

Abstract
Background
Due to its great sensitivity, the nucleic acid amplification test (NAAT) is widely used for detection of respiratory viruses (RV). However, few reports have described a direct comparison between multiplex RT‐PCR assays for RV. The objective of this study was to perform a direct comparison of three multiplex RT‐PCR assays for the detection of respiratory viruses.
Methods
A total of 201 respiratory samples (161 nasopharyngeal swab samples and 40 sputum samples) were tested with three commercial RV assays: Seegene Anyplex II RV16 (AP), LG AdvanSure RV (AD), and Biosewoom Real‐Q RV (RQ). The additional tests for the discrepant results were conducted by repeat RV assay or monoplex PCR coupled direct sequencing. Data analysis using percent agreement, kappa, and prevalence‐adjusted and bias‐adjusted kappa (PABAK) values was performed for comparisons among the three RV assays.
Results
Of the 201 samples, AP, AD, and RQ detected 105 (52.2%), 99 (49.3%), and 95 (47.3%) positive cases respectively. The overall agreement, kappa, and PABAK values for the three assays ranged between 97%‐98%, 0.76‐0.86, and 0.93‐0.96 respectively. The performance of the three assays was very similar, with 94%‐100% agreement for all comparisons, each virus types. The additional testing of samples showed discrepant results demonstrating that AD assay had the highest rate of concordance with original results.
Conclusions
We suggest that all multiplex assay would be suitable for the detection of for respiratory viruses in clinical setting.
Keywords: agreement, nucleic acid amplification test, respiratory virus
1. Introduction
Acute respiratory infections (ARI) are one of the major causes of mortality worldwide and approximately half of ARI are caused by respiratory viruses (RV).1 There are several types of tests for detection of RV. Among them, the nucleic acid amplification test (NAAT) has widely been accepted in recent years.2 NAAT has advantages in comparison to the classical methods of viral culture and direct fluorescent antibody tests. First, NAAT has superior sensitivity identifying RV cases not detected by classical methods. Second, RV testing results are available faster than those of viral cultures. Speed to result allows medical professionals to prescribe antiviral agents and perform appropriate infection control more rapidly. Lastly, NAAT does not need a strict transportation protocol for maintaining viability of RV.3 In addition, the application of multiplexing technology in NAAT allows better detection than classical methods of a broad range of viruses. For all of these reasons NAAT has replaced classical methods.
There are a number of commercial multiplex kits that can detect between 12 and 23 virus types.3, 4 Performance evaluation studies for newly developed multiplex RV kits are weak, do not establish the reference standard method and therefore do not sufficiently calculate sensitivity and specificity of each test. Instead, some studies have suggested the reference test with in‐house multiplex real‐time PCR or commercial duplex PCR tests.5, 6, 7, 8 We performed a direct comparison of three commercial multiplex assays and produced the values of the agreement and kappa instead of sensitivity and specificity with the reference tests. The objective of this study was to perform a direct comparison of three multiplex RT‐PCR assays for the detection of respiratory viruses.
2. Materials and Methods
2.1. Clinical samples
Respiratory samples used in this study were collected between December 2015 and March 2016 at Armed Forces Medical Research Institute and Armed Forces Daejeon hospital (Daejeon, Republic of Korea) from those submitted for the detection of respiratory viruses. Clinical samples from 201 young male soldiers (age range 18‐27 years, median 21 years) with acute respiratory illness were randomly selected for study without knowledge of previous results from Real‐Q RV or Anyplex II RV16 tests. They consisted of nasopharyngeal (NP) swab samples (n=161) and sputum samples (n=40) and were stored at −70°C until this study was conducted.
Nasopharyngeal swab samples were obtained using flocked swabs and transported in 3 mL universal transport medium (COPAN Diagnostics, Murrieta, CA, USA). Sputum samples were received in sterile plastic containers and treated in order to homogenize samples using a 1:1 ratio dithiothreitol, which was diluted 1:100 with distilled water because of it viscosity. This study was approved by the Ethics Committee of Armed Forces Medical Command (AFMC‐16‐IRB‐023).
