

Abstract
Epidemiological studies in humans and research in vertebrates indicates that developmental exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), a ubiquitous and biopersistent environmental toxicant, is associated with incidence of early congenital heart disease in the embryo and later in the adult. TCDD-mediated toxicity depends on the aryl hydrocarbon receptor (AHR) but the role of the TCDD-activated AHR in cardiac function is not well-defined. To characterize the mechanisms responsible for AHR-mediated disruption of heart function, we generated several mouse strains with cardiomyocyte-specific Ahr gene knockout. Here, we report results on one of these strains in which the Ahr gene was deleted by cre recombinase regulated by the promoter of the cardiomyocyte-specific Nkx2.5 gene. We crossed mice with loxP-targeted Ahrfx/fx alleles with Nkx2.5+/cre mice bearing a “knock-in” cre recombinase gene integrated into one of the Nkx2.5 alleles. In these mice, loss of one Nkx2.5 allele is associated with disrupted cardiac development. In males, Nkx2.5 hemizygosity resulted in cardiac haploinsufficiency characterized by hypertrophy, dilated cardiomyopathy, and impaired ejection fraction. Ahr ablation protected Nkx2.5+/cre haploinsufficient males from cardiac dysfunction while inducing a significant increase in body weight. These effects were absent or largely blunted in females. Starting at 3 months of age, mice were exposed by oral gavage to 1 μg/kg/week of TCDD or control vehicle for an additional 2 months. TCDD exposure restored cardiac physiology in aging males, appearing to compensate for the heart dysfunction caused by Nkx2.5 hemizygosity. Our findings underscore the conclusion that deletion of the Ahr gene in cardiomyocytes protects males from heart dysfunction due to NKX2.5 haploinsufficiency.
Keywords: aryl hydrocarbon receptor, cardiomyocytes, cardiac hypertrophy, Nkx2.5, haploinsufficiency
The aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor, mediates the teratogenic and carcinogenic effects of environmental planar aromatic xenobiotic hydrocarbons, among which the most potent agent is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). AHR is a member of the basic-region-helix-loop-helix PER/AHR nuclear translocator (ARNT)/SIM (bHLH-PAS) superfamily of transcription factors. Members of this superfamily function as sensors of extracellular signals and environmental stresses affecting growth and development (Gu et al., 2000). Activation by TCDD causes receptor translocation to the nucleus, dissociation from its cytosolic HSP90 chaperone and heterodimerization with its ARNT partner, also a member of the bHLH/PAS superfamily (Reyes et al., 1992). Binding of AHR-ARNT complexes to AHR-binding sites in the promoters of target genes recruits transcription cofactors and associated chromatin remodeling proteins and signals initiation of gene transcription (Schnekenburger et al., 2007). Well-known AHR target genes encode drug-metabolizing enzymes belonging to the cytochrome P450 CYP1 family and several phase II detoxification enzymes (Nebert et al., 2004). Emerging evidence indicates that in addition to the well-known roles of the receptor in mediating xenobiotic toxicity there are other AHR transcriptional targets, including genes involved in cell cycle regulation and morphogenetic processes that may play a vital function during embryonic development (Sartor et al., 2009). AHR is also involved in regulation of growth factor signaling, cell cycle proliferation, differentiation, arrest, and apoptosis that result from various forms of cross talk between AHR and other regulatory proteins (Ma et al., 2009).
TCDD-mediated AHR activation disrupts cardiovascular homeostasis in all species studied to date, including fish, avian, and mouse (Ivnitski-Steele et al., 2005). In humans, long-term epidemiologic studies have established a link between occupational exposure to high doses of TCDD and ischemic heart disease (Flesch-Janys et al., 1995). A higher incidence of hypertension, stroke, and coronary heart disease have been found in U.S. Army Chemical Corps veterans and Korean veterans who were occupationally exposed to Agent Orange in Vietnam, and exposed individuals from the accidental exposure in Seveso (Kang et al., 2006; Kim et al., 2003; Pesatori et al., 1998). A systematic analysis of all English-language epidemiologic studies of dioxin exposure and mortality from cardiovascular disease concluded that dioxin exposure in humans was associated with mortality from both ischemic heart disease and all cardiovascular disease (Humblet et al., 2008).
