

Abstract
Nintedanib is an intracellular inhibitor of tyrosine kinases used in the treatment of non–small cell lung cancer and idiopathic pulmonary fibrosis (IPF). This phase 1 open‐label study investigated the influence of mild and moderate hepatic impairment on the pharmacokinetics (PK), safety, and tolerability of nintedanib following oral administration of a single 100‐mg dose. Subjects with hepatic impairment classified as Child‐Pugh A (mild hepatic impairment) or Child‐Pugh B (moderate hepatic impairment) were eligible. The control group comprised healthy matched subjects. Primary end points were Cmax and AUC0–∞ of nintedanib. Thirty‐three subjects received nintedanib (8 in each of the Child‐Pugh A and Child‐Pugh B groups and 17 controls). The shape of the plasma concentration–time curve for nintedanib was similar between Child‐Pugh A or B and healthy subjects. Nintedanib exposure was ∼2‐fold higher in Child‐Pugh A subjects and ∼8‐fold higher in Child‐Pugh B subjects than in healthy subjects. Adverse events were reported in 3 Child‐Pugh B subjects (37.5%), no Child‐Pugh A subjects, and 3 healthy subjects (17.6%). In conclusion, exposure to nintedanib was higher in Child‐Pugh A and B subjects than in matched healthy subjects. A single dose of nintedanib 100 mg had an acceptable safety and tolerability profile in subjects with hepatic impairment. Results of this dedicated phase 1 study are in line with exploratory investigations into the PK of nintedanib in patients with advanced solid tumors or IPF and hepatic impairment.
Keywords: absorption, adverse drug event, biological availability, liver function tests, protein‐tyrosine kinases
Nintedanib (formerly known as BIBF 1120) is a potent intracellular inhibitor of tyrosine kinase receptors, including vascular endothelial growth factor receptors 1–3, fibroblast growth factor receptors 1–3, and platelet‐derived growth factor receptors α and β, and nonreceptor members of the Src family.1 By binding competitively to the adenosine triphosphate sites of these receptors, nintedanib blocks autophosphorylation and so inhibits the downstream intracellular signaling cascades necessary for the proliferation, migration, and survival of endothelial cells, pericytes, and fibroblasts.1, 2 Nintedanib is approved in the European Union in combination with docetaxel for the treatment of non–small cell lung cancer (NSCLC) of adenocarcinoma histology after first‐line chemotherapy and for the treatment of idiopathic pulmonary fibrosis (IPF) in several countries and regions, including the European Union and the United States.3, 4, 5 The recommended dose of nintedanib is 200 mg twice daily in combination with docetaxel in patients with NSCLC and 150 mg twice daily in patients with IPF, with each dose taken approximately 12 hours apart with food.3, 4, 5
The pharmacokinetic (PK) properties of nintedanib are comparable in healthy volunteers, patients with IPF, and patients with advanced solid tumors. Following oral administration, nintedanib is rapidly absorbed and reaches maximum plasma concentration after approximately 2–4 hours; steady state is reached within 7 days of dosing.3, 4 Nintedanib undergoes extensive first‐pass metabolism and displays at least biphasic disposition kinetics, with a terminal half‐life of 10–15 hours.3, 4, 6, 7 Nintedanib is metabolized by methyl ester cleavage to form the carboxylate derivative BIBF 1202 as the predominant metabolite, which is glucuronidated by UGT enzymes to form BIBF 1202 glucuronide.7 The absolute bioavailability of nintedanib 100 mg in healthy volunteers is approximately 5%.6 Nintedanib exposure is approximately 20% higher when it is administered after food intake, and its absorption is delayed (from approximately 2 to 4 hours) compared with fasted conditions.5 Nintedanib is eliminated primarily (>90%) through biliary/fecal excretion, with renal excretion playing a negligible role. It is a high‐clearance drug, with total body clearance in healthy subjects of 1390 mL/min.6 In patients with solid tumors8, 9, 10 or IPF,11 PK variables show moderate to high interindividual variability across dose groups. The PK properties of nintedanib are linear with respect to dose and time.4, 6, 8, 10
Hepatic impairment may increase plasma concentrations of nintedanib.3, 4 Patients with elevated aspartate aminotransferase (AST), alanine aminotransferase (ALT), or total bilirubin >1.5 × the upper limit of normal (ULN) at screening were excluded from clinical trials of nintedanib in patients with NSCLC or IPF10, 12, 13, 14, 15 except for in the LUME‐Lung 2 trial of nintedanib in patients with NSCLC, in which ALT or AST elevation up to 2.5 × ULN was permitted for patients with liver metastases.16
This dedicated study investigated the influence of mild and moderate hepatic impairment (Child‐Pugh A and B) on the PK, safety, and tolerability of nintedanib following oral administration of a single 100‐mg dose.