2.2. Nucleic acid extraction
The nucleic acid extraction system was used for each RV assay. For Anyplex II RV 16 (AP), nucleic acids were extracted from 500 μL of clinical samples using a MICROLAB Nimbus IVD workstation (Hamilton, Reno, NV, USA) with a STARMag 96 Virus Kit (Seegene, Seoul, Republic of Korea) and eluted into 80 μL of elution buffer. For AdvanSure RV real‐time PCR (AD), nucleic acids were extracted from 200 μL of clinical samples using the TANBead Smart LabAssist‐32 extraction system with a TANBead Viral Auto Plate kit (Taiwan Advanced Nanotech Inc., Taoyuan City, Taiwan) and eluted into 80 μL of elution buffer. For Real‐Q RV Detection assay (RQ), nucleic acids were extracted from 200 μL of clinical samples using a Maxwell™ 16 device with the Maxwell 16 Viral Total Nucleic Acid Purification Kit (Promega, Madison, WI, USA) and eluted into 80 μL of elution buffer.
2.3. Multiplex RT‐PCR assays
Three commercial multiplex RV assays were performed based on manufacturers’ protocols. Characteristics of the three multiplex assays are briefly reported in Table 1. The AP assay is composed of two‐step RT‐PCR. Before multiplex target PCR was performed, complementary DNA (cDNA) was synthesized from each sample's RNA through reverse transcriptase (RT) reaction with the cDNA Synthesis kit (Seegene). The mixture for CDNA synthesis is a 20 μL final volume, including 8 μL of the sample's nucleic acid, 8 μL of RT buffer, 2 μL of the RT enzyme Mix and 2 μL of random Hexamer with Veriti Thermal Cycler (Applied Biosystems, Foster City, CA, USA) under the following conditions: 25°C for 5 minutes, 37°C for 60 minutes and 95°C for 2 minutes. After cDNA synthesis, the 16 target viruses were multiplexed into two tubes according to target primer and probe. Multiplex real‐time PCR was conducted in a 20 μL mixture containing 8 μL of cDNA, 5 μL of 4X TOCE Oligo Mix primer, 5 μL of 4X PCR master mix, and 2 μL of RNase‐free water with the CFX96 Real‐time PCR detection system (Bio‐rad, Hercules, CA, USA). The thermal cycling conditions were: 50°C for 4 minutes; denaturation at 95°C for 15 minutes followed by 50 cycles of 95°C for 30 seconds, 60°C for 1 minutes and 72°C for 30 seconds. During the interval of the 50 cycles, melting temperature analysis was conducted by cooling the mixture to 55°C and maintaining for 15 seconds followed by heating the mixture from 55°C to 85°C (0.5°C/5 seconds) after 30, 40, and 50 cycles respectively. The fluorescence was continually detected during heating process and the data were converted to derive the melting temperature by plotting the negative derivative of fluorescence vs temperature (−dF/dT against T). Seegene viewer software can automatically interpret melting peaks derived and present the results as ‘+, ++, and +++ positive’ according to detection cycles of melting temperature analysis or ‘negative.’ Bacteriophage MS2 as an Internal Control (IC) was added to clinical specimens before nucleic acid extraction and was incorporated into the product as an exogenous whole process control in order to monitor during the period of time between nucleic acid isolation to result interpretation. The IC was co‐amplified with the target nucleic acids within the clinical specimens.
Table 1.
Summary of characteristics in multiplex assays for detection of respiratory viruses
| Assay | AP | AD | RQ |
|---|---|---|---|
| Target virus | Influenza virus A | Influenza virus A | Influenza virus A |
| Influenza virus B | Influenza virus B | Influenza virus B | |
| Respiratory syncytial virus A | Respiratory syncytial virus A | Respiratory syncytial virus A | |
| Respiratory syncytial virus B | Respiratory syncytial virus B | Respiratory syncytial virus B | |
| Parainfluenza virus 1 | Parainfluenza virus 1 | Parainfluenza virus 1 | |
| Parainfluenza virus 2 | Parainfluenza virus 2 | Parainfluenza virus 2 | |
| Parainfluenza virus 3 | Parainfluenza virus 3 | Parainfluenza virus 3 | |
| Parainfluenza virus 4 | — | — | |
| Coronavirus 229E | Corona Virus 229E | Corona Virus 229E | |
| Coronavirus OC43 | Corona Virus OC43 | Corona Virus OC43 | |
| Coronavirus NL63 | Corona Virus NL63 | Corona Virus NL63/HKU1 | |
| Bocavirus 1/2/3/4 | Bocavirus | Bocavirus | |
| Adenovirus | Adenovirus | Adenovirus | |
| Rhinovirus A/B/C | Rhinovirus A/B/C | Rhinovirus | |
| Enterovirus | — | — | |
| Metapneumovirus | Metapneumovirus | Metapneumovirus | |
| Technology | Melting curve analysis & Real‐time RT PCR | Real‐time RT PCR | Real‐time RT PCR |
| Tagging Oligonucleotide Cleavage and Extension (TOCE) | Taqman probe chemistry | Taqman probe chemistry | |
| Samples per run | 40 | 16 | 16 |
| Automated result presentation | Yes | Yes | Yes |
| Method step | 3 | 2 | 2 |
| Turn‐around time (h)a | 4.5 | 2 | 2 |
AP, Anyplex II RV16; AD, AdvanSure RV; RQ: Real‐Q RV.
