

Abstract
Laccase‐mediated grafting on lignocelluloses has gained considerable attention as an environmentally benign method to covalently modify wood, paper and cork. In recent decades this technique has also been employed to modify fibres with a polysaccharide backbone, such as cellulose or chitosan, to infer colouration, antimicrobial activity or antioxidant activity to the material. The scope of this approach has been further widened by researchers, who apply mediators or high redox potential laccases and those that modify synthetic polymers and proteins. In all cases, the methodology relies on one‐ or two‐electron oxidation of the surface functional groups or of the graftable molecule in solution. However, similar results can very often be achieved through simple deposition, even after extensive washing. This unintended adsorption of the active substance could have an adverse effect on the durability of the applied coating. Differentiating between actual covalent binding and adsorption is therefore essential, but proves to be challenging. This review not only covers excellent research on the topic of laccase‐mediated grafting over the last five to ten years, but also provides a critical comparison to highlight either the lack or presence of compelling evidence for covalent grafting.
Keywords: biomass, enzyme catalysis, grafting, polymers, surface chemistry
1. Introduction
Grafting of (bio)molecules on polymers and surfaces has gained substantial interest in recent years. Grafting concerns covalent‐bond formation between a macromolecule and a small molecule. In the context of polymer chemistry, grafting is defined as “a reaction in which one or more species of block are connected to the main chain of a macromolecule as side chains having constitutional or configurational features that differ from those in the main chain”.1 From a surface‐chemistry perspective, one needs to expand this definition to composites in which the main chain constitutes a diverse array of materials, ranging from brick2 and fibreglass3 to paper4 and wood.5 Grafting can be achieved via three different pathways: grafting‐to, grafting‐from and grafting‐through (Scheme 1).6 Grafting‐to is characterised by the attachment of polymer chains with a reactive moiety that covalently binds to a compatible reactive group present on the backbone of the composite or macromolecule. Functionalising polyethyleneimine (PEI) with isocyanate‐terminated poly(ethylene glycol) to enhance gene‐delivery systems is an example of a grafting‐to approach.7 In the grafting‐from approach, polymerisation of soluble monomers is initiated from the backbone of the composite or macromolecule. Surface‐initiated atom‐transfer radical polymerisation (SI‐ATRP) is often used to polymerise zwitterionic monomers via a radical initiator on the surface, so as to confer anti‐fouling properties to the surface.8 Lastly, grafting‐through involves the co‐polymerisation of bare monomers with monomers containing the graftable moiety to simultaneously form the graft and backbone.9
Scheme 1.
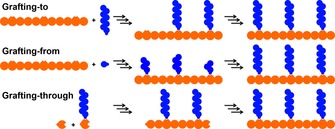
Three different types of grafting: grafting‐to, grafting‐from and grafting‐through.
Whichever method is chosen, very often (expensive) metal catalysis or the use of harsh conditions is required. In the previously mentioned ATRP process, copper(I) complexes are used that, in the presence of oxygen, lead to oxidised metal species, which are detrimental to the turnover of the catalysts.10 In such cases, stringent oxygen‐free conditions are essential, which make the grafting procedure more time‐consuming, more complex and more expensive. Similarly; although new developments allow photografting initiated by visible light,11 grafting acrylic monomers/polymers often requires high‐energy UV irradiation.12 Furthermore, the thus‐formed radicals are susceptible to quenching by oxygen.
In light of the growing world population and the concomitant scarcity of resources, more and more effort is being put forth to circumvent the use of expensive metal catalysts and high‐energy processes. One way of bypassing these disadvantages is the use of enzymes to initiate grafting. Enzymes operate under mild conditions, are highly tuneable and most often very selective. In the context of grafting, the catalytic use of oxidases, in particular, has gained considerable attention over the last ten years.13 Within this class, laccases (EC 1.10.3.2) are of particular interest because they can oxidise a substrate by using molecular oxygen as the oxidant to initiate radical grafting with water as the sole by‐product, whereas other oxidases, such as horseradish peroxidase (HRP), require hydrogen peroxide as an oxidant, which is hazardous at elevated concentrations.14 The earliest documentation of laccases dates back to the 19th century. In 1883, Yoshida described the isolation of this enzyme from the lacquer tree Rhus vernicifera.15 More than 100 years later, this research is still significant in the production of artificial Urushi lacquer by using laccase isolates.16 Apart from its presence in plants, laccases have also been found in insects17 and prokaryotes.18 They are, however, most ubiquitous in fungi, particularly in white rot fungi, such as Trametes versicolor.19 The role of laccases in the fungal biome is to assist in the formation of nutrients by degrading lignin to smaller molecules. Laccases act on the omnipresent phenolic moieties in lignin (Scheme 2); however, laccases exhibit a far larger substrate scope than only lignin‐derived phenols. Depending on the type of laccase, the scope ranges from catechols to phenols, and even non‐phenolic substrates can be directly converted by using laccase (although laccases form a family of enzymes, we typically use the singular because usually only one type of laccase is used in an experiment).20 By employing mediators, laccase is able to oxidise molecules that are normally too sterically congested to be oxidised directly, which further expands the scope of this already versatile enzyme.21
Scheme 2.
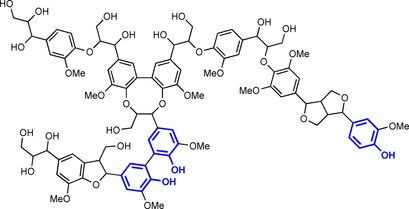
A small lignin segment with phenolic moieties highlighted in blue.
This wide scope under ambient conditions is also the reason for the widespread use of laccase in a variety of applications. Comparable to its native function, laccases are used to degrade lignin to obtain pharmaceutically interesting building blocks.22 They also have an enormous impact on environmental protection through the detoxification of dyes and pharmaceuticals from soil or wastewater.23 Because laccase‐assisted reactions involve radical chemistry, laccases not only aid in breaking down molecules, but also in building them up. Thus, laccases are used to synthesise small molecules, such as dyestuffs.20a, 24 Furthermore, the previously mentioned grafting is a field in which laccase plays an essential role. At the basis of all of these applications is the, among laccases, highly conserved four‐atom copper cluster (Scheme 3).19b In some laccase species, this cluster comprises less than four copper atoms or it contains other metals. In general, however, three different types of copper form the active site of laccase. At the type 1 site (T1), four substrate molecules are converted into their radical cations. The thus‐extracted electrons are then transferred to the type 2 and type 3 sites (T2 and T3, respectively), at which molecular oxygen is reduced to two molecules of water. The formed substrate radical cations quickly lose a proton to form reactive radical species, which can then participate in non‐enzymatic covalent‐bond formation or bond cleavage.
Scheme 3.
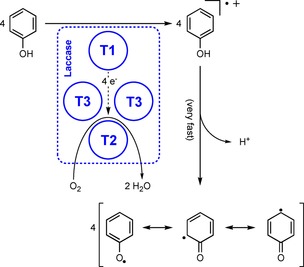
Generic mechanism for the conversion of four phenolic substrates to their radical cations, with concomitant reduction of one molecule of dioxygen, followed by the rapid (non‐enzymatic) loss of a proton to form the delocalised phenolic radical.
For small‐molecule synthesis, this bond formation can be easily studied by means of NMR spectroscopy or mass spectrometry. However, laccase‐mediated grafting often results in the formation of a bond between two components, of which at least one is a very complex matrix, for example, lignin. The formed bond is one of many in a very heterogeneous system; analysis of such an intricate architecture thus shows much resemblance to the infamous search for the needle in a haystack. In this search, a covalent bond can easily be mistaken for (non‐specific) adsorption. We postulate that covalent grafting by using laccase is often claimed without posing substantial scientific evidence.
To properly assess the likelihood of actual covalent bonding, the respective studies/methodologies are evaluated on the basis of five factors (F1–F5).
F1) Mechanistic rationale
Mixing two amino acids together at room temperature often does not result in peptide coupling; adding a carbodiimide, however, will bind the two.25 More related to grafting: laccase oxidises (mostly) phenols, if these cannot be oxidised directly, a mediator might be required.21 Is laccase‐mediated oxidation feasible with regard to the molecule's oxidation potential? Are the reactants reactive at the required pH? These are some of the questions that arise when estimating the validity of the purported modification. Of course, grafting does not always have to proceed through the envisioned mechanism, which makes finding the imaginary needle even more challenging.
F2) Pretreatment
Many of the materials discussed herein are of natural origin and could be contaminated with feculence or fats.26 Contaminants might shield the surface; thus hampering the intended grafting. Furthermore, (partially) soluble monomers or small oligomers of the material to be modified could be present in the material matrix.27 These molecules are likely to be more reactive than the molecules on the solid/liquid interface due to increased mobility and accessibility.28 Modification of these reactive species, followed by precipitation onto the surface, could unintentionally suggest grafting. In some cases, pretreatment involving cleaning or even extraction might therefore be required to prevent this.
F3) Control experiments
Laccase‐mediated grafting very often involves oligomerisation of phenols.13c Logically, the chemical and physical properties of the formed oligomers will be different from those of the parent monomer. Not observing the monomer in control experiments in which it is solely supplied to the surface in the absence of laccase does not necessarily confirm laccase‐mediated grafting. In a proper experimental design, the influence of oligomers on surface coating without laccase or with inactivated laccase should be assessed as well. Furthermore, lignin is rich in phenols, and therefore, also susceptible to laccase‐mediated oxidation if no additional phenols are present.29 Additionally, proteins are notorious for fouling;30 control experiments in which only laccase is applied on the surface (without additional monomers) are thus a necessity.
F4) Washing and cleaning
Proper washing after modification is at the essence of discrimination between adsorption and grafting, since this should remove the loosely bound adsorbents. The choice of washing solvent, additive and washing time might prove to be crucial, especially if oligomers are formed. These oligomers have a tendency to be less soluble in the aqueous reaction medium; more apolar solvents are thus often advisable for cleaning. The simplicity of these steps has often not been combined with a proper estimation of their importance.
F5) Analytical tools
Arguably the most important step of grafting is the evaluation afterwards. Spectra are easily misinterpreted or, worse, misused. A direct indication of grafting would be the formation of a new unique covalent bond between the surface and grafted molecule that has a distinct observational characteristic. Such indications are, however, typically rare. In most cases in which grafting is expected, simultaneous adsorption will also have occurred. Without the presence of the distinct observational characteristic, discrimination between adsorption and grafting can thus not (easily) be determined by techniques such as FTIR spectroscopy and X‐ray photoelectron spectroscopy (XPS). To conclude, without proper control experiments, the mere observation of a molecule on the surface does not have to indicate grafting because noncovalent adsorption often results in the same spectral alterations.
If all of the factors above have been considered and the resulting prerequisites have been met, it is very likely that the presented methodology has resulted in covalent‐bond formation. However, taking into account all of the above only results in a binary statement: grafting or no grafting. It is essential to quantify the degree of grafting, the modes of grafting and the degree of adsorption. Additionally, in some applications (unintentional), leaching of adsorbents could result in contamination of the environment.31 In these cases, strong (covalent) binding is thus essential and adsorption should be reduced to a minimum. Furthermore, covalent binding does not have to indicate irreversible binding, that is, a reaction such as imine formation yields strong covalent bonds, but is known to be fully reversible. If the application requires strong binding, the grafting methodology should be tuned to meet those requirements.
The studies reviewed herein, in general, employ grafting in heterogeneous systems: dispersions, solid surfaces, undissolved solids and so forth. These are often the most difficult systems to analyse due to the limited solubility of the resulting material, which induces identification errors. Additionally, all grafts discussed below arose from a grafting‐to or a grafting‐from approach. Laccase‐mediated grafting‐through strategies come very close to copolymerisation, which is beyond the scope of this review. Each subdomain is individually discussed on the basis of the material upon which grafting is performed: lignocelluloses, polysaccharides, proteins and synthetic polymers. For each subdomain, the mechanistic foundations for grafting, the state‐of‐the‐art regarding analysis of the corresponding grafts, and the pitfalls/challenges arising from grafting on these materials are discussed first. These are followed by specific examples of grafting that are, if possible, grouped according to application or employed methodology. For each example, we elaborate on the factors that determine the likelihood of grafting.
The goal of this review is thus not only to outline the large numbers of excellent studies on the topic of laccase‐mediated grafting, but also to compare it side by side to highlight either the lack or presence of considerable evidence for real covalent grafting.
2. Laccase‐Mediated Grafting on Lignocelluloses
2.1. Laccase‐mediated grafting validity
By appreciating that (fungal) laccases naturally act on lignin to convert it into useful feedstock for the host organism,19a it is not difficult to imagine that laccases are mainly employed in the modification of lignocellulosic material.13c Lignocelluloses comprise all natural fibres that are made up from lignin, hemicellulose and cellulose, albeit in varying ratios.32 This includes biomass, such as grasses and trees, value‐added materials, that is, paper, pulp, cork and wood, but also waste generated through processing the biomass into these materials. Although celluloses or other polysaccharides can also be modified (indirectly) by using laccase, the abundance of lignin in the lignocellulosic material is vital to the success of laccase‐mediated grafting on lignocelluloses.33 The traditional removal of lignin‐derived chromophores from lignocelluloses to enhance whiteness (bleaching) by chlorine and peroxides is gradually being replaced by enzymatic methods, which includes the use of laccase.34 The mild way in which laccases generate radicals is employed in lignin polymerisation/oligomerisation, but also in delignification;35 an essential part of the bleaching process. This topic is beyond the scope of this review and excellently reviewed by Meyer and co‐workers.29
The most common route towards lignocellulosic grafts, at first glance, provides a clear mechanistic rationale (Scheme 4): a laccase‐generated phenolic radical (initiation) reacts with an oxidisable moiety (propagation) or another radical (termination). However, the free radical, be it from a phenol in solution or from lignin, is delocalised over the aromatic system, which implicates multiple reactive sites. Coupling of such delocalised radicals thus results in diverse substrate–lignin, substrate–substrate and even lignin–lignin dimers and subsequent oligomers. Achieving mechanistic understanding of these processes is further complicated by the presence of phenolic extractives in the lignocellulosic matrix because these extractives could also act as laccase substrates. Additionally applying mediators to the system further alters the reaction pathway. Its highly diverse nature thus clearly indicates the complexity of laccase‐mediated modification of lignocelluloses.
Scheme 4.
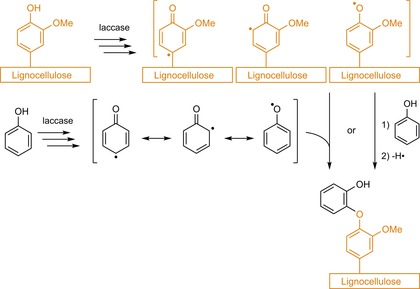
Generalised schematic of laccase‐mediated phenol grafting.
