

Abstract
Despite decades of accumulated knowledge about proteins and their post-translational modifications (PTMs), numerous questions remain regarding their molecular composition and biological function. One of the most fundamental queries is the extent to which the combinations of DNA-, RNA- and PTM-level variations explode the complexity of the human proteome. Here, we outline what we know from current databases and measurement strategies including mass spectrometry–based proteomics. In doing so, we examine prevailing notions about the number of modifications displayed on human proteins and how they combine to generate the protein diversity underlying health and disease. We frame central issues regarding determination of protein-level variation and PTMs, including some paradoxes present in the field today. We use this framework to assess existing data and to ask the question, “How many distinct primary structures of proteins (proteoforms) are created from the 20,300 human genes?” We also explore prospects for improving measurements to better regularize protein-level biology and efficiently associate PTMs to function and phenotype.
Proteins come in all shapes, sizes and forms. They are deeply involved in the major processes of life and comprise a large and enigmatic space between human genetics and diverse phenotypes of both wellness and disease. Assigning function and dysfunction to proteins is a major challenge for the coming era of basic and clinical research, so we take up the challenge of defining protein composition, including diverse contributions to its variation and the biological ramifications of this diversity.
The size of the human proteome is a matter of debate, and numbers in the literature range from as few as 20,000 to several million1,2. The huge discrepancy between these numbers is not a scientific controversy, but more a matter of definition. Thanks to the human genome project, we can now estimate the number of protein-coding genes to be in the range of 19,587–20,245 (refs. 1,3,4). Thus, if a single representative protein from every gene is used as the definition of the proteome, the estimated size is just ~20,000. This number may decrease somewhat, as it has been difficult to find an expressed protein encoded by some of these putative protein-coding genes5,6. However, if one considers that many genes are transcribed with splice variants, the number of human proteins increases to ~70,000 (per Ensembl3). In addition, many human proteins undergo PTMs that can strongly influence their function or activity. These PTMs include glycosylation, phosphorylation and acetylation, among a few hundred others (Fig. 1a), giving rise to many hundreds of thousands of additional protein variants5; furthermore, though many proteins are unmodified, some fraction of proteins are already annotated with multiple modifications (Fig. 1b). Finally, selected genes for proteins like immunoglobulins and T-cell receptors undergo somatic recombination to increase the number of potential protein variants into the billions in certain cell types across one’s lifetime7,8.
Figure 1. Two parsings of post-translational modifications from the SwissProt database of 20,245 human proteins.

(a) Histogram of PTMs in SwissProt for Homo sapiens (taxon identifier: 9606). Phosphorylation (phospho) is by far the most frequently annotated PTM at 38,030 (72%). Note that there are ~400 different types of PTMs known in biology (see: http://www.unimod.org). (b) Histogram of PTMs per SwissProt entry. Note that the distribution of PTMs is not uniform with 75% of entries containing two or fewer annotated PTMs; yet only five entries have >90 annotated PTMs.
Each individual molecular form of an expressed protein has come to be called a proteoform9. This term captures the disparate sources of biological variation that alter primary sequence and composition at the whole-protein level (Fig. 2). These include biological events that change single or multiple residues within the sequence of amino acids and the many modifications that can decorate the protein during its synthesis or after it is produced within a cell. These sources of variation produce the largely unmapped complexity of human proteoforms. At first glance, characterizing such diversity appears to be intractable, but closer inspection of the sources and limitations imposed upon proteoform diversity, as well as an examination of measurement techniques, can provide bounded estimates. In a few examples, proteoforms and their PTMs have been mapped, enabling early efforts to assign and understand their biological functions.
Figure 2. Graphical depiction of sources of protein variation that combine to make up proteoforms, each of which map back to a single human gene.

Depicted is a single human gene and two of its isoforms, which differ by the coding for several different amino acids of a protein primary sequence (at left); isoforms commonly arise from alternative splicing of RNA and from use of different promoters or translational start sites. Isoform variation combines with site-specific changes to generate human proteoforms (at right); three examples of site-specific changes include single-nucleotide polymorphisms (SNPs) and co- or post-translational modifications like N-glycosylation or phosphorylation, respectively.
Sources of proteoform diversity
Our aim is to help diverse communities better understand the composition and nature of human proteins in health and disease. We now assemble known information for the main sources of variation at the levels of DNA, RNA and protein that contribute to proteoform diversity. We then examine how these sources of diversity expand the number of theoretical human proteoforms (Fig. 3a) and contrast that with the number of observed proteoforms carrying multiple PTMs that are actually produced in biological systems (Fig. 3b).
Figure 3. Contrasting the potential sources of protein variability versus those that actually occur in combination as proteoforms detectable in actual human systems.

(a) Common sources of protein variability include alternative splicing of RNA, single-nucleotide polymorphisms (SNPs) in regions of genes coding for amino acids, and PTMs. Note that there are ~33,000 splice isoforms, ~78,000 site-specific amino acid variants (i.e., polymorphisms and mutations) and ~53,000 PTMs in the October 2017 release of the Human SwissProt database. (b) Depiction of two proteoforms from specific combinations of protein variability.
Estimates of DNA-level variation
Substantial sources of variation in human proteins include coding single-nucleotide polymorphisms (cSNPs) and mutations, with ~135,000 validated nonsynonymous cSNPs currently housed within SwissProt. In dbSNP, there are 4.7 million candidate cSNPs, yet only ~670,000 cSNPs have been validated in the 1,000-genomes set as nonsynonymous cSNPs that change the identity of an amino acid in a protein. However, the number of polymorphisms found in databases is reduced to only the two alleles actually harbored by any individual. Our adaptive immune system also presents a major source of somatic alteration in specific cell types. One can therefore divide these two kinds of variations into ‘population variation’, which exists across the human population, versus ‘individual variation’, which exists in an individual human being.
Main sources of RNA-level variation
Alternative splicing is a key contributor to transcriptome complexity and modulation of complex human traits10. RNA sequencing (RNA-seq) studies indicate that ~93% of human genes undergo alternative splicing and about 86% have a minor isoform frequency above 15% (ref. 11). Recently, functional consequences of alternative splicing were explored12, showing that the interacting partners for minor isoforms from a given human gene are as different as those for proteins encoded by entirely different genes. Alternative splicing often leads to the inclusion or exclusion of mitochondrial targeting sequences, leading to two mature proteins that have identical warheads but are localized to two different places13. Transcriptome diversity is further expanded through RNA editing. Though over 4.5 million adenosine- to-inosine editing events (the most common type) have been detected in human transcripts, only ~4,400 actually alter the corresponding amino acid14.
