016/j.clpt.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 16.St Helen G, Novalen M, Heitjan DF, Dempsey D, Jacob P, 3rd, Aziziyeh A, Wing VC, George TP, Tyndale RF, Benowitz NL. Reproducibility of the nicotine metabolite ratio in cigarette smokers. Cancer Epidemiol Biomarkers Prev. 2012;21(7):1105. doi: 10.1158/1055-9965.EPI-12-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bloom J, Hinrichs AL, Wang JC, von Weymarn LB, Kharasch ED, Bierut LJ, Goate A, Murphy SE. The contribution of common CYP2A6 alleles to variation in nicotine metabolism among European-Americans. Pharmacogenet Genomics. 2011;21(7):403. doi: 10.1097/FPC.0b013e328346e8c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benowitz NL, Swan GE, Jacob P, 3rd, Lessov-Schlaggar CN, Tyndale RF. CYP2A6 genotype and the metabolism and disposition kinetics of nicotine. Clin Pharmacol Ther. 2006;80(5):457. doi: 10.1016/j.clpt.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 19.Haberl M, Anwald B, Klein K, Weil R, Fuss C, Gepdiremen A, Zanger UM, Meyer UA, Wojnowski L. Three haplotypes associated with CYP2A6 phenotypes in caucasians. Pharmacogenet Genomics. 2005;15(9):609. doi: 10.1097/01.fpc.0000171517.22258.f1. [DOI] [PubMed] [Google Scholar]
- 20.Wortham M, Czerwinski M, He L, Parkinson A, Wan Y-JY. Expression of constitutive androstane receptor, hepatic nuclear factor 4 alpha, and P450 oxidoreductase genes determines interindividual variability in basal expression and activity of a broad scope of xenobiotic metabolism genes in the human liver. Drug Metab Dispos. 2007;35(9):1700. doi: 10.1124/dmd.107.016436. [DOI] [PubMed] [Google Scholar]
- 21.Ring HZ, Valdes AM, Nishita DM, Prasad S, Jacob P, 3rd, Tyndale RF, Swan GE, Benowitz NL. Gene-gene interactions between CYP2B6 and CYP2A6 in nicotine metabolism. Pharmacogenet Genomics. 2007;17(12):1007. doi: 10.1097/01.fpc.0000220560.59972.33. [DOI] [PubMed] [Google Scholar]
- 22.Bloom AJ, Martinez M, Chen L-S, Bierut LJ, Murphy SE, Goate A. CYP2B6 non-coding variation associated with smoking cessation is also associated with differences in allelic expression, splicing, and nicotine metabolism independent of common amino-acid changes. PLoS One. 2013;8(11):e79700. doi: 10.1371/journal.pone.0079700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bloom AJ, von Weymarn LB, Martinez M, Bierut LJ, Goate A, Murphy SE. The contribution of common UGT2B10 and CYP2A6 alleles to variation in nicotine glucuronidation among european americans. Pharmacogenet Genomics. 2013;23(12):706. doi: 10.1097/FPC.0000000000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bloom AJ, Murphy SE, Martinez M, von Weymarn LB, Bierut LJ, Goate A. Effects upon in-vivo nicotine metabolism reveal functional variation in FMO3 associated with cigarette consumption. Pharmacogenet Genomics. 2013;23(2):62. doi: 10.1097/FPC.0b013e32835c3b48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy SE, Park S-SL, Thompson EF, Wilkens LR, Patel Y, Stram DO, Le Marchand L. Nicotine n-glucuronidation relative to n-oxidation and c-oxidation and UGT2B10 genotype in five ethnic/racial groups. Carcinogenesis. 2014;35(11):2526. doi: 10.1093/carcin/bgu191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel YM, Stram DO, Wilkens LR, Park S-SL, Henderson BE, Le Marchand L, Haiman CA, Murphy SE. The contribution of common genetic variation to nicotine and cotinine glucuronidation in multiple ethnic/racial populations. Cancer Epidemiol Biomarkers Prev. 2015;24(1):119. doi: 10.1158/1055-9965.EPI-14-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanner J-A, Prasad B, Claw KG, Stapleton P, Chaudhry A, Schuetz EG, Thummel KE, Tyndale RF. Predictors of variation in CYP2A6 mRNA, protein, and enzyme activity in a human liver bank: Influence of genetic and nongenetic factors. J Pharmacol Exp Ther. 2017;360(1):129. doi: 10.1124/jpet.116.237594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pérez-Stable EJ, Herrera B, Jacob P, 3rd, Benowitz NL. Nicotine metabolism and intake in black and white smokers. JAMA. 1998;280(2):152. doi: 10.1001/jama.280.2.152. [DOI] [PubMed] [Google Scholar]
