

Abstract
Background and Aims
Take‐home naloxone can prevent death from heroin/opioid overdose, but pre‐provision is difficult because naloxone is usually given by injection. Non‐injectable alternatives, including naloxone nasal sprays, are currently being developed. To be effective, the intranasal (i.n.) spray dose must be adequate but not excessive, and early absorption must be comparable to intramuscular (i.m.) injection. We report on the pharmacokinetics (PK) of a specially produced concentrated novel nasal spray. The specific aims were to: (1) estimate PK profiles of i.n. naloxone, (2) compare early systemic exposure with i.n. versus i.m. naloxone and (3) estimate i.n. bioavailability.
Design
Open‐label, randomized, five‐way cross‐over PK study.
Setting
Clinical trials facility (Croydon, UK).
Participants
Thirty‐eight healthy volunteers (age 20–54 years; 11 female).
Intervention and comparator
Three doses of i.n. (1 mg/0.1 ml, 2 mg/0.1 ml, 4 mg/0.2 ml) versus 0.4 mg i.m. (reference) and 0.4 mg intravenous (i.v.) naloxone.
Measurements
Regular blood samples were taken, with high‐frequency sampling during the first 15 minutes to capture early systemic exposure. PK parameters were determined from plasma naloxone concentrations. Exploratory analyses involved simulation of repeat administration.
Findings
Mean peak concentration (Cmax) values for 1 mg (1.51 ng/ml), 2 mg (2.87 ng/ml) and 4 mg (6.02 ng/ml) i.n. exceeded 0.4 mg i.m. (1.27 ng/ml) naloxone. All three i.n. doses rapidly achieved plasma levels > 50% of peak concentrations (T50%) by 10 minutes, peaking at 15–30 minutes (Tmax). For comparison, the i.m. reference reached Tmax at 10 minutes. Mean bioavailability was 47–51% for i.n. relative to i.m. naloxone. Simulation of repeat dosing (2 × 2 mg i.n. versus 5 × 0.4 mg i.m. doses) at 3‐minute intervals showed that comparable plasma naloxone concentrations would be anticipated.
Conclusions
Concentrated 2 mg intranasal naloxone is well‐absorbed and provides early exposure comparable to 0.4 mg intramuscular naloxone, following the 0.4 mg intramuscular curve closely in the first 10 minutes post‐dosing and maintaining blood levels above twice the intramuscular reference for the next 2 hours.
Keywords: Antidote, drug overdose, intranasal, naloxone, nasal, opiate, opioids, pharmacokinetics
Introduction
Overdose deaths from heroin and other opioids represent a significant international public health concern, accounting for approximately 106 000 deaths annually 1. In 2014, the World Health Organization (WHO) launched guidelines 2 recommending that ‘people likely to witness an opioid overdose should have access to naloxone’ (p. x). However, pre‐provision has been difficult, because naloxone is usually available only in injectable form.
The development of nasal naloxone spray for layperson use was first mooted in the late 1990s 3. During the 2000s, various groups began introducing improvised nasal spray kits, consisting of a syringe pre‐filled with the most concentrated injectable naloxone solution available (1 mg/ml) and a Mucosal Atomisation Device 4. While their use has been criticized for the absence of supporting pharmacokinetic (PK) data 5, 6, it undoubtedly established the potential worth of nasal naloxone. In the United States, a step‐change occurred in 2012 with the joint initiative of the US Food and Drug Administration (FDA), Centers for Disease Control and Prevention (CDC) and National Institute on Drug Abuse (NIDA) to encourage non‐injectable naloxone formulations, alongside FDA clarification of the regulatory benchmark: one or multiple doses of non‐injectable formulation must result in similar or greater naloxone exposure than a 0.4 mg intramuscular (i.m.) reference 7; i.m. naloxone reverses opioid overdose rapidly, with adequate respiration resuming typically within 3–7 minutes 8. For new non‐injectable formulations, the early absorption curve is thus crucial, corresponding to the time‐frame during which i.m. naloxone reaches peak concentration.
The FDA and Health Canada approved a first nasal naloxone product in November 2015 9, 10, 11 and October 2016 12, respectively. However, only limited PK data for intranasal (i.n.) naloxone exist to date 11, 13, 14, 30, with bioavailability estimates ranging from 4% for dilute [2 mg/5 ml relative to intravenous (i.v.)] 11 to 46–54% for concentrated (0.8 mg/0.1 ml relative to i.v.; 4 mg/0.1 ml relative to i.m.) formulations 14, 30 [Correction added on 9 Feb 2018, after first online publication: Pharmacokinetics of a naloxone nasal spray formulation of intermediate concentration values were missing and are updated in this version].
Recent work in Europe has explored different i.n. formulations, with the objective of developing a nasal spray suitable for lay administration, aiming for rapid onset of action and adequate exposure, without risk of ‘over‐antagonism’ 7, 15, 16, 30. We now report on the PK characteristics of nasal naloxone spray at different concentrations and doses, with particular focus on early absorption. The primary aims of this study were to (1) estimate the PK profile of i.n. naloxone and (2) compare its early partial systemic exposure to the i.m. reference. The secondary aim was to (3) estimate i.n. bioavailability.
Methods
Study design
A randomized, open‐label, five‐way cross‐over study (EudraCT number: 2015–004493‐15) in healthy volunteers was conducted at Richmond Pharmacology (Croydon University Hospital, UK) to determine naloxone pharmacokinetics from highly concentrated nasal spray solution (10 mg/ml, 20 mg/ml; Summit Biosciences, Lexington, KY, USA) at three doses; i.n. naloxone was administered as atomized spray with the unit dose system (Aptar Pharma, Cary, IL, USA). Laser diffraction determined that the size of ≥ 94% of droplets was greater than 10 μm to ensure deposition of the spray in the nasal cavity.
The reference routes were i.m. (primary reference) and i.v. administration of proprietary naloxone hydrochloride solution (Braun Melsungen, Melsungen, Germany).
The five study sessions comprised: (a) 1 mg naloxone i.n. (1 mg/0.1 ml i.n. one nostril); (b) 2 mg naloxone i.n. (2 mg/0.1 ml i.n. one nostril); (c) 4 mg naloxone i.n. (2 mg/0.1 ml i.n. each nostril); (d) 0.4 mg naloxone i.m. (0.4 mg/ml into the deltoid) and (e) 0.4 mg naloxone i.v. (0.4 mg/ml into the ante cubital fossa).