To understand what happens in a complex system, such as wood, one often reverts to lignin model compounds; small “monomers” of the highly complex lignin structure that contain many of the functional groups present in lignin. These monomers, however, are generally soluble in aqueous buffer/organic solvent mixtures, and have limited complexity, so they can be separated and studied by both LC/MS and NMR spectroscopy. The groups of Nyanhongo and Guebitz (vide infra) have extensively studied lignin model compounds and their interaction with laccase. In early studies, beech wood, laccase and tyramine (Scheme 5 A) were reacted together to result in a depletion of tyramine;36 tyramine itself is not a laccase substrate due to its high oxidation potential. The influence of beech wood is confirmed when the lignin model adlerol (Scheme 5 B) is used instead of wood; adlerol is also not a laccase substrate, so no depletion of tyramine was observed. In all cases for which phenolic lignin model compounds were used, tyramine was converted (Scheme 5 C).
Scheme 5.
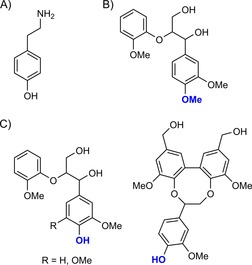
A) Tyramine. B) Non‐phenolic lignin model compound adlerol. C) Phenolic lignin model compounds guaiacylglycerol β‐guaiacyl ether (R=H), syringylglycerol β‐guaiacyl ether (R=OMe; left) and dibenzodioxocin (right).
“Grafting” on model compounds was later confirmed by studying the coupling of lignin models to several functional molecules by LC/MS.37 In all cases in which a phenolic molecule was reacted in the presence of the phenolic lignin model dibenzodioxocin (Scheme 5 C), the heterodimer [M+H]+ ion of the model/phenol conjugate was observed. In this way, the lignin model compound was conjugated to tyramine, tyramine derivatives and molecules containing a fluorine probe. The use of fluorine‐substituted phenols was further exploited towards the hydrophobisation of beech veneers.38 The authors clearly recognised the possible interference of extractives because the wood was Soxhlet‐extracted with acetone prior to modification. Depending on the fluorinated phenol that was grafted, the static water contact angle (SWCA; an indirect measurement of the hydrophobicity) increased from 54° for unmodified wood to up to 90° for grafted wood. Observation of the coupling between the fluorinated phenol and a model compound through NMR spectroscopy analysis was posed as additional theoretical evidence for the plausibility of grafting‐on lignocelluloses.
In additional studies, the efficiency of grafting by other laccases was examined. With laccase from Bacillus SF spores, it was shown that tyramine and syringylglycerol β‐guaiacyl ether (Scheme 5 C) coupled through a C−O bond because the ortho positions, with respect to the phenyl alcohol, were blocked (Scheme 6, P1).39 Further transformations included dehydroxylation (P2), oxidation (P3) and nucleophilic attack by either of the solvents water (P4) or methanol (P5); this has been shown to be rather common in laccase‐mediated oxidation.40
Scheme 6.
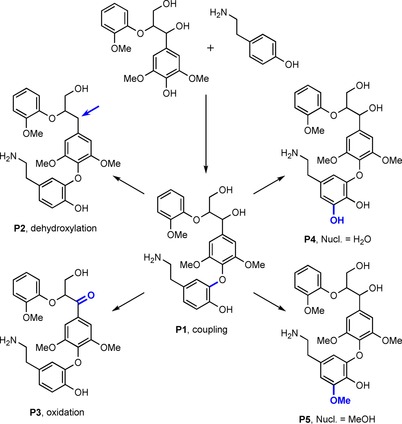
Coupling of syringylglycerol β‐guaiacyl ether and tyramine to form P1 followed by subsequent chemistries. Compounds P2–P5 are representative structures for the set of possibilities related to the mentioned reaction (based on Kudanga and co‐workers).39 Nucl.: nucleophile.
Apart from grafting phenolics from lignin, Kudanga and co‐workers also managed to graft and concomitantly hydrophobise beech veneers by using alkylamines.41 To support their hypothesis that covalent bonding, and not mere adsorption, was responsible for the increase in hydrophobicity, the authors again reverted back to simple phenolics and lignin model compounds (Scheme 7 A, B). Reacting either catechol or guaiacol with laccase in the presence of n‐dodecylamine resulted in mono‐, di‐ and trimeric species coupled to exactly two ndodecylamine molecules. A similar coupling pattern (phenol/amine=1:2, 2:2 and/or 3:2) was observed for di‐n‐hexylamine (Scheme 7 C). Upon coupling n‐dodecylamine with lignin model compounds, 1:1 coupling products between the guaiacol‐type lignin model compounds and the amine were observed (Scheme 7 D). As a follow‐up, the beech veneers were reacted with laccase in the presence of either amine to afford highly hydrophobic beech wood (increase in SWCA from 58° (unmodified beech) to 107 and 81°). Furthermore, extensive washing and rinsing with a good solvent (with respect to the amines) assured that any adsorbed material was washed off. Similar results were later found for the modification of jute fabrics with octadecylamine.26 The importance of extraction prior to laccase‐mediated modification was again exemplified because non‐extracted and unmodified jute exhibited SWCAs of 111°, due to the presence of lipophilic wax and fats.
Scheme 7.
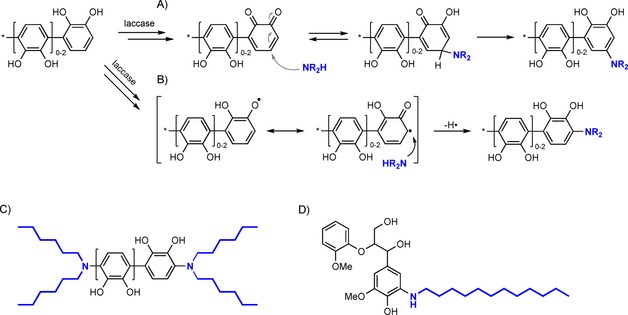
A) Alkylamine coupling on laccase‐generated quinone through Michael addition. B) C−N bond formation through an amine and phenolic carbon radical. C) Catechol/di‐n‐hexylamine 1:2, 2:2 and 3:2 conjugates. D) Guaiacylglycerol β‐guaiacyl ether/n‐dodecylamine 1:1 conjugate (based on Kudanga et al.).41
Although the use of model compounds to mimic bigger polymeric matrices provides a powerful tool to study grafting reactions on a molecular level, it also has its limitations. Lignin is a highly complex network composed of many types of functionalities. To simplify this system to an extent that NMR spectroscopy or LC/MS‐based techniques are viable, one is thus always limited to only a selection of functional groups per model compound. This implies that, to properly mimic lignin, a set of fragments is required. Additionally, approximately 10–20 % of lignin is comprised of phenolic subunits, whereas the model compounds used by Kudanga and co‐workers are 19–28 % phenol‐based, which means that any laccase reactivity will be an overestimation.42 This discrepancy is accentuated further by appreciating the fact that the model compounds are soluble and small enough for all phenols to be oxidised by laccase, whereas only some of the phenols on the exterior of the insoluble lignin will be accessible to laccase. Furthermore, in a homogenous mixture, as is the case for model compounds, the chances for the model compound and graftable molecule to react are far higher than those in the case of insoluble lignin. A phenolic lignin surface radical is likely to be quenched by a nearby lignin moiety or other unintentional scavengers present in the reaction mixture. Any representative set of lignin model compounds would thus not be complete without a non‐phenolic and insoluble molecule (that could be made soluble upon analysis).
Apart from the mentioned bottom‐up approach with lignin model compounds, several researchers aim to utilise pyrolysis coupled to GC/MS analysis (Py‐GC/MS) to study grafting in a top‐down fashion. Using this methodology, Vidal et al. studied the laccase‐mediated modification of flax and sisal pulps in the presence of a range of simple phenols.43 Initial measurements revealed that kappa numbers (an indication of lignin content) most notably increased for flax treated with p‐coumaric acid. Laccase‐induced sisal pulp treatment with guaiacyl‐type (G‐type) phenolics, such as ferulic acid (FA) or coniferaldehyde, resulted in higher kappa numbers than those with syringyl‐type (S‐type) phenolics (Scheme 8). This was rationalised by assuming more steric hindrance around the phenyl alcohol, which was surrounded by two methoxy substituents, rather than one. Additionally, the C2‐carbon without a methoxy substituent has a high electron density in the phenolic radical.45 G‐type phenolics thus have not one but two reactive sites, and therefore, the chance of covalent coupling is increased. Subsequently, the modified and acetone‐extracted sisal and flax fibres were subjected to Py‐GC/MS analysis in the presence of tetramethylammonium hydroxide (TMAH) as the base and methylating agent (Scheme 9). The observation of the 3,4‐dimethoxycinnamic acid methyl ester confirmed the presence of FA after laccase‐mediated sisal modification. Similar results were observed for the modification of flax fibres. Assuming that all adsorbed phenol had been extracted, this would thus imply that grafting had really taken place.
Scheme 8.
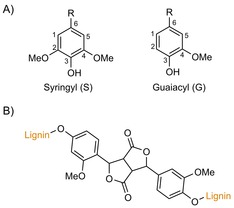
A) Syringyl/guaiacyl nomenclature. B) FA dilactone graft, as envisioned by Rencoret et al.44
Scheme 9.
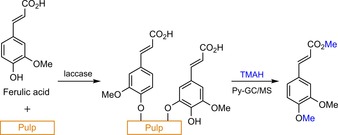
Simplified schematic representation of the laccase‐mediated grafting of FA from pulps followed by Py‐GC/MS analysis with the methylating agent TMAH (the depicted pulp–FA graft is merely an indication of the possible structure; many more structures possible, based on Vidal and co‐workers).43
As a follow‐up, the laccase‐mediated modification of sisal pulp with FA was studied in more detail.44 The formation of an etherified FA derivative is claimed on the basis of a 1 ppm shift in the 13C spectrum of isolated modified lignin. However, caution has to be exerted when interpreting NMR spectra of complex lignocellulosic grafts. Upon grafting phenolics from lignin, the formed bonds are so similar to those present in the native lignin that differentiation between the two is highly challenging. Nonetheless, a plausible dilactone originating from FA dimerisation could also be observed (Scheme 8 B). Although both grafting of the monomer and the dilactone are highly likely, (2D) NMR spectroscopy performed in this way could, in our opinion, not unambiguously confirm covalent binding to the lignin backbone. As mentioned, employing mediators further enlarges the scope of laccase‐mediated oxidation reactions. However, one should be aware of the type of chemistry that is introduced with the use of these mediators. A mediator, such as veratryl alcohol, for example, only acts as a one‐electron shuttle from laccase to a non‐laccase substrate, whereas N−O radicals, such as (2,2,6,6‐tetramethylpiperidin‐1‐yl)oxyl (TEMPO; Scheme 10), are oxidised by laccase to the corresponding oxoammonium cation, which can perform two‐electron oxidations.46 Using four different mediators (ABTS, HOBt, HPI and TEMPO; Scheme 10) and two types of lignin (wheat straw lignin (WSL) and beech organosolv lignin (BOL)), Munk et al. further exploited the use of Py‐GC/MS to study the effect of laccase‐mediator treatments on lignin.47 Grafting could only be observed for HOBt and HPI, whereas grafting of HPI was substantially more efficient (Table 1). This difference was explained by taking into account that the half‐lifetime of the HOBt radical was approximately 800 times shorter than that of the HPI radical. The HPI radical, therefore, has more time to “find” the insoluble lignin. Grafting from BOL was relatively less efficient than that of grafting from WSL. This could be rationalised by appreciating the fact that the S/G ratio of BOL was approximately five times higher than that of WSL. Additionally, the high lignin content of BOL implies that the core lignin is almost inaccessible. Furthermore, pretreatment of organosolv lignin during production is known to result in less reactive C−C bonds. Actual covalent grafting is made highly likely because no mediator was observed by Py‐GC/MS analysis in control experiments without laccase, and laccase‐mediated oligomerisation (and subsequent adsorption) of these mediators is not probable. Although unlikely, one should be aware of possible other adsorption mechanisms through mediator–oligomers or mediator/laccase complexes that would not occur in the control experiments mentioned above.
Scheme 10.
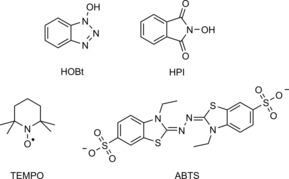
Mediators 1‐hydroxybenzotriazole (HOBt), N‐hydroxyphthalimide (HPI), TEMPO and 2,2′‐azino‐bis(3‐ethylbenzothiazoline‐6‐sulfonic acid (ABTS).
Table 1.
Grafting yields for mediator grafting on lignin.
| S/G[a] | Lignin [%][b] | HOBt [%][c] (t 1/2=100 s) | HPI [%][c] (t 1/2=7900 s) | |
|---|---|---|---|---|
| WSL | 0.6 | 43.7 | 5–6 | 32–35 |
| BOL | 2.7 | 87.4 | 1 | 11 |
[a] Syringyl‐type (S)/guaiacyl‐type (G) lignin ratio for unmodified lignin. [b] wt % lignin. [c] Grafting yield in percentages (half‐lifetime of mediator radical).
These studies have shown that Py‐GC/MS is a promising tool to study laccase‐mediated grafting because it can directly detect the grafted moiety. Unfortunately, one has to rely on the efficiency of the washing procedure to ensure substantial removal of noncovalently bound material. If pyrolysis, or any breakdown technique for that matter, can be balanced in such a way that internal lignin bonds between the grafted moiety and lignin are also broken, it might provide an even better view of the molecular nature of the lignin graft. In addition, 2D NMR spectroscopy has proven to aid lignin characterisation to a huge extent.48 If the grafted molecules are chosen in the right way, 2D NMR spectroscopy may also be the optimal tool to study grafting reactions on lignin.
In laccase‐mediated grafting, control experiments have to be designed with care. Very often laccase‐mediated modification is only compared with the simple addition of the monomer (without laccase). However, apart from laccase‐induced grafting on the surface, laccase also oligomerises phenolic monomers in solution. The physical and chemical properties, such as solubility and adsorption tendency, of these oligomers will be different to those of the monomeric species itself. In earlier work by Kim and co‐workers, this issue was partially addressed by comparing in situ coloured flax fibres to those that were coloured with ex situ generated oligomers (from morin and quercetin; Scheme 11) in a two‐step one‐pot process.49 Although laccase was not deactivated prior to the second step, laccase activity will be limited because most monomers and short oligomers small enough to enter the active site will have reacted in the first step. However, in the case of quercetin, flax fibres were equally well coloured with both procedures. This could indicate two things: either the formed oligomers are still small enough to be oxidised by laccase and are thus able to graft from the surface, or colouration is mainly affected by adsorption of oligomers. However, with morin, different results were obtained. In this case, the two‐step colouration was less effective than that of the one‐step process. Because morin has a higher oxidation potential (more difficult to oxidise/initiate oligomerisation) than quercetin, adsorption phenomena are more likely to contribute to colouration. Furthermore, colour strength still increased as the temperature in the second step was increased to 80 or 90 °C, under which conditions laccase is inactive.50 This is thus an extra indication that adsorption is likely to play a role. On the other hand, flax fibres subjected to a bleaching treatment prior to modification showed less colouration. This might indicate that lignin content was further depleted and less sites were available for the covalent attachment of phenolics. These results specify that adsorption phenomena play a role during laccase‐mediated grafting, and that proper control experiments can indicate to what extent.