- 29.Benowitz NL, Pérez-Stable EJ, Herrera B, Jacob P., 3rd Slower metabolism and reduced intake of nicotine from cigarette smoking in Chinese-Americans. J Natl Cancer Inst. 2002;94(2):108. doi: 10.1093/jnci/94.2.108. [DOI] [PubMed] [Google Scholar]
- 30.Fagan P, Moolchan ET, Pokhrel P, Herzog T, Cassel KD, Pagano I, Franke AA, Kaholokula JK, Sy A, Alexander LA, Trinidad DR, Sakuma K-L, Johnson CA, Antonio A, Jorgensen D, Lynch T, Kawamoto C, Clanton MS Addictive Carcinogens Work-group. Biomarkers of tobacco smoke exposure in racial/ethnic groups at high risk for lung cancer. Am J Public Health. 2015;105(6):1237. doi: 10.2105/AJPH.2014.302492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Park SL, Stram DO, Haiman CA, Wilkens LR, Hecht SS, Kolonel LN, Murphy SE, Le Marchand L. Associations between genetic ancestries and nicotine metabolism biomarkers in the multiethnic cohort study. Am J Epidemiol. 2015;182(11):945. doi: 10.1093/aje/kwv138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chenoweth MJ, Novalen M, Hawk LW, Jr, Schnoll RA, George TP, Cinciripini PM, Lerman C, Tyndale RF. Known and novel sources of variability in the nicotine metabolite ratio in a large sample of treatment-seeking smokers. Cancer Epidemiol Biomarkers Prev. 2014;23(9):1773. doi: 10.1158/1055-9965.EPI-14-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lerman C, Jepson C, Wileyto EP, Patterson F, Schnoll R, Mroziewicz M, Benowitz N, Tyndale RF. Genetic variation in nicotine metabolism predicts the efficacy of extended-duration transdermal nicotine therapy. Clin Pharmacol Ther. 2010;87(5):553. doi: 10.1038/clpt.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnoll RA, Patterson F, Wileyto EP, Tyndale RF, Benowitz N, Lerman C. Nicotine metabolic rate predicts successful smoking cessation with transdermal nicotine: a validation study. Pharmacol Biochem Behav. 2009;92(1):6. doi: 10.1016/j.pbb.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patterson F, Schnoll RA, Wileyto EP, Pinto A, Epstein LH, Shields PG, Hawk LW, Tyndale RF, Benowitz N, Lerman C. Toward personalized therapy for smoking cessation: a randomized placebo-controlled trial of bupropion. Clin Pharmacol Ther. 2008;84(3):320. doi: 10.1038/clpt.2008.57. [DOI] [PubMed] [Google Scholar]
- 36.Ho MK, Mwenifumbo JC, Al Koudsi N, Okuyemi KS, Ahluwalia JS, Benowitz NL, Tyndale RF. Association of nicotine metabolite ratio and CYP2A6 genotype with smoking cessation treatment in African-American light smokers. Clin Pharmacol Ther. 2009;85(6):635. doi: 10.1038/clpt.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L-S, Bloom AJ, Baker TB, Smith SS, Piper ME, Martinez M, Saccone N, Hatsukami D, Goate A, Bierut L. Pharmacotherapy effects on smoking cessation vary with nicotine metabolism gene (CYP2A6) Addiction. 2014;109(1):128. doi: 10.1111/add.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lerman C, Schnoll RA, Hawk LW, Jr, Cinciripini P, George TP, Wileyto EP, Swan GE, Benowitz NL, Heitjan DF, Tyndale RF PGRN-PNAT Research Group. Use of the nicotine metabolite ratio as a genetically informed biomarker of response to nicotine patch or varenicline for smoking cessation: a randomised, double-blind placebo-controlled trial. Lancet Respir Med. 2015;3(2):131. doi: 10.1016/S2213-2600(14)70294-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wells QS, Freiberg MS, Greevy RA, Jr, Tyndale RF, Kundu S, Duncan MS, King S, Scoville E, Beaulieu D, Gatskie V, Tindle HA. Nicotine metabolism-informed care for smoking cessation: A pilot precision RCT. Nicotine Tob Res. doi: 10.1093/ntr/ntx235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-attributable Disease : a Report of the Surgeon General. U.S. Department of Health and Human Services, Public Health Service, Office of the Surgeon General; 2010. [PubMed] [Google Scholar]