There is yet a major gap between the number of alternative transcripts asserted by RNA-seq and that detectable by proteomics (e.g., <0.1% of putative novel splice junctions in cancer xenografts)15. This discrepancy is due, in part, to the limited sensitivity and coverage of the current proteomic platforms. Although deep proteomic analyses can identify the majority of gene expression, sequence coverage for most proteins remains low, particularly for low-abundance genes. For example, the aggregated NCI-60 proteomics data set16 covers only 12% of the whole encoded proteome, and only ~5% of the genes had sequence coverage >50% of their protein coding regions17.
Because small size is a confounding factor in gene prediction, small open reading frames (smORFs) in stretches of RNA previously assumed to be noncoding have only recently been annotated as protein coding regions owing to advances in sequencing and proteomics18. Although the total number of novel human small proteins encoded in smORFs is still unclear, with estimates ranging from hundreds to thousands, roles of specific small proteins in fundamental biological processes have been established (for example, control of genome maintenance)19.
Errors in translation
Errors during protein translation provide one very large source of potential proteome expansion, particularly in aging or stressed cells. Error frequencies of 0.01–0.1% per amino acid (AA) have been estimated for misincorporating structurally similar amino acids in vivo20. This source of low-level protein heterogeneity is apparent in characterized recombinant proteins expressed in Escherichia coli, for which misincorporations can range from 0.5 to 5%. Mistranslation events have also been identified in recombinant monoclonal antibodies expressed in mammalian cell lines, wherein asparagine is substituted for serine at 0.01–0.2% of AGC codons21.
Post-translational modifications
The exponential increase in the potential number of proteoforms due to PTMs generates an open question in the field that lies at the heart of this Perspective. To help inform and frame the question, one could divide co- and post-translational modifications in different ways (for example, based on their chemical structures or whether or not they are reversible). A structural view would divide PTMs into subtypes based on whether proteins are cleaved or site-specifically modified with ‘simple’ PTMs (for example, phospho, acetyl, methyl, O-GlcNAc, etc.). The structures of other PTMs are highly complex in nature (for example, glycosylation, polyubiquitinylation, etc., as addressed below). The structural view of protein complexity is linked to how PTMs make the number of proteoforms increase and how difficult it is to characterize them precisely. A functional view focuses more on the way that proteoforms and their combinations of PTMs underlie cellular decision making and contribute to overall phenotypes (as has been shown for the histone code22).
Complex post-translational modifications
In contrast to the linear assembly of amino acids in polypeptide chains, the ten common monosaccharide building blocks of human glycans can be linked at multiple positions, resulting in highly branched structures. Taking into account the other structural features of oligosaccharides like linear sequence, linkage position and anomeric configuration, the number of possible glycan structures is large. However, one may also conclude that although glycan biosynthesis is untemplated, that does not mean it is unrestricted. Nature may only access a limited number of protein glycoforms. Thus far, on the order of a few tens23 to more than one hundred24,25 glycoform compositions above the ~1% detection threshold are readily measured by current technologies, with some examples provided in Table 1.
Table 1.
Examples of mapping proteoforms and correlating them to function and phenotype in complex systems
| System | Number of proteoforms mapped | Proteoform→PTM→function |
|---|---|---|
| 14-3-3 proteins83 | 11a | Phosphorylation mediates protein–protein interaction |
| α-Synuclein; human brain in Parkinson’s disease60 | 11 | Phosphorylation→weak correlation to Parkinson’s disease pathology |
| Amyloid-β; human brain in Alzheimer’s disease59 | 23 | Diversity of proteoforms is not captured by traditional ELISA assays |
| Angiotensin converting enzyme; human84 | 24a | Multiple isozymes with multiple functions |
| Apolipoprotein C-III; human high-density lipoprotein particles65 | 4 | Branched glycoproteoforms on Thr104 correlate with HDL-C levels |
| B-type natriuretic peptide; heart failure66,85 | 7–24 | Multiple PTMs and proteolysis correlate with heart failure |
| Cardiac troponin I; heart failure64,86 | 17 | Altered in phosphoproteoforms associated with cardiac disease |
| Chorionic gonadotropin; α/β subunits, human23 | 10 and 24b | Sialic acid content influences receptor binding activity and clearance |
| Erythropoietin; recombinant fusion protein expressed in CHO cells | >230c | Modulation of receptor binding kinetics during red blood cell production |
| Etanercept; human24 | >80 | Galactosylation and fucosylation modulate immunogenic potency |
| Histone H2B; human46 | 15 | Many gene family members possible→few observed proteoforms |
| Histone H3; human87 | >250d | Low dosage of H3.3K27M (<10%) associated with pediatric diffuse intrinsic pontine gliomas (DIPG)52 |
| Histone H4; human27,88 | 75 | Associated with both gene repression and activation |
| Interferon β-1a; commercial recombinant protein (Avonex)89 | 138 | Loss of N-terminal Met correlated with multiple sites of deamidation and loss of potency (used clinically to treat multiple sclerosis) |
| Myosin regulatory light chain; swine heart failure90 | 4 | Decreased phosphorylation correlates with myocardial infarction |
| Outer membrane proteins in C. glutamicum68 | 30 | O-mycoloylation→localization to the outer membrane |
| PilE, pilin proteins in N. meningitidis infection69 | 18 | Phosphoglycerylation→increased in vivo dissemination and virulence |
| Reactive cysteines in S. typhimurium infection67 | 34 | S-glutathionylation and S-cysteinylation→infection-like conditions |
| Transthyretin; familial amyloidosis55 | 25 | Genetic mutation alters PTM profiles |
Proteoforms and their PTMs have been mapped on selected microbial, pig, mouse and human proteins.
Estimated.
Human chorionic gonadotropin (hCG) is another prominent example for which glycosylation strongly regulates its biological function. The hCG protein contains more than 40 N- and O-glycan structures on two glycosylated subunits. Combinatorial analysis for the α-subunit and β-subunit predicts ~16,000 theoretical glycoforms; however, only 10 and 24 could be assigned for each subunit, respectively.
Although glycosylation is an untemplated process, when one takes into account multiple functional and biosynthetic arguments, it has been estimated that fewer than 3,000 N- and O-linked glycan monomers exist in humans91.