The i.n. dose range was based on our earlier analysis, which identified 1–4 mg i.n. as producing potentially similar early naloxone exposure to 0.4–0.8 mg i.v. 13.
With sequence assigned randomly in a cross‐over design, each subject received all five study treatments, with a single naloxone dose per session (see Supporting information, Fig. S1). Each session was separated by a minimum 4‐day washout period. Dosing occurred in the fasting state. Subjects were in a fully supine position, remaining supine for at least 1 hour post‐dose and semi‐supine thereafter until at least 4 hours post‐dose.
Blood sampling and chemical analysis
Given the special interest in early absorption, blood collection included intense sampling during the first 15 minutes, with a total of 19 samples per session (pre‐dose, at 1, 2, 4, 6, 8, 10, 12.5, 15, 30 and 45 minutes and 1, 1.5, 2, 4, 6, 8, 10 and 12 hours after dosing). Blood samples were centrifuged (1500 × g, 4°C, 15 minutes) within 30 minutes of collection, with plasma stored (−20°C) within 1 hour. Naloxone plasma concentrations were quantified by liquid chromatography–tandem mass spectrometry (LC–MS/MS) methodology using a previously validated assay.
Pharmacokinetic analysis
Individual subject PK parameters for naloxone were derived using non‐compartmental analysis in Phoenix WinNonlin version 6.4 (Certara LP, Princeton, NJ, USA), a validated PK analysis program. The area under the concentration–time curve (AUCt) was determined using the linear‐up/log‐down trapezoidal method from dosing (0 h) to the final observed plasma concentration (Clast) for AUCt. The ratio of Clast to LambdaZ was used to estimate the area between the last measured time‐point and infinity and added to AUCt to yield AUCINF. The maximum observed plasma concentration (Cmax) and the time to Cmax (Tmax) were obtained directly from plasma concentration data. LambdaZ was estimated using points in the terminal log‐linear phase, and terminal phase half‐life (t1/2Z) was determined from the ratio of the natural logarithm of 2 to LambdaZ.
Bioavailability
Dose‐adjusted AUC data (per mg) from i.n. administration were compared against the 0.4 mg i.m. and i.v. reference doses. Mean bioavailability estimates were determined for subjects for whom paired data were available.
Exploratory analyses
Exploratory analyses were conducted additionally to consider early exposure relative to the i.m. reference and repeat naloxone administration.
Early naloxone absorption
Given that Tmax may not describe the early absorption curve fully 5, three exploratory PK parameters were introduced to assess early exposure from i.n. relative to 0.4 mg i.m. naloxone: t50% [defined as time taken to achieve blood levels of 50% of Cmax (C50%)] 5; AUCp (partial AUC from time of dosing to median Tmax of i.m. naloxone); and, for i.n. administration, T50%ref (time taken to reach the C50% of the primary reference 0.4 mg i.m.).
Simulation of repeat administration
In emergency medicine, naloxone doses may be repeated to achieve overdose reversal. Therefore, simulations of repeat administrations were performed using the superposition approach. Superposition relies upon linear pharmacokinetics and employs a simple overlay technique, assuming that observed plasma concentrations after a single dose can be used to predict plasma concentrations after multiple dosing 17. Opioid overdose management guidelines typically recommend initial administration of 0.4 mg naloxone, repeated every 2–3 minutes if necessary 7, 16, 18, 19. Consequently, we simulated repeat administration of five doses of the 0.4 mg i.m. reference at 3‐minute intervals (simulating the upper limit of the recommended dose range, 2 mg i.m.) versus two doses of 2 mg i.n. at 3‐minute intervals (simulating similar naloxone exposure, assuming 50% i.n. bioavailability).
Simulation of immediate administration of the full dose
In the crisis situation, non‐medical first responders may forget or ignore instructions and administer the full available dose. We therefore also scaled the observed PK curves to doses of 5 × 0.4 mg i.m. and 2 × 2 mg i.n., the total doses that would be available for a first responder to administer.
Protection of human subjects
Approval was given by South Central–Berkshire B Research Ethics Committee (Reading, UK). All subjects provided written informed consent.
Sample size
The sample size was calculated on the basis that 28 evaluable subjects would provide approximately 80–90% power for detecting 90% confidence intervals of the ratio between treatment in pharmacokinetic parameters with bounds lying completely between 80–125%, assuming a true ratio of 1 and a standard deviation between 0.35–0.4 for the period differences on log scale. It was assumed that up to 20% of subjects might not be part of the primary analysis population or yield valid PK parameters for analysis of each of the five study treatments. Hence, at least 35 subjects were planned to be enrolled and analysed.
Subject eligibility
Eligible volunteers (female and male) had to be aged 18–55 years, body weight 55–100 kg, BMI ≥ 18.5 and ≤ 30.0 and healthy, i.e. free of significant abnormal findings as determined by medical history, physical examination, vital signs, laboratory tests and electrocardiograph (ECG). Volunteers were excluded if they had abnormal nasal anatomy, nasal symptoms (e.g. polyps, blocked/runny nose) or history of hayfever/seasonal allergy/rhinitis. Female volunteers were excluded if pregnant or lactating.
Statistical analysis
Randomization to one of five treatment sequences was performed by the study sponsor (Mundipharma Research Ltd, Cambridge, UK) using validated software (Random Patient Allocation System, Episys Ltd, Letchworth, UK) that automates the random assignment of treatment sequences to randomization numbers. The randomization scheme was reviewed by the sponsor's statistics department and locked after approval. Randomization was completed once eligibility was verified and occurred between day −1 and prior to first dosing (day 1). Randomization order was determined on a central randomization list held at site.
Descriptive statistics for pharmacokinetic outcomes were calculated using SAS software.
Results
Study participants
Thirty‐eight subjects (age 20–54 years) were randomized, 27 of whom were male and 11 were female. Prior to period 2 dosing there were four subject discontinuations (three subjects tested positive for drugs of abuse, one subject withdrew consent). As the protocol allowed for replacement subjects, it was agreed to randomize three additional subjects to the next available treatment sequence in the randomization schedule, and therefore the original enrolment target of 35 subjects was exceeded. In total, six subjects did not complete the study (see Supporting information, Fig. S1): six missed the 0.4 mg i.m. session; five missed the 1 mg i.n. session, four missed the 4 mg i.n. and 0.4 mg i.v. sessions, and two missed 2 mg i.n. session. These 21 sessions were handled as missing data. Consequently, values reported below refer to sample sizes of n = 32 (0.4 mg i.m.), n = 33 (1 mg i.n.), n = 34 (4 mg i.n., 0.4 mg i.v.) and n = 36 (2 mg i.n.), unless specified otherwise.