Turn‐around time excludes nucleic acid extraction time.
The AD assay performs both reverse‐transcription(RT) reaction with the extracted RNA from specimen and multiplex PCR reaction simultaneously in a single tube (1‐step RT‐PCR). It uses human RNase P gene, as an internal control, to give information for validity of the RNA extraction procedure and to prevent misjudgment from sampling error and RT‐PCR reaction error. Briefly, the assay was multiplexed into five tubes targeting 14 RVs and an IC gene. This was conducted in a 20 μL mixture containing 5 μL of nucleic acid, 5 μL of primer/probe mixture and 10 μL of RT‐PCR premix with the SLAN Real‐time PCR detection system (Shanghai Hongshi Medical Technology Co., Shanghai, China). The thermal cycling conditions for the RT step were: 50°C for 10 minutes and 95°C for 30 seconds followed by 40 cycles of 95°C for 15 seconds; 53°C for 30 seconds and 60°C for 30 seconds.
The RQ assay is also a one‐step RT‐ PCR method. Briefly, the assay was multiplexed into five tubes targeting 14 RVs and 1 IC gene, which was human RNase P, like in the AD assay. RQ was conducted in a 22 μL mixture containing 5 μL of nucleic acid, 12.5 μL of 2X PCR reaction mixture, 1 μL of RT‐PCR enzyme, and 3.5 μL of water with CFX96 Real‐time PCR detection system (Bio‐rad). The RT‐PCR reaction was performed with the following conditions for the RT step: 50°C for 30 minutes; 95°C for 15 minutes, followed by 45 cycles of 95°C for 15 seconds and 62°C for 45 seconds.
2.4. Discrepant result analysis
A flow diagram of discrepant analysis is described in Figure 1. In summary, 167 samples (83.1%) with positive results from at least two RV assays or negative results from all three assays were not tested with the additional assay. However, the other 34 samples (16.9%), containing 36 discordant results showing a single positive result in any assay, were tested with the following additional assay. A single positive result from AP and RQ assay was retested individually with RV assay. Whereas a single positive result from AD assay was performed with monoplex PCR followed by direct sequencing. If the results between the original and additional tests were concordant, they were considered to be a consensus of positive results. Conversely, if the results between original and additional tests showed a discordance, they were considered to be a negative consensus. Because both AD and RQ assay cannot detect PIV4 and EV, the results about PIV4 and EV were excluded from the discrepant analysis.
Figure 1.
