

Abstract
Background
Cell-free circulating DNA (cfDNA) is becoming a useful biopsy for noninvasive diagnosis of diseases. Microbial sequences in plasma cfDNA may provide important information to improve prognosis and treatment. We have developed a stringent method to identify microbial species via microbial cfDNA in the blood plasma of early-onset breast cancer (EOBC) patients and healthy females. Empirically, microbe-originated sequence reads were identified by mapping non-human PE reads in cfDNA libraries to microbial databases. Those mapped concordantly to unique microbial species were assembled into contigs, which were subsequently aligned to the same databases. Microbial species uniquely aligned were identified and compared across all individuals on MCRPM (Microbial CfDNA Reads Per Million quality PE reads) basis.
Results
The predominant microbial cfDNAs in all plasma samples examined are originated from bacteria and these bacteria were limited to only a few genera. Among those, Acinetobacter johnsonii XBB1 and low levels of Mycobacterium spp. were commonly found in all healthy females, but also present in an EOBC patient. Compared to those in healthy counterparts, bacterial species in EOBC patients are more diverse and more likely to present at high levels. Among these three EOBC patients tested, a patient who has record high titer (2,724 MCRPM) of Pseudomonas mendocina together with 8.82 MCRPM of Pannonibacter phragmitetus has passed away; another patient infected by multiple Sphingomonas species remains alive; while the third patient who has similar microbial species (Acinetobacter johnsonii XBB1) commonly seen in normal controls is having a normal life.
Conclusions
Our preliminary data on the profiles of microbial cfDNA sequences suggested that it may have some prognostic value in cancer patients. Validation in larger number of patients is warranted.
Keywords: Cell-free circulating DNA (cfDNA), Microbial cfDNA reads per million quality PE reads (MCRPM), Microbial cfDNA
Background
It is recently established that human blood contains cell-free circulating DNA (cfDNA) resulted from apoptosis and necrosis of normal and diseased human cells or inhabited microbes [1]. Microbial genomic DNA can be detected in plasma [2, 3], and the level of bacterial sequences in cfDNA was found to be higher for diseased and immunocompromised individuals than healthy counterparts [4]. A number of studies have also indicated that a low level of pleomorphic bacteria may inhabit normal blood circulation [5–7], and bacteria were also found in tumor tissues [8]. Data have also shown that these microbes may regulate a broad range of host physiological activities including metabolism, inflammation, immunity and hematopoiesis, which may in turn influence tumorigenesis and/or cancer development [9]. These data suggest that microbiota may play a key role to orchestrate cancer therapy and many more cancer-associated microbes need to be identified.
It was estimated that 15–20% of human cancers were caused by viral infections [10, 11]. The best-known and best-studied carcinogenic microbes are viruses. These include hepatitis B (HBV) and C (HCV) viruses, human papillomavirus (HPV), human T-cell leukemia virus and Epstein-Barr virus (EBV) [12–16]. The discoveries of viral roles in tumorigenesis were facilitated by the fact that viruses are absolute parasites and that viral genomes are generally smaller and with fewer genes compared to bacterial and fungal genomes. Although reports have shown the presence of microbes in human breast cancer tissues and lateral gene transfer of microbial genetic sequences into the human genome, especially in cancer tissues [17, 18]. Bacteria and fungi have complex genomes and their interactions with hosts are more intricate than that of viruses, making it more difficult to justify their roles in tumorigenesis [18].
With the robust speed and yield of next-generation sequencing (NGS) [19], in combination with the noninvasiveness and easy accessibility of liquid biopsies and the availability of microbial genome databases, NGS-based cfDNA sequencing is a powerful approach for studying microbiota in body fluids. Recently, Grumaz and colleagues have demonstrated that NGS-coupled microbial cfDNA analysis is strongly correlated with the results of blood cultures [3]. Besides the Gram-negative bacteria which exactly matched the results from corresponding blood culturing, they also found microbial species showing false negative in blood culture, indicating the superiority of the approach. Their work represents a proof-of-concept pioneer work for direct detection of bacterial species using plasma cfDNA.
Here, we present another NGS-based approach which is able to identify microbes of a broad range. Our method relies on a stringent mapping/alignment procedure for specific association of microbial cfDNA sequences with potential pathogens and rare microbial species which may present in cancer patients and/or healthy individuals. Empirically, paired-end (PE) reads unmappable to hg19 were collected and mapped to microbial databases composed of bacterial, fungal and viral/phage genomes. Those mapped concordantly to unique microbial species were assembled into contigs, which were subsequently used to BLAST the same microbial databases. The microbial species aligned uniquely by contigs were considered as the real hits. Microbes associated with each person were portrayed with their relative microbial cfDNA levels on a MCRPM (Microbial CfDNA Reads Per Million quality PE reads) basis.