Scheme 11.
Structures of A) morin and B) quercetin.
To summarise: the tools mentioned above (lignin model studies and Py‐GC/MS), together with a reliable mechanistic background, adequate pretreatment, appropriate control experiments and sufficient washing procedures, have provided a solid foundation for the ability of laccase to graft phenolics, alkylamines and certain mediators covalently on lignin. In the studies discussed below, we thus often assume that covalent grafting is possible. We do, however, highlight those instances in which it is unclear whether covalent grafting is the main mode of functionalisation, as in the case of insufficient washing, for example.
2.2. Grafting examples
Mechanical strength, hydrophobicity, antimicrobial activity or antioxidant activity are the properties studied most frequently to improve chemical or mechanical stability of lignocelluloses by laccase‐assisted treatments. Enriched paper hand sheets made from softwood Kraft pulp subjected to a laccase treatment in the presence of several phenolic monomers, as developed by Orlandi and co‐workers, is one of many examples.51 In almost all cases, the modified hand sheets were proven to diminish bacterial growth, in comparison to that of unmodified paper. The most notable effects were found with caffeic acid, 4‐hydroxybenzoic acid (4‐HBA) and dopamine oligomers, which induced bactericidal activity against multiple bacterial strains. Although grafting is highly likely to contribute, simple adsorption could not be ruled out because the modified sheets were only washed with water after the enzymatic treatment.
Laccase‐mediated functionalisation of wood veneer and pulp using tannins (natural polyphenols) conferred antibacterial properties to the wood.52 Although simple phenols proved to reduce bacterial growth to a higher extent, tannic acid modified lignocelluloses almost completely minimised the growth of Staphylococcus aureus. Additionally, chestnut tannin and cationic tannin, in particular, induced a substantial antibacterial effect against Escherichia coli. Coupling between tannin base units (catechins) and simple phenolics as lignin model compounds mainly resulted in 1:1 coupled conjugates.
Oligomers originating from laccase‐mediated coupling of phenols are known to be strongly coloured due to their extended π systems. Schroeder et al. coloured flax fibres through in situ laccase‐assisted oligomerisation of several phenolic monomers.53 However, most likely due to limited washing with water only, colour fastness was low. Modification did, nonetheless, result in growth inhibition of Bacillus subtilis and S. aureus (at 6 and 4 mm, respectively) by the orange‐coloured FA‐coated surface. The minimum inhibitory concentration for the white hydroquinone surface against these bacterial strains was even below the detection limit.
To minimise biodeterioration and oxidation of linen, Silva and co‐workers pretreated flax fibres with laccase, followed by coupling with catechin or chitosan (CS), or firstly CS, followed by catechin (Scheme 12).54 Prior oxidation with laccase proved to be essential to enhance colour strength if this was followed by a catechin/laccase treatment; without pretreatment, the colour strength was two to five times lower. The authors indicate more grafting on the surface as the cause of higher colour strength due to the presence of, pretreatment‐induced, reactive quinones that are susceptible to radical coupling. Grafting of CS on the pretreated linen in the presence of laccase, followed by laccase‐mediated grafting of catechin, however, resulted in poor fixation of catechin. Amine groups of CS most likely covalently bound the preformed quinones generated from bare flax lignin through the previously mentioned radical coupling, Michael addition (Scheme 7) or imine formation. In all cases, fewer amine groups are available to couple with oxidised catechin in the subsequent step. Both CS/catechin and catechin‐modified linen showed excellent antioxidant properties compared with those of control linen only treated with CS and/or catechin (no laccase).
Scheme 12.
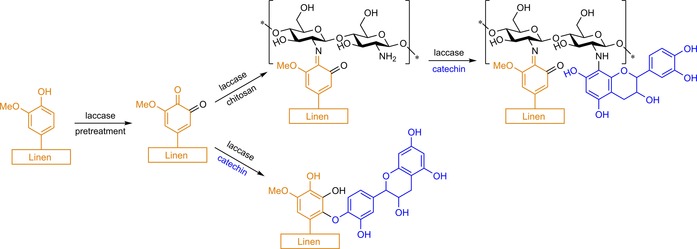
Reaction scheme for the modification of linen with a laccase‐mediated CS, catechin or CS/catechin treatment with or without laccase pretreatment (depicted structures are merely an indication of the molecular nature of the graft; many more structures are possible, based on Silva and co‐workers).54
Roncero and co‐workers treated simple phenols directly with laccase to confer antibacterial properties to flax pulp.55 Almost complete growth reduction of several bacterial strains was achieved for paper made from pulps treated with laccase and p‐coumaric acid at 15 mm initial phenol concentrations. However, laccase‐mediated modification with syringaldehyde or acetosyringone only resulted in substantial growth inhibition of Klebsiella pneumoniae at concentrations above 20 mm. Once again, the treated pulp was only washed with water, so deposition of less soluble phenol oligomers could be responsible for the effects obtained.
Traditionally, conferring antimicrobial activity to lignocellulosics by using laccase is performed through the grafting of phenolic compounds. Schubert et al., however, employed the ability of laccase to oxidise iodide to iodinate lignin to enhance the antimicrobial activity of spruce wood.56 The growth of E. coli, S. aureus and Saccharomyces cerevisiae was completely inhibited after iodination at an initial iodide concentration of 50 mm with the addition of the mediator ABTS. Additionally, anti‐fungal activity was assessed after iodination by comparing the mass loss of treated and untreated samples caused by the wood‐digesting fungi Oligoporus placenta and T. versicolor. Wood treated with an initial iodide concentration of 50 mm in the presence of laccase (with or without mediator acetosyringone) was completely resistant to mass loss caused by both fungi. Antifungal activity was thereby as high as with commercial fungicide VP 7/260a. Applying iodide only was significantly less effective against both bacterial growth and fungal degradation in all cases. Substantial and repeated leaching procedures with water were applied in all mentioned cases, which should be sufficient to leach out all formed and unbound I3 −. FTIR analysis was used to assess chemical alterations in the modified wood. Apart from observing structural changes after grafting, no direct proof of iodination was obtained. FTIR spectroscopy could have been used more effectively in this case because C−I stretching vibrations, which are not present in unmodified wood, would be observed as a strong signal at =500 cm−1. As a benchmark for their iodination strategy, Schubert and co‐workers also assessed the antibacterial effect induced by phenol grafting rather than iodination.56 It was observed that wood treated with thymol or isoeugenol alone resulted in a significant growth reduction of multiple bacterial strains. However, if laccase was added to the reaction mixtures, only the laccase/isoeugenol mix conferred antibacterial activity to the wood, and only for one bacterial strain (S. aureus). Apparently grafting, and/or oligomerisation and adsorption of thymol, leads to a loss of the antibacterial properties. The authors have subjected wood to extensive washing with water, but it is unclear how effective this is considering the poor solubility of the phenols and its oligomers in water. Orlandi and co‐workers, however, obtained different results:51 in their hands, isoeugenol was not an effective antibacterial agent against S. aureus and thymol only moderately so, but wood pulp treated with laccase/isoeugenol was bactericidal. In all cases, the effect is less than that with the iodination approach of Schubert et al.,56 which shows that iodination is a more efficient, but underappreciated, method to confer antimicrobial activity to lignocellulosic material.
Tzanov and co‐workers developed a silver nanoparticle (NP)–cork composite as an antibacterial adsorbent for wastewater treatment (Scheme 13).57 AgNO3 was firstly reduced to Ag in the presence of an amine‐functionalised carbohydrate (chitosan (CS) or a 6‐deoxy‐6‐(ω‐aminoethyl)amino cellulose derivative (AC)), followed by laccase‐assisted fixation of the NP to cork. Cork consists of lignin, tannins and suberin (a polyester containing long‐chain fatty acids, hydroxy fatty acids and phenolic acids), which are susceptible to laccase‐mediated oxidation.58 The cork–AgNP–CS composite fully inhibited the growth of E. coli and S. aureus, whereas the AC composite was only able to completely inhibit the growth of S. aureus. Although substantial grafting can be expected, washing after modification was performed with water, which might not have removed all adsorbed insoluble NPs. To test the antibacterial efficiency during use, water filtration cartridges were filled with the cork–AgNP–CS composite material.59 All bacteria could be removed from the effluent at 8 h residence time. However, approximately 25 % of the total silver content leached out after five disinfection cycles. This would contaminate the water and diminish the antibacterial effect of the composite, but, additionally, the question arises to what extent leached silver contributes to the antibacterial effect.
Scheme 13.
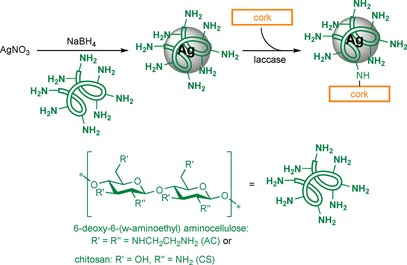
Reaction scheme for the synthesis of an antibacterial silver NP–cork composite. Based on Francesko et al.57
Natural sources are exploited more and more as a basis for composite materials. Unfortunately, these materials often suffer from low compatibility with man‐made plastics due to their high hydrophilicity. To enhance its hydrophobicity, Fan et al. modified jute with laccase in the presence of dodecyl gallate (Scheme 14).60 Soxhlet extraction with acetone was expected to ensure removal of any unbound material after grafting. SWCA measurements revealed an increase from 30° for unmodified material to 111° for modified material. Furthermore, although control experiments with laccase or gallate only, also resulted in higher SWCAs, these droplets wetted the fibres within 1 min, whereas the SWCA for the laccase/gallate‐modified jute stayed practically unchanged for the first 5 min. FTIR analysis was used to indicate whether gallate or gallate oligomers were covalently bound to the jute. The appearance of new bands, however, does not necessarily indicate the formation of a bond between the surface and graftable moiety. To aid analysis, one should at least perform control experiments in which the phenolic oligomers are formed ex situ and subsequently deposited on the surface. But, even then, the formed bonds are very similar to those in the lignin matrix, especially when grafting phenols, which makes FTIR analysis at best highly complicated, and often an unreliable tool. Modified jute/polypropylene composites exhibited higher breaking strengths than those of the composites with native jute. As a follow‐up, propyl and octyl gallate were used as jute modifiers in the presence of laccase.61 As expected, these molecules were less efficient at conferring hydrophobicity to the jute. These new materials thus showed non‐optimal compatibility with polypropylene, which was apparent from a lower tensile strength for these composites, than that of the dodecyl gallate jute/polypropylene composite.
Scheme 14.
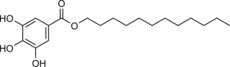
Structure of the common hydrophobisation agent dodecyl gallate.
Lignocelluloses with higher lignin contents, such as beech wood, could also be made hydrophobic through in situ laccase‐assisted dodecyl gallate oligomerisation and grafting.62 Apart from the wood being hydrophobic (SWCA>90°), the presence of dodecyl gallate (oligomers) after acetone extraction was also confirmed by FTIR and XPS analysis. On the basis of a decrease in the surface oxygen/carbon ratio, covalent attachment was claimed; however, noncovalent deposition of gallate oligomers would cause a similar decrease. So, although XPS analysis can be a powerful tool, as with FTIR measurements, one should be careful with its interpretation.
Garcia‐Ubasart and co‐workers developed a simple method to make paper more resistant to wetting by reacting pulp with dodecyl gallate and laccase.63 Although other phenols were also investigated, dodecyl gallate was the only compound that could substantially increase the wetting time of the paper. Unfortunately, the mechanical strength of the treated paper suffered from the conditions under which the highest degree of grafting could be expected (higher laccase/gallate concentrations and longer reaction times). Later studies further confirmed wetting resistance by SWCA measurements.64 Furthermore, an increase in kappa number (an indication of the degree of lignin/phenolics) was observed after modification and subsequent acetone extraction. The exact mechanism behind laccase‐mediated functionalisation of lignocelluloses in the presence of gallates has, however, yet to be clarified. Studies have shown that under some conditions the kappa number increases after laccase/gallate treatment, but drops again after extraction. After modification of the lignocellulosic material with a similar lignin content (>5 %) to that of jute and wood mentioned above, Reynaud et al. observed that after extraction a drop in kappa number coincided with a decrease in SWCA and an increase in water absorption rate (back to the level of unmodified material).65 During lignocellulosic processing, some xylan monomers are converted into hexenuronic acids (HexAs; Scheme 15); undesired olefinic chromophores that cause aging of the lignocelluloses. Laccase/mediator treatments are known to decrease the HexA content of the material; therefore, Cadena and co‐workers tested p‐coumaric acid and dodecyl gallate as mediators for the removal of HexA.33 The use of dodecyl gallate in the presence of Trametes villosa laccase was indeed very effective in removing HexA (82 % decrease), whereas a Pycnoporus cinnabarinus/p‐coumaric acid treatment removed only 2.8 μmol g−1 (22 %) of the HexA present in the unbleached flax pulp (lignin content 1.4 %). Unlike what Reynaud and co‐workers observed, the kappa number was approximately twice as high after modification and subsequent acetone extraction, even though a low lignin content flax was used.65 Moreover, if lignin and HexA‐free pulp were used, almost no change in kappa number was detected. However, if lignin‐free pulp with high HexA content was modified, an approximate twofold increase in kappa number was again observed. It is thus postulated that grafting does not occur through lignin, but through coupling with HexA. These remarkable results have yet to be confirmed, but laccase‐assisted modification of gallates in the presence of a HexA model compound might reveal more details on the grafting process.
Scheme 15.
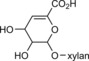
Structure of hexenuronic acid (HexA).
Witayakran and Ragauskas oxidised softwood Kraft pulp by using laccase to functionalise it with amino acids.66 Imine formation, radical coupling or Michael addition on the generated quinones would result in covalent grafting. If histidine was used, the tensile strength, tear strength and wet tensile strength of paper made from these pulps improved. Measurement of the carboxylic acid content was used to demonstrate the presence of amino acids. However, treating the pulp with laccase or amino acids only also resulted in an increase in carboxylic acid content. Although the carboxylic acid content after laccase/amino acid treatment was higher than that of the samples treated with either laccase or amino acids alone, this could simply result from the adsorption of both.