Methods
Study Design
The study was approved by local ethics committees and was carried out in compliance with the protocol, the principles of the Declaration of Helsinki, International Conference on Harmonization Good Clinical Practice guidelines, and applicable regulatory requirements. All subjects provided written informed consent before study entry. The study was registered on clinicaltrials.gov (NCT02191865). This was a phase 1 open‐label, single‐dose, parallel‐group, matched‐group study. Subjects received a single dose of nintedanib 100 mg, administered as an oral soft‐gelatin capsule with 240 mL of water under fed conditions. There was a 28‐day posttreatment follow‐up period.
Subjects
Individuals aged 18–79 years with a body mass index (BMI) of 18.5–34 kg/m2 were eligible to participate. Subjects with hepatic impairment were classified as Child‐Pugh A or Child‐Pugh B.17 The Child‐Pugh criteria assess the severity of hepatic impairment taking into account measures beyond elevations in hepatic enzymes, such as prothrombin time, the degree of ascites, and the grade of hepatic encephalopathy. Each measure is assigned a score of 1–3, with higher score indicating worse hepatic impairment. Child‐Pugh A (total score, 5–6) indicates mild hepatic impairment, whereas Child‐Pugh B (total score, 7–9) indicates moderate hepatic impairment. For safety reasons, Child‐Pugh A subjects were dosed prior to Child‐Pugh B subjects.
Child‐Pugh A or B subjects had to have hepatic insufficiency diagnosed ≥ 3 months before screening and an estimated glomerular filtration rate (eGFR) > 40 mL/min/1.73 m2, according to the Modification of Diet in Renal Disease (MDRD) formula, at screening. Healthy subjects were matched by age (±10 years), weight (±10%), race, and smoking habits (current versus former or never smokers), as these factors were known to influence the PK of nintedanib4, 5, 18, 19 and also by sex. Healthy subjects had to have an eGFR > 70 mL/min/1.73 m2 (MDRD) at screening.
Subjects with significant or recent acute gastrointestinal disorders with diarrhea as a major symptom or history of gastrointestinal bleeding within the past 3 months were excluded from the study. Subjects who were moderate or heavy smokers (>10 cigarettes or 3 cigars or 3 pipes per day) were excluded. As nintedanib is a substrate of P‐glycoprotein,6 patients who used potent P‐glycoprotein inhibitors or inducers were excluded. Subjects with hepatic impairment who had significant diseases other than the underlying diagnosis causing hepatic impairment and diseases related to it were excluded. Subjects with hepatic impairment and severe cerebrovascular or cardiac disorders (eg, myocardial infarction < 6 months prior to administration of study drug, congestive heart failure of New York Heart Association grade III or IV, or severe arrhythmia) were excluded.
End Points
The primary end points were Cmax (maximum concentration in plasma) and AUC0–∞ (area under concentration–time curve in plasma from time 0 extrapolated to infinity) of nintedanib. Secondary end points were AUC0–tz (area under the concentration–time curve in plasma from time 0 to last quantifiable plasma concentration) of nintedanib and the proportion of subjects with adverse events (AEs) between administration of nintedanib and the end of the 28‐day posttreatment follow‐up period. PK samples were taken until day 8 after the administration of nintedanib.