For ubiquitinylation, homo- and mixed polymers of the 8.5 kDa protein ubiquitin can reach ~25 monomers in length, adding >200 kDa in molecular weight. These modifications exert profound influence on the subcellular location, function and degradation of (apparently) all cellular proteins26 through complex mechanisms that may include crosstalk with other PTMs (for example, phosphorylation or acetylation). For poly(ADP-ribose), linear chains of 20–50 units combine to form branched polymers that are over 300 residues long. Though a comprehensive understanding of the composition of branched, polymeric PTMs may lie outside immediately available technologies, progress is being made with polyubiquitins, and the growth in native mass spectrometry for high-mass distributions will continue to make inroads and help elucidate the relationship between protein composition, function and disease phenotypes.
How does proteoform number scale with simple PTMs?
The simplistic answer to how PTM number translates to scaling of the number of possible proteoforms is 2n, where n is the number of PTMs. This refers to site-specific PTMs that are ‘binary’, like phosphorylation and acetylation (i.e., either on or off). However, consider that a lysine on a protein can exist in at least five different states, taking into account both acetylation and methylation (for example, Kunmod, Kme1, Kme2, Kme3 and Kac). The general formula describing how proteoform number grows with protein variation is shown in Box 1. A specific example is human histone H4 (UniProt accession: P62805), in which a combinatorial explosion of its 58 SwissProt-annotated PTMs at 17 known sites gives rise to >1010 theoretical proteoforms. Use of just the most common 13 PTM sites from the literature and the E64Q variant (its minor allele frequency is ~0.001%) creates 98,304 possible proteoforms (see Box 1). However, recent analyses of intact H4 proteoforms by seven participating labs reported just 75 proteoforms observed at >0.01% relative abundance27. The dramatic, orders-of-magnitude difference between actual and theoretical proteoforms aligns with a view wherein proteoform diversity is limited by a high degree of control over the enzymatic writing and maintenance of PTMs (see section below on proteoform diversity and function).
Box 1. Calculating the number of theoretical proteoforms.
Recent analyses of human histone H4 (P62805) mapped 75 proteoforms in human cells (see main text). Considering the 13 most common PTMs (acetylation, methylation and phosphorylation, shown below) and a single SNP, 98,304 theoretical proteoforms are possible.
26 (K5/8/12/16/31/91 ac) × 31 (R3 me1/2) × 41 (K20 me1/me2/me3) × 25 (S1/S46/Y51/T79/Y87 ph) × 2 (N-term ac) × 2 (E63Q cSNP) = 98,304 proteoforms

In protein databases, the number of PTM sites on a single protein can range from 0 to over 90 (see PTM distributions in Fig. 1). Considering only binary modifications makes the number of theoretical proteoforms astronomically large (i.e., 290 = 1 × 1027). Here is where two paradoxes arise. The first one is rooted in technologies used to measure protein molecules, whereas the other is one of perspective. Use of technologies that either do, or do not, capture complete compositional information about whole proteoforms drastically changes what is measured and perceived by the scientists using them. Today’s perceptions about the diversity of human proteins can be in two extremes: that a majority of the possible variations exist on proteins or that only a minority of possible PTMs actually co-exist on the same protein (see Fig. 2). These different perspectives are central to understanding why protein-level biology is enigmatic, and authors on this Perspective offer a continuum of viewpoints and some data to help frame and inform this open question.
Limits on proteoform diversity
The exponential increase in possible proteoform number due to PTMs creates an explosion in the number of possible protein compositions populated by human biology. There are both natural and technological limits to this ‘proteoform explosion’, and we deal with each of these in turn.
Copy numbers limit protein complexity in single cells
One limit to protein complexity is copy number. Consider a protein present at 1,000 copies per cell; 1,000 proteoforms is then the maximal number in that cell at a given time. Of course, in a population of 1 million cells, the cell-to-cell diversity could significantly increase that number, especially as cells respond to stimuli by PTM remodeling over time. Such lines of thinking trigger questions regarding how post-translational diversity arose, its function and its range of variation in single states or in response to diverse stimuli.
Handling the proteoform explosion via abundance thresholding
Another valuable point of reference comes from consideration of just how many genes are expressed into a protein in a given cell type. Estimates from deep proteomics and transcript profiling suggest that about half the human genome is expressed in proteins at over 20 copies per cell in a given cell type (i.e., about 10,000 of the 20,000 human genes)28. Assuming this expression threshold of 10,000 genes and allowing for detection of ~100 proteoforms for each gene product, one then multiplies these two to arrive at a measurement target of 1,000,000 distinct proteoforms in a given cell type. A 2016 estimate based on trends in databases indicated that the number may be ~6 million proteoforms29. Better estimates of this proteoform diversity are needed, and are analogous to the extrapolations of the number of human genes using expressed sequence tags (ESTs) in the year 2000 (ref. 30).
The question of how many proteoforms exist may prove impossible to answer fully (i.e., down to single copy proteins or across a billion-fold dynamic range of the most-to-least-abundant proteoforms). Errors in transcription and translation or exposure to toxic chemicals can produce numerous low-abundance proteoforms, perhaps even at the single-molecule level in a large population of cells. However, this issue may be more philosophical than practical, as current technologies for identifying and tracking proteoforms (for example, chromatography, mass spectrometry and antibody-based measurements) are constrained to operate above a given abundance level (i.e., the number of detectable proteoforms rather than all proteoforms per se). Through this lens, the number and variety of proteoforms expressed in biological systems appears to be well below the theoretical combinatorial possibilities31, with several examples providing a glimpse into this open question (Table 1). However, should new technologies emerge that relieve these constraints (for example, single-molecule proteoform detection32), this comfortable myopia may prove fragile.
Challenges in measuring proteoforms
Inference versus direct readout of proteoforms
The dominant paradigm of modern proteomics is the ‘bottom-up’ strategy, in which protein mixtures are digested with a protease, typically trypsin, to yield complex mixtures of peptides (Box 2). These peptides are analyzed by LC–MS/MS and identified by comparison of their MS/MS fragmentation spectra with theoretical spectra produced from the known genome sequence of the organism under study or customized protein sequence databases derived from matched DNA- or RNA-sequencing data from the same sample. The presence of a given protein in the sample is inferred from identification of the peptides it contains, in a process known as ‘protein inference’32. Although protein inference is a widely employed cornerstone of bottom-up proteomics, it is not generally possible to identify proteoforms in the same manner, as different proteoforms often share most of their peptides with one another. Instead, it is necessary to use ‘top-down’ proteomic methods, in which the entire proteoform is analyzed by LC–MS/MS without prior digestion to peptides (Box 2). Ideally, the complete amino acid sequence and localized PTMs are obtained; for proteins that are especially large or those harboring many PTMs, there are often ambiguities in the complete description of related proteoforms. Addressing these limitations of top-down proteomics in both denatured and native modes is a frontier area of current research.
Box 2. Bottom-up and top-down strategies for the analysis ofprotein sequence and composition.