Apart from the common nucleophilic coupling of amines or the grafting of phenols, functionalisation with methacrylate or acrylamide is a popular strategy for the modification of lignocelluloses. Fan and co‐workers employed laccase‐initiated grafting using acrylamide and tert‐butylhydroperoxide to functionalise jute fibres.67 Earlier studies confirmed that the presence of hydroperoxide was essential because the use of only laccase did not result in substantial grafting, but the necessity of laccase was dependent on the type of lignin.68 Modification resulted in a 10° drop in SWCA and a somewhat enhanced colouration of the jute. These modest results indicate minimal grafting of acrylamide. Additionally, the initial SWCA of the jute proved to be exceptionally high (110°), which was likely to have been caused by contamination of the jute with fats and waxes.26 The presence of these compounds could also have influenced possible grafting, which makes these results hard to interpret. Ko and co‐workers used a similar laccase‐initiated grafting approach with N‐isopropylacrylamide (NIPAM) as monomer, but in a two‐step sequence.69 Prior to grafting, they functionalised lignin with an ATRP initiator (α‐bromoisobutyryl bromide). Grafting subsequently resulted in 800 to 1000 nm long poly(NIPAM) brushes with thicknesses up to approximately 100 nm and polydispersity indexes as low as 1.3; this is common for brushes synthesised through ATRP. The downside of this type of polymerisation is inhibition by molecular oxygen. However, laccase requires oxygen as an electron acceptor; thus, exclusion of oxygen causes laccase not to act as a catalyst, but as a stoichiometric oxidant. The previously mentioned grafting with acrylamide was not performed under oxygen‐deprived conditions; this might explain the modest functionalisation of jute.
Most of the work discussed so far had been conducted on the laboratory scale, but laccase‐mediated grafting has also been applied in more practical settings. Schubert and co‐workers used industrial process water from a wood fibreboard plant directly as the phenol source in a laccase‐mediated treatment to enhance the mechanical strength of wood fibre insulation boards.70 The compression strength and internal bond strength of boards made from laccase/process water treated pulp were significantly improved relative to treatments with fresh water in the presence of laccase. This effect was enhanced even further if conventional latex binder was used as an additive. Pulp treated with a low laccase dose (1.7 U mg−1) and 3 % latex, and pulp treated with 5 % latex only, were equally strong; thus, the fossil‐fuel‐based binder dosage could be lowered by 40 %. For the production of medium‐density fibreboards, Kharazipour et al. applied a laccase/4‐HBA treatment to replace traditional binders.71 By doing so, some grafting was likely, but most pronounced was rapid delignification; the used lignin pulp was deprived of approximately 40 % of its aromatic compounds within 30 min. This is remarkable because 4‐HBA is a very unreactive mediator due to the presence of a conjugated carboxylic acid, with only one ring‐bound hydroxyl functionality.72 Nonetheless, fibreboards produced in this way complied with European standards regarding bending strength, internal bond strength and thickness swelling, whereas pulps treated with laccase only and two other laccase‐mediator systems did not. Although a substantial contribution of adsorbed material can be expected because in both studies the pulp has not been washed afterwards, this example does show the potential of laccase‐assisted modifications in an industrial setting.
Aracri and co‐workers used laccase‐mediated grafting in a completely different, but application‐oriented, adhesive formulation.73 Several commercial isolated lignins were reacted in a laccase/mediator (acetosyringone) system to generate quinones, followed by grafting of gallic acid, tannic acid and dopamine, which is known to have strong adhesion properties. Further grafting at 50 °C generated phenol‐ and quinone‐enriched lignin. The quinones in this paste could further undergo oligomerisation, nucleophilic attack and imine‐bond formation if the paste was smeared on the backing of a wool carpet sample that naturally contained protein‐based amine groups. Thus, the paste could act as a carpet adhesive through both covalent and noncovalent interactions. The force required to pull a loop of fabric from the carpet was used to assess adhesion. The resulting adhesion was only approximately 10 % lower than that of commercial latex‐based adhesives, while being almost completely constructed from renewable sources.
To conclude: laccase‐mediated grafting has proven to be a cheap, mild and eco‐friendly way of modifying lignocelluloses. Whether grafting or adsorption is the main mechanism for modification of the material is highly dependent on process parameters and washing procedures. The latter requires extra attention in many cases, especially if noncovalent binding, and thus, leaching results in a health risk. Although Py‐GC/MS and NMR spectroscopy are powerful techniques to study possible grafting, their use has not reached its full potential. Furthermore, the use of model compounds is essential in understanding the molecular details of the grafting process. It is, however, advisable to also employ both insoluble and non‐phenolic lignin models to mimic lignin to a greater extent.
3. Laccase‐Mediated Grafting on Polysaccharides
Modification of polysaccharides is the second biggest subdomain within laccase‐mediated grafting. To some degree, this topic has already been discussed, as applying laccase‐mediator systems sometimes also results in oxidation of the cellulose present in lignocelluloses. For some fibres, cellulose and hemicellulose are by far the most abundant constituents, for example, 97 % for flax fibres.74 Thus, all cellulose considered herein is assumed to constitute less than 2 % lignin.
Whereas the mechanism behind lignin modification is highly multifaceted due to its intrinsic complexity, laccase‐mediated polysaccharide treatments are, in general, easier to comprehend. Two main modes of action could be considered dependent on the polysaccharide of choice: 1) laccase‐mediator system‐assisted oxidation of carbohydrate hydroxyl groups to aldehydes or carboxylic acids followed by coupling with a suitable reagent, or 2) laccase‐mediated oxidation of a phenol that can be sequentially coupled to an amine‐functionalised carbohydrate similar to the previously discussed functionalisation of cork with CS (Scheme 13). Although laccase‐assisted carbohydrate modification could be considered to be mechanistically less complex than that of lignin modification, the same guidelines apply and similar challenges remain. The intended grafting strategy should be mechanistically feasible and proper control experiments and washing should be conducted. Adsorption might also occur during carbohydrate modification, so correct analysis and characterisation is sometimes puzzling.
In the mid‐1990s, Viikari and co‐workers filed a patent that described, for the first time, the laccase‐mediated oxidation of cellulose in the presence of TEMPO.75 This strategy resulted in chemoselective oxidation of the cellulose C6 hydroxy groups. As mentioned earlier, laccase converts TEMPO through a one‐electron oxidation to the corresponding oxoammonium ion that can selectively oxidise primary alcohols through a two‐electron oxidation due to intrinsic steric bulk around the cation (Scheme 16).76
Scheme 16.
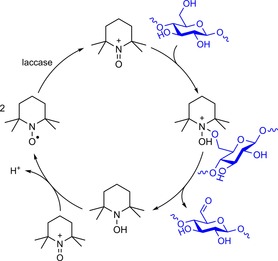
Laccase‐mediated oxidation of TEMPO followed by TEMPO‐mediated oxidation of cellulose. Based on the proposed mechanism of laccase/TEMPO‐mediated oxidation by Tromp et al.76
Whereas chemical oxidation of cellulose with bleach is known to cause depolymerisation of cellulose, laccase‐mediated TEMPO treatments minimise this depolymerisation to a certain extent.80 The use of TEMPO as a mediator is essential because hydroxylamine‐type mediators are not able to oxidise cellulose to a significant degree. Furthermore, applying TEMPO without laccase does not result in substantial oxidation.80 Later studies showed that whether selectivity towards the aldehyde with respect to the carboxylic acid was observed was highly dependent on the conditions (Table 2). Evaluation of the carboxyl/carbonyl ratio by Xu and co‐workers on cellulose oxidation and Yu et al. on β‐cyclodextrin (a cellulose model) oxidation revealed a general trend that temperatures above 30 °C, longer reaction times, higher laccase dosage and higher TEMPO concentrations favoured carboxylic acid formation.78, 83 Apart from introducing additional functionality, paper made from oxidised cellulose is stronger, most markedly due to an increase in wet strength.81, 84
Table 2.
Variability in laccase/TEMPO‐mediated cellulose oxidation: results from experiments with highest degree of oxidation.
| Author[a] | Laccase | Type of pulp | Concentration [μmol g−1][b] | COOH vs. | |||
|---|---|---|---|---|---|---|---|
| source | COOHi | COOHf | CHOi | CHOf | CHO[c] | ||
| Jiang77 | T. versicolor | bleached acacia Kraft (80 % α‐cellulose) | 98 | 596 | 16 | 241 | 69:31 |
| Xu78 | Aspergillus oryzae | bleached softwood Kraft | 30 | 138 | 32 | 104 | 60:40 |
| Aracri79 | T. villosa | sisal soda anthraquinone (79 % glucan, 1 % lignin) | 110 | 266 | 1 | 191 | 45:55 |
| Patel80 | Trametes pubescens | cotton linters | 7 | 31 | 8 | 120 | 18:82 |
| Aracri81 | T. villosa | sisal soda anthraquinone (79 % glucan, 1 % lignin) | 110 | 126 | 1 | 107 | 13:87 |
| Jaušovec82 | T. versicolor | cellulose nanofibres (95 %) | 10 | 30 | 750 | 1523 | 3:97 |
[a] First author and reference to the corresponding paper. [b] Initial and final concentrations of carboxyl and carbonyl groups. [c] Formed carboxyl to carbonyl ratio.
Yu and co‐workers used laccase/TEMPO‐mediated oxidation in the presence of octadecylamine to increase the hydrophobicity of cotton.85 Initial modifications were performed on glucans (water‐soluble short‐chain glucose polymers) to enhance reactivity and ease characterisation. FTIR analysis of the modified glucan revealed (almost) full conversion of the aldehyde and the appearance of a new band at =1640 cm−1, which corresponds to the formed imine. However, due to the strong absorbance of cotton at that wavelength, no increased intensity of the band at =1640 cm−1 was observed after cotton modification. Nonetheless, SWCA revealed that cellulose was rendered hydrophobic (SWCA>110°). Washing fastness was high because the SWCA was barely altered for up to eight washing cycles, even though washing was performed in the presence of a commercial detergent.
The importance of washing steps, a proper understanding of adsorption/grafting mechanisms and appropriate analytical tools was nicely accentuated by Guimarães and co‐workers, who overdyed denim fabrics with in situ laccase‐generated catechin or catechol oligomers.86 Dyeing in this case does not result in covalent grafting due to the lack of reactivity between cellulose and (oxidised/oligomerised) catechin or catechol. Nonetheless, intense colouration of the fabrics with sufficient to good fastness levels was achieved. It was hypothesised that less soluble oligomers had reasonably strong interactions with the denim surface. This thus indicates that non‐specific adhesion can be very resilient. Even when grafting is possible, mere adsorption cannot always be ignored.
Whereas Py‐GC/MS analysis proves to be very promising in the field of lignocellulosics, Díaz Blanco and co‐workers envisioned an elegant coupling of breakdown, LC/MS/MS and NMR spectroscopy to fulfil a similar role in the analysis of polysaccharide grafts.87 In their approach, cellulose dyeing was achieved by firstly tosylating the cellulose, followed by nucleophilic displacement of the tosylate with 2,5‐diaminobenzenesulfonic acid (2,5‐DABSA). Further functionalisation was thereafter accomplished through laccase‐mediated grafting from immobilised 2,5‐DABSA with catechol. Removal of unbound catechol was achieved by extensive washing of the modified cellulose in boiling water in the presence of a surfactant. Indirectly, grafting was confirmed by showing that aminated cellulose dyed with ex situ generated catechol oligomers exhibited a far lower wet rubbing fastness. MS/MS analysis of the modified cellulose hydrolysate revealed the presence of glucose dimers covalently attached to one molecule of 2,5‐DABSA, which bound two to six units of catechol (Scheme 17). Similar dimers were found where a tosylate was still present; this indicated that nucleophilic displacement by 2,5‐DABSA was not complete. Breakdown of the graft followed by analysis proves to be a powerful tool to study grafting on cellulose.
Scheme 17.
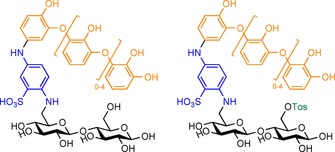
Possible covalently bound structures of 2,5‐DABSA/catechol‐modified cellulose observed by MS, as proposed by Díaz Blanco and co‐workers.87 Tos: tosyl.
For many grafting applications, the outermost layer of the material should be activated, but its internal consistency should not be altered. Because TEMPO‐mediated oxidations of cellulose decrease the degree of polymerisation, Liu and co‐workers developed an immobilised TEMPO–poly(vinylamine) complex (PVAm‐T) and used this as a mediator in the laccase‐mediated oxidation of cellulose.88 It was proposed that some surface C6 hydroxy groups were oxidised to the aldehyde first, which would then form imine bonds with the remaining amine groups of PVAm‐T (Scheme 18). Since PVAm‐T is still positively charged and laccase is slightly negatively charged, they formed a PVAm‐T/laccase complex on the surface. It was further hypothesised that this immobilised laccase would oxidise proximal TEMPO units to their corresponding oxoammonium ions. Because oxidation was only observed at the exterior of the cellulose membranes, it was assumed that TEMPO did not act as a direct shuttle for oxidising cellulose, but that it oxidised neighbouring TEMPO units on PVAm‐T. These neighbouring TEMPO units would do the same until the TEMPO moiety closest to the surface would oxidise the C6 primary alcohol of cellulose. Since only the exterior of cellulose is oxidised (Figure 1), lower immobilised TEMPO concentrations (with respect to that of free TEMPO) are required for oxidation.
Scheme 18.
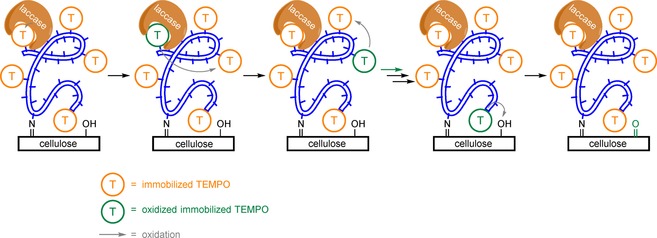
PVAm‐T/laccase complex grafted on cellulose for the laccase‐mediated oxidation of TEMPO followed by “oxidation‐state transfer” to oxidise cellulose. Based on work by Liu et al.88
Figure 1.
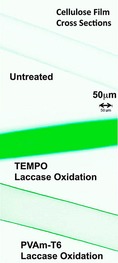
Distribution of fluorescein‐labelled aldehyde groups in cellulose membrane cross sections. Aldehydes were labelled with green fluorescein‐5‐thiosemicarbazide. Reproduced with permission from Liu et al.88 Copyright American Chemical Society, 2013.
Furthermore, the PVAm‐T/laccase complex, together with non‐functionalised polyvinylamine, was used as a cellulose‐to‐cellulose adhesive. In later studies, the use of poly(acrylic acid)–TEMPO instead of PVAm‐T as the TEMPO source did not result in cellulose‐to‐cellulose adhesion.89 Adhesion could be restored by adding polyvinylamine again, thereby indirectly indicating the importance of grafting.
Similar to the hydrophobisation of lignocelluloses, cellulosic material can also be modified with laccase and dodecyl gallate.90 These celluloses have low lignin (≈1 %; kappa number <6.3) and HexA contents, which make grafting unlikely. Nonetheless, increases in kappa number, although limited, are observed after laccase‐mediated modification and extraction with acetone. This means that either the graft consists of long gallate oligomers, or acetone extraction is simply not sufficient to remove all unbound gallate and gallate oligomers.