In this study, we identified microbial species in both healthy and diseased females. One of the most striking features is that the microbial inhabitants in breast cancer patients are not only much more diverse than that of normal females, but also can be tightly correlated to the health conditions of the patients. Taken together, this investigation indicates that NGS-based microbial cfDNA sequencing has a strong potential to improve disease diagnosis and treatment.
Results and discussion
Microbial databases
Genomic sequences of bacteria, fungi and viruses were downloaded from NCBI (April 6, 2017). Plasmids were subsequently removed. The database contain 9,336, 913 and 9,050 genomes of bacteria, fungi and viruses/phages, respectively (Table 1).
Table 1.
Microbial databases employed in the study
| Bacteria | Fungi | Viruses | |
|---|---|---|---|
| #Contigs | 39,434 | 20 | 0 |
| #Scaffold | 36,076 | 170 | 3 |
| #Chromosome | 978 | 38 | 22 |
| #Complete genome | 6,711 | 7 | 7,175 |
| #Subtotal | 83,199 | 235 | 7,200 |
| #Species | 7,689 | 45 | 7,197 |
| #Sequences | 15,849 | 913 | 9,050 |
| Total #, after plasmid sequences excluded | 9,336 | 913 | 9,050 |
RefSeq genome: April 6, 2017
Library statistics
Five plasma DNA libraries including BBC (normal), EJC (normal), BC0145 (EOBC), BC0190 (EOBC) and CGBC025 (EOBC) were sequenced by Illumina HiSeq X Ten to produce at least 384 million PE reads per library (eq. ~ 38-fold coverage) (Table 2). Mapping against human genome assembly hg19 resulted in mappable ratios of 94.62%, 96.79%, 92.13%, 95.99% and 95.16% (eq. to 5.38%, 3.21%, 7.87%, 4.01% and 4.84% of unmappable ratios) for BBC, EJC, BC0145, BC0190 and CGBC025 libraries, respectively.
Table 2.
Library statistics
| Library | BBC (normal) | EJC (normal) | BC0145 (EOBC) | BC0190 (EOBC) | CGBC025 (EOBC) |
|---|---|---|---|---|---|
| Raw PE reads | 384,623,309 | 420,790,943 | 392,322,204 | 434,928,103 | 628,712,403 |
| Quality PE reads | 371,837,085 | 388,551,037 | 375,255,962 | 426,187,686 | 579,192,276 |
| Normalization factor | 372 | 389 | 375 | 426 | 579 |
| hg19-mapped reads (%) | 351,841,885 (94.62%) | 376,097,300 (96.79%) | 345,714,693 (92.13%) | 409,083,110 (95.99%) | 551,181,421 (95.16%) |
| hg19-unmappable PE reads (%) | 19,995,200 (5.38%) | 12,453,737 (3.21%) | 29,541,269 (7.87%) | 17,104,576 (4.01%) | 28,010,855 (4.84%) |
Most of the microbial cfDNA sequences were originated from bacteria, instead of fungi or viruses/phages
Non-human PE reads (i.e., reads unmappable to hg19) were mapped against the microbial databases. 0.08% - 4.85% (per library) were found to be originated from bacteria, 0.00% - 0.01% from fungi, and 0.00% - 0.16% from viruses/phages (Table 3, upper part). PE reads uniquely and concordantly mapped to single species were then used to build contigs. When contigs were aligned to the same microbial databases, numbers of contigs aligned to bacteria were also significantly higher than that aligned to either fungi or viruses/phages for all libraries (Table 3, lower part).
Table 3.