- 41.Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. doi: 10.1371/journal.pcbi.1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baurley JW, Edlund CK, Pardamean CI, Conti DV, Krasnow R, Javitz HS, Hops H, Swan GE, Benowitz NL, Bergen AW. Genome-Wide association of the Laboratory-Based nicotine metabolite ratio in three ancestries. Nicotine Tob Res. 2016;18(9):1837. doi: 10.1093/ntr/ntw117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel YM, Park SL, Han Y, Wilkens LR, Bickeböller H, Rosenberger A, Caporaso N, Landi MT, Brüske I, Risch A, Wei Y, Christiani DC, Brennan P, Houlston R, McKay J, McLaughlin J, Hung R, Murphy S, Stram DO, Amos C, Le Marchand L. Novel association of genetic markers affecting CYP2A6 activity and lung cancer risk. Cancer Res. 2016;76(19):5768. doi: 10.1158/0008-5472.CAN-16-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chenoweth MJ, Ware JJ, Zhu AZX, Cole CB, Cox LS, Nollen N, Ahluwalia JS, Benowitz NL, Schnoll RA, Hawk LW, Jr, Cinciripini PM, George TP, Lerman C, Knight J, Tyndale RF PGRN-PNAT Research Group. Genome-wide association study of a nicotine metabolism biomarker in african american smokers: impact of chromosome 19 genetic influences. Addiction. doi: 10.1111/add.14032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whirl-Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, Thorn CF, Altman RB, Klein TE. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neter J, Kutner MH, Nachtsheim CJ, Wasserman W GTEx Consortium. Applied linear statistical models. Vol. 4. Irwin Chicago: 1996. [Google Scholar]
- 47.Hesterberg T, Choi NH, Meier L, Fraley C, et al. Least angle and l1 penalized regression: A review. Statistics Surveys. 2008;2:61. [Google Scholar]
- 48.Kyung M, Gill J, Ghosh M, Casella G, et al. Penalized regression, standard errors, and bayesian lassos. Bayesian Analysis. 2010;5(2):369. [Google Scholar]
- 49.Tibshirani R. Regression shrinkage and selection via the lasso. Journal of the Royal Statistical Society Series B (Methodological) 1996:267–288. [Google Scholar]
- 50.Hoerl A, Kennard R. Ridge regression, in ‘encyclopedia of statistical sciences’. 1988;8 [Google Scholar]
- 51.Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2005;67(2):301. [Google Scholar]
- 52.Friedman J, Hastie T, Tibshirani R. Springer series in statistics. Vol. 1. New York: 2001. The elements of statistical learning. [Google Scholar]
- 53.Bergen Andrew W, Edlund Christopher K, Pardamean Carissa I, Baurley James W. Smokescreen translational analysis platform: Prediction of nicotine metabolism and smoking cessation. NIDA Genetics Consortium meeting; 2016. [Google Scholar]
- 54.Hoeting JA, Madigan D, Raftery AE, Volinsky CT. Bayesian model averaging: a tutorial. Statistical science. 1999:382–401. [Google Scholar]
- 55.Caruana R, Niculescu-Mizil A, Crew G, Ksikes A. Ensemble selection from libraries of models. Proceedings of the twenty-first international conference on Machine learning; ACM; 2004. p. 18. [Google Scholar]
- 56.Wasserman L. Bayesian model selection and model averaging. Journal of mathematical psychology. 2000;44(1):92. doi: 10.1006/jmps.1999.1278. [DOI] [PubMed] [Google Scholar]
- 57.Gelman A. Encyclopedia of environmetrics. Prior distribution. [Google Scholar]
- 58.Baurley JW, Conti DV, Gauderman WJ, Thomas DC. Discovery of complex pathways from observational data. Stat Med. 2010;29(19):1998. doi: 10.1002/sim.3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gelman A, Carlin JB, Stern HS, Rubin DB. Bayesian data analysis [Google Scholar]
- 60.Patkar AA, Rozen S, Mannelli P, Matson W, Pae C-U, Krishnan KR, Kaddurah-Daouk R. Alterations in tryptophan and purine metabolism in cocaine addiction: a metabolomic study. Psychopharmacology. 2009;206(3):479. doi: 10.1007/s00213-009-1625-1. [DOI] [PubMed] [Google Scholar]
- 61.Cho YU, Lee D, Lee J-E, Kim KH, Lee DY, Jung Y-C. Exploratory metabolomics of biomarker identification for the internet gaming disorder in young korean males. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1057:24. doi: 10.1016/j.jchromb.2017.04.046. [DOI] [PubMed] [Google Scholar]
- 62.Oreland L, Lagravinese G, Toffoletto S, Nilsson KW, Harro J, Robert Cloninger C, Comasco E. Personality as an intermediate phenotype for genetic dissection of alcohol use disorder. J Neural Transm. doi: 10.1007/s00702-016-1672-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Foulds J, Newton-Howes G, Guy NH, Boden JM, Mulder RT. Dimensional personality traits and alcohol treatment outcome: a systematic review and meta-analysis. Addiction. 2017;112(8):1345. doi: 10.1111/add.13810. [DOI] [PubMed] [Google Scholar]