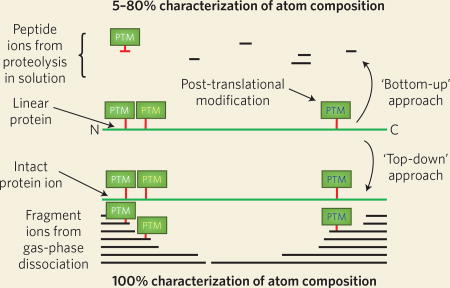
With the ‘bottom-up’ proteomics workflow, preanalytical processing of proteins (and corresponding proteoforms) is performed with proteases (e.g., trypsin) to generate analytically manageable peptides (top) that are sequenced in order to determine protein identity. The ‘top-down’ approach avoids the digestion step and characterizes proteoform microheterogeneity directly through tandem mass spectrometry techniques (bottom). Analysis at the intact level is advantageous because 100% of the proteoform’s primary structure is present in the top-down workflow, contrasting with bottom-up methods in which incomplete sampling of peptides across the protein backbone may cloud actual proteoform determination.
Mapping protein composition with complete molecular specificity
The next stage of proteomic investigation goes beyond identification of peptides and individual PTMs to reach for complete protein characterization through proteoform-resolved measurement34,35. For elucidating functions of proteoforms, complete knowledge of their molecular composition and that of their interacting partners is preferred. A potential confounding factor in this endeavor can arise from artifacts of sample preparation of tissues, cells and their extracts via enzymatic or chemical modification or degradation (for example, oxidation and chain cleavage). As a result, proteoforms can be proteolytically truncated, thereby forming new proteoforms with a loss of correlative power and relationship to their function. Enzymatic conversions of proteoforms also occur in body fluids in vitro, further complicating their quantification. For example, brady-kinin, a proteoform of kininogen-1 and a vasoactive peptide hormone, is degraded faster in vitro than in vivo (half-life of 17 s). In addition, other enzymes like phosphatases can convert proteoforms in homogenates unless they are inhibited. With respect to these problems, new sampling procedures like direct mass-spectrometric imaging36,37 of tissues yielding the spatial distribution of proteoforms and Picosecond InfraRed Laser technology (PIRL) are promising for providing higher fidelity readouts of whole proteoforms38. Tissue samples collected with PIRL by cold, soft and very fast ablation show more intact proteoforms than those obtained by conventional protein extraction39.
Prospects for mapping the majority of human proteoforms
With proteins being dynamic and so dependent on their context, it is critical to frame the dimensions of their measurements. Analysis of protein molecules can be performed at different levels in the hierarchical organization of the human body (Fig. 4). Mapping of proteins can also mean determining their spatial distribution in a solid tissue or deducing their atom composition. The question arises as to what level of understanding is needed to obtain a holistic view of the human proteome and how that would augment our biomedical goals for science, technology and society. Recent efforts to describe the composition and spatial distribution of proteins have advanced in draft maps of the human proteome40,41 and the Human Protein Atlas2, respectively. This year, a major endeavor called the Human Cell Atlas has been launched to define the cell types that comprise the human body42. This effort will expand with a variety of consortia and take on the definition of cell types in diverse organs, the immune system of the blood and bone marrow, and even the brain.
Figure 4. Levels of organization in the human body.

Starting from protein primary structure (proteoforms), the complexity of organ systems is built up in layers. A key concept is that diverse measurement approaches in proteomics seeks analysis of protein molecules at the various levels and contexts represented. Proteoform membership in protein complexes and localization within organelles, cells and tissues are all aspirations of measurement technologies to map protein molecules more precisely in molecular composition, across space and through time.
How much proteoform variation exists between cell types?
Recent advances in single-cell RNA sequencing (scRNA-seq) technology allow comprehensive and data-driven characterization of major cell types within a tissue42. For some tissues, estimates based upon the sum total of previous (pre-single-cell) studies provide a good estimate of the number of cell types, whereas for other tissues there are many cell types that remain to be classified. One could envision that analysis of cellular proteoforms would complement the scRNA-seq gene expression data and add power to robust classification of cell types and states. Additionally, with the availability of the Human Cell Atlas, an effort focused on compositional mapping of proteoforms in each human cell type could become feasible, as outlined in a separate publication43. This cell-based approach to compositional mapping of human proteins was framed for a depth of 250,000 proteoforms per cell type43, with a focus on defining normal variation in health and wellness; such a project would require establishing cost effective approaches to cell- and proteoform-specific measurements.
Mapping proteoforms and their kinetics in health and disease
With the overview above regarding sources of combinatorial variation, what are some functional implications arising from this protein-level diversity? Once a PTM present only on a specific splice variant can be asserted precisely, how does it vary across cell type and disease? Such questions are being addressed using a common approach of mapping proteoforms, determining their composition (including any new ones resulting from mutation or aberrant PTM patterning), and then correlating proteoform-level dynamics to functional readouts and phenotype (Fig. 5). Several examples from the past few years are summarized in Table 1, with reviews available to highlight early examples44. In the domain of microbiology and infectious disease, the process of assigning proteoform function and obtaining clinical value is farthest along. In more complex human diseases across the spectrum of neurodegeneration, oncology and cardiovascular disease, functional assignment for combinations of events detected at the proteoform level are accruing, albeit at a slower rate.
Figure 5. Proteoforms and their families underlie complex traits and molecular mechanisms operative in living systems.

In nature, individual proteoforms (left), arising from variable sources of biological variation like PTMs, often exist in groups of related proteoforms. These dynamic ‘proteoform families’ (middle left) are the true protein products from the same human gene that convey information within signaling and regulatory networks (middle right) that underlie complex traits in wellness and disease (right). Discrete proteoforms and their families offer challenging, high-value targets for direct measurement by top-down proteomics.