Statistics of contigs and alignment
| BBC | EJC | BC0145 | BC0190 | CGBC025 | ||
|---|---|---|---|---|---|---|
| hg19-unmapped PE reads | 19,995,200 | 12,453,737 | 29,541,269 | 17,104,576 | 28,010,855 | |
| Bacteria | Mapped PE reads (%) | 15,504 (0.08%) | 31,653 (0.25%) | 1,432,423 (4.85%) | 50,310 (0.29%) | 45,858 (0.16%) |
| Fungi | Mapped PE reads (%) | 451 (0.00%) | 456 (0.00%) | 590 (0.00%) | 1,153 (0.01%) | 996 (0.00%) |
| Viruses/Phages | Mapped PE reads (%) | 1,367 (0.01%) | 677 (0.01%) | 1,528 (0.01%) | 26,749 (0.16%) | 1,267 (0.00%) |
| Bacteria | #Contigs | 894 | 1,495 | 7,971 | 2,609 | 2,588 |
| Max contig length | 900 | 2,287 | 16,248 | 2,035 | 3,911 | |
| Min contig length | 64 | 64 | 64 | 64 | 64 | |
| Median contig length | 211 | 227 | 231 | 214 | 220 | |
| N50 | 215 | 266 | 1,616 | 219 | 237 | |
| #contigs w/ size ≥250 bp (%) | 80 (9.0%) | 558 (37.3%) | 3,602 (45.2%) | 333 (12.8%) | 748 (28.9%) | |
| #aligned contigs (also see Table 4) | 54 | 381 | 2,456 | 225 | 451 | |
| Fungi | #contigs | 44 | 38 | 215 | 92 | 78 |
| Max contig length | 315 | 235 | 352 | 427 | 262 | |
| Min contig length | 65 | 64 | 64 | 64 | 64 | |
| Median contig length | 71 | 84 | 71 | 84 | 73.5 | |
| N50 | 99 | 192 | 78 | 192 | 127 | |
| #contigs w/ size ≥250 bp (%) | 2 (4.6%) | 0 (0.0%) | 3 (1.4%) | 3 (3.3%) | 1 (1.3%) | |
| #aligned contigs | 2 | 0 | 0 | 2 | 1 | |
| Viruses/ phages | #Contigs | 75 | 62 | 287 | 155 | 72 |
| Max contig length | 655 | 546 | 336 | 695 | 274 | |
| Min contig length | 64 | 64 | 64 | 64 | 64 | |
| Median contig length | 68 | 84 | 70 | 73 | 74 | |
| N50 | 194 | 210 | 85 | 189 | 126 | |
| #contigs w/ size ≥250 bp (%) | 9 (12.0%) | 7 (11.3%) | 10 (3.5%) | 13 (8.4%) | 1 (1.4%) | |
| #aligned contigs | 7 | 5 | 2 | 10 | 0 | |
Most bacterial cfDNA in healthy females are derived from Acinetobacter spp., while high titers of bacterial cfDNA in EOBC patients are derived from Pseudomonas or Sphingomonas spp.
To compare the levels (or titers) of microbial cfDNA fragments between individuals, the constituent sequence reads of all contigs were summed up for each microbe and normalized on per-million basis (see Normalization in Methods section). This strategy is able to reflect the relative levels for the cfDNA fragment compositions in the blood, because each read was supposed to be resulted from a cfDNA fragment.
Alignment with contigs maximizes the accuracy of microbial species identification, while read-level normalization aims to achieve the best approximation of relative titers for microbial species identified. Microbial species mapped with single contigs were also recorded because the number of constituent reads may vary significantly and can be very high for long contigs. Notice that each read represents a cfDNA fragment either in the original plasma sample or produced by PCR amplification. To minimize the impact of nonspecific alignment, bacterial species with < 0.1 MCRMP were removed from the list.
Bacterial species identified are significantly different between healthy and diseased individuals (except BC0190) (Table 4). As indicated by non-parametric Mann-Whitney-Wilcoxon test, bacterial species found between normal and diseased individuals are different (p-value 0.020). Healthy females tend to harbor Acinetobacter species, especially Acinetobacter johnsonii XBB1, although the level may fluctuate (ranging between 0.27–6.00 MCRPM). Mycobacterium spp. and Ralstonia mannitolilytica were also present in healthy females (and BC0190 as well), but less significant. On the other hand, EOBC patients tend to harbor more diverse bacterial species, presumably influenced by secondary infection. The highest titer (2742.72 MCRPM) was found to be Pseudomonas mendocina ymp in patient BC0145, while the second highest titer (13.64 MCRPM) was Sphingomonas melonis in patient CGBC025. Patient BC0145, who was diagnosed with bilateral breast cancer and passed away a year after surgery in 2007 (see Plasma cfDNA preparation in Methods section), was also infected by other Pseudomonas spp. (e.g., P. alcaliphila of 2.84 MCRPM and P. aeruginosa of 0.89 MCRPM) and Pannonibacter phragmitetus (8.82 MCRPM). In contrast to patients BC0145 and CGBC025 but similar to normal females, patient BC0190 had Acinetobacter johnsonii XBB1 as the major bacterial inhabitant. She had surgery in February of 2007 and since then has been living a healthy life.
Table 4.