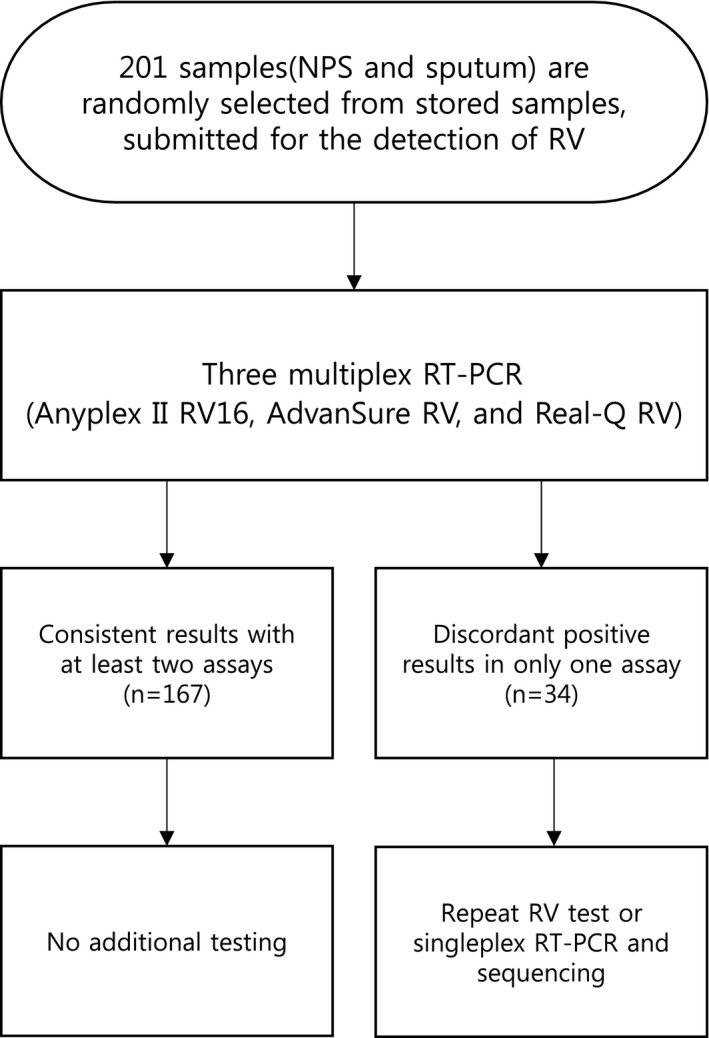
Flow diagram for evaluation of three commercial multiplex assays
2.5. Statistical analysis
Statistical methods of inter‐rater agreement, including percent agreement and kappa statistics, were calculated to compare the three RV assays. In addition, prevalence‐adjusted and bias‐adjusted kappa (PABAK) was calculated to compensate for underestimation of kappa values caused by low prevalence of each respiratory type.9 The kappa value can be interpreted as follows: <0.20 as poor; 0.21‐0.40 as fair; 0.41‐0.6 as moderate; 0.61‐0.8 as good and 0.81‐1 as very good agreement.10 Statistical analyses were performed using Microsoft Excel (Microsoft, Redmond, WA, USA) and MedCalc version 14.8.1 (MedCalc, Mariakerke, Belgium).
3. Results
Statistical analysis using percent agreement between RV assays in each virus type were high, ranging from 94% to 100%. Overall agreement between three RV assays ranged from 97% to 98%. The total kappa values were 0.76 between AP and AD, 0.82 between AP and RQ, and 0.86 between AD and RQ respectively. The kappa values ranged from 0.49 for PIV (AP vs AD and AD vs RQ) to 1.0 for PIV (AP vs RQ). In addition, PABAK values, which were adopted to compensate for the underestimation of kappa value when viruses show at a low prevalence rate, demonstrated very good agreement with ranges from 0.88 to 1.0 in comparison with kappa values.11 Table 2 shows the above results.
Table 2.
Analysis of agreement among three multiplex assays
| Virus | Agreement(95% CI) | Kappa value (PABAK) | |||
|---|---|---|---|---|---|
| Assays | AD | RQ | AD | RQ | |
| INFA | AP | 97 (93.9‐99.1) | 99 (96.8‐100) | 0.86 (0.93) | 0.93 (0.97) |
| AD | 97 (94.7‐99.4) | 0.87 (0.94) | |||
| INFB | AP | 98 (96.1‐99.9) | 99 (97.6‐100) | 0.70 (0.96) | 0.85 (0.98) |
| AD | 99 (97.6‐100) | 0.85 (0.98) | |||
| PIVa | AP | 99 (97.6‐100) | 100 | 0.49 (0.98) | 1.00 (1.00) |
| AD | 99 (97.6‐100) | 0.49 (0.98) | |||
| CoVb | AP | 96 (93.3‐98.7) | 98 (95.4‐99.7) | 0.79 (0.92) | 0.84 (0.95) |
| AD | 95 (91.4‐97.7) | 0.69 (0.89) | |||
| HRV | AP | 97 (94.0‐99.1) | 96 (93.3‐98.7) | 0.74 (0.93) | 0.69 (0.92) |
| AD | 99.5 (98.5‐100) | 0.97 (0.99) | |||
| MPV | AP | 98 (95.4‐99.7) | 99 (97.6‐100) | 0.77 (0.95) | 0.90 (0.98) |
| AD | 99 (96.8‐100) | 0.86 (0.97) | |||
| RSVc | AP | 94 (90.8‐97.3) | 95 (92.0‐98.0) | 0.57 (0.88) | 0.64 (0.90) |
| AD | 99 (97.6‐100) | 0.90 (0.98) | |||
| ADV | AP | 96 (93.3‐98.7) | 97 (94.0‐99.1) | 0.78 (0.92) | 0.82 (0.93) |
| AD | 99 (96.8‐100) | 0.91 (0.97) | |||
| Total | AP | 97 (95.8‐97.6) | 98 (97.0‐98.4) | 0.76 (0.93) | 0.82 (0.95) |
| AD | 98 (97.5‐98.8) | 0.86 (0.96) | |||
PIV including type 1‐4.