Safety
No severe adverse events (AEs) occurred. In total, 17 (of n = 38) subjects experienced 24 AEs (see Supporting information, Table S1), of which 11 AEs in nine subjects were assessed as naloxone‐related. AE‐occurrence did not seem to be dose‐related: seven subjects experienced AEs after 2 mg i.n. dose, while only three subjects experienced AEs after 4 mg i.n. dose.
Pharmacokinetic profiles
PK parameters are shown in Table 1. Mean plasma naloxone concentrations during the first 2 hours post‐dosing are displayed in Fig. 1 (left‐hand side), including expanded depiction of the first 20 minutes (right‐hand side).
Table 1.
Pharmacokinetic parameters.
| Parameter | Unit | 1 mg i.n. | 2 mg i.n. | 4 mg i.n. | 0.4 mg i.m. | 0.4 mg i.v. |
|---|---|---|---|---|---|---|
| AUCt | ||||||
| Geometric mean (CV%) | Hour × ng/ml | 2.56 (43.2) | 4.86 (39.4) | 10.01 (35.8) | 2.01 (17.7) | 2.01 (22.5) |
| Arithmetic mean (SD) | Hour × ng/ml | 2.78 (1.2) | 5.21 (2.0) | 10.57 (3.5) | 2.04 (0.4) | 2.05 (0.4) |
| Range (min, max) | Hour × ng/ml | 1.15, 5.78 | 2.33, 11.23 | 3.96, 21.08 | 1.31, 2.92 | 1.00, 3.12 |
| AUCINF | ||||||
| Geometric mean (CV%) | Hour × ng/ml | 2.69 (40.5) | 4.97 (38.5) | 10.07 (35.8) | 2.12 (16.6) | 2.10 (21.1) |
| Arithmetic mean (SD) | Hour × ng/ml | 2.90 (1.2) | 5.31 (2.0) | 10.64 (3.5) | 2.15 (0.3) | 2.14 (0.4) |
| Range (min, max) | Hour × ng/ml | 1.23, 5.88 | 2.44, 11.35 | 4.05, 21.12 | 1.37, 3.05 | 1.06, 3.35 |
| Cmax | ||||||
| Geometric mean (CV%) | ng/ml | 1.51 (50.2) | 2.87 (49.6) | 6.02 (54.5) | 1.27 (55.8) | 5.94 (92.9) |
| Arithmetic mean (SD) | ng/ml | 1.27 (0.7) | 3.18 (1.5) | 6.84 (4.0) | 1.42 (0.6) | 7.93 (6.0) |
| Range (min, max) | ng/ml | 0.42, 3.49 | 1.24, 7.50 | 1.72, 24.60 | 0.35, 2.38 | 1.17, 21.9 |
| LambdaZa | 1/hour | 0.55 (0.1) | 0.53 (0.1) | 0.44 (0.1) | 0.53 (0.1) | 0.57 (0.1) |
| t1/2Z a | Minute | 80 (23) | 84 (30) | 102 (28) | 81 (16) | 75 (13) |
| HVDa | Minute | 79 (40) | 76 (33) | 75 (38) | 65 (67) | 8 (12) |
| Tmax b | Minute | 15 (10, 60) | 30 (8, 60) | 15 (10, 60) | 10 (4, 90) | 2 (1, 15) |
AUCt = area under the curve (AUC) up to last measurable time point; AUCINF = AUC up to infinity; Cmax = maximum observed plasma concentration; LambdaZ = terminal phase rate constant; t1/2Z = terminal phase half‐life; HVD = half‐value duration; Tmax = time to Cmax.; i.n. = intranasal; i.m. = intramuscular.
Arithmetic mean [standard deviation (SD)];
median (min, max). CV = coefficient of variation.
Figure 1.
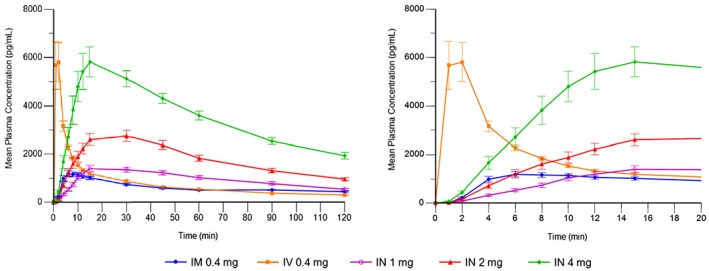
Mean plasma naloxone concentrations (observed values): dosing to 120 minutes (left) and dosing to 20 minutes (right)
Plasma concentration following i.v. administration (0.4 mg) spiked and reached an early peak (geometric mean Cmax 5.94 ng/ml, median Tmax 2 minutes), followed by a rapid decline during the next 10 minutes and a gradual decline thereafter.
The i.m. administration (0.4 mg) showed more gradual early uptake, with lower and later peak (geometric mean Cmax 1.27 ng/ml, median Tmax 10 minutes), and flatter and slower decline thereafter. Overall AUCt‐based exposure was comparable for i.m. and i.v. All i.n. doses (1 mg, 2 mg, 4 mg) achieved maximum plasma levels within 15–30 minutes (median Tmax). Geometric mean Cmax values for 1 mg i.n. (1.51 ng/ml), 2 mg i.n. (2.87 ng/ml) and 4 mg i.n. (6.02 ng/ml) were greater than for i.m..
For all three i.n. doses, geometric mean AUCt values (2.56–10.01 h × ng/ml) exceeded those of 0.4 mg i.m. and i.v. (both: 2.01 h × ng/ml). Of the three i.n. doses, the 2 mg dose followed the 0.4 mg i.m. curve most closely during the first 10 minutes post‐dose, reached blood levels at twice the 0.4 mg i.m. dose by 15 minutes and maintained blood levels at more than twice the 0.4 mg i.m. dose for the next 2 hours. Individual profiles for the 2‐mg dose are also provided to display individual variation (see Fig. 2; see Supporting information, Fig. S2 for other doses).