Bacterial species identified
| ID | Species | No. of contigs | Total aligned length | Total no. of associated PE reads | Total no. of associated SE reads | Sum of PE and SE reads | MCRPM |
|---|---|---|---|---|---|---|---|
| BBC ctl | (sum/372) | ||||||
| Acinetobacter johnsonii XBB1 | 17 | 5,428 | 69 | 30 | 99 | 0.27 | |
| Bacillus cereus | 2 | 708 | 6 | 35 | 41 | 0.11 | |
| Mycobacterium sp. | 4 | 2,124 | 73 | 70 | 143 | 0.38 | |
| Ralstonia mannitolilytica | 4 | 1,227 | 31 | 12 | 43 | 0.12 | |
| EJC ctl | (sum/389) | ||||||
| Acinetobacter haemolyticus | 12 | 4,345 | 59 | 30 | 89 | 0.23 | |
| Acinetobacter johnsonii XBB1 | 231 | 99,403 | 1,719 | 616 | 2,335 | 6.00 | |
| Acinetobacter junii | 4 | 1,948 | 30 | 7 | 37 | 0.10 | |
| Acinetobacter lwoffii | 17 | 6,186 | 87 | 31 | 118 | 0.30 | |
| Acinetobacter schindleri | 4 | 1,770 | 43 | 16 | 59 | 0.15 | |
| Bacillus cereus | 4 | 1,314 | 31 | 51 | 82 | 0.21 | |
| Corynebacterium pseudotuberculosis | 2 | 588 | 2 | 48 | 50 | 0.13 | |
| Methylobacterium populi | 18 | 5,102 | 54 | 18 | 72 | 0.19 | |
| Mycobacterium abscessus | 4 | 2,238 | 53 | 14 | 67 | 0.17 | |
| Mycobacterium avium | 8 | 2,751 | 30 | 15 | 45 | 0.12 | |
| Mycobacterium sp. | 8 | 2,620 | 36 | 63 | 99 | 0.25 | |
| Mycoplasma mycoides | 1 | 276 | 17 | 28 | 45 | 0.12 | |
| Porphyrobacter sp. | 7 | 2,478 | 20 | 34 | 54 | 0.14 | |
| Ralstonia mannitolilytica | 8 | 2,899 | 37 | 40 | 77 | 0.20 | |
| Sphingomonas sp. | 11 | 3,533 | 27 | 28 | 55 | 0.14 | |
| BC 0145 | (sum/375) | ||||||
| Pannonibacter phragmitetus | 714 | 230,779 | 1,999 | 1,309 | 3,308 | 8.82 | |
| Pseudomonas aeruginosa | 2 | 1,824 | 254 | 78 | 332 | 0.89 | |
| Pseudomonas alcaliphila | 6 | 5,093 | 896 | 170 | 1,066 | 2.84 | |
| Pseudomonas mendocina ymp | 1,675 | 2,678,493 | 918,733 | 109,787 | 1,028,520 | 2,742.72 | |
| Pseudomonas sp. | 3 | 1,195 | 41 | 36 | 77 | 0.21 | |
| Pseudomonas stutzeri | 3 | 1,717 | 220 | 21 | 241 | 0.64 | |
| Rhizobium sp. | 34 | 12,418 | 144 | 65 | 209 | 0.56 | |
| BC 0190 | (sum/426) | ||||||
| Acinetobacter baumannii | 2 | 1,078 | 29 | 17 | 46 | 0.11 | |
| Acinetobacter johnsonii XBB1 | 71 | 24,930 | 366 | 168 | 534 | 1.25 | |
| Bacillus cereus | 4 | 1,172 | 24 | 76 | 100 | 0.23 | |
| Methylobacterium populi | 38 | 10,970 | 123 | 72 | 195 | 0.46 | |
| Mycobacterium abscessus | 5 | 2,599 | 67 | 28 | 95 | 0.22 | |
| Mycobacterium sp. | 7 | 2,432 | 52 | 88 | 140 | 0.33 | |
| Porphyrobacter sp. | 6 | 2,195 | 38 | 31 | 69 | 0.16 | |
| Ralstonia mannitolilytica | 7 | 2,370 | 19 | 53 | 72 | 0.17 | |
| Sphingomonas sp. | 17 | 5,030 | 63 | 44 | 107 | 0.25 | |
| CGBC 025 | (sum/579) | ||||||
| Acinetobacter baumannii | 24 | 6,818 | 118 | 48 | 166 | 0.29 | |
| Bradyrhizobium sp. | 7 | 2,059 | 41 | 16 | 57 | 0.10 | |
| Enterobacter cloacae | 8 | 3,140 | 108 | 33 | 141 | 0.24 | |
| Sphingobium japonicum | 8 | 5,088 | 262 | 43 | 305 | 0.53 | |
| Sphingomonas melonis | 336 | 157,346 | 5,397 | 2,503 | 7,900 | 13.64 | |
| Sphingomonas panacis | 4 | 1,261 | 36 | 30 | 66 | 0.11 | |
| Sphingomonas sp. | 51 | 21,410 | 720 | 271 | 991 | 1.71 |
Only microbial species with MCRPM ≥ 0.1 are listed. Those with MCRPM ≥ 1 are listed in bold.
Ctl control; MCRPM, microbial cfDNA per million quality PE reads; sp. (same as spp.), species with unspecified species name
Acinetobacter johnsonii (Gram-negative (G-)) is a ubiquitous bacterium commonly found in human and fish [20–22]. However, it is becoming an emerging threat to human health [23]. A. johnsonii XBB1 was first isolated from a hospital in Chengdu City, China. Both Mycobacteria and Ralstonia mannitolilytica are also ubiquitous in nature. R. mannitolilytica (G-), which was ever named as Pseudomonas thomasii, Pseudmonas pickettii or Ralstonia pickettii, is a soil bacterium. In 1989, this bacterium caused a nosocomial outbreak in Taiwan through sodium chloride contamination [24]. Sphingomonas melonis was first isolated from melon. Strain Sphingomonas melonis sp. nov. is becoming a common pathogen in melon fruits [25]. This species was detected in both healthy and diseased females and a high titer (13.64 MCRPM) was observed in patient CGBC025.