Further end points included other PK parameters of nintedanib, its metabolites BIBF 1202 and BIBF 1202 glucuronide, including tmax (time from last dosing to maximum concentration in plasma), t1/2 (terminal half‐life in plasma), and renal clearance. Safety was assessed via clinical laboratory tests, vital signs, 12‐lead electrocardiogram (ECG) and physical examination. AEs were coded according to the Medical Dictionary for Regulatory Activities, version 17.1. Investigators reported the possible relationship between nintedanib and AEs based on their own judgment.
In addition, the plasma protein binding of nintedanib and BIBF 1202 was determined in predose plasma samples after ex vivo spiking of 100 ng/mL [14C]‐radiolabeled nintedanib or [14C]‐radiolabeled BIBF 1202. Protein binding was measured in vitro using equilibrium dialysis and quantification of radioactivity by liquid scintillation counting. The mean protein‐bound fractions of nintedanib and BIBF 1202 were calculated for each group of subjects.
Statistical and Pharmacokinetic Methodology
Pharmacokinetic analysis was performed using WinNonlin (Cetera, Princeton, NJ). An analysis of variance model on the logarithmic scale was used for the analysis of the AUC0–∞, Cmax, and AUC0–tz of nintedanib in Child‐Pugh A or B subjects compared with healthy controls. The model included hepatic status as a fixed effect and matched pair as a random effect. SAS version 9.2 (SAS, Cary, NC) was used for statistical analysis. Other parameters were analyzed descriptively.
The treated set comprised subjects documented to have taken the dose of study drug. The PK set comprised subjects documented to have taken the dose of study drug who provided at least 1 observation for at least 1 primary end point that was judged as evaluable and was not affected by important protocol violations relevant to the evaluation of PK.
A sample size of 24 to 32 subjects was regarded as adequate to fulfill the study objectives. This was not based on a statistical power calculation, but was assessed as adequate to attain reliable results and to fulfill the objectives and requirements of the study, in line with regulatory guidance on PK studies in patients with hepatic impairment.20
Results
Baseline Characteristics and Subject Disposition
A total of 34 subjects were screened, and 33 received nintedanib (8 subjects in each of the Child‐Pugh A and Child‐Pugh B groups and 17 matched healthy subjects). Of the 33 subjects included in the treated set, 30 subjects (90.9%) were included in the PK set. One healthy subject vomited shortly after nintedanib administration, and 2 healthy subjects were excluded because of protocol violations. Baseline characteristics of the treated set are shown in Table 1. The majority of subjects were male (60.6%), mean age was 58.1 years, and mean BMI was 27.3 kg/m2. All subjects were white. There were no relevant differences in demographic characteristics between the Child‐Pugh A, Child‐Pugh B, and healthy control groups. All subjects completed the planned observation time.
Table 1.
Baseline Characteristics (Treated Set)
| Child‐Pugh A (n = 8) | Child‐Pugh B (n = 8) | Healthy (n = 17) | Total (n = 33) | |
|---|---|---|---|---|
| Sex, n (%) | ||||
| Male | 5 (62.5) | 5 (62.5) | 10 (58.8) | 20 (60.6) |
| Female | 3 (37.5) | 3 (37.5) | 7 (41.2) | 13 (39.4) |
| Age (years), mean ± SD | 60.6 ± 8.1 | 56.6 ± 6.5 | 57.6 ± 8.4 | 58.1 ± 7.8 |
| Weight (kg), mean ± SD | 87.5 ± 20.2 | 87.0 ± 14.0 | 84.3 ± 15.3 | 85.7 ± 15.9 |
| BMI (kg/m2), mean ± SD | 27.7 ± 4.0 | 28.6 ± 4.5 | 26.6 ± 3.9 | 27.3 ± 4.0 |
| Smoking status, n (%) | ||||
| Current smoker | 5 (62.5) | 6 (75.0) | 11 (64.7) | 22 (66.7) |
| Former/never smoker | 3 (37.5) | 2 (25.0) | 6 (35.3) | 11 (33.3) |
| Child‐Pugh score, n (%) | ||||
| 5 | 6 (75.0) | 0 | — | 6 (18.2) |
| 6 | 2 (25.0) | 0 | — | 2 (6.1) |
| 7 | 0 | 5 (62.5) | — | 5 (15.2) |
| 8 | 0 | 2 (25.0) | — | 2 (6.1) |
| 9 | 0 | 1 (12.5) | — | 1 (3.0) |
All subjects were white. BMI, body mass index; SD, standard deviation.