Figure 2.
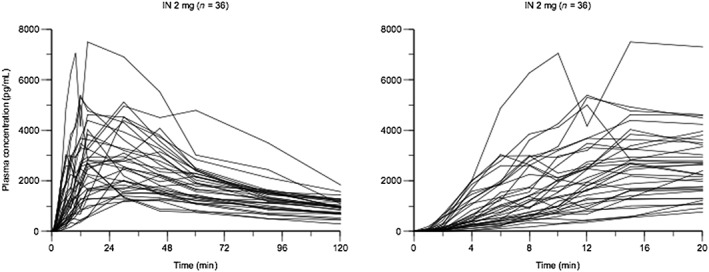
Plots of plasma naloxone concentrations over time (left: 120 minutes; right: 20 minutes) for individual subjects after 2 mg intranasal (i.n.) dose
Intranasal bioavailability
The mean absolute bioavailability (F%) estimates for i.n. naloxone (i.e. relative to i.v.) from dosing to last measureable concentration (AUCt) were 50.2% (1 mg i.n.; n = 32), 46.8% (2 mg i.n.; n = 33) and 48.1% (4 mg i.n.; n = 33); see Table 2; i.n. administration had a mean bioavailability relative to i.m. (Fi.m.%) of 50.8% (from 1 mg i.n.), 47.1% (2 mg i.n.) and 48.3% (4 mg i.n.), also determined from AUCt data (all n = 32).
Table 2.
Absolute (F%) and relative (Fi.m.%) mean bioavailability (90% confidence interval) based on area under the curve up to last measurable time‐point.
| Reference | n | 1 mg i.n. | 2 mg i.n. | 4 mg i.n. | |
|---|---|---|---|---|---|
| Fi.m.% | 0.4 mg i.m. | 32 | 50.8 (45.2, 57.1) | 47.1 (41.5, 53.5) | 48.3 (43.2, 54.1) |
| F% | 0.4 mg i.v. | 32–33 | 50.2 (44.6, 56.6) | 46.8 (41.7, 52.6) | 48.1 (43.3, 53.5) |
i.n. = intranasal; i.m. = intramuscular.
Exploratory analyses
As it followed the 0.4 mg i.m. reference most closely (see above), the 2 mg i.n. dose was chosen as comparator against the i.m. reference in exploratory analyses.
Early naloxone absorption
Given our special interest in early uptake, we examined AUCp, T50% and T50%ref for the early part of the plasma concentration–time profiles for i.n. naloxone relative to the i.m. reference (see Table 3).
AUCp: the rounded partial AUC values, measured from dosing to Tmax of the i.m. reference, were identical for both 0.4 mg i.m. and 2 mg i.n. (geometric mean: 0.11 h× ng/ml).
T50%: i.m. achieved plasma levels > 50% of Cmax (C50%) at 4 minutes. The 2 mg i.n. dose took 9 minutes to reach C50%.
T50% ref: the 2 mg i.n. dose achieved concentrations equivalent to C50% of the i.m. reference at 6 minutes, i.e. within 2 minutes of the i.m. reference, suggesting that early i.n. and i.m. naloxone plasma concentrations did not differ greatly.
Table 3.
Exploratory pharmacokinetic parameters.
| Parameter | Unit | 1 mg i.n. | 2 mg i.n. | 4 mg i.n. | 0.4 mg i.m. | 0.4 mg i.v. |
|---|---|---|---|---|---|---|
| AUCp | ||||||
| Geometric mean (CV%) | Hour × ng/ml | 0.05 (112.2) | 0.11 (105.1) | 0.27 (98.6) | 0.11 (67.9) | 0.44 (56.2) |
| Arithmetic mean (SD) | Hour × ng/ml | 0.07 (0.1) | 0.15 (0.1) | 0.36 (0.3) | 0.13 (0.1) | 0.50 (0.2) |
| Range (min, max) | Hour × ng/ml | 0.00, 0.21 | 0.01, 0.56 | 0.04, 1.52 | 0.03, 0.28 | 0.07, 1.12 |
| T50%a | Minute | 10 (5) | 9 (4) | 9 (5) | 4 (1) | 1 (1) |
| T50%ref a | Minute | 10 (8) | 6 (4) | 4 (2) | (4 (1)) | 1 (2) |
Arithmetic mean (standard deviation).
AUCp = partial area under the curve with cut‐off at Tmax of reference; T50% = time to 50% of Cmax; T50%ref = time to C50% of 0.4 mg i.m. (primary reference); i.n. = intranasal; i.m. = intramuscular; i.v. = intravenous; SD = standard deviation; CV% = coefficient of variation.
Simulation (1) repeat administration
To explore the pharmacokinetics of repeated administration, i.n. bioavailability (relative to i.m., as reported above) in the range of 47–51%, hence a 2 : 1 dose ratio (i.n. : i.m.) was assumed for the simulations, comparing cumulative 2 × 2 mg i.n. doses at 3‐minute intervals with five cumulative 0.4 mg i.m. doses at 3‐minute intervals (total 2 mg i.m.).
Figure 3 shows the peak plasma level from the observed 2 mg i.n. dose occurring between those from the observed i.m. 0.4 mg dose and the simulated 2 mg i.m. dose (5 × 0.4 mg administered 3 minutes apart). This simulation suggests that, in a hypothetical overdose scenario, a second dose of 2 mg i.n. administered after 3 minutes would expose the patient to approximately the same plasma naloxone levels, in terms of both initial rise and peak, as five consecutive i.m. 0.4 mg doses (also 3 minutes apart), i.e. 2 mg in total.
Figure 3.