Most of the bacteria on the list (even Pseudomonas spp.) are known as opportunistic bacterial pathogens. For example, P. aeruginosa (G-) is a bacterium commonly found in skin of healthy people [26]. Both R. mannitolilytica, P. aeruginosa are innocuous bacteria, but can be acquired from hospital where antibiotics are commonly used. Through constant challenge by novel and multiple antibiotics, they might have become multidrug resistant through horizontal transfer.
Fungal viral and phage species
When aligned to fungal and viral/phage databases, PE reads of all five libraries showed no significant hit for all individuals, either healthy or diseased. Thus, for all these categories, results are much less significant than that of bacterial species.
Conclusions
As revealed by previous investigations, microbes may inhabit a number of human tissues which are technically inaccessible for direct analysis. Fortunately, their genomic sequences are released into blood circulation and are thus become available for consecutive experimentation for diagnostic or prognostic purposes. As such, NGS-based cfDNA sequencing has emerged to be one of the most suitable choices.
Here, we empirically demonstrate the feasibilities of using NGS-based cfDNA sequencing for direct detection of microbial species in human body. The feasibility is made possible by NGS, the noninvasiveness and easy accessibility of plasma cfDNA samples, and a stringent protocol for mapping and alignment.
The process is guarded by multiple layers of stringency. Firstly, it started only with PE reads which map concordantly to unique microbial genomes; both discordant hits and single-end hits were excluded in this step, although single-end matches may be included during contig assembly. Secondly, since microbes share significant degree of sequence similarity, contigs were built and used to align to the same microbial genome assemblies for better accuracy. Thirdly, contig-level alignment was reinforced with stringent parameters. Fourthly, only those microbial species “uniquely” aligned by contigs were considered as real hits. Moreover, microbial spp. with less than 0.1 MCRPM were excluded.
Although only five samples were analyzed, this study reveals a number of interesting phenomena: 1) microbial cfDNA can also be detected in the blood plasma of healthy individuals; 2) bacteria constitute the major microbial species in human body; 3) these bacteria belong to only a limited number of genera and species, no matter for healthy or diseased individuals; 4) most microbial species in healthy human bodies show only low levels of cfDNA; 5) Acinetobacter (esp., A. johnsonii XBB1) cfDNA is ubiquitous for healthy individuals, but it may also present in EOBC patient with near normal heath conditions; 6) on the other hand, Pseudomonas and Sphingomonas cfDNA can be found in the plasma of breast cancer patients with severe symptoms; 7) microbial spp. in breast cancer patients tend to be more diverse compared to that in healthy counterparts.
Pseudomonas species are well known opportunistic bacteria commonly found on the surface of human body. It is thus not surprising to detect Pseudomonas in cancer patients with symptoms severe enough to compromise the immune system. On the other hand, the presence of Sphingomonas melonis in a cancer patient is somewhat surprising. In natural environment, this bacterium is known to cause brown spots on yellow Spanish melons. Its pathogenicity upon human was first described only 15 years ago [25]. Its quick spreading is a warning sign to human communities.
For cancer or other diseases resulted from viruses living constantly in human body (e.g., HPV and HSV which cause cervical cancer and herpes respectively), the presence of corresponding viral cfDNA sequences in plasma are expected. However, the viral cfDNA titer is expected to fluctuate with its proliferation status, with relatively higher titer during active stage and lower titer during dormant stage. For breast cancer, no clear correlation with viral pathogens has ever been unambiguously demonstrated. This partially explains why no significant viral cfDNA were detected among these EOBC patients.
Furthermore, the microbial species detected in EOBC patients may not be responsible for the tumorigenesis of the disease, because, as influenced by health conditions as well as environmental factors, the composition of microbial species in human body can be very dynamic and the cfDNA sequences of the causative agents may no longer present or may present only at an insignificant level when the disease is eventually established. However, our results indicate a correlation between disease status and the composition of microbial cfDNA in plasma, and suggest the feasibility of using NGS-based microbial cfDNA sequencing for personalized treatment of diseases.
Although this study allows us to extrapolate certain interesting phenomena from microbial cfDNA in plasma, the capacity of our approach is limited by sample size. We intend to survey large number of healthy and diseased individuals in the future, including patients with different types of cancer and/or different cancer stages. Besides cancer, it would also be interesting to expand the approach to other diseases, especially chronic diseases such as Type I and Type II diabetes. Through analysis of microbial cfDNA in plasma, we should be able to gain further insight into the role played by microbial inhabitants during the progression of a disease. Knowledge of this kind should be able to facilitate the effectiveness of personalized treatment of diseases.