In cancer epigenetics, there are several examples in which PTM crosstalk has been mapped definitively (see also the top rows of Table 1)22,45–49. It has been estimated that ~1,000 H3 proteoforms above a 0.1% abundance threshold exist for each of three histone H3 genes50. Such examples have been mapped in the context of multiple myeloma51 and diffuse intrinsic pontine glioma (DIPG)52. In each case, a global decrease in trimethylation of histone H3 on lysine 27 (H3K27me3; normally ~20% abundance) could result in hundreds of dysregulated histone codes in the diseased epigenome. In other disease areas, including organ fibrosis, several examples exist in which a mutation at one site can affect PTM profiles elsewhere on the protein53–55. In neurological disease and aging, modified proteins are the histopathological hallmarks of a number of diseases, such as SOD1 in amyotrophic lateral sclerosis (ALS)56,57, and a class of diseases long referred to as the proteinopathies57, including tauopathies in Alzheimer’s disease58 and inclusions of amyloid-β59, α-synuclein in Parkinson’s60,61 and multiple secondary ubiquitinopathies62,63. In heart disease, proteoform dynamics have been observed on proteins such as cardiac troponin I64, apolipoprotein C-III65, and B-type natriuretic peptide, the latter a key regulator of blood pressure and also the gold standard biomarker for clinically assessing heart failure66. Within the field of infectious diseases, proteoform-resolved approaches have been instrumental for understanding infectivity and dissemination of Salmonella typhimurium67, Corynebacterium glutamicum68 and Neisseria meningitidis69. Finally, the clinically deployed use of whole-protein MALDI–TOF MS for rapid identification of the species and strain of pathogenic bacteria has been adopted by thousands of hospitals and clinics worldwide70,71.
Deciphering the functions of proteoforms and their PTMs
With a far more precise understanding of protein composition and distribution in human biology, several advances can be anticipated. For compositional proteomics, the assignment of proteoform function and their combinations of PTMs can be made more efficient, as this is a holy grail in both basic and translational research. The use of proteoforms as protein-based biomarkers of disease is in its infancy (Table 1). To assign biological functions to proteoforms and their PTMs, a more precise map of protein composition would be the basis for the creation of new reagents and tools, such as the two examples outlined below.
Proteoform synthesis for functional studies
Understanding the language of site-specific PTMs remains a challenge, in part, because endogenous proteins are complex mixtures of related compositions, depending on their biosynthesis, functional regulation and subcellular distribution. Tools and technologies for precision proteoform synthesis (i.e., the ability to produce useful quantities of proteins with defined post-translational decorations for biochemical, mechanistic and structural studies) have advanced recently via two main approaches. First, the installation of genetically encoded chemistry by co-translationally incorporating noncanonical amino acids site specifically into proteins has afforded new advances (for example, phosphorylated amino acids)72–74. Furthermore, precision installation of glycans affords chemically defined glycoforms to study their structure and function. Recent efforts in glycoengineering of cellular systems have also expanded our ability to reliably synthesize chemically defined glycoforms75. Complementing these cell-based strategies (and emerging cell-free alternatives76) are well-established protein chemical synthesis and semisynthesis strategies for preparing proteoforms containing a wide repertoire of PTMs77. For example, histone proteoforms harboring multiple PTMs have been generated for functional studies via semisynthesis.
Affinity reagents and assays
The need to understand and assert PTM function benefits from antibody and mass-spectrometric methods working in a complementary and proteoform-informed fashion. For the development of affinity reagents, full-length proteoforms or domains decorated with PTMs are needed as antigens for production and validation of high-quality affinity reagents using methods like phage display78. The use of multiple antibodies, created using full-length antigens, can be deployed for cell-type-resolved or spatial mapping using frontier methods like mass cytometry (CyTOF) or for targeted analysis of a few dozen epitopes using single-cell proteomics79. Combining these methods with proteoform information by creating affinity reagents based upon precise knowledge of protein composition would enable efforts to map the spatial information of proteoforms in distinct cell types within human tissues. In the long-term, it is crucial to generate recombinant antibody tools as monospecific, permanent and renewable reagents to replace perishable animal-derived polyclonal or even monoclonal antibodies. Moreover, to detect proteins in their natural state, it is important that recombinant antibodies be generated to intact and folded proteins, because most high-affinity and specific antibodies recognize tertiary, not primary, sequence determinants. To this end, the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium’s (NCI–CPTAC) Antibody Portal (http://antibodies.cancer.gov) provides well-characterized, renewable antibodies against full-length protein antigens that are useful in development of targeted assays (e.g., immuno-MRM; http://assays.cancer.gov).
On the roles of PTM complexity in human biology
Below, we provide some thoughts on the possible roles of proteoform diversity, first through the evolution of complex traits and then on information processing for complex systems like human cells. Although the potential complexity afforded to proteins by PTMs is enormous, the few studies available suggest that only a small amount of this complexity is accessed in any given biological context. At the same time, different contexts may elicit diverse parts of proteoform complexity, so large swaths of PTM combinatorial space could have been explored over evolutionary timescales. One way to think about the role of this complexity is that it offers an ‘escape’ from the central dogma by accessing a far broader ensemble of protein compositions than can be realized from the genetic level alone80. PTMs thereby create capabilities that would not be accessible through protein translation or diverse splicing mechanisms. This viewpoint interprets PTMs as providing protein conformational states that may subsequently be exploited to modulate effector pathways in response to physiological conditions. For instance, PTM codes on central hub proteins like histones can be seen as a form of molecular ‘weak linkage’, which can facilitate evolution of higher complex traits81.
Another perspective is that biological systems select for proteoform diversity to improve robustness by having a distribution of forms and activities. This point of view actually argues for some level of promiscuity of PTM transferases in creating proteoform diversity. Natural selection must constantly wrestle with the tradeoff between fidelity and variability. High fidelity of biochemical processes would seem, at first blush, to be advantageous for a tightly orchestrated biological system; however, the higher the fidelity, the more amenable the organism is to the mutation and selection processes that are central to evolution. Moreover, although natural selection cannot look ahead, variation within proteins in a population allows more opportunities for later exploitation and adaptation as selective conditions change.
Protein compositions play a central role in cellular information processing47. For information ‘coding’ in cellular signaling, the ‘histone code’ is perhaps the best-known example22. It is usually implied that some highly modified proteins can act as hubs to integrate signals and orchestrate complex cellular functions82. In this area of chromatin biology, individual PTMs are sometimes called ‘marks’; combinations of PTM marks make up ‘codes’ (which are captured through proteoform measurement). Different combinatorial patterns of PTMs are ‘written’ on these hubs by the combined activity of forward-modifying and reverse-demodifying enzymes in response to varied physiological conditions. This framework is being extended to other PTM marks like phosphorylation, methylation, acetylation, ubiquitination, etc., and they combine to regulate responses to physiological conditions80 and to fine-tune individual molecular interactions. Such interactions can themselves be formidably intricate. More than 5% of the protein complement in a cell are enzymes (for example, ~500 kinases and ~140 protein phosphatases are encoded in the human genome), which can both compensate for and compete with each other at individual amino acids (for example, O-GlcNAcylation on canonical phosphorylation sites) and PTMs can be clustered in ‘hotspots’ at which different PTMs can influence each other. The resulting combinatorial patterns of PTMs convey information through ‘PTM crosstalk’ on the protein. The resulting PTM codes can then be ‘read’ by downstream interacting proteins in effector pathways. In this way, multiple upstream processes can collectively orchestrate a variety of downstream processes in various ways depending on conditions while working through one or a few hub proteins. The tumor suppressor p53, which can be modified on over 100 sites47, serves as a clear example of this case; however, what is unknown is how many of these PTMs coexist on the same proteoform.