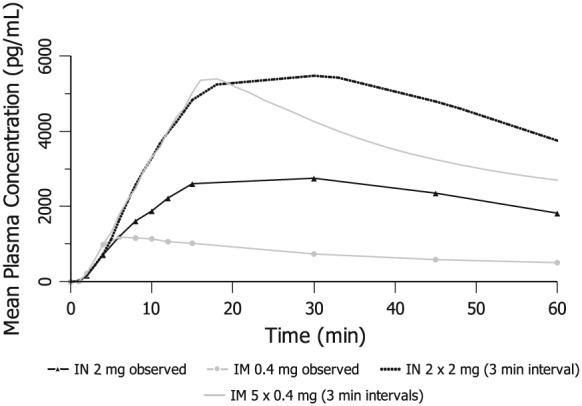
Scaled mean plasma naloxone concentrations after repeat administration at 3‐minute intervals (versus mean observed profiles of 0.4 mg intramuscular (i.m.) and 2 mg intranasal (i.n.) doses)
In addition, the plasma naloxone levels from the simulated 2 × 2 mg i.n. dose declined more slowly than the simulated × 0.4 mg i.m. administrations, indicating that plasma naloxone concentrations would be sustained for longer than those from i.m. dosing.
Simulation (2) immediate multiple dose administration of the full dose
The observed PK profiles, scaled in dose, were used to explore the pharmacokinetics of possible unintended immediate administration of the full 2 mg i.m. injection (i.e. all five 0.4 mg doses up to the top of approved therapeutic dose range) versus two simultaneous 2 mg i.n. doses (see Fig. 4). The scaled concentration data indicate that the 2 mg i.m. dose would have the fastest speed of uptake. In addition, the scaled 4‐mg profile (two 2 mg i.n. doses) was compared with the observed data from the 4 mg i.n. dose (administered as 2 mg per nostril) and lagged only slightly behind the observed 4 mg i.n. profile. However, in terms of dose‐adjusted AUC, the three administrations (2 mg i.m., 2 × 2 mg i.n., 4 mg i.n.) were roughly equivalent.
Figure 4.
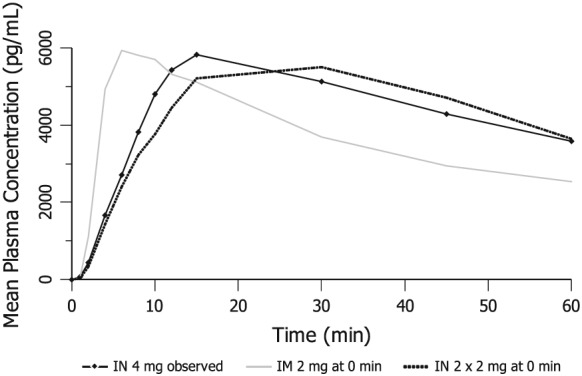
Scaled mean plasma naloxone concentrations after immediate administration of multiple doses at 0 minutes (versus mean observed profile of 4 mg intranasal (i.n.) dose)
Discussion
Principal findings
Our recent report of analysis of previously unpublished PK data established proof‐of‐concept for i.n. administration of concentrated naloxone formulations 13. The results of this new investigation confirm i.n. naloxone bioavailability of approximately 50% (relative to i.m.), thus markedly different from the 4% absolute bioavailability reported by Dowling et al. 14, yet very similar to the 46% relative bioavailability reported by Krieter et al. for the FDA/Health Canada‐approved product (4 mg/0.1 ml) 11. Our results suggest feasibility of concentrated naloxone nasal spray at three doses (1 mg/0.1 ml; 2 mg/0.1 ml; 4 mg as 2 × 2 mg/0.1 ml) for use by non‐medical first‐responders. Across these doses, naloxone plasma concentrations increased rapidly, with median peak concentrations between 15 and 30 minutes. This is consistent with Tmax values of 18–30 minutes reported for concentrated i.n. naloxone (also 0.1 ml volume) by Krieter et al. and by Tylleskar et al. 11, 30, and confirms that i.n. naloxone is absorbed rapidly into the systemic circulation via the nasal mucosa [Correction added on 9 Feb 2018, after first online publication: Tmax values are updated in this version to include those reported by Tylleskar et al. (2017).]. In the present study, the 2 mg/0.1 ml i.n. dose resulted in early uptake and exposure similar to the 0.4 mg i.m. reference, reaching concentrations equivalent to C50% of 0.4 mg i.m. within 2 minutes of the i.m. reference (T50%ref: 4 versus 6 minutes; see Table 3), suggesting that i.n. naloxone may be a feasible alternative to an i.m. injection.
The low bioavailability reported by Dowling et al. 14 was due probably to use of dilute (2 mg/5 ml) naloxone solution for injection which led to administration of high volumes—far in excess of what can be absorbed nasally without significant loss from the nasal cavity or post‐nasal drip. Low bioavailability of dilute nasal sprays is also seen in a recent conference report 20 and data contained in patent registrations 21.
As for the shape of the PK curves following all i.n. naloxone doses, there is reasonably rapid early absorption, followed by good maintenance of plasma levels throughout the period of time reported. This is in sharp contrast to i.v. (where the sudden rise in plasma level is followed by rapid decline) and i.m. administration (which mainly differs from the i.v. curve by lacking the initial spike). If i.m. is the reference route of administration then, after dose‐adjustment, concentrated nasal naloxone appears to offer comparable early onset followed by better maintenance of plasma levels during the intermediate period.
The 2‐mg i.n. dose produced speed of onset and early exposure comparable to 0.4 mg i.m. dose, while maintaining plasma levels for the next 2 hours at twice the level of the i.m. reference. Plasma concentrations from 2 mg in exceeded those from 0.4 mg i.m. within 6 minutes post‐dosing on average. The focus on this 2 mg i.n. dose may be particularly applicable to emergency administration using a dose‐escalation schedule as recommended with i.m. doses 16, 18, 19, starting at 0.4 mg and increased at intervals (e.g. 3‐minute intervals) to a total of 2 mg. A comparable i.n. dose‐escalation schedule would involve an initial 2 mg dose to achieve onset comparable to i.m. 0.4 mg, followed by a second 2 mg i.n. dose 3 minutes later; this is the hypothetical schedule examined in the repeat‐dosing simulation. As the second i.n. dose is given to the unused nostril, absorption from both administrations should be equal, and the similarity between the simulation and the 4 mg tested dose supports this assumption. Furthermore, the sustained plasma concentrations for the 2 mg i.n. dose compared with the i.m. reference may benefit post‐resuscitation care. Given that the half‐life of some opioids exceeds substantially that of naloxone (1–1.5 h) 2, sustained plasma naloxone concentrations from i.n. administration would probably reduce the risk of rebound toxicity when naloxone concentrations drop following i.v. or i.m. administration.