Cov including type 229E, OC43 and NL63.
RSV including type A and B.
A total of 167 samples (83.1%) showed concordant results. There was a total of 34 samples with a single positive result by one of the RV assays and two of these samples had discrepant results of two viruses. The 34 samples included 36 discordant results, 16, 18, and 2 viruses were positive only by AP, AD, and RQ respectively. Eighteen of the 36 discordant results from AP and RQ were resolved by repeat testing, with AD by monoplex PCR followed by direct sequencing. For the 36 discrepant virus results, additional testing revealed that 18.8% (3/16), 55.6% (10/18), and 50.0% (1/2) were consistent with the original virus result of AP, AD, and RQ assays respectively. These results were considered positive (Table 3).
Table 3.
Discordant analysis of positive results by only one RV assay
| Type of discrepant results | Result from AP | Result from AD | Result from RQ | Consensus | No. of samples |
|---|---|---|---|---|---|
| 1 | RSVB | Negative | Negative | Negative | 9 |
| 2 | RSVB | PIV2 | Negative | Negative | 1 |
| 3 | RSVB | OC43 | Negative | OC43 | 1 |
| 4 | RSVA | Negative | Negative | Negative | 1 |
| 5 | Negative | RSVB | Negative | Negative | 1 |
| 6 | Negative | RSVB | Negative | RSVB | 1 |
| 7 | Negative | 229E | Negative | Negative | 4 |
| 8 | Negative | 229E | Negative | 229E | 1 |
| 9 | Negative | Negative | NL63 | Negative | 1 |
| 10 | Negative | MPV | Negative | MPV | 1 |
| 11 | Negative | NL63 | Negative | NL63 | 1 |
| 12 | Negative | Negative | ADV | ADV | 1 |
| 13 | Negative | OC43 | Negative | OC43 | 1 |
| 14 | Negative | INFA | Negative | Negative | 1 |
| 15 | Negative | INFA | Negative | INFA | 3 |
| 16 | Negative | MPV | Negative | MPV | 1 |
| 17 | Negative | MPV | Negative | Negative | 1 |
| 18 | MPV | Negative | Negative | MPV | 1 |
| 19 | INFA | Negative | Negative | INFA | 1 |
| 20 | ADV | Negative | Negative | Negative | 1 |
| 21 | ADV | Negative | Negative | ADV | 1 |
| Total no. | 34 |
Of the 201 samples, AP, AD, and RQ detected 103 (51.2%), 99 (49.3%), and 95 (47.3%) positive cases respectively (Table 4). Viral co‐infection samples were identified in AP assay for 24 (11.9%) patients, in AD assay for 17 (8.5%) patients, and in RQ assay for 11 (5.5%) patients. In the co‐infected samples: two viruses were detected in 24 patients by AP assay; two viruses were detected in 14 patients and three viruses in three patients by AD assay; two viruses were detected in 11 patients by RQ assay. INF A was the most commonly detected virus by co‐infected samples, followed by ADV, RSVB, MPV and etc. Overall distribution of respiratory viruses from 3 RV assays is presented in Table 4. There are indications of the predominance of INFA, ADV, HRV, and MPV. Excluding these four major types, the rest of RV types accounted for only 0.8% to 8.9% of total viruses identified.
Table 4.