Whether in the context of human evolution or cellular signaling, this tension arises for proteoforms: are PTMs and other differences between proteoforms carefully controlled and regulated or are they subject to high levels of stochastic ‘noise’? The answer may well be both, with different strategies being appropriate in different cellular or developmental contexts. For histones, a relatively strict doctrine of PTM writing and maintenance appears to limit the combinatorial explosion of proteoforms. Outside of histone biology, it is not well known whether systems for protein-based coding in the language of PTMs are prone to ‘loose constructionism’, defined here as a high tolerance for imprecision and noise in writing and erasing PTMs in cells of a living organism. How much of each strategy is operative and in which contexts? For unravelling such questions regarding the fidelity of information encoding and transfer, it will be essential to quantify the distribution of modification patterns on proteins, to develop mathematical and statistical frameworks for analyzing these distributions and to experimentally demonstrate how protein ‘readers’, ‘writers’ and ‘erasers’ interact with these distributions. Addressing these challenges in distinct areas of biology, even with improved tools for precise determination of protein composition at the proteoform level, will take several years to sort out47.
Summary and future prospects
From the many considerations above, a precise estimate of the number of human proteoforms is still difficult to provide. Finding ways to sample and better estimate proteoform number would assist in bounding the breadth and depth of the human proteome. For a given cell type, the depth of proteome coverage needed to detect the majority of human proteoforms above a specified threshold can serve as a protein-level analog of the 5× genome coverage employed for sequencing the first human genomes. For example, the 1,000,000 proteoform mark for cells of a given type would allow for mapping of ~100 proteoforms for each expressed gene. Compositional proteomics is maturing to the point whereby such depth may become possible to better decipher conserved, functional PTMs relative to biochemical noise. At whatever depth, building proteoform-informed measurement modalities to translate absolute molecular knowledge for proteins (and their combinatorial sources of modification) into deep functional insight will assist efforts to regularize and even domesticate the human proteome in the years ahead. Whether a large-scale endeavor to compositionally map cellular proteomes is launched depends on the perceived feasibility, endpoint(s) and value of such a project, and we hope this Perspective allows diverse communities to better frame the open questions about the composition and nature of the human proteome in both health and disease.
Acknowledgments
This article was enabled through generous funding of the Paul G. Allen Frontiers Program (Award 11715 to N.L.K.), which supports the curation of a human proteoform atlas (http://allen.kelleher.northwestern.edu). N.L.K. also acknowledges the NIH (P41 GM108569) and H. Thomas, M. Mullowney and S. Bratanch for their support and assistance in constructing this collaborative manuscript.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Gaudet P, et al. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 2017;45:D177–D182. doi: 10.1093/nar/gkw1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uhlén M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 3.Aken BL, et al. Ensembl 2017. Nucleic Acids Res. 2017;45:D635–D642. doi: 10.1093/nar/gkw1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. This manuscript introduces UniProt, a centralized, authoritative resource for protein sequences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duek P, Bairoch A, Gateau A, Vandenbrouck Y, Lane L. Missing protein landscape of human chromosomes 2 and 14: progress and current status. J. Proteome Res. 2016;15:3971–3978. doi: 10.1021/acs.jproteome.6b00443. [DOI] [PubMed] [Google Scholar]
- 6.Paik YK, et al. The chromosome-centric human proteome project for cataloging proteins encoded in the genome. Nat. Biotechnol. 2012;30:221–223. doi: 10.1038/nbt.2152. [DOI] [PubMed] [Google Scholar]
- 7.Hood L, Kronenberg M, Hunkapiller T. T cell antigen receptors and the immunoglobulin supergene family. Cell. 1985;40:225–229. doi: 10.1016/0092-8674(85)90133-3. [DOI] [PubMed] [Google Scholar]
- 8.Glanville J, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. USA. 2009;106:20216–20221. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith LM, Kelleher NL, The Consortium for Top Down Proteomics Proteoform: a single term describing protein complexity. Nat. Methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. This manuscript introduces and defines the term “Proteoform” The proteomics community has adopted this term, which regularizes the description of whole-protein molecules. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li YI, et al. RNA splicing is a primary link between genetic variation and disease. Science. 2016;352:600–604. doi: 10.1126/science.aad9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang X, et al. Widespread expansion of protein interaction capabilities by alternative splicing. Cell. 2016;164:805–817. doi: 10.1016/j.cell.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu. Rev. Genomics. Hum. Genet. 2010;11:25–44. doi: 10.1146/annurev-genom-082509-141720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Picardi E, D’Erchia AM, Lo Giudice C, Pesole G. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017;45:D750–D757. doi: 10.1093/nar/gkw767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruggles KV, et al. An analysis of the sensitivity of proteogenomic mapping of somatic mutations and novel splicing events in cancer. Mol. Cell. Proteomics. 2016;15:1060–1071. doi: 10.1074/mcp.M115.056226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gholami AM, et al. Global proteome analysis of the NCI-60 cell line panel. Cell Reports. 2013;4:609–620. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, et al. proBAMsuite, a bioinformatics framework for genome-based representation and analysis of proteomics data. Mol. Cell. Proteomics. 2016;15:1164–1175. doi: 10.1074/mcp.M115.052860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saghatelian A, Couso JP. Discovery and characterization of smORF-encoded bioactive polypeptides. Nat. Chem. Biol. 2015;11:909–916. doi: 10.1038/nchembio.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnoult N, et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature. 2017;549:548–552. doi: 10.1038/nature24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loftfield RB, Vanderjagt D. The frequency of errors in protein biosynthesis. Biochem. J. 1972;128:1353–1356. doi: 10.1042/bj1281353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu XC, et al. Identification of codon-specific serine to asparagine mistranslation in recombinant monoclonal antibodies by high-resolution mass spectrometry. Anal. Chem. 2009;81:9282–9290. doi: 10.1021/ac901541h. [DOI] [PubMed] [Google Scholar]