Strengths and limitations of the study
Strengths
Our particular focus has been the study of concentrated naloxone nasal spray to identify a dose and formulation comparable to i.m. 0.4 mg and similarly appropriate for dose‐titration, as recommended currently for the i.m. dose. Our exploratory dose simulations indicate that a 2‐mg i.n. dose provides opportunity for titration, with administration of a second 2‐mg i.n. dose resulting in naloxone exposure similar to a series of five i.m. 0.4 mg doses (i.e. 2 mg i.m. in total). Such an i.n. schedule would straddle the overall dose range of an ‘initial [injectable] dose between 0.4 mg–2 mg’, as recommended by WHO 2.
The possibility of incremental dose titration presents distinct advantages, as it could reduce significantly the risk of adverse reactions. High initial naloxone doses may trigger severe sudden‐onset opioid withdrawal 2, 22. A recent qualitative analysis of cases of heroin/opioid overdose reversals identified apparent excessive naloxone dosing (‘over‐antagonism’), sometimes triggering patient self‐discharge and active further drug‐seeking 15. In addition to pharmacological toxicity, such ‘behavioural toxicity’ needs to be considered. Withdrawal symptoms can be particularly challenging for overdose witnesses to manage in the community setting. Simulation of repeat administration of the 2 mg i.n. dose produced roughly equivalent plasma naloxone levels to a single 4‐mg i.n. dose. Giving a single 2 mg i.n. dose at first and following up with a second 2 mg i.n. dose only if needed could lower the risk of naloxone ‘over‐antagonism’ and improve the safety of the overdose victim and those attending the overdose scene. In addition, if naloxone doses trigger frequent severe withdrawal symptoms, then there is a real danger that, despite its life‐saving value, naloxone may be viewed as a punitive medication that is to be avoided, as was the perspective of many overdose patients interviewed by Neale 15. This might be regarded as ‘reputational toxicity’.
Limitations
While our findings support good early absorption and overall bioavailability in healthy subjects, concentrated naloxone nasal spray has yet to be tested formally in the target population of opioid users. Nasal naloxone might be absorbed differently by opioid users due to compromised nasal mucosa (e.g. chronic ulceration from drug snorting 23) or nasal obstruction from either mucus or vomit during overdose 5. At a practical level, it is challenging to quantify the impact of nasal congestion and compromised nasal mucosa, both in terms of possible lack or potentiation of absorption 24. Preliminary clinical evidence from an Australian ambulance‐based randomized trial 25 suggests that risk of modified absorption of i.n. naloxone may be small: in patients with suspected opioid overdose (n = 155) assigned to treatment of 2 mg/5 ml i.n. versus 2 mg/2 ml i.m. naloxone, there was no significant group difference regarding the need for a rescue injection of naloxone. However, statistical power for this comparison was limited, pointing to the potential need for replication of this study in a larger sample size.
Implications of results for practice and health policy
In recent years, international clinical practice has seen a shift from i.v. to i.m. naloxone for greater ease of administration, given that venous access can be difficult to establish in long‐term injecting drug users 18; i.v. administration leads to immediate increase of plasma naloxone concentrations with early Tmax, high Cmax and rapid decline thereafter. By comparison, i.m. administration produces slower absorption (i.e. later Tmax) and lower Cmax. However, the i.m. profile is now increasingly considered to be therapeutically beneficial: it avoids the extreme spike of i.v. naloxone but still attains efficacious plasma levels within the first minutes post‐dosing; we have additionally studied the T50% parameter for this very reason. Furthermore, the longer duration of effect of i.m. naloxone is considered beneficial 26.
Overall, the results of this study point to the worth of concentrated i.n. spray for opioid overdose reversal. The 2 mg i.n. dose provided comparable rate and extent of early exposure to the 0.4 mg i.m. reference, but maintained plasma levels at the approximate level of two 0.4 mg i.m. doses for more than 2 hours post‐dosing.
The use of i.n. may offer advantages over injectable naloxone formulations. First, i.n. naloxone removes the risk of needle‐stick injury and contraction of blood‐borne diseases (e.g. hepatitis C, HIV), which are highly prevalent among this patient group 27. Secondly, as i.n. naloxone does not require training in needle‐and‐syringe assembly, it enables laypeople to administer naloxone safely and could thus be pre‐provided to a much wider intervention work‐force (including hostel staff, outreach workers, police). Thirdly, injectable medicinal products are subject to prescription according to Article 71 of the EU Medicinal Products Directive (2001/83), 18. Development of licensed injection‐free naloxone products including nasal sprays may thus enable future re‐classification to over‐the‐counter medication 18.
Questions for future research
In our exploratory analysis of early absorption, the 2‐mg i.n. dose took, on average, 2 minutes longer than the i.m. reference (6 versus 4 minutes) to reach 50% Cmax of the i.m. reference (T50%ref). It is unclear what implications this 2‐minute delay may have in clinical practice, because speed of administration of the nasal spray may be quicker so that, for example, the 2‐minute delay could be offset partially or fully.
Measurement of time involved in preparing and administering the device is largely absent from current considerations of different naloxone formulations. An early exception is Wagner and colleagues’ ambulance study 28 of subcutaneous naloxone: the time from arrival at the patient's side to dose administration was 4 minutes (subcutaneous) versus 6 minutes (i.v.). Inclusion of these time intervals is essential for consideration of the merits of different routes of administration, especially in case of layperson administration. Future studies should examine the time to naloxone administration for different devices when used by laypeople without medical training. Such studies could assess whether the slightly earlier onset of i.m. administration observed in this study may be offset by better usability of the nasal spray.
A separate question for future research concerns the extent to which naloxone pharmacokinetics give accurate understanding of real‐world overdose reversals. A direct nose‐to‐brain connection has been proposed (whereby nasally administered drugs might bypass systematic circulation), but human evidence of direct drug transport from the nose to the cerebrospinal fluid is lacking 29. Laboratory‐based study of low‐dose naloxone reversal of opioid effects could elucidate the relationship between naloxone pharmacokinetics and pharmacodynamics.