Distribution of respiratory viruses detected by each assay
| Target virus | AP (%) | AD (%) | RQ (%) | Consensus (%) |
|---|---|---|---|---|
| Influenza A | 26 (20.2) | 29 (24.4) | 25 (23.6) | 30 (24.4) |
| Influenza B | 6 (4.7) | 7 (5.9) | 7 (6.4) | 7 (5.7) |
| Parainfluenza 1 | 1 (0.8) | 0 (0.0) | 1 (0.9) | 1 (0.8) |
| Parainfluenza 2 | 1 (0.8) | 2 (1.7) | 1 (0.9) | 1 (0.8) |
| Parainfluenza 4 | 2 (1.6) | NT | NT | |
| Coronavirus 229E | 7 (5.4) | 12 (10.1) | 7 (6.6) | 8 (6.5) |
| Coronavirus OC43 | 6 (4.7) | 9 (7.6) | 5 (4.7) | 9 (7.3) |
| Coronavirus NL63 | 3 (2.3) | 4 (3.4) | 3 (2.8) | 4 (3.3) |
| Human rhinovirus | 12 (9.3) | 17 (14.3) | 16 (15.1) | 17 (13.8) |
| Metapnuemovirus | 12 (9.3) | 12 (10.1) | 11 (10.4) | 14 (11.4) |
| Respiratory syncytial virus A | 2 (1.6) | 1 (0.8) | 1 (0.9) | 1 (0.8) |
| Respiratory syncytial virus B | 23 (17.8) | 10 (8.4) | 10 (9.4) | 11 (8.9) |
| Adenovirus | 21 (16.3) | 16 (13.4) | 19 (17.9) | 20 (16.3) |
| Enterovirus | 7 (5.4) | NT | NT | |
| Total viruses detected | 129 (100) | 119 (100) | 106 (100) | 123 (100) |
| No virus | 96 | 102 | 106 | 98 |
| 1 virus | 81 | 82 | 84 | 87 |
| 2 virus | 24 | 14 | 11 | 16 |
| 3 virus | 0 | 3 | 0 | |
| Total samples | 201 (100) | 201 (100) | 201 (100) | 201 (100) |
NT, Not tested for target virus.
4. Discussion
The current study was performed for direct comparison between commercial multiplex RT‐PCR for detection of respiratory viruses. A high degree agreement was found between AP, AD, and RQ assays. The agreement between 3 assays for INF A, INF B, PIV, CoV, HRV, MPV, RSV, and ADV ranged from 94% to 100% (Table 2). The agreement for PIV demonstrated the highest percent ranges, between 99% and 100%. While the kappa values varied according to virus types detected, PIV between AD vs AP and RQ was the lowest value (κ=0.49). This result was caused by low frequency of PIV in samples making them eligible for PABAK. The value of PABAK increased to 0.98. RSV between AP vs AD and RQ show a low kappa value; however, PABAK for RSV did not show a sharp increase as seen with PIV because of the number of discrepant results between three assays.
In real circumstances of a clinical laboratory, the introduction of multiplex RT‐PCR assay for detection of RV was considered based on the assay's performance data and user friendliness.2, 3, 12 The assay characteristics of 3 RV assays related to end‐users was briefly summarized in Table 1. AP assay can detect 16 virus types and AD and RQ assays can detect 14 virus types. According to our study, the AP assay can test 40 samples in a single run whereas AD and RQ assay can simultaneously test 16 samples per run. However, the AP assay required the longest turn‐around time compared with other assays: 4.5 hours for the AP assay vs 2 hours for both the AD and RQ assays. In addition, the AP assay consisted of two steps, reverse transcription for cDNA synthesis and real‐time PCR coupled melting temperature analysis. As such it required end‐users to spend more hands‐on time to handle the reagents than other assays did. To overcome the time‐consuming step in the AP assay, an automated liquid handling system of mixing and handling reagents for PCR mater mix setup could be automated using MICROLAB Nimbus IVD (Hamilton). The characteristics between AD and RQ assays were very similar. Both AD and RQ assays were used based on TaqMan probe and 1‐step multiplex RT‐PCR combined with reverse transcription followed by real‐time PCR in a closed system. The test result of all assays was automatically presented by free analysis software offered from each company. The selection of multiplex RT‐PCR assay for RV detection requires consideration of each laboratory's facility, human resources and the number of tests.
To the best of our knowledge there is no study that compares Anyplex II RV16, AdvanSure RV, and Real‐Q RV in young adult patients with acute respiratory illness. Among the three assays, performance evaluation of AP assay was discussed in a number of articles. A first evaluation of AP assay against a combined standard of AP, xTAG Respiratory Viral Panel and Seeplex RV15 reported 95.2% sensitivity and ≥98.6% specificity rate.13 About the same time, Cho et al.14 reported that the performance of AP was superior in comparison with viral culture and Seeplex RV15. Huh et al.15 analyzed agreement between AP and Seeplex RV12 without standard method and found that the AP assay produced an equivalent performance against Seeplex RV12. The result of present study also showed that AP assay produced good agreements in performance comparable with AD and RQ assay in performance. In a previous study, AD showed good agreement (98%) against conventional multiplex RT‐PCR.16 Similarly, the performance evaluation of AD vis‐à‐vis a composite standard method revealed the most sensitive performance compared with viral culture and Seeplex RV15.17 The performance study of RQ assay was not found in the pubmed database, but evaluation of a previous version (1‐step RV real‐time PCR) against direct sequencing as a standard method reported 94.1% sensitivity and 96.6% specificity.18 We demonstrated that the agreement between three assays was excellent. Moreover, the end‐user in clinical laboratories selecting the multiplexed RT‐PCR for RV will need to consider the benefits of each assay in terms of both performance and user‐friendliness.