Pharmacokinetics
Median Cmax was reached 3–4 hours after nintedanib administration (range, 1–8 hours), with no clear difference between Child‐Pugh A, Child‐Pugh B, and healthy subjects. After reaching Cmax, plasma concentrations declined in an at least biphasic manner. The shape of the plasma concentration–time curve for nintedanib was similar between Child‐Pugh A or B and healthy subjects (Figure 1).
Figure 1.
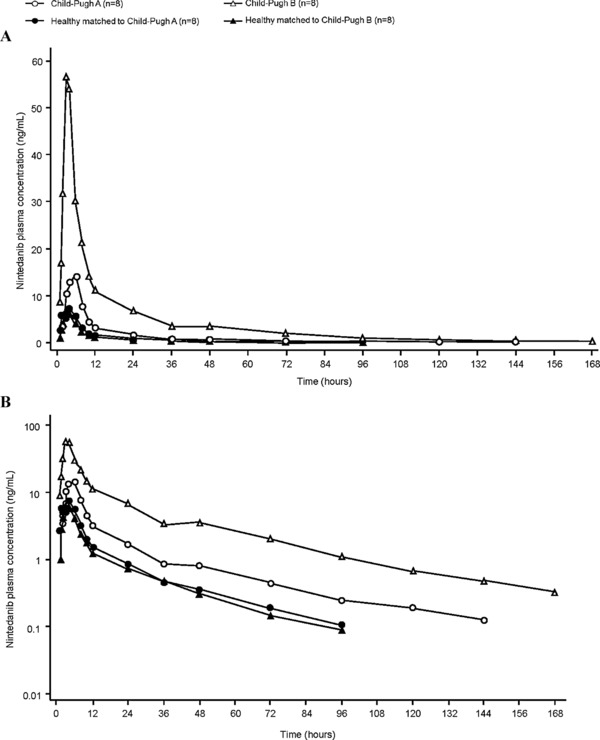
Geometric mean plasma concentration–time profiles of nintedanib after a single oral dose of nintedanib 100 mg in Child‐Pugh A and Child‐Pugh B subjects compared with healthy subjects: (a) linear scale; (b) semilog scale (PK set).
Nintedanib exposure was ∼2‐fold higher in Child‐Pugh A subjects and ∼8‐fold higher in Child‐Pugh B subjects than in healthy subjects (Figure 1, Table 2). Compared with healthy subjects, in Child‐Pugh A subjects, the adjusted gMean ratio was 215.4% (90%CI, 121%–384%) for AUC0–∞ and 221.8% (90%CI, 135%–365%) for Cmax, and in Child‐Pugh B subjects, the adjusted gMean ratio was 867.1% (90%CI, 573%–1312%) for AUC0–∞ and 761.0% (90%CI, 439%–1319%) for Cmax.
Table 2.
Relative Bioavailability of Nintedanib (PK Set)
| Adjusted gMean | Adjusted gMean | |||||||
|---|---|---|---|---|---|---|---|---|
| Child‐Pugh A (n = 8) | Child‐Pugh A Matched Healthy Subjects (n = 8) | Ratio (%) | 90%CI (%) | Child‐Pugh B (n = 8) | Child‐Pugh B matched Healthy Subjects (n = 8) | Ratio (%) | 90%CI (%) | |
| AUC0–∞ (ng∙h/mL) | 199.7 | 92.7 | 215.4 | 121–384 | 674.3 | 77.8 | 867.1 | 573–1312 |
| Cmax (ng/mL) | 20.5 | 9.3 | 221.8 | 135–365 | 59.4 | 7.8 | 761.0 | 439–1319 |
| AUC0–tz (ng∙h/mL) | 193.4 | 89.2 | 216.8 | 120–390 | 651.6 | 74.8 | 870.7 | 576–1316 |
AUC, area under concentration–time curve in plasma; AUC0–∞, AUC from time 0 extrapolated to infinity; AUC0–tz, AUC from time 0 to last quantifiable plasma concentration; Cmax, maximum measured concentration in plasma; CI, confidence interval; gMean, geometric mean.