- 22.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. This manuscript describes the ‘histone code’, a complex set of PTMs that govern gene transcription. [DOI] [PubMed] [Google Scholar]
- 23.Toll H, et al. Glycosylation patterns of human chorionic gonadotropin revealed by liquid chromatography-mass spectrometry and bioinformatics. Electrophoresis. 2006;27:2734–2746. doi: 10.1002/elps.200600022. [DOI] [PubMed] [Google Scholar]
- 24.Wohlschlager T, et al. Proteomic Forum 2017. Deutsche Gesellschaft für Proteomforschung e.V.; Potsdam, Germany: 2017. Native mass spectrometry for the revelation of highly complex glycosylation in protein therapeutics. [Google Scholar]
- 25.Yang Y, et al. Hybrid mass spectrometry approaches in glycoprotein analysis and their usage in scoring biosimilarity. Nat. Commun. 2016;7:13397. doi: 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 27.Dang X, et al. The first pilot project of the consortium for top-down proteomics: a status report. Proteomics. 2014;14:1130–1140. doi: 10.1002/pmic.201300438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beck M, et al. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ponomarenko EA, et al. The size of the human proteome: the width and depth. Int. J. Anal. Chem. 2016;2016:7436849. doi: 10.1155/2016/7436849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ewing B, Green P. Analysis of expressed sequence tags indicates 35,000 human genes. Nat. Genet. 2000;25:232–234. doi: 10.1038/76115. [DOI] [PubMed] [Google Scholar]
- 31.Skinner OS, et al. Top-down characterization of endogenous protein complexes with native proteomics. Nat. Chem. Biol. 2018;14:36–41. doi: 10.1038/nchembio.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rissin DM, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010;28:595–599. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Chen B, Brown KA, Lin Z, Ge Y. Top-down proteomics: ready for prime time? Anal. Chem. 2018;90:110–127. doi: 10.1021/acs.analchem.7b04747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toby TK, Fornelli L, Kelleher NL. Progress in top-down proteomics and the analysis of proteoforms. Annu. Rev. Anal. Chem. (Palo Alto, Calif.) 2016;9:499–519. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aichler M, Walch A. MALDI Imaging mass spectrometry: current frontiers and perspectives in pathology research and practice. Lab. Invest. 2015;95:422–431. doi: 10.1038/labinvest.2014.156. [DOI] [PubMed] [Google Scholar]
- 37.Schey KL, Grey AC, Nicklay JJ. Mass spectrometry of membrane proteins: a focus on aquaporins. Biochemistry. 2013;52:3807–3817. doi: 10.1021/bi301604j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dilillo M, et al. Ultra-high mass resolution MALDI imaging mass spectrometry of proteins and metabolites in a mouse model of glioblastoma. Sci. Rep. 2017;7:603. doi: 10.1038/s41598-017-00703-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwiatkowski M, et al. Homogenization of tissues via picosecond-infrared laser (PIRL) ablation: Giving a closer view on the in-vivo composition of protein species as compared to mechanical homogenization. J. Proteomics. 2016;134:193–202. doi: 10.1016/j.jprot.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim MS, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilhelm M, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 42.Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA. The Human Cell Atlas: from vision to reality. Nature. 2017;550:451–453. doi: 10.1038/550451a. [DOI] [PubMed] [Google Scholar]
- 43.Kelleher NL. A cell-based approach to the human proteome project. J. Am. Soc. Mass Spectrom. 2012;23:1617–1624. doi: 10.1007/s13361-012-0469-9. This manuscript framed a project to define the human proteome by mapping the composition of ~1 billion proteoforms within all the different types of human cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savaryn JP, Catherman AD, Thomas PM, Abecassis MM, Kelleher NL. The emergence of top-down proteomics in clinical research. Genome Med. 2013;5:53. doi: 10.1186/gm457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benayoun BA, Veitia RA. A post-translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol. 2009;19:189–197. doi: 10.1016/j.tcb.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Dang X, et al. Label-free relative quantitation of isobaric and isomeric human histone H2A and H2B variants by fourier transform ion cyclotron resonance top-down MS/MS. J. Proteome Res. 2016;15:3196–3203. doi: 10.1021/acs.jproteome.6b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008;9:702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 48.Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- 49.Verhey KJ, Gaertig J. The tubulin code. Cell Cycle. 2007;6:2152–2160. doi: 10.4161/cc.6.17.4633. [DOI] [PubMed] [Google Scholar]
- 50.Sidoli S, Lin S, Karch KR, Garcia BA. Bottom-up and middle-down proteomics have comparable accuracies in defining histone post-translational modification relative abundance and stoichiometry. Anal. Chem. 2015;87:3129–3133. doi: 10.1021/acs.analchem.5b00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng Y, et al. Unabridged analysis of human histone H3 by differential top-down mass spectrometry reveals hypermethylated proteoforms from MMSET/NSD2 overexpression. Mol. Cell. Proteomics. 2016;15:776–790. doi: 10.1074/mcp.M115.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piunti A, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017;23:493–500. doi: 10.1038/nm.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Connors LH, et al. Heterogeneity in primary structure, post-translational modifications, and germline gene usage of nine full-length amyloidogenic kappa1 immunoglobulin light chains. Biochemistry. 2007;46:14259–14271. doi: 10.1021/bi7013773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klimtchuk ES, Prokaeva TB, Spencer BH, Gursky O, Connors LH. In vitro co-expression of human amyloidogenic immunoglobulin light and heavy chain proteins: a relevant cell-based model of AL amyloidosis. Amyloid. 2017;24:115–122. doi: 10.1080/13506129.2017.1336996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim A, et al. Characterization of transthyretin variants in familial transthyretin amyloidosis by mass spectrometric peptide mapping and DNA sequence analysis. Anal. Chem. 2002;74:741–751. doi: 10.1021/ac010780+. [DOI] [PubMed] [Google Scholar]
- 56.Bradley WG. Possible therapy for ALS based on the cyanobacteria/BMAA hypothesis. Amyotroph. Lateral Scler. 2009;10(Suppl 2):118–123. doi: 10.3109/17482960903285951. [DOI] [PubMed] [Google Scholar]
- 57.Schmitt ND, Agar JN. Parsing disease-relevant protein modifications from epiphenomena: perspective on the structural basis of SOD1-mediated ALS. J. Mass Spectrom. 2017;52:480–491. doi: 10.1002/jms.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dickson DW. Neuropathology of non-Alzheimer degenerative disorders. Int. J. Clin. Exp. Pathol. 2009;3:1–23. [PMC free article] [PubMed] [Google Scholar]