To create a conductive form of cellulose, Zhang and co‐workers performed in situ laccase‐mediated synthesis of polyaniline to generate cotton, which showed good conductivity and increased anti‐static properties.91 By using sodium dodecylbenzenesulfonate as a micellar soft template, polyaniline was synthesised in a linear head‐to‐tail fashion (Scheme 19). Although no covalent bonding to the cellulose is claimed, some Schiff bases between the polyaniline and oxidised cellulose may have formed. This will, however, be limited because the ability of laccase to oxidise cellulose is minor if TEMPO is not applied as a mediator.
Scheme 19.
Micellar soft‐template‐assisted head‐to‐tail synthesis of polyaniline.
In more recent work, sodium dodecylbenzenesulfonate was replaced by perfluorooctanesulfonic acid potassium salt (PFOS), which provided cellulose with the possibility to perform surface energy switching, and thus, also wettability switching (Scheme 20).92 In this elegant approach, the functionalised surface displayed SWCA>120° due to the presence of PFOS, which not only acted as a template, but also as a dopant. Dedoping could subsequently be achieved by using ammonia or ethylenediamine to result in a fully wettable (SWCA<15°) surface; this process was repeatable up to more than ten times. Concomitantly, the cellulose appeared blue/purple in the dedoped state (green when PFOS‐doped) and was no longer conductive. The equivalent of 25 repeated water washing cycles resulted in a 30° decrease of the SWCA in the doped state. Although this is substantial, the SWCA is still far higher than that of bare cellulose, which indicates that polyaniline and/or PFOS is still present on the surface, which, in turn, could indicate covalent bonding between polyaniline and cellulose.
Scheme 20.
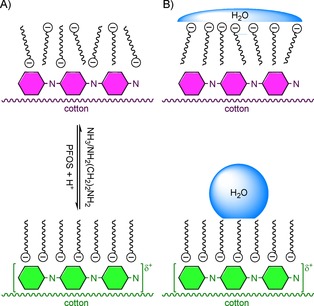
A) Wettability switching of polyaniline‐functionalised cotton due to doping with PFOS and dedoping with amines.92 B) Schematic depiction of SWCA on the corresponding surface.
Apart from cellulose itself, its etherified derivative, ethyl cellulose, is also used in laccase‐mediated transformations. Iqbal and co‐workers used several approaches towards the development of ethyl cellulose composites.93 By mixing ethyl cellulose and keratin in the presence of laccase, composites with SWCAs and glass transition temperatures higher than those of either parent material were developed. The exact role of laccase is, however, debatable. Because most of the hydroxyl functionalities of the cellulose backbone are etherified and no mediator is applied, oxidation of cellulose towards reactive aldehydes is unlikely. The authors propose amide formation through oxidation of the scarce hydroxyl moieties of ethyl cellulose, which further couple with amine groups of keratin. However, to the best of our knowledge, this type of laccase‐assisted amide‐bond formation has not been observed before. In a later study, this material was further functionalised through a laccase‐mediated approach that involved several simple phenols.94 Cellulose–keratin‐based composites treated in the presence of caffeic acid, gallic acid, thymol or 4‐HBA showed complete bacteriostatic activity towards the gram‐negative strains E. coli and Pseudomonas aeruginosa. Furthermore, similar activity was observed for thymol and 4‐HBA modified composites against B. subtilis and S. aureus. Additionally human keratinocyte‐like (HaCaT) skin cells proved to be fully viable in the presence of these composites after five days of growth, while maintaining properly stretched morphologies. Grafting most likely occurs through C−N or C=N bond formation between the amine moieties of keratin and oxidised phenols. However, a substantial contribution of adsorbed phenolic oligomers can also be expected, since the composites were washed only after being dried. Drying of the swollen ethyl cellulose will cause compression of the material. During compression, deposition of unbound phenolics still present is likely to occur. This will impede their removal through washing afterwards. Nonetheless, the phenol‐functionalised ethyl cellulose/keratin composites might prove to be easily accessible materials for several biotechnological applications.
The same authors used a similar approach to develop ethyl cellulose/poly(3‐hydroxybutyrate) (P(3‐HB)) composites.95 Again composites generated in the presence of laccase exhibited higher glass transition temperatures than those of either of their parent compounds. Although claimed, laccase‐mediated grafting is unlikely due to the absence of a mediator and a deficiency in (nucleophilic) coupling partners in P(3‐HB). Nonetheless, composites generated in the absence of laccase did not show an increase in glass transition temperature. Incorporation of laccase itself within the composite might be a reason for this. Further functionalisation of these materials was achieved by modification with either 4‐HBA or FA in the presence of laccase.96 Both modifications resulted in surfaces with bacteriostatic activity against E. coli. Of these, 4‐HBA‐functionalised ethyl cellulose/P(3‐HB) proved to be biocompatible with HaCaT cells. Any property enhancement has likely occurred due to deposition of phenolic oligomers because there is no basis for reactivity between (oxidised) phenolics and ethyl cellulose or P(3‐HB). Similar results were found when thymol, caffeic acid or gallic acid were used as the phenolic modifier, although caffeic acid and gallic acid modification did not result in equally efficient antibacterial composites.97 Iqbal and co‐workers also employed lipase instead of laccase to generate composites from ethyl cellulose and P(3‐HB) through esterification.98 Although ethyl cellulose possesses a limited number of hydroxy moieties, this seems a more viable way of covalently coupling ethyl cellulose and P(3‐HB).
Of all amine‐bearing carbohydrates, chitosan (CS) is most commonly employed in grafting. CS consists of β‐(1–4)‐linked d‐glucosamine units and is obtained by deacetylation of chitin.99 Chitin is the second‐most abundant natural carbohydrate polymer, but generally regarded as waste; thus, recycling it as CS is highly desirable. The solubility of CS is strongly dependent on pH; at a pH lower than 6.1, enough amine groups are protonated to dissolve CS.100 For the sake of consistency, we mainly discuss heterogeneous reactions with insoluble CS.
Božič and co‐workers and Aljawish et al. (vide infra) more or less simultaneously pioneered the field of laccase‐mediated modification of CS. The former initially modified CS by functionalising it with caffeic acid and gallic acid oligomers to confer antioxidant and antimicrobial properties to the material.101 By using T. versicolor laccase, CS was modified at several pH values. Of these, only laccase/gallic acid treated CS at pH 4.5 showed improved antibacterial activity (E. coli) relative to CS treated at that pH without the presence of laccase. ABTS radical cation scavenging activity, a measure of the antioxidant activity, was excellent for both CS derivatives, modified at all acidities. Due to limited washing of the resulting grafts, the degree of actual covalent grafting is hard to determine. Laccase‐mediated treatments with other phenolics (quercetin and tannic acid) to modify CS resulted in no or limited increases in antimicrobial activity.102 By employing Suberase, a Myceliophthora thermophila laccase, Aljawish et al. modified CS with FA and ethyl ferulate under heterogeneous reaction conditions followed by extensive washing with buffer, but also with methanol, ethanol and acetone, to remove moderately soluble oligomers.103 Similarly to the above‐mentioned CS derivatives, a pronounced antioxidant effect was observed, although FA‐modified CS exhibited EC50 values that were approximately three times higher than those of the ethyl ferulate modified derivatives. The extensive washing procedure allowed for better characterisation of the CS derivatives. FTIR analysis revealed two new bands at =1620 and 1640 cm−1 for both grafted phenolics. The presence of these two bands might indicate covalent grafting, since these could correspond to C=N stretching vibrations of Schiff bases. Although only speculative, additional evidence for the presence of Schiff bases might be found in partially overlooked, but valuable, 13C NMR spectra of derivatised CS. Any imines present would be formed from a reaction between an amine and a quinone. The peak at approximately δ=150 ppm could represent the carbon atom of a quinone‐based imine.104 This is further substantiated by the absence of this signal in the spectrum of hydrated CS derivatives, since the imine would be in equilibrium with its parent amine and quinone. It was furthermore shown that bacterial growth on both ethyl ferulate and FA‐modified CS was comparable to that of unmodified CS. By combining these results with those of Božič and co‐workers, it could thus be concluded that laccase‐mediated functionalisation is not a viable way to improve the antibacterial behaviour of CS. Modification additionally decreased water and oxygen permeability of the material, which indicated its potential as (food) packaging material.105 Due to its increased hydrophobicity, modified CS adsorbed more protein from cell growth media than CS itself did, which is essential for the viability of cells.106 Cell growth of human umbilical vein endothelial cells (HUVECs) and mesenchymal stem cells was thus improved and mainly influenced by grafting time and (derivatised) CS film thickness. An approximate 20–40 % increase in cell viability was achieved relative to that of non‐modified CS, while also improving cell morphology from round to stretched.
Yang et al.107 employed the methodology established by Aljawish et al.;103 thus including extensive washing, to modify CS with cinnamic acid derivatives to minimise bacterial growth of several Ralstonia solanacearum (RS) species. Of main interest was the growth inhibition of RS‐5, which is responsible for mulberry wilt disease. Of all functionalised CS derivatives, the caffeic acid modified CS proved to be the most promising, with an IC50 value of 0.23 mg mL−1, which was similar to that of the oligomerised caffeic acid itself.
In summary, laccase‐mediated covalent polysaccharide functionalisation is a growing field that allows easy and eco‐friendly access to highly complex and functional materials. Promising biotechnological applications, such as antibacterial and biodegradable plastics that enhance cell growth, rapidly emerge. However, the chemistry behind this technology has thus far not been substantially studied. Many of the developed procedures for laccase‐mediated grafting lack proper washing cycles, which makes differentiation between adsorption and covalent grafting challenging. Studies with polysaccharide model compounds, such as simple glucoses and glucosamines, might aid in establishing a better understanding of the underlying chemistry. Furthermore, most of these modifications are based on imine‐bond formation, which is reversible in nature. For numerous applications, this is not desirable, and chemical or enzymatic reduction of the imine bond would introduce a more stable graft. Overall, it is therefore a fair characterisation of the state‐of‐the‐art in this field that laccase‐mediated grafting on polysaccharides is promising, but that there is a lot to be learned.
4. Laccase‐Mediated Grafting on Proteins
Apart from grafting on textiles such as jute and cotton, laccase is also commonly employed to functionalise wool fibres. The high abundance of nucleophiles in wool keratin allows covalent bonding to phenols oxidised by enzymes.108 Laccase‐mediated grafting is regularly used for in situ colouration, enhancement of hydrophobicity or to confer antibacterial properties to the material. An important factor in wool modification is prior extraction and cleaning because raw wool is often contaminated with feculence and is abundant in fats and waxes.109
Jeon and co‐workers studied laccase‐mediated in situ colouration of yak wool with a comprehensive set of phenols and phenolic mixtures.110 Strongly coloured oligomers were synthesised that covered a wide range of tints. From this, mixtures displaying colours that resembled those of natural human hair were selected and further tested for their dyeing ability. Modification with gallic acid and syringic acid resulted in brown wool, catechol and catechin gave black wool, and FA/syringic acid modification resulted in a faint reddish colour (Figure 2). Washing with detergents commonly encountered in shampoo did not result in loss of colour. Although this was not elaborated on, the good washing fastness might have resulted from covalent binding between keratin and the oxidised mono‐ or oligomers, although the poor solubility of phenolic oligomers might also have played a role.
Figure 2.

A) Virgin yak wool. B) Yak wool dyed with gallic acid plus syringic acid. C) Yak wool dyed with catechin plus catechol. D) Yak wool dyed with FA plus syringic acid (reproduced with permission from Jeon and co‐workers).110
Yuan and co‐workers used a similar approach to colour wool with catechin or gallic acid (Scheme 21 A).111 To impede shrink resistance to the fabric, a polymer coating can be applied to smooth the wool cuticle surface. The addition of PEI to the laccase/phenol mixture in a one‐step process, however, prevented colouration of the fabric (Scheme 21 B). It was hypothesised that the amine groups of the PEI competed with nucleophilic moieties of wool for covalent binding to the oxidised phenols. Indeed, implementation of a two‐step/one‐pot process, in which laccase‐mediated modification was followed by the addition of PEI, did result in successful colouration and an average 15 % increase in shrink resistance (of alkali pretreated samples) after four washing cycles (Scheme 21 C). Furthermore, similar levels of colouration in the one‐ (without PEI) and two‐step processes imply that colouration has substantially occurred through covalent grafting because otherwise PEI would also have been covalently bound to loosely adsorbed phenolics in the two‐step process. Covalent binding is further suggested by the fact that the fabrics remain equally strongly coloured after four washing cycles. Shrink resistance could further be increased by the addition of amine crosslinking agent glycerol diglycidyl ether in the second step to increase PEI polymer length. Addition of the crosslinker also further increased the colour strength of the fabric, most likely because dissolved coloured P(EI) could be crosslinked to the surface‐immobilised amines (Scheme 21 D).
Scheme 21.
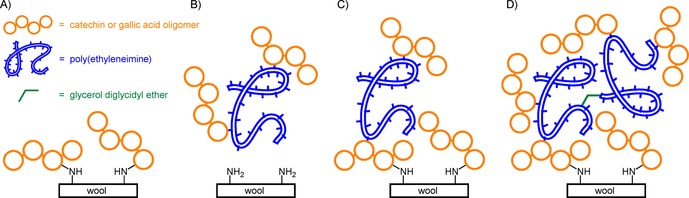
Schematic representations of laccase‐mediated colouration of wool with catechin or gallic acid oligomers. A) Colouration of wool with laccase‐generated oligomers only. B) One‐step attempt at colouration in the presence of PEI. C) Two‐step process: colouration followed by the addition of PEI. D) Two‐step: colouration followed by the addition of PEI and glycerol diglycidyl ether (based on Yuan et al.).111
Other work by the same group involved the in situ laccase‐mediated synthesis of poly(2,5‐DABSA) to confer pH‐dependent colouration and conductivity to wool.112 Poly(2,5‐DABSA)‐coated wool doped at pH 1.8 was conductive and dark purple; after dedoping at pH 10, the modified wool was rendered electrochemically inactive and became yellowish/brown. Although no covalent bonding between wool and the coating was claimed, this is likely to occur because 2,5‐DABSA could also be oxidised by laccase to generate a radical, which is similar to radical generation on phenols. This makes this moiety also susceptible to attack by amines of the wool protein.
To enhance laccase‐mediated colouration of wool even further, Zhang and co‐workers pretreated wool with 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride (EDC) to activate carboxylic acids present on wool.113 Next to direct coupling of wool to oxidised phenolics, phenols could now bind to wool through ester‐bond formation. This methodology indeed resulted in a 50 % colour strength increase relative to that of wool that had not been pretreated. Additionally, substantial improvements in wet rubbing fastness and washing fastness were achieved. To further examine the degree to which adsorbed oligomers contribute to colouration, washing with more apolar solvents would be desirable.