Other PK parameters are shown in Table 3. The fraction of the nintedanib dose excreted in urine was higher in Child‐Pugh A and B subjects than in healthy subjects; however, renal clearance was comparable across groups. The t1/2 was slightly prolonged in Child‐Pugh A and B subjects compared with healthy subjects, with no difference between the Child‐Pugh A and Child‐Pugh B groups.
Table 3.
PK Parameters of Nintedanib and Its Metabolites (PK Set)
| gMean (gCV%) | ||||
|---|---|---|---|---|
| Child‐Pugh A (n = 8) | Child‐Pugh A Matched Healthy Subjects (n = 8) | Child‐Pugh B (n = 8) | Child‐Pugh B Matched Healthy Subjects (n = 8) | |
| Nintedanib | ||||
| tmax a (h) | 4.0 (1.5–6.0) | 4.0 (1.0–6.0) | 3.5 (3.0–8.0) | 3.0 (1.0–8.0) |
| t1/2 (h) | 41.1 (36.5) | 35.0 (31.4) | 42.1 (23.1) | 32.1 (24.5) |
| fe0–72 (%) | 0.204 (96.2) | 0.102 (37.9) | 0.543 (31.7)b | 0.0945 (28.7) |
| CLR0–‐72 (mL/min) | 19.4 (54.6) | 17.2 (39.8)c | 17.6 (47.3)b | 19.5 (26.5)c |
| BIBF 1202 | ||||
| Cmax (ng/mL) | 17.4 (47.1) | 7.55 (56.2) | 60.7 (75.2) | 6.47 (57.5) |
| AUC0–∞ (ng∙h/mL) | 216 (81.8) | 86.8 (48.8) | 1190 (86.4) | 70.4 (38.7) |
| t1/2 (h) | 39.1 (53.8) | 24.7 (65.1) | 46.9 (16.6) | 23.6 (50.1) |
| BIBF 1202 glucuronide | ||||
| Cmax (ng/mL) | 15.1 (113) | 11.4 (62.0) | 19.6 (201) | 12.7 (76.2) |
| AUC0–∞ (ng∙h/mL) | 1040 (95.9) | 657 (66.8) | 2650 (285) | 761 (69.1) |
| t½ (h) | 41.1 (45.5) | 31.9 (34.1) | 57.4 (114) | 28.3 (21.9) |
AUC, area under concentration–time curve in plasma; AUC0–tz, AUC from time 0 to last quantifiable plasma concentration; AUC0–∞, AUC in plasma from time 0 extrapolated to infinity; CLR,t1–t2, renal clearance over time interval from t1 to t2; fet1–t2, fraction of administered drug excreted unchanged in urine over time interval from t1 to t2; gMean, geometric mean; gCV, geometric coefficient of variation; t1/2, terminal half‐life in plasma; tmax, time from last dosing to maximum measured concentration in plasma.
Median (min–max); bn = 7; cn = 6.
Similar to nintedanib, BIBF 1202 exposure was ∼2‐fold higher in the Child‐Pugh A group than in healthy subjects (Table 3). AUC0–∞ and Cmax were ∼17‐fold and 9‐fold higher, respectively, in Child‐Pugh B subjects than in healthy subjects. For BIBF 1202 glucuronide, Cmax was comparable across groups, whereas AUC0–∞ was ∼1.5‐fold and ∼3.5‐fold higher in Child‐Pugh A and B subjects, respectively, compared with healthy subjects (Table 3). The t1/2 values for BIBF 1202 and BIBF 1202 glucuronide were longer in Child‐Pugh A and B subjects than in healthy subjects.
Protein Binding
Plasma protein binding of nintedanib and BIBF 1202 was similar in spiked predose plasma samples taken from Child‐Pugh A, Child‐Pugh B, and healthy subjects (Table 4). The mean ± SD protein‐bound fraction of nintedanib was 99.5% ± 0.1%, 99.1% ± 0.7%, and 99.1% ± 0.5% in Child‐Pugh A, Child‐Pugh B, and healthy subjects, respectively.