- 59.Wildburger NC, et al. Diversity of amyloid-beta proteoforms in the Alzheimer’s disease brain. Sci. Rep. 2017;7:9520. doi: 10.1038/s41598-017-10422-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kellie JF, et al. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci. Rep. 2014;4:5797. doi: 10.1038/srep05797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCann H, Stevens CH, Cartwright H, Halliday GM. α-Synucleinopathy phenotypes. Parkinsonism Relat. Disord. 2014;20(Suppl 1):S62–S67. doi: 10.1016/S1353-8020(13)70017-8. [DOI] [PubMed] [Google Scholar]
- 62.Dickson DW. Chapter 7 Ubiquitinopathies. Blue Books of Neurology. 2007;30:165–185. [Google Scholar]
- 63.Kabashi E, Durham HD. Failure of protein quality control in amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 2006;1762:1038–1050. doi: 10.1016/j.bbadis.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 64.Zhang J, et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J. Proteome Res. 2011;10:4054–4065. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mazur MT, et al. Quantitative analysis of intact apolipoproteins in human HDL by top-down differential mass spectrometry. Proc. Natl. Acad. Sci. USA. 2010;107:7728–7733. doi: 10.1073/pnas.0910776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang S, Raedschelders K, Santos M, Van Eyk JE. Profiling B-type natriuretic peptide cleavage peptidoforms in human plasma by capillary electrophoresis with electrospray ionization mass spectrometry. J. Proteome Res. 2017;16:4515–4522. doi: 10.1021/acs.jproteome.7b00482. [DOI] [PubMed] [Google Scholar]
- 67.Ansong C, et al. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella typhimurium in response to infection-like conditions. Proc. Natl. Acad. Sci. USA. 2013;110:10153–10158. doi: 10.1073/pnas.1221210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carel C, et al. Identification of specific posttranslational O-mycoloylations mediating protein targeting to the mycomembrane. Proc. Natl. Acad. Sci. USA. 2017;114:4231–4236. doi: 10.1073/pnas.1617888114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chamot-Rooke J, et al. Posttranslational modification of pili upon cell contact triggers N. meningitidis dissemination. Science. 2011;331:778–782. doi: 10.1126/science.1200729. [DOI] [PubMed] [Google Scholar]
- 70.van Belkum A, Welker M, Erhard M, Chatellier S. Biomedical mass spectrometry in today’s and tomorrow’s clinical microbiology laboratories. J. Clin. Microbiol. 2012;50:1513–1517. doi: 10.1128/JCM.00420-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lévesque S, et al. A side by side comparison of Bruker Biotyper and VITEK MS: utility of MALDI-TOF MS technology for microorganism identification in a public health reference laboratory. PLoS One. 2015;10:e0144878. doi: 10.1371/journal.pone.0144878. This manuscript describes the use of intact mass measurement to provide a specific, orthogonal method for microorganism identification in the clinical research lab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoppmann C, et al. Site-specific incorporation of phosphotyrosine using an expanded genetic code. Nat. Chem. Biol. 2017;13:842–844. doi: 10.1038/nchembio.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luo X, et al. Genetically encoding phosphotyrosine and its nonhydrolyzable analog in bacteria. Nat. Chem. Biol. 2017;13:845–849. doi: 10.1038/nchembio.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang A, et al. A chemical biology route to site-specific authentic protein modifications. Science. 2016;354:623–626. doi: 10.1126/science.aah4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baker JL, Çelik E, DeLisa MP. Expanding the glycoengineering toolbox: the rise of bacterial N-linked protein glycosylation. Trends Biotechnol. 2013;31:313–323. doi: 10.1016/j.tibtech.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 76.Oza JP, et al. Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nat. Commun. 2015;6:8168. doi: 10.1038/ncomms9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Müller MM, Muir TW. Histones: at the crossroads of peptide and protein chemistry. Chem. Rev. 2015;115:2296–2349. doi: 10.1021/cr5003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hornsby M, et al. A high through-put platform for recombinant antibodies to folded proteins. Mol. Cell. Proteomics. 2015;14:2833–2847. doi: 10.1074/mcp.O115.052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Porpiglia E, et al. High-resolution myogenic lineage mapping by single-cell mass cytometry. Nat. Cell Biol. 2017;19:558–567. doi: 10.1038/ncb3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prabakaran S, Lippens G, Steen H, Gunawardena J. Post-translational modification: nature’s escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012;4:565–583. doi: 10.1002/wsbm.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kirschner M, Gerhart J. Evolvability. Proc. Natl. Acad. Sci. USA. 1998;95:8420–8427. doi: 10.1073/pnas.95.15.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edwards AV, Schwämmle V, Larsen MR. Neuronal process structure and growth proteins are targets of heavy PTM regulation during brain development. J. Proteomics. 2014;101:77–87. doi: 10.1016/j.jprot.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 83.Sluchanko NN, Gusev NB. Moonlighting chaperone-like activity of the universal regulatory 14-3-3 proteins. FEBS J. 2017;284:1279–1295. doi: 10.1111/febs.13986. [DOI] [PubMed] [Google Scholar]
- 84.Howard TE, Shai SY, Langford KG, Martin BM, Bernstein KE. Transcription of testicular angiotensin-converting enzyme (ACE) is initiated within the 12th intron of the somatic ACE gene. Mol. Cell. Biol. 1990;10:4294–4302. doi: 10.1128/mcb.10.8.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schellenberger U, et al. The precursor to B-type natriuretic peptide is an O-linked glycoprotein. Arch. Biochem. Biophys. 2006;451:160–166. doi: 10.1016/j.abb.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 86.Zhang P, et al. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods. 2007;4:487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 88.Pesavento JJ, Bullock CR, LeDuc RD, Mizzen CA, Kelleher NL. Combinatorial modification of human histone H4 quantitated by two-dimensional liquid chromatography coupled with top down mass spectrometry. J. Biol. Chem. 2008;283:14927–14937. doi: 10.1074/jbc.M709796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bush DR, Zang L, Belov AM, Ivanov AR, Karger BL. High resolution CZE-MS quantitative characterization of intact biopharmaceutical proteins: proteoforms of interferon-b1. Anal. Chem. 2016;88:1138–1146. doi: 10.1021/acs.analchem.5b03218. [DOI] [PubMed] [Google Scholar]
- 90.Peng Y, et al. Top-down proteomics reveals concerted reductions in myofilament and Z-disc protein phosphorylation after acute myocardial infarction. Mol. Cell. Proteomics. 2014;13:2752–2764. doi: 10.1074/mcp.M114.040675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cummings RD. The repertoire of glycan determinants in the human glycome. Mol. Biosyst. 2009;5:1087–1104. doi: 10.1039/b907931a. [DOI] [PubMed] [Google Scholar]
- 92.Sidoli S, et al. Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics. 2014;14:2200–2211. doi: 10.1002/pmic.201400084. [DOI] [PubMed] [Google Scholar]