Methods
Plasma cfDNA preparation
Detailed information about the EOBC patients: 1) Patient BC0145 (ER-, PR+ and HER-2+) was diagnosed at age 39.5 to have infiltrating ductal carcinoma at grade III in both breasts, had surgery on August 22, 2006 and passed away on September 01, 2007 (about 1 year after surgery). 2) Patient BC0190 (ER+, PR+ and HER-2+) was diagnosed at age 25.1 to have infiltrating ductal carcinoma at grade III, had her surgery on February 16, 2007 and was reported to remain healthy in April of 2017. 3) Patient CGBC025 (ER+, PR+ and HER-2+) was diagnosed at age 34.8 to have invasive ductal carcinoma at grade II, had her surgery on August 28, 2015 and remains alive and healthy.
Blood samples were taken from EOBC patients BC0145, BC0190 and CGBC025 at age 39.5, 25.1 and 34.8, respectively, and normal blood samples were prepared from healthy females BBC and EJC, both less than 30 years old. All plasma cfDNA samples were isolated from blood using Quick-cfDNA Serum and Plasma Kit (Zymo Research, D4076) following manufacturer’s instructions.
Preparation of microbial reference databases
Microbial reference databases were downloaded from NCBI reference genome database (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/, April 6, 2017). For each category, only “Complete Genome” and “Chromosome” from assembly_summary.txt according guide line from NCBI web site (https://www.ncbi.nlm.nih.gov/genome/doc/ftpfaq/#allcomplete) were used in mapping/alignment. The final reference databases contain 9,336 bacterial sequences, 9,050 viral sequences, and 913 fungal sequences (Table 1).
Sequencing and sequence processing
Sequences were determined by 150 × 150 PE sequencing (30×) using Illumina HiSeq X Ten (Macrogen Corp).
Illumina sequencing adapter was trimmed by cutadapt. Both base quality and read quality were checked after contaminant reads were removed. PRINseq [27] was employed for base quality checking following the sequential steps: removal of low quality bases from both 5′- and 3′-ends, removal of reads having ≥ 3 ambiguous (N) bases, and removal of reads with read length < 30 bp. NGS QC Toolkit [28] was then used to select high quality reads each of which has base score ≥ 20 in ≥ 70% of the contained bases. A generalized workflow for data analysis is shown in Fig. 1.
Fig. 1.
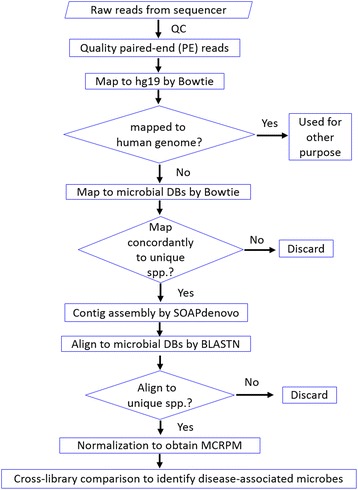
Workflow showing the stepwise procedure of sequence data processing leading to the identification of microbes in the body
Mapping of PE reads against microbial databases
Quality PE reads were mapped to UCSC hg19 human genome assembly using bowtie2 under default parameters (maximum insert size defined as 800 bp). The unmapped PE reads were then used to map against bacterial, viral and fungal databases separately with bowtie2 under multiple hit output option (parameters: --end-to-end --no-mixed --no-discordant –k 2). The option “-k 2” defines the maximum number of hits as 2 (species) per PE read [29]. Since there exists a high degree of similarity among microbial sequences, we chose only PE reads that hit concordantly to a unique microbial species for contig assembly.
Contig assembly and alignment for microbial species identification
Contigs were assembled with SOAPdenovo using Kmer 63 and then used to BLAST (BLASTN) against the same microbial databases with the following criteria: i) sequence identity ≥ 95%; ii) contig length ≥ 250 bp; and iii) contig aligned ratio (aligned length/contig length) = 100% (i.e., full range aligned). Strains belonging to the same genus/species were merged and number of contigs and total contig length were summed up accordingly.
Normalization
Normalization is essential for intra- and inter-library comparison of microbial cfDNA level. It is calculated based on the number of reads, instead of the number of contigs associated with each microbial species. Notice that, the numbers of reads per contig may vary, and only the total read count, which correlates the total number of cfDNA fragments in plasma, is able to reflect the level of cfDNA fragments in plasma.
Thus, the numbers of sequence reads associated with all contigs that aligned to each species were summed up and normalized on “per-million quality reads” basis. This approach would not only reflect the relative levels of microbial cfDNA fragments in the original sample, but also allow us to weigh the relative significance among microbial cfDNA across all libraries.
Statistical analysis
To compare the amount of microbial cfDNA in plasma between cancer patients and normal females, the average value of titer in MCRPM for each microbial species was calculated separately for normal females and diseased patients. Then, the averages for all bacteria were compared across these two groups using the non-parametric Mann-Whitney-Wilcoxon test.