Conclusions
Taken together with other recently published data 11, 13, 30, the results of this study lend strong support to the potential value of concentrated i.n. naloxone spray for opioid overdose reversal. Across all three i.n. doses, naloxone exposure was dose‐proportional, with approximately 50% absolute bioavailability. We identified the 2 mg/0.1 ml i.n. dose as most similar to the 0.4 mg i.m. reference, producing comparable early naloxone concentrations. The 2 mg i.n. dose had the added feature of effective maintenance of plasma levels for the next 2 hours. (In September 2017, following acceptance of this manuscript but pre‐publication, the European Medicines Agency gave a positive opinion to the application for product license for a 1.8mg/0.1mL concentrated naloxone nasal spray under the trade name Nyxoid; 1.8mg naloxone in Nyxoid is equivalent to 2mg naloxone hydrochloride).
Clinicians and policymakers will need to consider the potential merits of the different time–course profiles (including speed of onset and duration of effect) of i.n. versus injectable naloxone and may also see implementation advantages with i.n. naloxone for broad‐based take‐home naloxone provision.
Clinical trial registration details
EudraCT: 2015–004493‐15.
Declaration of interests
This research was supported financially by Mundipharma Research Ltd, Cambridge, UK, by whom it was designed in consultation with J.S., and was conducted at Richmond Pharmacology Ltd, Croydon, UK, under contract from Mundipharma Research Ltd. Data were evaluated jointly by the authors and the sponsor. U.L. is an employee of Richmond Pharmacology Ltd and was the Principal Investigator for this study. J.W., H.J., G.M. and K.S. are employees of Mundipharma Research Ltd. B.B. is an employee of Mundipharma Research GmbH & Co. KG, an independent associated company of Mundipharma Research Limited. J.S. and R.M. are employed by the University King's College London (KCL), UK. J.S. is a researcher and clinician who has worked with a range of governmental and non‐governmental organizations and with pharmaceutical companies to seek to identify new or improved treatments (including naloxone products), and from whom he and his employer (KCL) have received research funding, honoraria, travel costs and/or consultancy payments, including from Mundipharma Research Ltd to KCL for J.S.'s time and input to the study reported above. J.S. has also been named as an inventor in an earlier patent application filed by Euro‐Celtique S.A. (an independent associated company of Mundipharma Research Ltd) and entitled ‘Intranasal Pharmaceutical Dosage Forms comprising Naloxone’. For fuller account, see www.kcl.ac.uk/ioppn/depts/addictions/people/hod.aspx. J.S. is an NIHR Senior Investigator and is also supported by the NIHR Biomedical Research Centre for Mental Health at South London and Maudsley NHS Foundation Trust and KCL. R.M. has undertaken an unpaid student industry placement with Mundipharma Research Ltd, with focus on the analysis of naloxone nasal spray formulations. R.M. and J.S. are working as consultants for the United Nations Office on Drugs and Crime (UNODC), supporting a feasibility study of community‐based opioid overdose prevention strategies in the framework of the UNODC–WHO Programme on Drug Dependence Treatment and Care (GLOK32). The views expressed in this paper are those of the authors and do not necessarily reflect the position of the United Nations. KCL (employer for both J.S. and R.M.) has applied separately to register intellectual property on a novel buccal naloxone formulation with which J.S. and R.M. are involved.
Supporting information
Figure S1 Study disposition.
Figure S2 Plots of plasma naloxone concentrations during 20 minutes for individual subjects.
Table S1 Adverse events.
Acknowledgements
The authors would like to thank the study participants for taking part in the study, as well as Drs Evelin Kozma and Femi Adekunle for their valuable comments on the manuscript. This research was financially supported by Mundipharma Research Ltd, Cambridge, UK.
McDonald, R. , Lorch, U. , Woodward, J. , Bosse, B. , Dooner, H. , Mundin, G. , Smith, K. , and Strang, J. (2018) Pharmacokinetics of concentrated naloxone nasal spray for opioid overdose reversal: Phase I healthy volunteer study. Addiction, 113: 484–493. doi: 10.1111/add.14033.
This study was performed according to the Declaration of Helsinki (1964), International Conference on Harmonization and Good Clinical Practice (CPMP/ICH/135/95) guidelines of the EMA and European Union Clinical Trials Directive 2001/20/EC. [Correction added on 9 Feb 2018, after first online publication: Tylleskar et al. (2017) was missing from the reference list and is updated in this version.]
References
- 1. United Nations Office on Drugs and Crime (UNODC) . World Drug Report. 2016. Available at: https://www.unodc.org/wdr2016/ (accessed 1 December 2016) (Archived at http://www.webcitation.org/6u56T1R41 on 9 October 2017).
- 2. World Health Organization (WHO) . Community Management of Opioid Overdose 2014. Available at: http://apps.who.int/iris/bitstream/10665/137462/1/9789241548816_eng.pdf?ua=1&ua=1 (accessed 25 January 2016) (Archived at http://www.webcitation.org/6qHBtnGWj on 7 May 2017).
- 3. Strang J. Take‐home naloxone: the next steps. Comments on Strang et al.'s ‘Preventing opiate overdose fatalities with take‐home naloxone: pre‐launch study of possible impact and acceptability’. Addiction 1999; 94: 206–207. [DOI] [PubMed] [Google Scholar]
- 4. Doe‐Simkins M., Walley A. Y., Epstein A., Moyer P. Saved by the nose: bystander‐administered intranasal naloxone hydrochloride for opioid overdose. Am J Public Health 2009; 99: 788–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Strang J., McDonald R., Tas B., Day E. Clinical provision of improvised nasal naloxone without experimental testing and without regulatory approval: imaginative shortcut or dangerous bypass of essential safety procedures? Addiction 2016; 111: 574–582. [DOI] [PubMed] [Google Scholar]
- 6. Dale O. Ethical issues and stakeholders matter. Addiction 2016; 111: 587–589. [DOI] [PubMed] [Google Scholar]
- 7. Hertz S. Naloxone for Outpatient Use: Data Required to Support an NDA. Silver Spring, MD: Food and Drug Administration; 2012. [Google Scholar]
- 8. United Nations Office on Drugs and Crime/World Health Organization (UNODC/WHO) . Opioid overdose: preventing and reducing opioid overdose mortality Vienna: United Nations, 2013. Available at: http://www.unodc.org/docs/treatment/overdose.pdf (accessed 10 March 2016) (Archived at http://www.webcitation.org/6qH7oiNbN on 7 May 2017).
- 9. Adapt . NARCAN® (naloxone hydrochloride) nasal spray—patient information leaflet, 2015. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208411lbl.pdf (accessed 4 April 2016) (Archived at http://www.webcitation.org/6qHAYi0jQ on 7 May 2017).