Hossain and co‐workers employed laccase‐mediated oligomerisation of nordihydroguaiaretic acid (Scheme 22) for the functionalisation of wool fibres.114 This modification resulted in an increased UV resistance, improved antioxidant activity, a 25 % tensile strength increase and better shrink resistance. The improvement of the last two physical parameters was ascribed to crosslinking of fibres due to the bicatechol moiety of nordihydroguaiaretic acid. Unfortunately, no distinctive characteristics of covalent bonding, such as imine formation, were reported.
Scheme 22.
Structure of nordihydroguaiaretic acid.
As with cotton and lignocellulosic fibres, dodecyl gallate has the potential to also graft on wool through radical coupling, Michael addition or Schiff base formation. By doing so, wool was made hydrophobic (SWCA>110°), while, at the same time, conferring upon it moderate antioxidant and antibacterial activity.115 Additional washing with aqueous solutions of ethanol and at higher temperature most likely assured proper removal of any adsorbed material. It was claimed that laccase‐mediated modifications with higher concentrations (>1 mm) did, however, result in substantial adsorption. Further studies revealed that laccase‐mediated modifications with gallates containing shorter alkyl chains were less efficient at conferring antibacterial activity, antioxidant activity and hydrophobicity to the wool.116
By laccase/TEMPO‐mediated oxidation of β‐cyclodextrin, wool could be functionalised with this cyclic heptasugar through imine‐bond formation.83 MALDI‐TOF MS analysis revealed that, in initial tests with tyrosine as a wool model compound, all seven sugar moieties in one molecule of β‐cyclodextrin could be simultaneously functionalised. Wool functionalisation was claimed to be confirmed by an increase in the C−O, O−H and N−H stretching bands in FTIR. Furthermore, a new band at =1502 cm−1 was attributed to the C=N stretching of newly formed imines. However, it is well known that imine formation results in a new band between =1600 and 1700 cm−1.85, 103, 117 Although likely, imine formation after cellulose modification could thus not be unambiguously confirmed.
Mussel‐inspired modification of surfaces is a hot topic because dopamine derivatives provide easily applicable and highly adhesive coatings.119 Oxidation of dopamine furthermore results in melanin polymers that provide resistance against UV light by absorbing it and dissipating it as heat. Jia and co‐workers used laccase to oxidise dopamine and make silk UV resistant.120 Polydopamine coatings were applied in two ways: 1) firstly, by adsorbing it on the silk surface followed by dipping the silk in a solution of laccase, or 2) through in situ laccase‐mediated dopamine polymerisation in the presence of silk. Although method two provided higher colour strength, washing fastness was inferior. Further washing was attempted by treating the samples with DMF at 90 °C. The negligible drop in colour strength for silk modified with either method was ascribed to result from covalent binding of melanin to silk. Interestingly, XPS analysis revealed an increase in surface nitrogen from 3 % for unmodified silk to 19 % for polydopamine‐modified silk. Elemental nitrogen only accounts for approximately 9–10 % of all elements in melanin, any increase above that would thus indicate the presence of other substances, for example, laccase. However, it is more likely that surface nitrogen is more abundant in unmodified silk than initially indicated.121
To conclude: much of the work presented above focused on applying laccase‐mediated strategies to modify wool. However, only limited fundamental knowledge on the grafting mechanism has been gained since the first laccase‐mediated dyeing process was developed by Shin and co‐workers.122 To properly identify whether actual grafting plays a role, the functionalisation mechanisms should be investigated. Questions such as “does Schiff base formation occur?” and “do amino acid thiols perform nucleophilic attack on laccase‐oxidised phenols?” need to be answered. Furthermore, to this point, the scarcity in studies performing proper washing and control experiments hamper further discrimination between grafting and adsorption. Nonetheless, laccase‐mediated functionalisation proves to be, among others, an easily applicable and eco‐friendly way to colour wool fabrics.
5. Laccase‐Mediated Grafting on Synthetic Polymers
Functionalising synthetic polymers form a unique subdomain in the field of laccase‐mediated grafting, since these types of modifications are not based on a common grafting mechanism. To overcome the inherent inertness of polypropylene, Schroeder and co‐workers used plasma‐initiated radical polymerisation in the presence of an amine‐functionalised methacrylate monomer.123 The amine‐functionalised copolymer was subsequently used in laccase‐mediated grafting of guaiacol sulfonic acid. Laccase‐mediated grafting efficiency was directly related to the type of amine‐functionalised methacrylate present on the surface; only primary amines reacted with the oxidised phenolics, whereas tertiary and quaternary amines did not. MS analysis of the laccase‐mediated coupling of guaiacol sulfonic acid and hexylamine (an amine‐functionalised polypropylene model compound) revealed the presence of a signal at m/z 301.7. This signal was attributed to hexylamine coupled to guaiacol sulfonic acid, thereby presenting a basis for the possible grafting of the phenolic monomer on the functionalised polymer.
Díaz Blanco and co‐workers also employed oxygen plasma to hydroxylate flat poly(dimethylsiloxane) (PDMS; Scheme 23).118 The hydroxylated surface was then treated with APTES to generate amine‐functionalised PDMS (PDMS‐NH2). A two‐step laccase‐assisted grafting strategy was thereafter employed to couple gallic acid oligomers to PDMS‐NH2, followed by further modification with SBMA. XPS analysis allowed for the discrimination of several types of chemical species on the surface. In this way, the presence of sp2‐hybridised nitrogen and quaternary nitrogen could be proven with reasonable reliability. Unfortunately, this is one of the few studies regarding laccase‐mediated grafting that harnesses the power of high‐resolution XPS narrow scans; many other grafting studies would greatly benefit from its use. This functionalisation strategy (silanisation followed by laccase‐mediated grafting) was also applied to PDMS‐based urinary catheters (PDMS–SBMA). Initially, PDMS–SBMA and SBMA‐coated catheters that did not undergo plasma treatment (PDMS–SBMAx) exhibited similarly good anti‐fouling properties against synthetic urine. However, fouling was still only minimal for PDMS‐SBMA after seven days of incubation, whereas PDMS–SBMAx was severely fouled in that time frame. Furthermore, the coating proved to only induce a minimal reduction in human foreskin fibroblast viability (≈10 %) compared with cells grown only in medium. Static growth of P. aeruginosa and S. aureus on PDMS–SBMA was reduced by approximately 60 % relative to growth on PDMS. However, in a dynamic setup with an artificial bladder, this effect proved to be less pronounced.
Scheme 23.
Schematic representation of laccase‐mediated N‐(3‐sulfopropyl)‐N‐(methacryloxyethyl)‐N,N‐dimethylammonium betaine (SBMA) functionalisation of PDMS. Based on Díaz Blanco et al.;118 APTES: (3‐aminopropyl)triethoxysilane.
Gonçalves and co‐workers utilised a simpler strategy to functionalise catheters. Their approach involved an alkaline pretreatment of polyurethane and PDMS catheters followed by laccase‐mediated modification with catechin.124 Modification of both materials, either with or without pretreatment, led to coating of the surface, as indicated by an increase in colour strength. Furthermore, pretreated samples exhibited higher colour strength than those that did not undergo pretreatment. This could be rationalised by appreciating that alkaline pretreatment hydrolysed some of the urethane moieties to expose amines that could covalently bind to oxidised catechin. Water washing did not result in a substantial reduction of colour strength; however, washing with a detergent in all cases resulted in a drop in colour strength. This caused the colour strength of samples that were alkaline pretreated not to be any different to those of samples that were not pretreated. Because, without pretreatment, only a limited amount of amines are able to covalently bind catechin (oligomers), this indicates that a substantial part of the colouration is caused by mere deposition of the catechin oligomers. In the case of silicone catheters, a considerable loss of colour was subsequently observed after incubation in synthetic urine. Nonetheless, substantial antimicrobial activity against E. coli was observed in many cases and in some cases against S. epidermidis.
To graft FA from polyamides, Acero et al. employed a two‐step enzymatic cascade.125 Polyamidase was first used to partially hydrolyse the polyamide to expose amines that, in the second step, covalently bound to laccase‐oxidised FA. The use of butyl amine as a model compound in a laccase‐mediated coupling with FA resulted in a 1:1 covalently bound conjugate between FA and the amine, as analysed by MS. This result makes grafting likely, but more in‐depth analysis of the graft is required.
Fouling is not only an issue that concerns medical devices, but it also induces problems in the shipping and water filtration industries, for example.126 Water filtration membranes are made out of inert polymers to reduce chemical deterioration.127 However, their chemical inertness also induces biofouling because inert membranes tend to be hydrophobic, and therefore, induce protein adsorption followed by biofilm formation. Previous work within our group by Nady and co‐workers, involved the modification of inert poly(ethersulfone) (PES) water filtration membranes through laccase‐mediated coating with simple phenolics.128 To improve hydrophilicity of the membrane, polar hydroxybenzoic acids, such as 4‐HBA and gallic acid, were employed. Subsequent colouration of the membranes indicated coating. To test whether, and to what extent, this change was caused by adsorption, the membranes were also incubated with a solution of phenolic oligomer in which laccase was deactivated by using dilute sodium hydroxide. In the case of gallic acid, adsorption accounted for more than 87 % of the colour change. The colouration of membranes caused by pre‐oligomerised 4‐HBA, however, diminished by approximately 35 % compared with colouration induced through in situ laccase‐mediated oligomerisation. It was therefore assumed that covalent grafting from the membrane took place. By employing quantum chemical calculations, it was made probable that the graft consisted of both C−O and C−C coupled oligomers (Scheme 24). Later studies confirmed this C−O and C−C bond formation and it was further observed that the initially formed dimers rapidly oligomerised onwards; thus indicating that the membrane coating was likely to be composed of highly diverse and relatively long oligomers.72
Scheme 24.
Result of laccase‐mediated grafting from PES membranes, as proposed by Nady and co‐workers.128
Initial studies towards the anti‐fouling behaviour of these modified membranes focused on the adsorption of bovine serum albumin (BSA) as a protein model foulant.129 In particular, grafted 4‐HBA was very effective in minimising BSA adsorption, even to levels that were below the detection limit. Importantly, modification with 4‐HBA did not alter the water flux substantially. Studies on spin‐coated PES further revealed a sharp decrease in fouling towards BSA, dextrin (polysaccharide) and tannin (polyphenol), especially at short modification times (<2 h).130 This effect was more pronounced for 4‐HBA than for gallic acid. Finally, repellence of Listeria monocytogenes was tested by using the commonly employed EGD strain and the LR‐991 strain, which is known for its tendency to form biofilms.131 Initial attachment of both strains on surfaces modified with 4‐HBA, FA and gallic acid was all reduced by approximately 60 % compared to attachment on bare PES. Further biofilm formation was equally well reduced in the cases of FA and gallic acid modification. Attachment of both strains on modified surfaces under dynamic flow conditions resulted in an even greater reduction in all cases (70–95 %).
However, recent unpublished results from our group have shone new light on the plausibility of grafting. Laccase‐mediated oligomerisation of 4‐HBA was performed in the presence of insoluble (Scheme 25 A) and soluble (Scheme 25 B) PES model compounds to determine the nature of the covalent bond between PES and the phenolic compound. However, no covalent conjugate could be observed in either case. Additionally, the use of a more reactive phenol also did not result in the observation of phenol/model PES adducts. To mimic the architecture of PES more accurately, a PES model compound was immobilised on a resin, which was subsequently used as a PES mimic in laccase‐mediated oligomerisation, and successively cleaved off from the resin to be analysed by LC/MS and NMR spectroscopy (Scheme 25 C). However, this approach also did not result in any observation of a phenol/model PES conjugate. Finally, to impart additional functionality to PES membranes, laccase was reacted with sulfonated phenolics in the presence of the membrane (Scheme 25 D). Although extensive colouration of the medium and substantial conversion of the starting material were observed, the membrane did not get coloured. The current hypothesis is that the PES coating is based on laccase‐generated phenolic oligomers that adsorb strongly to the PES membrane due to their poor solubility and hydrophobicity. If completely soluble oligomers are generated (like those from sulfonated monomers), these stay in solution and do not adhere.
Scheme 25.
Compounds used in the discrimination between grafting and adsorption on PES membranes. A) Insoluble PES model compound. B) Water‐soluble PES model compound. C) PES model for on‐resin immobilisation with variable spacer length. D) Highly water‐soluble sulfonated phenols: potassium guaiacolsulfonate (left) and Tiron (right).
To summarise: due to the high diversity of synthetic oligomers used in laccase‐mediated functionalisation, it is difficult to draw general conclusions. However, it is clear that modification of synthetic polymers and subsequent analysis can be interpreted in multiple ways. Any discrimination between grafting and adsorption should not be dealt with lightly, and, even if grafting seems likely, one has to conduct suitable control experiments to verify the results. Furthermore, many of the mentioned plastics could not be functionalised directly, but needed pre‐activation. This severely increases the carbon footprint of the functionalisation approach.
6. Summary and Outlook
It has become clear that laccase‐mediated modification of surfaces can benefit many applications. Properties that are altered or conferred by doing so include colour, mechanical strength, antioxidant effects and antibacterial effects. The power of laccase clearly lies in its ability to generate radicals in a mild way, and to produce water as the only by‐product, while possessing a huge substrate scope and high solvent tolerance. If laccases start being produced on a larger scale, they will become cheaper, and therefore, can also be proposed as eco‐friendly alternatives to inorganic or oil‐based catalysts. Furthermore, because many of the materials susceptible to laccase‐mediated modification are now being considered as waste, recycling through functionalisation by laccase is highly attractive.
Grafting, nonetheless, is not the main origin of alteration of the material in all mentioned applications. Often grafting coincides with adsorption; this results in a material that is altered due to both types of modification. Strong, persistent binding without the influence of adsorbents is, however, frequently required in these applications. If the appropriate washing procedure is chosen, simple washing might already be enough to remove any loosely bound molecules. Additionally, one has to consider the influence of reactions in solution: potentially formed oligomers might precipitate; as a result, control experiments in which only monomer is employed will not ensure a fair comparison. Grafting might also result in reversible covalent bonding, as in the case of imine formation. For many applications, irreversibility, and thus, stability is crucial; therefore, one has to tune the grafting strategy to the desired application. Discrimination between grafting and adsorption is not easy, so the analytical tools employed to do so should be chosen wisely. FTIR analysis, for example, is much more valuable for examining grafting on polysaccharides than it is for grafting on lignocelluloses, as in the former functional group transformation (as in the formation of imines) is employed more frequently. Ideally, all modified surfaces are examined with a range of surface‐sensitive techniques, including XPS, FTIR, SWCA, Py‐GC/MS or complementary techniques. Furthermore, the use of ambient MS to study covalently bound molecules on a surface is a promising technique that would certainly be of great use in the characterisation of materials generated through laccase‐mediated grafting.132
Among all materials studied, lignocelluloses currently provide the best platform for grafting. However, this conclusion can only be drawn because this chemistry is well understood. Burton et al. set a benchmark approximately seven years ago by indicating that laccase‐mediated grafting (and coupling) is indeed a promising technique to mildly modify several materials for a plethora of applications.13c This prediction has indeed shown to be true. Given its rapidly increasing potential, it is therefore now time to think again beyond the applications and study its chemistry in much more detail, with many more techniques. This will provide new insights into how to make sure that functionalisation is really covalent, and can persistently yield the desired surface modification.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Sjoerd Slagman is a PhD student at the Laboratory of Organic Chemistry of Wageningen University & Research. His work encompasses the (bio)synthetic modification of polymer surfaces. The biocatalytic use of laccase plays a central role in his research. His main interest is in expanding the role of enzymes in synthetic transformations. He finished his undergraduate education in 2012, during which he was trained as a synthetic organic chemist in the laboratory of van Maarseveen and Hiemstra (University of Amsterdam).