- 64.Volkow ND, Koob GF, Croyle RT, Bianchi DW, Gordon JA, Koroshetz WJ, Pérez-Stable EJ, Riley WT, Bloch MH, Conway K, Deeds BG, Dowling GJ, Grant S, Howlett KD, Matochik JA, Morgan GD, Murray MM, Noronha A, Spong CY, Wargo EM, Warren KR, Weiss SRB. The conception of the ABCD study: From substance use to a broad NIH collaboration. Dev Cogn Neurosci. doi: 10.1016/j.dcn.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bogdan R, Salmeron BJ, Carey CE, Agrawal A, Calhoun VD, Garavan H, Hariri AR, Heinz A, Hill MN, Holmes A, Kalin NH, Goldman D. Imaging genetics and genomics in psychiatry: A critical review of progress and potential. Biol Psychiatry. 2017;82(3):165. doi: 10.1016/j.biopsych.2016.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramsey A. Integration of technology-based behavioral health interventions in substance abuse and addiction services. Int J Ment Health Addict. 2015;13(4):470. doi: 10.1007/s11469-015-9551-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Agrawal A, Edenberg HJ, Gelernter J. Meta-Analyses of Genome-Wide association data hold new promise for addiction genetics. J Stud Alcohol Drugs. 2016;77(5):676. doi: 10.15288/jsad.2016.77.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Acion L, Kelmansky D, van der Laan M, Sahker E, Jones D, Arndt S. Use of a machine learning framework to predict substance use disorder treatment success. PLoS One. 2017;12(4):e0175383. doi: 10.1371/journal.pone.0175383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suchting R, Hébert ET, Ma P, Kendzor DE, Businelle MS. Using elastic net penalized cox proportional hazards regression to identify predictors of imminent smoking lapse. Nicotine Tob Res. doi: 10.1093/ntr/ntx201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chih M-Y, Patton T, McTavish FM, Isham AJ, Judkins-Fisher CL, Atwood AK, Gustafson DH. Predictive modeling of addiction lapses in a mobile health application. J Subst Abuse Treat. 2014;46(1):29. doi: 10.1016/j.jsat.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vivanti R, Szeskin A, Lev-Cohain N, Sosna J, Joskowicz L. Automatic detection of new tumors and tumor burden evaluation in longitudinal liver CT scan studies. Int J Comput Assist Radiol Surg. doi: 10.1007/s11548-017-1660-z. [DOI] [PubMed] [Google Scholar]
- 72.Zong N, Kim H, Ngo V, Harismendy O. Deep mining heterogeneous networks of biomedical linked data to predict novel drug-target associations. Bioinformatics. 2017;33(15):2337. doi: 10.1093/bioinformatics/btx160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Institute of Medicine. Cancer Biomarkers: The Promises and Challenges of Improving Detection and Treatment. National Academies Press; 2007. Committee on Developing Biomarker-Based Tools for Cancer Screening, Diagnosis, and Treatment. [Google Scholar]
- 74.National Academies of Sciences, Engineering, and Medicine, Institute of Medicine, Board on Health Care Services. Biomarker Tests for Molecularly Targeted Therapies: Key to Unlocking Precision Medicine. National Academies Press; 2016. Committee on Policy Issues in the Clinical Development and Use of Biomarkers for Molecularly Targeted Therapies. [DOI] [PubMed] [Google Scholar]
- 75.National Academies of Sciences, Engineering, and Medicine, Institute of Medicine, Board on Health Care Services. Biomarker Tests for Molecularly Targeted Therapies: Key to Unlocking Precision Medicine. National Academies Press; 2016. Committee on Policy Issues in the Clinical Development and Use of Biomarkers for Molecularly Targeted Therapies. [PubMed] [Google Scholar]
- 76.Committee on the Review of Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials, Board on Health Care Services, Board on Health Sciences Policy, Institute of Medicine. Evolution of Translational Omics: Lessons Learned and the Path Forward. National Academies Press; 2012. [PubMed] [Google Scholar]