Table 4.
Protein Binding of Nintedanib and BIBF 1202 in Plasma Samples (Treated Set)
| Child‐Pugh A (n = 8) | Child‐Pugh B (n = 8) | Healthy (n = 17) | |
|---|---|---|---|
| Protein‐bound fraction of nintedanib (%), mean ± SD | 99.5 ± 0.1 | 99.1 ± 0.7 | 99.1 ± 0.5 |
| Protein‐bound fraction of BIBF 1202 (%), mean ± SD | 81.2 ± 3.6 | 82.4 ± 5.5 | 85.0 ± 3.0 |
SD, standard deviation.
Safety
AEs were reported in 3 Child‐Pugh B subjects (37.5%), no Child‐Pugh A subjects, and 3 healthy subjects (17.6%); see Table 5. The most frequently reported AE was nausea, reported by 1 Child‐Pugh B subject (12.5%) and 2 healthy subjects (11.8%). All AEs were assessed as possibly drug related by the investigator. No clinically relevant abnormal findings and no AEs related to laboratory data, ECG data, or vital signs were reported. No severe or serious AEs were reported.
Table 5.
Adverse Events (Treated Set)
| n (%) | Child‐Pugh A (n = 8) | Child‐Pugh B (n = 8) | Healthy (n = 17) |
|---|---|---|---|
| Subjects with any AE | 0 | 3 (37.5) | 3 (17.6) |
| Headache | 0 | 0 | 1 (5.9) |
| Diarrhea | 0 | 1 (12.5) | 0 |
| Nausea | 0 | 1 (12.5) | 2 (11.8) |
| Vomiting | 0 | 0 | 1 (5.9) |
| Pruritus | 0 | 1 (12.5) | 0 |
| Skin irritation | 0 | 1 (12.5) | 0 |
Adverse events (AEs) were reported according to MedDRA version 17.1. AEs with onset between administration of trial medication and the end of the 28‐day posttreatment follow‐up period are shown.
Discussion
In this study, after a single dose of nintedanib 100 mg, the shape of the nintedanib plasma concentration–time curve was similar between subjects with mild or moderate hepatic impairment and healthy subjects, but exposure to nintedanib was ∼2‐fold higher in Child‐Pugh A subjects and ∼8‐fold higher in Child‐Pugh B subjects than in matched healthy controls. The protein‐bound fraction of nintedanib was >99% across the groups. PK observations point toward an increase in the bioavailable fraction of nintedanib in subjects with hepatic impairment (ie, higher exposure in subjects with hepatic impairment than in healthy subjects, with comparable plasma concentration–time profiles). This is in line with nintedanib being a high‐clearance drug with high first‐pass metabolism.7 Renal clearance of nintedanib was not influenced by impaired hepatic elimination. As observed for nintedanib, Cmax and AUC0–∞ for the metabolites BIBF 1202 and BIBF 1202 glucuronide demonstrated increased exposure with increasing hepatic impairment. The effect of hepatic impairment on exposure was more pronounced for BIBF 1202, but less pronounced for BIBF 1202 glucuronide than for nintedanib. This can be interpreted as a consequence of first‐pass metabolism and the different metabolic pathways of nintedanib (metabolized via ester cleavage), BIBF 1202 (metabolized via glucuronidation), and BIBF 1202 glucuronide (fecal excretion), as hepatic impairment may interfere mostly with the glucuronidation process.
The design of this study was in line with regulatory guidance on PK studies in patients with hepatic impairment, which recommends that PK studies should be carried out when hepatic impairment is likely to significantly alter the PK of a drug and/or its active metabolites, and a posology adjustment may be needed to ensure the efficacy and safety of the drug in these patients.18, 20 A single‐dose study is sufficient when a drug and its active metabolites exhibit linear and time‐independent pharmacokinetics.18, 20 Categorization of hepatic impairment using the Child‐Pugh classification is appropriate in this setting.20 In this study, a single dose of 100 mg was used for tolerability reasons; however, the PK results are transferable to multiple doses of nintedanib because of its linear PK properties with respect to dose and time.6 In all groups, the single dose of nintedanib 100 mg was well tolerated.