Biographical Information
Han Zuilhof's interests focus on surface‐bound (bio)organic chemistry and bio‐nanotechnology. After completing a PhD in organic chemistry (Leiden University), and postdoctoral work at the University of Rochester, NY, and Columbia University, he joined the faculty at Wageningen University. He has been a Professor of Organic Chemistry since 2007. He is a Distinguished Adjunct Professor of Chemical Engineering at the King Abdulaziz University in Jeddah, Saudi Arabia, and a Perennial Distinguished Guest Professor of Molecular Science and Medicinal Chemistry at the School of Pharmaceutical Science and Technology (SPST) at Tianjin University, P.R. China. He is a founder of Surfix and co‐founder of Biomarque.

Biographical Information
Maurice Franssen's main interests are the application of surface‐bound enzymes and biocatalytic processes in organic chemistry. He obtained his PhD in organic chemistry from Wageningen University. In 1987, he was appointed Assistant Professor at the Laboratory of Organic Chemistry, and he has been an Associate Professor of Enzymes and Surfaces since 1999. In 1988, he won the Royal/Dutch Shell Award and in 1989 he was a visiting scientist at the Laboratory of Prof. A. M. Klibanov (MIT). He is a member of the Permanent Steering Committee of the large European biocatalysis symposium series “Biotrans”.

Acknowledgements
This project was funded by the NanoNextNL project 04A.01 “Biomimetic selective layers”.
S. Slagman, H. Zuilhof, M. C. R. Franssen, ChemBioChem 2018, 19, 288.
References
- 1. Horie K., Baron M., Fox R. B., He J., Hess M., Kahovec J., Kitayama T., Kubisa P., Marechal E., Mormann W., Stepto R. F. T., Tabak D., Vohlidal J., Wilks E. S., Work W. J., Allegra G., Fradet A., Hatada K., Jenkins A. D., Jin J. I., Jones R. G., Kratochvil P., Marcechal E., Meisel I., Metanomski W. V., Moad G., Penczek S., Rebelo L. P., Rinaudo M., Schopov I., Schubert M., Shibaev V. P., Slomkowskj S., Dorfner K., Frechet M. J., Harris W. I., Hodge P., Nishikubo T., Ober C. K., Reichmanis E., Sherrington D. C., Tomoi M., Wohrle D., Pure Appl. Chem. 2004, 76, 889–906. [Google Scholar]
- 2. Liu X., Wu Q., Polymer 2001, 42, 10013–10019. [Google Scholar]
- 3. Zang D., Liu F., Zhang M., Niu X., Gao Z., Wang C., Chem. Eng. J. 2015, 262, 210–216. [Google Scholar]
- 4. Carlmark A., Malmström E. E., Biomacromolecules 2003, 4, 1740–1745. [DOI] [PubMed] [Google Scholar]
- 5. Hüttermann A., Mai C., Kharazipour A., Appl. Microbiol. Biotechnol. 2001, 55, 387–394. [DOI] [PubMed] [Google Scholar]
- 6. Zhao B., Brittain W. J., Prog. Polym. Sci. 2000, 25, 677–710. [Google Scholar]
- 7. Petersen H., Fechner P. M., Martin A. L., Kunath K., Stolnik S., Roberts C. J., Fischer D., Davies M. C., Kissel T., Bioconjugate Chem. 2002, 13, 845–854. [DOI] [PubMed] [Google Scholar]
- 8. Lange S. C., van Andel E., Smulders M. M. J., Zuilhof H., Langmuir 2016, 32, 10199–10205. [DOI] [PubMed] [Google Scholar]
- 9. Neugebauer D., Zhang Y., Pakula T., Sheiko S. S., Matyjaszewski K., Macromolecules 2003, 36, 6746–6755. [Google Scholar]
- 10. Matyjaszewski K., Macromolecules 2012, 45, 4015–4039. [Google Scholar]
- 11.
- 11a. Zoppe J. O., Ataman N. C., Mocny P., Wang J., Moraes J., Klok H.-A., Chem. Rev. 2017, 117, 1105–1318; [DOI] [PubMed] [Google Scholar]
- 11b. Chen W.-L., Cordero R., Tran H., Ober C. K., Macromolecules 2017, 50, 4089–4113; [Google Scholar]
- 11c. Niu J., Lunn D. J., Pusuluri A., Yoo J. I., O'Malley M. A., Mitragotri S., Soh H. T., Hawker C. J., Nat. Chem. 2017, 9, 537–545. [DOI] [PubMed] [Google Scholar]
- 12. Zhong P. S., Widjojo N., Chung T.-S., Weber M., Maletzko C., J. Membr. Sci. 2012, 417–418, 52–60. [Google Scholar]
- 13.
- 13a. Nyanhongo G. S., Kudanga T., Prasetyo E. Nugroho, Guebitz G. M. in Biofunctionalization of Polymers and Their Applications (Eds.: G. S. Nyanhongo, W. Steiner, G. Guebitz), Springer, Berlin, 2011, pp. 47–68; [Google Scholar]
- 13b. Mikolasch A., Schauer F., Appl. Microbiol. Biotechnol. 2009, 82, 605–624; [DOI] [PubMed] [Google Scholar]
- 13c. Kudanga T., Nyanhongo G. S., Guebitz G. M., Burton S., Enzyme Microb. Technol. 2011, 48, 195–208; [DOI] [PubMed] [Google Scholar]
- 13d. Guebitz G. M., Cavaco-Paulo A., Trends Biotechnol. 2008, 26, 32–38. [DOI] [PubMed] [Google Scholar]
- 14. Madhavi V., Lele S. S., Bioresources 2009, 4, 1694–1717. [Google Scholar]
- 15. Yoshida H., J. Chem. Soc. Trans. 1883, 43, 472–486. [Google Scholar]
- 16. Kobayashi S., Uyama H., Ikeda R., Chem. Eur. J. 2001, 7, 4755–4760. [DOI] [PubMed] [Google Scholar]
- 17. Coy M. R., Salem T. Z., Denton J. S., Kovaleva E. S., Liu Z., Barber D. S., Campbell J. H., Davis D. C., Buchman G. W., Boucias D. G., Scharf M. E., Insect Biochem. Mol. Biol. 2010, 40, 723–732. [DOI] [PubMed] [Google Scholar]
- 18. Santhanam N., Vivanco J. M., Decker S. R., Reardon K. F., Trends Biotechnol. 2011, 29, 480–489. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Rivera-Hoyos C. M., Morales-Álvarez E. D., Poutou-Piñales R. A., Pedroza-Rodríguez A. M., Rodríguez-Vázquez R., Delgado-Boada J. M., Fungal Biol. Rev. 2013, 27, 67–82; [Google Scholar]
- 19b. Baldrian P., FEMS Microbiol. Rev. 2006, 30, 215–242; [DOI] [PubMed] [Google Scholar]
- 19c. Kumar S. V. S., Phale P. S., Durani S., Wangikar P. P., Biotechnol. Bioeng. 2003, 83, 386–394. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Witayakran S., Ragauskas A. J., Adv. Synth. Catal. 2009, 351, 1187–1209; [Google Scholar]
- 20b. Jeon J. R., Baldrian P., Murugesan K., Chang Y. S., J. Microb. Biotechnol. 2012, 5, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mogharabi M., Faramarzi M. A., Adv. Synth. Catal. 2014, 356, 897–927. [Google Scholar]
- 22. Camarero S., Ibarra D., Martínez Á. T., Romero J., Gutiérrez A., del Río J. C., Enzyme Microb. Technol. 2007, 40, 1264–1271. [Google Scholar]
- 23.
- 23a. Sarma R., Islam M. S., Miller A.-F., Bhattacharyya D., ACS Appl. Mater. Interfaces 2017, 9, 14858–14867; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Zhang X., Hua M., Lv L., Pan B., Sci. Rep. 2015, 5, 8253; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23c. Lloret L., Eibes G., Lú-Chau T. A., Moreira M. T., Feijoo G., Lema J. M., Biochem. Eng. J. 2010, 51, 124–131. [Google Scholar]
- 24. Polak J., Jarosz-Wilkolazka A., Process Biochem. 2012, 47, 1295–1307. [Google Scholar]
- 25. Nakajima N., Ikada Y., Bioconjugate Chem. 1995, 6, 123–130. [DOI] [PubMed] [Google Scholar]
- 26. Dong A., Fan X., Wang Q., Yu Y., Cavaco-Paulo A., Int. J. Biol. Macromol. 2015, 79, 353–362. [DOI] [PubMed] [Google Scholar]
- 27. Vassilev S. V., Baxter D., Andersen L. K., Vassileva C. G., Morgan T. J., Fuel 2012, 94, 1–33. [Google Scholar]
- 28. Lecoanet H. F., Bottero J.-Y., Wiesner M. R., Environ. Sci. Technol. 2004, 38, 5164–5169. [DOI] [PubMed] [Google Scholar]
- 29. Munk L., Sitarz A. K., Kalyani D. C., Mikkelsen J. D., Meyer A. S., Biotechnol. Adv. 2015, 33, 13–24. [DOI] [PubMed] [Google Scholar]
- 30. Banerjee I., Pangule R. C., Kane R. S., Adv. Mater. 2011, 23, 690–718. [DOI] [PubMed] [Google Scholar]
- 31. Kaegi R., Sinnet B., Zuleeg S., Hagendorfer H., Mueller E., Vonbank R., Boller M., Burkhardt M., Environ. Pollut. 2010, 158, 2900–2905. [DOI] [PubMed] [Google Scholar]
- 32. Pérez J., Muñoz-Dorado J., de la Rubia T., Martínez J., Int. Microbiol. 2002, 5, 53–63. [DOI] [PubMed] [Google Scholar]
- 33. Cadena E. M., Du X., Gellerstedt G., Li J., Fillat A., García-Ubasart J., Vidal T., Colom J. F., Bioresour. Technol. 2011, 102, 3911–3917. [DOI] [PubMed] [Google Scholar]
- 34. Camarero S., García O., Vidal T., Colom J., del Río J. C., Gutiérrez A., Gras J. M., Monje R., Martínez M. a. J., Martínez Á. T., Enzyme Microb. Technol. 2004, 35, 113–120. [Google Scholar]
- 35. Fillat Ú., Ibarra D., Eugenio M., Moreno A., Tomás-Pejó E., Martín-Sampedro R., Fermentation 2017, 3, 17. [Google Scholar]
- 36. Kudanga T., Prasetyo E. N., Sipilä J., Nousiainen P., Widsten P., Kandelbauer A., Nyanhongo G. S., Guebitz G., Eng. Life Sci. 2008, 8, 297–302. [Google Scholar]
- 37. Kudanga T., Prasetyo E. N., Sipilä J., Nyanhongo G. S., Guebitz G. M., Enzyme Microb. Technol. 2010, 46, 272–280. [Google Scholar]
- 38. Kudanga T., Prasetyo E. N., Widsten P., Kandelbauer A., Jury S., Heathcote C., Sipilä J., Weber H., Nyanhongo G. S., Guebitz G. M., Bioresour. Technol. 2010, 101, 2793–2799. [DOI] [PubMed] [Google Scholar]
- 39. Kudanga T., Nugroho Prasetyo E., Sipilä J., Eberl A., Nyanhongo G. S., Guebitz G. M., J. Mol. Catal. B 2009, 61, 143–149. [Google Scholar]
- 40. Manda K., Gördes D., Mikolasch A., Hammer E., Schmidt E., Thurow K., Schauer F., Appl. Microbiol. Biotechnol. 2007, 76, 407–416. [DOI] [PubMed] [Google Scholar]
- 41. Kudanga T., Prasetyo E. N., Sipilä J., Guebitz G. M., Nyanhongo G. S., J. Biotechnol. 2010, 149, 81–87. [DOI] [PubMed] [Google Scholar]
- 42. Schoemaker H. E., Harvey P. J., Bowen R. M., Palmer J. M., FEBS Lett. 1985, 183, 7–12. [Google Scholar]
- 43. Aracri E., Fillat A., Colom J. F., Gutiérrez A., del Río J. C., Martínez Á. T., Vidal T., Bioresour. Technol. 2010, 101, 8211–8216. [DOI] [PubMed] [Google Scholar]
- 44. Rencoret J., Aracri E., Gutiérrez A., del Río J. C., Torres A. L., Vidal T., Martínez A. T., Biochem. Eng. J. 2014, 86, 16–23. [Google Scholar]
- 45. Russell W. R., Forrester A. R., Chesson A., Burkitt M. J., Arch. Biochem. Biophys. 1996, 332, 357–366. [DOI] [PubMed] [Google Scholar]
- 46. Zhao M., Li J., Mano E., Song Z., Tschaen D. M., Grabowski E. J. J., Reider P. J., J. Org. Chem. 1999, 64, 2564–2566. [Google Scholar]
- 47. Munk L., Punt A. M., Kabel M. A., Meyer A. S., RSC Adv. 2017, 7, 3358–3368. [Google Scholar]
- 48. Capanema E. A., Balakshin M. Y., Kadla J. F., J. Agric. Food Chem. 2004, 52, 1850–1860. [DOI] [PubMed] [Google Scholar]
- 49. Kim S., López C., Güebitz G., Cavaco-Paulo A., Eng. Life Sci. 2008, 8, 324–330. [Google Scholar]
- 50. Forde J., Tully E., Vakurov A., Gibson T. D., Millner P., Ó'Fágáin C., Enzyme Microb. Technol. 2010, 46, 430–437. [DOI] [PubMed] [Google Scholar]
- 51. Elegir G., Kindl A., Sadocco P., Orlandi M., Enzyme Microb. Technol. 2008, 43, 84–92. [Google Scholar]
- 52. Widsten P., Heathcote C., Kandelbauer A., Guebitz G., Nyanhongo G. S., Prasetyo E. N., Kudanga T., Process Biochem. 2010, 45, 1072–1081. [Google Scholar]
- 53. Schroeder M., Aichernig N., Guebitz G. M., Kokol V., Biotechnol. J. 2007, 2, 334–341. [DOI] [PubMed] [Google Scholar]
- 54. Silva C., Matamá T., Kim S., Padrão J., Nugroho Prasetyo E., Kudanga T., Nyanhongo G. S., Guebitz G. M., Casal M., Cavaco-Paulo A., React. Funct. Polym. 2011, 71, 713–720. [Google Scholar]
- 55. Fillat A., Gallardo O., Vidal T., Pastor F. I. J., Díaz P., Roncero M. B., Carbohydr. Polym. 2012, 87, 146–152. [DOI] [PubMed] [Google Scholar]
- 56. Schubert M., Engel J., Thöny-Meyer L., Schwarze F. W. M. R., Ihssen J., Appl. Environ. Microbiol. 2012, 78, 7267–7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Francesko A., Blandón L., Vázquez M., Petkova P., Morató J., Pfeifer A., Heinze T., Mendoza E., Tzanov T., ACS Appl. Mater. Interfaces 2015, 7, 9792–9799. [DOI] [PubMed] [Google Scholar]
- 58. Silva S. P., Sabino M. A., Fernandes E. M., Correlo V. M., Boesel L. F., Reis R. L., Int. Mater. Rev. 2005, 50, 345–365. [Google Scholar]
- 59. Garcia Peña L. V., Petkova P., Margalef-Marti R., Vives M., Aguilar L., Gallegos A., Francesko A., Perelshtein I., Gedanken A., Mendoza E., Casas-Zapata J. C., Morató J., Tzanov T., Ind. Eng. Chem. Res. 2017, 56, 3599–3606. [Google Scholar]
- 60. Dong A., Yu Y., Yuan J., Wang Q., Fan X., Appl. Surf. Sci. 2014, 301, 418–427. [Google Scholar]
- 61. Ni X., Dong A., Fan X., Wang Q., Yu Y., Cavaco-Paulo A., Fibers Polym. 2015, 16, 2276–2283. [Google Scholar]
- 62. Fernández-Fernández M., Sanromán M. Á., Moldes D., J. Wood Chem. Technol. 2015, 35, 156–165. [Google Scholar]
- 63. Garcia-Ubasart J., Esteban A., Vila C., Roncero M. B., Colom J. F., Vidal T., Bioresour. Technol. 2011, 102, 2799–2803. [DOI] [PubMed] [Google Scholar]
- 64. Garcia-Ubasart J., Colom J. F., Vila C., Hernández N. G., Blanca Roncero M., Vidal T., Bioresour. Technol. 2012, 112, 341–344. [DOI] [PubMed] [Google Scholar]
- 65. Reynaud C., Tapin-Lingua S., Elegir G., Petit-Conil M., Baumberger S., J. Biotechnol. 2013, 167, 302–308. [DOI] [PubMed] [Google Scholar]
- 66. Witayakran S., Ragauskas A. J., Enzyme Microb. Technol. 2009, 44, 176–181. [Google Scholar]
- 67. Dong A., Yuan J., Wang Q., Fan X., J. Appl. Polym. Sci. 2014, 131, 40387. [Google Scholar]
- 68. Mai C., Majcherczyk A., Hüttermann A., Enzyme Microb. Technol. 2000, 27, 167–175. [DOI] [PubMed] [Google Scholar]
- 69. Gao G., Karaaslan M. A., Kadla J. F., Ko F., Green Chem. 2014, 16, 3890–3898. [Google Scholar]
- 70. Schubert M., Ruedin P., Civardi C., Richter M., Hach A., Christen H., PLoS One 2015, 10, e0128623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Euring M., Rühl M., Ritter N., Kües U., Kharazipour A., Biotechnol. J. 2011, 6, 1253–1261. [DOI] [PubMed] [Google Scholar]
- 72. Slagman S., Escorihuela J., Zuilhof H., Franssen M. C. R., RSC Adv. 2016, 6, 99367–99375. [Google Scholar]
- 73. Aracri E., Díaz Blanco C., Tzanov T., Green Chem. 2014, 16, 2597–2603. [Google Scholar]
- 74. Love G. D., Snape C. E., Jarvis M. C., Morrison I. M., Phytochemistry 1994, 35, 489–491. [Google Scholar]
- 75. Viikari L., Kruus K., Buchert J. (Valtion Teknillinen Tutkimuskeskus), WO 023117 A1, 1999.