In the analysis of discrepant samples, 18.8% (3/16) from AP, 55.6% (10/18) from AD, and 50.0% (1/2) from RQ were consistent with the original virus results (Table 3). Of note, when repeat AP assay was done all RSVB samples (n=11) with an initial positive result by single AP identified as negative. Original test results of these samples were presented as + positive as detected by a melting temperature analysis after 50 cycles because they were supposed to contain low virus concentration. Unfortunately, the manufacturing company does not provide target range for virus detection and discloses that AP is a qualitative test for the detection of RV.
Likewise, samples of CoV 229E identified as positive by single AD assay identified as negative (80%, 4/5) from result of monoplex PCR followed by direct sequencing. Negative samples had a high C t (threshold cycle) value (mean±SD, 25.17±1.19) close to 27.0 as a cut‐off value, whereas one identified as positive using AD assay had a low C t value (23.1). These findings suggest that the discordant results were frequently reported in samples containing low viral copy. Other studies also indicated that discrepant results between RV assay was quite associated with sample's viral load.7, 8, 19, 20, 21 One samples by AP and three samples by AD identified as INF A positive by additional testing. This may be caused by the difference of target region in influenza genome in each assay. Other studies also indicated that detection ability of the influenza virus between commercial RV assay was varied according to subtype.22, 23 Unfortunately, we did not perform the Influenza A subtyping on targeted discrepant samples so any assay in this study cannot differentiate between subtypes of INF A. Further evaluation of performance between assay with regard to influenza A subtype is needed.
There were some limitations to this study. First, this study used samples from only young adults for a winter season. Thus, we could not obtain adequate samples per target virus, such as PIV type 1‐4, CoV 229E, OC43 and NL63, and RSV A. Those viruses could not be subjected to measure agreement values for each target due to low numbers. Second, another limitation was the use of stored frozen samples. This could skew results of RV detection based on storage conditions and the defrosting procedure. However, since all specimens were stored at the same storage condition and were processed by one skilled technician, we presume that comparison results between three assays was not significantly influenced by this characteristic of the sample.
In conclusion, the agreement of the three assays were very good, with 94%‐100% agreement for all comparisons. We suggest that all multiplex assay would be suitable for the detection of for respiratory viruses in clinical setting.
Acknowledgments
This study was partly supported by LG Life science, Korea and Biosewoom, Korea through supplying AdvanSure RV and Real‐Q RV kits. The sponsors had no involvement in the design, data interpretation and making of the manuscript. We thank LG Life Science, Biosewoom and Seegene for their technical assistance in the validation of the results. The authors also acknowledge the assistance of master sergeant Hyun‐Ki Moon and warrant officer Kwon‐Young Lee in Armed Forces Daejeon Hospital.