- 10. Food and Drug Administration(FDA) . FDA moves quickly to approve easy‐to‐use nasal spray to treat opioid overdose, 2015. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm473505.htm (accessed 18 March 2016) (Archived at http://www.webcitation.org/6qH9DSNq5 on 7 May 2017).
- 11. Krieter P., Chiang N., Gyaw S., Skolnick P., Crystal R., Keegan F. et al Pharmacokinetic properties and human use characteristics of an FDA approved intranasal naloxone product for the treatment of opioid overdose. J Clin Pharmacol 2016; 56: 1243–1253. [DOI] [PubMed] [Google Scholar]
- 12. CBC News . Health Canada OK's non‐prescription nasal spray overdose antidote, 2016. Available at: http://www.cbc.ca/news/health/naloxone‐nasal‐spray‐1.3789643 (accessed 5 October 2016) (Archived at http://www.webcitation.org/6qH9uGq0R on 7 May 2017).
- 13. Mundin G., McDonald R., Smith K., Harris S., Strang J. Pharmacokinetics of concentrated naloxone nasal spray over first 30 minutes post‐dosing: analysis of suitability for opioid overdose reversal. Addiction 2017; 112: 1647–1652. [DOI] [PubMed] [Google Scholar]
- 14. Dowling J., Isbister G. K., Kirkpatrick C. M. J., Naidoo D., Graudins A. Population pharmacokinetics of intravenous, intramuscular, and intranasal naloxone in human volunteers. Ther Drug Monit 2008; 30: 490–496. [DOI] [PubMed] [Google Scholar]
- 15. Neale J., Strang J. Naloxone—does over‐antagonism matter? Evidence of iatrogenic harm after emergency treatment of heroin/opioid overdose. Addiction 2015; 110: 1644–1652. [DOI] [PubMed] [Google Scholar]
- 16. UK Medicines Information (UKMi) . Naloxone products for emergency opiate overdose reversal in non‐medical settings, 2016. Available at: www.ukmi.nhs.uk/filestore/ukmiaps/Naloxone%20product%20safety%20review_FINAL.pdf (accessed 1 December 2016) (Archived at http://www.webcitation.org/6u5AQgHQe on 9 October 2017).
- 17. Gilbaldi M., Perrier D. Pharmacokinetics, 2nd edn. New York: CRC Press; 1982. [Google Scholar]
- 18. Strang J., McDonald R., editors, European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) . EMCDDA insights: preventing overdose deaths from heroin and other opioids: pre‐provision of emergency naloxone (take‐home naloxone). Lisbon: EMCDDA; 2016. [Google Scholar]
- 19. World Health Organization (WHO) . IMAI District Clinician Manual: Hospital Care for Adolescents and Adults: Guidelines for the Management of Illnesses with Limited Resources. Geneva: WHO; 2011. [Google Scholar]
- 20. Edwards E. S., Kessler C., Kelley G., Gapasin A., Mardari G., Goldwater R. Pharmacokinetics of 2.0 mg intranasal and intramuscular naloxone HCl administration and the impact of vasoconstrictor use on the bioavailability of intranasal naloxone HCl. Poster presented at the 2016 AAPM Annual Meeting (Washington, DC; 31 July–4 August) 2016. Available at: http://www.painmed.org/2016posters/posterlb002.pdf (accessed 9 October 2017) (Archived at http://www.webcitation.org/6u56kICPc on 9 October 2017).
- 21. McDonald R., Glende Ø. D., Dale O., Strang J. International patent applications for non‐injectable naloxone for opioid overdose reversal: exploratory search and retrieve analysis of the PatentScope database. Drug Alcohol Rev 2017; https://doi.org/10.1111/dar.12571. [DOI] [PubMed] [Google Scholar]
- 22. Buajordet I., Næss A. C., Jacobsen D., Brørs O. Adverse events after naloxone treatment of episodes of suspected acute opioid overdose. Eur J Emerg Med 2004; 11: 19–23. [DOI] [PubMed] [Google Scholar]
- 23. Peyrière H., Léglise Y., Rousseau A., Cartier C., Gibaja V., Galland P. Necrosis of the intranasal structures and soft palate as a result of heroin snorting: a case series. Subst Abuse 2013; 34: 409–414. [DOI] [PubMed] [Google Scholar]
- 24. Arora P., Sharma S., Garg S. Permeability issues in nasal drug delivery. Drug Discov Today 2002; 7: 967–975. [DOI] [PubMed] [Google Scholar]
- 25. Kelly A.‐M., Kerr D., Dietze P., Patrick I., Walker T., Koutsogiannis Z. Randomised trial of intranasal versus intramuscular naloxone in prehospital treatment for suspected opioid overdose. Med J Aust 2005; 182: 24–27. [DOI] [PubMed] [Google Scholar]
- 26. Vilke G. M., Sloane C., Smith A. M., Chan T. C. Assessment for deaths in out‐of‐hospital heroin overdose patients treated with naloxone who refuse transport. Acad Emerg Med 2003; 10: 893–896. [DOI] [PubMed] [Google Scholar]
- 27. Degenhardt L., Charlson F., Stanaway J., Larney S., Alexander L. T., Hickman M. et al Estimating the burden of disease attributable to injecting drug use as a risk factor for HIV, hepatitis C, and hepatitis B: findings from the global burden of disease study 2013. Lancet Infect Dis 2016; 16: 1385–1398. [DOI] [PubMed] [Google Scholar]
- 28. Wanger K., Brough L., Macmillan I., Goulding J., MacPhail I., Christenson J. M. Intravenous vs subcutaneous naloxone for out‐of‐hospital management of presumed opioid overdose. Acad Emerg Med 1998; 5: 293–299. [DOI] [PubMed] [Google Scholar]
- 29. Djupesland P. G., Messina J. C., Mahmoud R. A. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther Deliv 2014; 5: 709–733. [DOI] [PubMed] [Google Scholar]
- 30. Tylleskar I., Skulberg A. K., Nilsen T., Skarra S., Jansook P., Dale O. Pharmacokinetics of a new, nasal formulation of naloxone. European Journal of Clinical Pharmacology 2017; 73: 555–562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study disposition.
Figure S2 Plots of plasma naloxone concentrations during 20 minutes for individual subjects.
Table S1 Adverse events.