- 76. Tromp S. A., Matijošytė I., Sheldon R. A., Arends I. W. C. E., Mul G., Kreutzer M. T., Moulijn J. A., de Vries S., ChemCatChem 2010, 2, 827–833. [Google Scholar]
- 77. Jiang J., Ye W., Liu L., Wang Z., Fan Y., Saito T., Isogai A., Biomacromolecules 2017, 18, 288–294. [DOI] [PubMed] [Google Scholar]
- 78. Xu S., Song Z., Qian X., Shen J., Cellulose 2013, 20, 2371–2378. [Google Scholar]
- 79. Aracri E., Vidal T., Cellulose 2012, 19, 867–877. [Google Scholar]
- 80. Patel I., Ludwig R., Haltrich D., Rosenau T., Potthast A., Holzforschung 2011, 65, 475–481. [Google Scholar]
- 81. Aracri E., Valls C., Vidal T., Carbohydr. Polym. 2012, 88, 830–837. [Google Scholar]
- 82. Jaušovec D., Vogrinčič R., Kokol V., Carbohydr. Polym. 2015, 116, 74–85. [DOI] [PubMed] [Google Scholar]
- 83. Yu Y., Wang Q., Yuan J., Fan X., Wang P., Carbohydr. Polym. 2016, 153, 463–470. [DOI] [PubMed] [Google Scholar]
- 84. Aracri E., Vidal T., Ragauskas A. J., Carbohydr. Polym. 2011, 84, 1384–1390. [Google Scholar]
- 85. Yu Y., Wang Q., Yuan J., Fan X., Wang P., Cui L., Carbohydr. Polym. 2016, 137, 549–555. [DOI] [PubMed] [Google Scholar]
- 86. Guimarães C., Kim S., Silva C., Cavaco-Paulo A., Biotechnol. J. 2011, 6, 1272–1279. [DOI] [PubMed] [Google Scholar]
- 87. Díaz Blanco C., González M. D., Monmany J. M. D., Tzanov T., Enzyme Microb. Technol. 2009, 44, 380–385. [Google Scholar]
- 88. Liu J., Pelton R., Obermeyer J. M., Esser A., Biomacromolecules 2013, 14, 2953–2960. [DOI] [PubMed] [Google Scholar]
- 89. Shi S., Pelton R., Fu Q., Yang S., Ind. Eng. Chem. Res. 2014, 53, 4748–4754. [Google Scholar]
- 90. Garcia-Ubasart J., Vidal T., Torres A. L., Rojas O. J., Biomacromolecules 2013, 14, 1637–1644. [DOI] [PubMed] [Google Scholar]
- 91. Zhang Y., Dong A., Wang Q., Fan X., Cavaco-Paulo A., Zhang Y., Appl. Biochem. Biotechnol. 2014, 174, 820–831. [DOI] [PubMed] [Google Scholar]
- 92. Zhang Y., Fan X., Wang Q., Cavaco-Paulo A., RSC Adv. 2016, 6, 49272–49280. [Google Scholar]
- 93. Iqbal H. M. N., Kyazze G., Tron T., Keshavarz T., Macromol. Mater. Eng. 2015, 300, 712–720. [Google Scholar]
- 94. Iqbal H. M. N., Kyazze G., Locke I. C., Tron T., Keshavarz T., Green Chem. 2015, 17, 3858–3869. [Google Scholar]
- 95. Iqbal H. M. N., Kyazze G., Tron T., Keshavarz T., Cellulose 2014, 21, 3613–3621. [Google Scholar]
- 96. Iqbal H. M. N., Kyazze G., Locke I. C., Tron T., Keshavarz T., Int. J. Biol. Macromol. 2015, 81, 552–559. [DOI] [PubMed] [Google Scholar]
- 97.
- 97a. Iqbal H. M. N., Kyazze G., Locke I. C., Tron T., Keshavarz T., Carbohydr. Polym. 2015, 131, 197–207; [DOI] [PubMed] [Google Scholar]
- 97b. Iqbal H. M. N., Kyazze G., Locke I. C., Tron T., Keshavarz T., eXPRESS Polym. Lett. 2015, 9, 764–772. [Google Scholar]
- 98. Iqbal H. M. N., Kyazze G., Tron T., Keshavarz T., Polym. Chem. 2014, 5, 7004–7012. [Google Scholar]
- 99. Rinaudo M., Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar]
- 100. Shepherd R., Reader S., Falshaw A., Glycoconjugate J. 1997, 14, 535–542. [DOI] [PubMed] [Google Scholar]
- 101. Božič M., Gorgieva S., Kokol V., Carbohydr. Polym. 2012, 87, 2388–2398. [DOI] [PubMed] [Google Scholar]
- 102. Božič M., Gorgieva S., Kokol V., Carbohydr. Polym. 2012, 89, 854–864. [DOI] [PubMed] [Google Scholar]
- 103. Aljawish A., Chevalot I., Piffaut B., Rondeau-Mouro C., Girardin M., Jasniewski J., Scher J., Muniglia L., Carbohydr. Polym. 2012, 87, 537–544. [DOI] [PubMed] [Google Scholar]
- 104. Yuan M., Tanabe I., Bernard-Schaaf J.-M., Shi Q.-Y., Schlegel V., Schurhammer R., Dowben P. A., Doudin B., Routaboul L., Braunstein P., New J. Chem. 2016, 40, 5782–5796. [Google Scholar]
- 105. Aljawish A., Muniglia L., Klouj A., Jasniewski J., Scher J., Desobry S., Food Hydrocolloids 2016, 60, 551–558. [Google Scholar]
- 106.
- 106a. Aljawish A., Muniglia L., Chevalot I., Biotechnol. Prog. 2016, 32, 491–500; [DOI] [PubMed] [Google Scholar]
- 106b. Aljawish A., Muniglia L., Jasniewski J., Klouj A., Scher J., Chevalot I., Process Biochem. 2014, 49, 863–871. [Google Scholar]
- 107. Yang C., Zhou Y., Zheng Y., Li C., Sheng S., Wang J., Wu F., Int. J. Biol. Macromol. 2016, 87, 577–585. [DOI] [PubMed] [Google Scholar]
- 108. Asquith R. S., Chemistry of Natural Protein Fibers, Plenum Press, New York, 1977. [Google Scholar]
- 109. Moldovan Z., Jover E., Bayona J. M., Anal. Chim. Acta 2002, 465, 359–378. [Google Scholar]
- 110. Jeon J.-R., Kim E.-J., Murugesan K., Park H.-K., Kim Y.-M., Kwon J.-H., Kim W.-G., Lee J.-Y., Chang Y.-S., Microb. Biotechnol. 2010, 3, 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.M. Yuan, Q. Wang, J. Shen, E. Smith, R. Bai, X. Fan, Text. Res. J DOI: https://dx.doi.org/10.1177/0040517517712096.
- 112.T. Zhang, R. Bai, J. Shen, Q. Wang, P. Wang, J. Yuan, X. Fan, Text. Res. J, DOI: https://dx.doi.org/10.1177/0040517517720497.
- 113. Zhang T., Bai R., Wang Q., Fan X., Wang P., Yuan J., Yu Y., Color. Technol. 2017, 133, 65–72. [Google Scholar]
- 114. Hossain K. M. G., González M. D., Juan A. R., Tzanov T., Enzyme Microb. Technol. 2010, 46, 326–330. [Google Scholar]
- 115. Hossain K. M. G., González M. D., Lozano G. R., Tzanov T., J. Biotechnol. 2009, 141, 58–63. [DOI] [PubMed] [Google Scholar]
- 116. Gaffar Hossain K. M., Díaz González M., Monmany J. M. D., Tzanov T., J. Mol. Catal. B: Enzym. 2010, 67, 231–235. [Google Scholar]
- 117. Ledbetter J. W., J. Phys. Chem. 1977, 81, 54–59. [Google Scholar]
- 118. Diaz Blanco C., Ortner A., Dimitrov R., Navarro A., Mendoza E., Tzanov T., ACS Appl. Mater. Interfaces 2014, 6, 11385–11393. [DOI] [PubMed] [Google Scholar]
- 119. Lee H., Dellatore S. M., Miller W. M., Messersmith P. B., Science 2007, 318, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jia W., Wang Q., Fan X., Dong A., Yu Y., Wang P., RSC Adv. 2017, 7, 12977–12983. [Google Scholar]
- 121. Shao J., Liu J., Zheng J., Carr C. M., Polym. Int. 2002, 51, 1479–1483. [Google Scholar]
- 122. Shin H., Guebitz G., Cavaco-Paulo A., Macromol. Mater. Eng. 2001, 286, 691–694. [Google Scholar]
- 123. Schroeder M., Fatarella E., Kovač J., Guebitz G. M., Kokol V., Biomacromolecules 2008, 9, 2735–2741. [DOI] [PubMed] [Google Scholar]
- 124. Gonçalves I., Matamá T., Cavaco-Paulo A., Silva C., Biocatal. Biotransform. 2014, 32, 2–12. [Google Scholar]
- 125. Acero E. H., Ribitsch D., Rodriguez R. D., Dellacher A., Zitzenbacher S., Marold A., Greimel K. J., Schroeder M., Kandelbauer A., Heumann S., Nyanhongo G. S., Schwab H., Guebitz G. M., J. Mol. Catal. B: Enzym. 2012, 79, 54–60. [Google Scholar]
- 126. Flemming H.-C., Appl. Microbiol. Biotechnol. 2002, 59, 629–640. [DOI] [PubMed] [Google Scholar]
- 127. Chang I.-S., Clech P. L., Jefferson B., Judd S., J. Environ. Eng. 2002, 128, 1018–1029. [Google Scholar]
- 128. Nady N., Schroën K., Franssen M. C. R., van Lagen B., Murali S., Boom R. M., Mohyeldin M. S., Zuilhof H., ACS Appl. Mater. Interfaces 2011, 3, 801–810. [DOI] [PubMed] [Google Scholar]
- 129. Nady N., Schroën K., Franssen M. C. R., Mohyeldin M. S., Zuilhof H., Boom R. M., J. Membr. Sci. 2012, 394, 69–79. [Google Scholar]
- 130. Nady N., Schroën K., Franssen M. C. R., Fokkink R., Mohyeldin M. S., Zuilhof H., Boom R. M., J. Colloid Interface Sci. 2012, 378, 191–200. [DOI] [PubMed] [Google Scholar]
- 131. van der Veen S., Nady N., Franssen M. C. R., Zuilhof H., Boom R. M., Abee T., Schroën K., J. Appl. Polym. Sci. 2015, 132, 41576. [Google Scholar]
- 132.
- 132a. Manova R. K., Joshi S., Debrassi A., Bhairamadgi N. S., Roeven E., Gagnon J., Tahir M. N., Claassen F. W., Scheres L. M. W., Wennekes T., Schroën K., van Beek T. A., Zuilhof H., Nielen M. W. F., Anal. Chem. 2014, 86, 2403–2411; [DOI] [PubMed] [Google Scholar]
- 132b. Sen R., Escorihuela J., Smulders M. M. J., Zuilhof H., Langmuir 2016, 32, 3412–3419. [DOI] [PubMed] [Google Scholar]