In addition to this dedicated study, supportive data on the PK of nintedanib in patients with hepatic impairment have been collected as part of the clinical development programs in oncology and IPF. Despite some differences in the classification of hepatic impairment, data on the PK of nintedanib in individuals with mild hepatic impairment are aligned across data sets. The PK data from this study are in accordance with 2 phase 1 dose‐escalation studies of open‐label nintedanib in Asian (n = 39; NCT00987935) and European (n = 32; NCT01004003) patients with impaired hepatic function and advanced hepatocellular carcinoma (HCC), in whom nintedanib was rapidly absorbed, with maximum plasma concentrations achieved ∼2–3 hours after administration and at least biphasic disposition kinetics (BI, data on file). Patients in these studies were stratified into groups according to ALT, AST, and Child‐Pugh score at baseline: group I comprised patients with ALT and AST ≤ 2 × ULN and Child‐Pugh score 5–6, whereas group II comprised patients with ALT or AST >2 to ≤5 × ULN or Child‐Pugh score 7. Group II criteria were chosen such that the group could comprise Child‐Pugh A or B patients. The majority of patients recruited for group II were Child‐Pugh A. In both studies, nintedanib exposure was ∼2‐fold higher in patients in group II compared with group I (BI, data on file). When PK data from the study in European patients with advanced HCC and impaired hepatic function (Child‐Pugh A; n = 32) were compared with PK data from patients with renal cell carcinoma and normal hepatic function (n = 64),21 a 1.6‐ to 1.7‐fold‐higher exposure to nintedanib was observed in patients with advanced HCC and impaired hepatic function (BI, data on file).
Two population PK analyses were performed to characterize the PK of nintedanib and to evaluate the effect of intrinsic and extrinsic patient factors on the PK of nintedanib (BI, data on file). One analysis included 849 patients with NSCLC and 342 patients with IPF; the second included 933 patients with IPF. No Child‐Pugh categorization was available for these patients. Therefore, in both analyses, patients were defined as having mild hepatic impairment if AST or ALT or bilirubin levels were >ULN, but AST and ALT were ≤10 × ULN and bilirubin was ≤1.5 × ULN at the start of treatment. The number of patients with mild hepatic impairment was 116 in the first analysis and 44 in the second. A trend toward elevated nintedanib exposure of up to 1.4‐fold was observed in patients with mild hepatic impairment compared with patients with normal hepatic function. Because of missing information on underlying hepatic disease, a robust assessment of the effect of hepatic impairment defined by elevation of transaminases or bilirubin on nintedanib exposure was not possible.
Treatment with nintedanib is not recommended for patients with moderate or severe hepatic impairment (Child‐Pugh B or C).3, 4, 5 No adjustment of the starting dose of nintedanib is recommended for patients with mild hepatic impairment (Child‐Pugh A) and NSCLC,3 but the recommended dose of nintedanib for patients with IPF and mild hepatic impairment (Child‐Pugh A) is 100 mg twice daily.4, 5 The key data from the current study are included in the latest EU and US prescribing information for nintedanib.3, 4, 5
Conclusions
In conclusion, this dedicated study provides the most robust data on the PK of nintedanib in patients with mild or moderate hepatic impairment. In subjects with mild and moderate hepatic impairment (Child‐Pugh A and B), exposure to a single dose of nintedanib 100 mg was higher in both groups than in matched healthy subjects. Nintedanib had an acceptable safety and tolerability profile in subjects with hepatic impairment. The PK of nintedanib has not been investigated in patients with severe hepatic impairment (Child‐Pugh C).
Acknowledgments
This study was performed at CRS Clinical Research Services Kiel GmbH, under the supervision of Dr. Atef Halabi. Writing assistance, supported financially by Boehringer Ingelheim, was provided by Julie Fleming and Wendy Morris of FleishmanHillard Fishburn, London, UK, during the preparation of this article. The authors were fully responsible for all content and editorial decisions and were involved at all stages of manuscript development and approved the final version.