Yun SG, Kim MY, Choi JM, et al. Comparison of three multiplex PCR assays for detection of respiratory viruses: Anyplex II RV16, AdvanSure RV, and Real‐Q RV. J Clin Lab Anal. 2018;32:e22230 10.1002/jcla.22230
References
- 1. Legand A, Briand S, Shindo N, et al. Addressing the public health burden of respiratory viruses: the Battle against Respiratory Viruses (BRaVe) initiative. Future Virol. 2013;8:953‐968. [Google Scholar]
- 2. Buller RS. Molecular detection of respiratory viruses. Clin Lab Med. 2013;33:439‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Somerville LK, Ratnamohan VM, Dwyer DE, Kok J. Molecular diagnosis of respiratory viruses. Pathology. 2015;47:243‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hanson KE, Couturier MR. Multiplexed molecular diagnostics for respiratory, gastrointestinal, and central nervous system infections. Clin Infect Dis. 2016;63:1361‐1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gharabaghi F, Hawan A, Drews SJ, Richardson SE. Evaluation of multiple commercial molecular and conventional diagnostic assays for the detection of respiratory viruses in children. Clin Microbiol Infect. 2011;17:1900‐1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pabbaraju K, Tokaryk KL, Wong S, Fox JD. Comparison of the Luminex xTAG respiratory viral panel with in‐house nucleic acid amplification tests for diagnosis of respiratory virus infections. J Clin Microbiol. 2008;46:3056‐3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pierce VM, Hodinka RL. Comparison of the GenMark Diagnostics eSensor respiratory viral panel to real‐time PCR for detection of respiratory viruses in children. J Clin Microbiol. 2012;50:3458‐3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pierce VM, Elkan M, Leet M, McGowan KL, Hodinka RL. Comparison of the Idaho Technology FilmArray system to real‐time PCR for detection of respiratory pathogens in children. J Clin Microbiol. 2012;50:364‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Byrt T, Bishop J, Carlin JB. Bias, prevalence and kappa. J Clin Epidemiol. 1993;46:423‐429. [DOI] [PubMed] [Google Scholar]
- 10. Altman DG. Practical Statistics for Medical Research. London: Chapman & Hall/CRC; 1991. [Google Scholar]
- 11. Mak HK, Yau KK, Chan BP. Prevalence‐adjusted bias‐adjusted kappa values as additional indicators to measure observer agreement. Radiology. 2004;232:302‐303. [DOI] [PubMed] [Google Scholar]
- 12. Anderson TP, Werno AM, Barratt K, Mahagamasekera P, Murdoch DR, Jennings LC. Comparison of four multiplex PCR assays for the detection of viral pathogens in respiratory specimens. J Virol Methods. 2013;191:118‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim HK, Oh SH, Yun KA, Sung H, Kim MN. Comparison of Anyplex II RV16 with the xTAG respiratory viral panel and Seeplex RV15 for detection of respiratory viruses. J Clin Microbiol. 2013;51:1137‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cho CH, Chulten B, Lee CK, et al. Evaluation of a novel real‐time RT‐PCR using TOCE technology compared with culture and Seeplex RV15 for simultaneous detection of respiratory viruses. J Clin Virol. 2013;57:338‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huh HJ, Park KS, Kim JY, et al. Comparison of the Anyplex(TM) II RV16 and Seeplex((R)) RV12 ACE assays for the detection of respiratory viruses. Diagn Microbiol Infect Dis. 2014;79:419‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rheem I, Park J, Kim TH, Kim JW. Evaluation of a multiplex real‐time PCR assay for the detection of respiratory viruses in clinical specimens. Ann Lab Med. 2012;32:399‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho CH, Lee CK, Nam MH, et al. Evaluation of the AdvanSure real‐time RT‐PCR compared with culture and Seeplex RV15 for simultaneous detection of respiratory viruses. Diagn Microbiol Infect Dis. 2014;79:14‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim H, Hur M, Moon HW, Yun YM, Cho HC. Comparison of two multiplex PCR assays for the detection of respiratory viral infections. Clin Respir J. 2014;8:391‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gadsby NJ, Hardie A, Claas EC, Templeton KE. Comparison of the Luminex Respiratory Virus Panel fast assay with in‐house real‐time PCR for respiratory viral infection diagnosis. J Clin Microbiol. 2010;48:2213‐2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salez N, Vabret A, Leruez‐Ville M, et al. Evaluation of four commercial multiplex molecular tests for the diagnosis of acute respiratory infections. PLoS ONE. 2015;10:e0130378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Popowitch EB, O'Neill SS, Miller MB. Comparison of the Biofire FilmArray RP, Genmark eSensor RVP, Luminex xTAG RVPv1, and Luminex xTAG RVP fast multiplex assays for detection of respiratory viruses. J Clin Microbiol. 2013;51:1528‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Wesenbeeck L, Meeuws H, Van Immerseel A, et al. Comparison of the FilmArray RP, Verigene RV+, and Prodesse ProFLU+/FAST+ multiplex platforms for detection of influenza viruses in clinical samples from the 2011‐2012 influenza season in Belgium. J Clin Microbiol. 2013;51:2977‐2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cho HJ, Jang JW, Ko SY, Choi SH, Lim CS, An SS. Evaluation and verification of the nanosphere Verigene RV+ assay for detection of influenza A/B and H1/H3 subtyping. J Med Virol. 2015;87:18‐24. [DOI] [PubMed] [Google Scholar]