- 77.McShane LM, Cavenagh MM, Lively TG, Eberhard DA, Bigbee WL, Williams PM, Mesirov JP, Polley M-YC, Kim KY, Tricoli JV, Taylor JMG, Shuman DJ, Simon RM, Doroshow JH, Conley BA. Criteria for the use of omics-based predictors in clinical trials. Nature. 2013;502(7471):317. doi: 10.1038/nature12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McShane LM, Cavenagh MM, Lively TG, Eberhard DA, Bigbee WL, Williams PM, Mesirov JP, Polley M-YC, Kim KY, Tricoli JV, Taylor JMG, Shuman DJ, Simon RM, Doroshow JH, Conley BA. Criteria for the use of omics-based predictors in clinical trials: explanation and elaboration. BMC Med. 2013;11:220. doi: 10.1186/1741-7015-11-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Olsen L, Aisner D, McGinnis J. Roundtable on evidence-based medicine, institute of medicine: learning healthcare system: workshop summary. Institute of Medicine of The National Academies; Washington DC: [Google Scholar]
- 80.McGinnis JM, Stuckhardt L, Saunders R, Smith M, et al. Best care at lower cost: the path to continuously learning health care in America. National Academies Press; 2013. [PubMed] [Google Scholar]
- 81.Berger AC, Olson S, Beachy SH, et al. Genomics-enabled learning health care systems: Gathering and using genomic information to improve patient care and research: Workshop summary. National Academies Press; 2015. [PubMed] [Google Scholar]
- 82.Kranzler HR, Smith RV, Schnoll R, Moustafa A, Greenstreet-Akman E. Precision medicine and pharmacogenetics: what does oncology have that addiction medicine does not? Addiction. 2017;112(12):2086. doi: 10.1111/add.13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salgado R, Moore H, Martens JWM, Lively T, Malik S, McDermott U, Michiels S, Moscow JA, Tejpar S, McKee T, Lacombe D. IBCD-Faculty, Societal challenges of precision medicine: Bringing order to chaos. Eur J Cancer. 2017;84:325. doi: 10.1016/j.ejca.2017.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Amur S, LaVange L, Zineh I, Buckman-Garner S, Woodcock J. Biomarker qualification: Toward a multiple stakeholder framework for biomarker development, regulatory acceptance, and utilization. Clin Pharmacol Ther. 2015;98(1):34. doi: 10.1002/cpt.136. [DOI] [PubMed] [Google Scholar]
- 85.Sauer J-M, Porter AC. Biomarker Programs, Predictive Safety Testing Consortium, Preclinical biomarker qualification. Exp Biol Med. 2017 1535370217743949. [Google Scholar]
- 86.Amur SG, Sanyal S, Chakravarty AG, Noone MH, Kaiser J, McCune S, Buckman-Garner SY. Building a roadmap to biomarker qualification: challenges and opportunities. Biomark Med. 2015;9(11):1095. doi: 10.2217/bmm.15.90. [DOI] [PubMed] [Google Scholar]
- 87.Bough KJ, Lerman C, Rose JE, McClernon FJ, Kenny PJ, Tyndale RF, David SP, Stein EA, Uhl GR, Conti DV, Green C, Amur S. Biomarkers for smoking cessation. Clin Pharmacol Ther. 2013;93(6):526. doi: 10.1038/clpt.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bough KJ, Amur S, Lao G, Hemby SE, Tannu NS, Kampman KM, Schmitz JM, Martinez D, Merchant KM, Green C, Sharma J, Dougherty AH, Moeller FG. Biomarkers for the development of new medications for cocaine dependence. Neuropsychopharmacology. 2014;39(1):202. doi: 10.1038/npp.2013.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dobson AJ, Barnett A. An Introduction to Generalized Linear Models. 3. Taylor & Francis; 2008. [Google Scholar]
- 90.Zou H. The adaptive lasso and its oracle properties. Journal of the American statistical association. 2006;101(476):1418. [Google Scholar]
- 91.Fan J, Li R. Variable selection via nonconcave penalized likelihood and its oracle properties. Journal of the American statistical Association. 2001;96(456):1348. [Google Scholar]
- 92.Zhang C-H, et al. Nearly unbiased variable selection under minimax concave penalty. The Annals of statistics. 2010;38(2):894. [Google Scholar]
- 93.Breheny P, Huang J. Coordinate descent algorithms for nonconvex penalized regression, with applications to biological feature selection. The annals of applied statistics. 2011;5(1):232. doi: 10.1214/10-AOAS388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Candes E, Tao T. The dantzig selector: Statistical estimation when p is much larger than n. The Annals of Statistics. 2007:2313–2351. [Google Scholar]
- 95.Li X, Zhao T, Yuan X, Liu H. The flare package for high dimensional linear regression and precision matrix estimation in r. The Journal of Machine Learning Research. 2015;16(1):553–557. [PMC free article] [PubMed] [Google Scholar]