Acknowledgements
This work was supported by the Summit Project for early-onset breast cancer research, funding for biosignature research, and funding for Genomics Research Center principal investigators, all provided by Academia Sinica. We would like to thank Yen-Chi Chiu for her help on sample collection.
Funding
Publication charge for this article is funded by Genomics Research Center of Academia Sinica.
Availability of data and materials
Microbial sequence reads related to this report are available at http://snowmountainlabs.com/main/microbial-cfdna/.
About this supplement
This article has been published as part of BMC Medical Genomics Volume 11 Supplement 1, 2018: Selected articles from Belyaev Conference 2017: medical genomics. The full contents of the supplement are available online at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-11-supplement-1.
Abbreviations
- cfDNA
Cell-free DNA
- CTCs
Circulating tumor cells
- EOBC
Early-onset breast cancer
- MCRPM
Microbial cfDNA reads per million quality PE reads
- NGS
Next-Generation Sequencing
- PE
Paired-End
Authors’ contributions
YFH co-developed methods, analyzed data and prepared tables. Yen-Ju Chen provided normal blood sample and reviewed manuscript. TCF and NCC acquired and managed patients’ tissue samples. MKM reviewed manuscript and provided ideas. Yi-Jie Chen provided healthy blood sample. THC managed all tissue samples and sequencing. HHY analyzed part of the data. YTW helped sequence data analysis. ALY acquired patients’ tissue samples, provided facilities and co-supervised the progress. KPC developed the concept, provided facilities, supervised the progress, co-developed methods, interpreted data and wrote the paper. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
All methods were performed in accordance with relevant guidelines and regulations.
All experimental protocols were approved by IRB on Biomedical Science Research/IRB-BM, Academia Sinica (AS-IRB03–105025), issued to Taiwan Biosignature Project for Breast Cancer: Investigate the Presence of Circulating DNA with Specific Mutations Identified by Whole Exome Sequencing of Early-onset Breast Cancer.
Consent for publication
Written informed consents for publication of clinical details were obtained from patients and from healthy individuals as well. No images are involved in this report.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yu-Feng Huang, Email: yfh1202@sinica.edu.tw.
Yen-Ju Chen, Email: yenju123@gate.sinica.edu.tw.
Tan-Chi Fan, Email: tcfan@cgmh.org.tw.
Nai-Chuan Chang, Email: logome@gmail.com.
Yi-Jie Chen, Email: ejchen1218@gate.sinica.edu.tw.
Mohit K. Midha, Email: mohitkmidha@sinica.edu.tw
Tzu-Han Chen, Email: thc01@gate.sinica.edu.tw.
Hsiao-Hsiang Yang, Email: vul4790127@gate.sinica.edu.tw.
Yu-Tai Wang, Email: yutaiwang@narlabs.org.tw.
Alice L. Yu, Email: a1yu@ucsd.edu
Kuo-Ping Chiu, Phone: (886) 2-2787-1257, Email: chiukp@gate.sinica.edu.tw.
References
- 1.Nikkari S, McLaughlin IJ, Bi W, Dodge DE, Relman DA. Does blood of healthy subjects contain bacterial ribosomal DNA? J Clin Microbiol. 2001;39(5):1956–1959. doi: 10.1128/JCM.39.5.1956-1959.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li SK, Leung RK, Guo HX, Wei JF, Wang JH, Kwong KT, Lee SS, Zhang C, Tsui SK. Detection and identification of plasma bacterial and viral elements in HIV/AIDS patients in comparison to healthy adults. Clin Microbiol Infect. 2012;18(11):1126–1133. doi: 10.1111/j.1469-0691.2011.03690.x. [DOI] [PubMed] [Google Scholar]
- 3.Grumaz S, Stevens P, Grumaz C, Decker SO, Weigand MA, Hofer S, Brenner T, von Haeseler A, Sohn K. Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med. 2016;8(1):73. doi: 10.1186/s13073-016-0326-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinakaran V, Rathinavel A, Pushpanathan M, Sivakumar R, Gunasekaran P, Rajendhran J. Elevated levels of circulating DNA in cardiovascular disease patients: metagenomic profiling of microbiome in the circulation. PLoS One. 2014;9(8):e105221. doi: 10.1371/journal.pone.0105221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tedeschi GG, Amici D, Sprovieri G, Vecchi A. Staphylococcus epidermidis in the circulating blood of normal and thrombocytopenic human subjects: immunological data. Experientia. 1976;32(12):1600–1602. doi: 10.1007/BF01924475. [DOI] [PubMed] [Google Scholar]