Declaration of Conflicting Interests
All authors are employees of Boehringer Ingelheim.
References
- 1. Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–4782. [DOI] [PubMed] [Google Scholar]
- 2. Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. BI . Vargatef (nintedanib) Summary of Product Characteristics. 2017. http://www.ema.europa.eu/ema/. Accessed June 21, 2017.
- 4. BI . Ofev (nintedanib) Summary of Product Characteristics. 2017. http://www.ema.europa.eu/ema/. Accessed June 21, 2017.
- 5. BI . Ofev® (nintedanib) Prescribing Information. 2017. https://www.ofev.com/. Accessed June 21, 2017.
- 6. Dallinger C, Trommeshauser D, Marzin K, Liesener A, Kaiser R, Stopfer P. Pharmacokinetic properties of nintedanib in healthy volunteers and patients with advanced cancer. J Clin Pharmacol. 2016;56:1387–1394. [DOI] [PubMed] [Google Scholar]
- 7. Stopfer P, Rathgen K, Bischoff D, et al. Pharmacokinetics and metabolism of BIBF 1120 after oral dosing to healthy male volunteers. Xenobiotica. 2011;41:297–311. [DOI] [PubMed] [Google Scholar]
- 8. Mross K, Stefanic M, Gmehling, D , et al. Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res. 2010;16:311–319. [DOI] [PubMed] [Google Scholar]
- 9. Okamoto I, Kaneda H, Satoh T, et al. Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors. Mol Cancer Ther. 2010;9:2825–2833. [DOI] [PubMed] [Google Scholar]
- 10. Reck M, Kaiser R, Eschbach C, et al. A phase II double‐blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non‐small‐cell lung cancer. Ann Oncol. 2011;22:1374–1381. [DOI] [PubMed] [Google Scholar]
- 11. Ogura T, Taniguchi H, Azuma A, et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1382–1392. [DOI] [PubMed] [Google Scholar]
- 12. Ellis PM, Kaiser R, Zhao Y, Stopfer P, Gyorffy S, Hanna N. Phase I open‐label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non‐small cell lung cancer patients. Clin Cancer Res. 2010;16:2881–2889. [DOI] [PubMed] [Google Scholar]
- 13. Reck M, Kaiser R, Mellemgaard A, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non‐small‐cell lung cancer (LUME‐Lung 1): a phase 3, double‐blind, randomised controlled trial. Lancet Oncol. 2014;15:143–155. [DOI] [PubMed] [Google Scholar]
- 14. Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–1087. [DOI] [PubMed] [Google Scholar]
- 15. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. [DOI] [PubMed] [Google Scholar]
- 16. Hanna NH, Kaiser R, Sullivan RN, et al. Nintedanib plus pemetrexed versus placebo plus pemetrexed in patients with relapsed or refractory, advanced non‐small cell lung cancer (LUME‐Lung 2): A randomized, double‐blind, phase III trial. Lung Cancer. 2016;102:65–73. [DOI] [PubMed] [Google Scholar]
- 17. Pugh RN, Murray‐Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. [DOI] [PubMed] [Google Scholar]
- 18. European Medicines Agency . 2005. Committee for Medicinal Products for Human Use (CHMP). Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003122.pdf. Accessed March 7, 2017.
- 19. Food and Drug Administration . 2014. Center for Drug Evaluation and Research. Clinical Pharmacology and Biopharmaceutics Review(s). http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205832Orig1s000ClinPharmR.pdf. Accessed March 7, 2017.
- 20. Food and Drug Administratiation . Guidance for Industry. Pharmacokinetics in patients with impaired hepatic function – study design, data analysis, and Impact on dosing and labeling. 2003. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072123.pdf. Accessed March 7, 2017.
- 21. Eisen T, Shparyk Y, Macleod N, et al. Effect of small angiokinase inhibitor nintedanib (BIBF 1120) on QT interval in patients with previously untreated, advanced renal cell cancer in an open‐label, phase II study. Invest New Drugs. 2013;31:1283–1293. [DOI] [PubMed] [Google Scholar]