- 6.McLaughlin RW, Vali H, Lau PC, Palfree RG, De Ciccio A, Sirois M, Ahmad D, Villemur R, Desrosiers M, Chan EC. Are there naturally occurring pleomorphic bacteria in the blood of healthy humans? J Clin Microbiol. 2002;40(12):4771–4775. doi: 10.1128/JCM.40.12.4771-4775.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potgieter M, Bester J, Kell DB, Pretorius E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev. 2015;39(4):567–591. doi: 10.1093/femsre/fuv013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cummins J, Tangney M. Bacteria and tumours: causative agents or opportunistic inhabitants? Infect Agent Cancer. 2013;8(1):11. doi: 10.1186/1750-9378-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy S, Trinchieri G. Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer. 2017;17(5):271–285. doi: 10.1038/nrc.2017.13. [DOI] [PubMed] [Google Scholar]
- 10.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118(12):3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 11.McLaughlin-Drubin ME, Munger K. Viruses associated with human cancer. Biochim Biophys Acta. 2008;1782(3):127–150. doi: 10.1016/j.bbadis.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egi Y, Ito M, Tanaka S, Imagawa S, Takata S, Yoshihara M, Haruma K, Chayama K. Role of Helicobacter pylori infection and chronic inflammation in gastric cancer in the cardia. Jpn J Clin Oncol. 2007;37(5):365–369. doi: 10.1093/jjco/hym029. [DOI] [PubMed] [Google Scholar]
- 13.Blaser MJ. Understanding microbe-induced cancers. Cancer Prev Res (Phila) 2008;1(1):15–20. doi: 10.1158/1940-6207.CAPR-08-0024. [DOI] [PubMed] [Google Scholar]
- 14.Butel JS. Viral carcinogenesis: revelation of molecular mechanisms and etiology of human disease. Carcinogenesis. 2000;21(3):405–426. doi: 10.1093/carcin/21.3.405. [DOI] [PubMed] [Google Scholar]
- 15.Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78(21):11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elgui de Oliveira D. DNA viruses in human cancer: an integrated overview on fundamental mechanisms of viral carcinogenesis. Cancer Lett. 2007;247(2):182–196. doi: 10.1016/j.canlet.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Riley DR, Sieber KB, Robinson KM, White JR, Ganesan A, Nourbakhsh S, Dunning Hotopp JC. Bacteria-human somatic cell lateral gene transfer is enriched in cancer samples. PLoS Comput Biol. 2013;9(6):e1003107. doi: 10.1371/journal.pcbi.1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xuan C, Shamonki JM, Chung A, Dinome ML, Chung M, Sieling PA, Lee DJ. Microbial dysbiosis is associated with human breast cancer. PLoS One. 2014;9(1):e83744. doi: 10.1371/journal.pone.0083744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guardabassi L, Dalsgaard A, Olsen JE. Phenotypic characterization and antibiotic resistance of Acinetobacter spp. isolated from aquatic sources. J Appl Microbiol. 1999;87(5):659–667. doi: 10.1046/j.1365-2672.1999.00905.x. [DOI] [PubMed] [Google Scholar]
- 21.Seifert H, Dijkshoorn L, Gerner-Smidt P, Pelzer N, Tjernberg I, Vaneechoutte M. Distribution of Acinetobacter species on human skin: comparison of phenotypic and genotypic identification methods. J Clin Microbiol. 1997;35(11):2819–2825. doi: 10.1128/jcm.35.11.2819-2825.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berlau J, Aucken H, Malnick H, Pitt T. Distribution of Acinetobacter species on skin of healthy humans. Eur J Clin Microbiol Infect Dis. 1999;18(3):179–183. doi: 10.1007/s100960050254. [DOI] [PubMed] [Google Scholar]
- 23.Visca P, Seifert H, Towner KJ. Acinetobacter infection--an emerging threat to human health. IUBMB Life. 2011;63(12):1048–1054. doi: 10.1002/iub.534. [DOI] [PubMed] [Google Scholar]
- 24.Pan HJ, Teng LJ, Tzeng MS, Chang SC, Ho SW, Luh KT, Hsieh WC. Identification and typing of Pseudomonas pickettii during an episode of nosocomial outbreak. Zhonghua Min Guo Wei Sheng Wu Ji Mian Yi Xue Za Zhi. 1992;25(2):115–123. [PubMed] [Google Scholar]
- 25.Buonaurio R, Stravato VM, Kosako Y, Fujiwara N, Naka T, Kobayashi K, Cappelli C, Yabuuchi E. Sphingomonas melonis sp. nov., a novel pathogen that causes brown spots on yellow Spanish melon fruits. Int J Syst Evol Microbiol. 2002;52(Pt 6):2081–2087. doi: 10.1099/00207713-52-6-2081. [DOI] [PubMed] [Google Scholar]
- 26.Cogen AL, Nizet V, Gallo RL. Skin microbiota: a source of disease or defence? Br J Dermatol. 2008;158(3):442–455. doi: 10.1111/j.1365-2133.2008.08437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2):e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Microbial sequence reads related to this report are available at http://snowmountainlabs.com/main/microbial-cfdna/.