A number of physiological roles of the AHR have been recognized from the study of AHR null mice. Mice with a homozygous ablation of the Ahr gene are viable and resistant to TCDD toxicity (Fernandez-Salguero et al., 1996) although numerous age-related pathologies were observed in several organs, including reduced liver size, diminished reproductive capabilities, immunosuppression, epidermal hyperplasia, and an impaired cardiovascular phenotype (Fernandez-Salguero et al., 1995). Whole-body knockout mice showed a gross enlargement of the heart size and histologically demonstrated chronic, focal inflammation, and fibrosis in the ventricular and atrial myocardia (Fernandez-Salguero et al., 1997). Interference with endogenous AHR developmental functions, either by gene ablation or by in utero exposure to TCDD, disrupts stem cell differentiation and alters expression of homeobox transcription factors that control cardiomyogenesis (Wang et al., 2013), causing a phenotype of structural, molecular, and physiological cardiac abnormalities that progresses into the adults (Carreira et al., 2015a).
Cardiogenesis is a highly concerted process that requires precise orchestration of multiple gene networks regulating developmental commitments toward cellular differentiation, migration, proliferation, and death (Rana et al., 2013; Srivastava and Olson, 2000; Van Vliet et al., 2012). A key regulator of heart differentiation is the homeodomain-containing transcription factor NKX2.5, an evolutionarily conserved transcription factor that regulates cardiac myogenesis and heart morphogenesis from fly to man (Harvey, 1996). NKX2.5 is the earliest known cardiogenesis marker, expressed as early as E7.5 in mouse embryos (Schwartz and Olson, 1999). Targeted disruption of the Nkx2.5 gene in mice caused abnormal heart morphogenesis at E8.5 and early embryonic death, whereas hemizygous mice with only one functional Nkx2.5 allele suffer from hemodynamic insufficiency (Lyons et al., 1995; Tanaka et al., 1999). NKX2.5 mutations in mice and humans cause congenital cardiac malformations and atrioventricular conduction defects (Biben et al., 2000; Jay et al., 2004; Schott et al., 1998; Tanaka et al., 2002), suggesting that NKX2.5 controls cardiac gene programming and specifies heart lineage while working in combination with other regulators to maintain cardiac homeostasis. Interactions between NKX2.5 and AHR have just begun to be recognized; genome-wide studies in mouse embryonic stem cells have shown that AHR activation by TCDD represses Nkx2.5 mRNA and protein expression in a dose-dependent manner (Wang et al., 2010). Furthermore, in vivo studies have shown a significant decrease in the number of NKX2.5 positive nuclei in embryonic hearts of TCDD-exposed Ahr+/+ mice (Carreira et al., 2015b). However, Nkx2.5 regulation by AHR appears to be indirect or dependent on interactions between AHR and other factors, for a screen of the Nkx2.5 promoter region between − 10,000 and + 1000 nucleotides from the transcription start site failed to find canonical AHR-binding motifs (Wang et al., 2013).
To characterize the mechanisms responsible for AHR-mediated disruption of heart function, we have generated several mouse strains with cardiomyocyte-specific Ahr gene knockout. We previously reported on one of these, in which loxP-targeted Ahrfx/fx alleles were deleted by cardiomyocyte-specific expression of cre recombinase driven by the promoter of the βMhc (myosin heavy chain-beta) gene (Kurita et al., 2016). In those mice, AHR expression protected adult female mice from TCDD exposure-induced heart dysfunction, we further wished to test the hypothesis that gene-environment combinatorial interactions between AHR, TCDD exposure, and NKX2.5 might aggravate the cardiac insufficiency resulting from Nkx2.5 hemizygosity. To this end, we took advantage of the availability of haploinsufficient Nkx2.5+/cre mice, bearing a “knock-in” cre recombinase gene integrated into the Nkx2.5 locus, to knockout the cardiomyocyte Ahr gene simultaneously with the initiation of NKX2.5 expression and heart development at E7.5 in mice. Our findings underscore the conclusion that deletion of the AHR protects males against heart dysfunction due to NKX2.5 haploinsufficiency.
MATERIALS AND METHODS
Mice, genotyping, TCDD treatment, and determination of Ahr allele excision
We housed C57BL/6J mice in the Experimental Laboratory Animals Medical Services at the University of Cincinnati under controlled conditions of temperature, humidity, and lighting and provided standard mouse chow and water ad libitum. We conducted all experiments following the highest standards of humane care in accordance with the NIH Guide for the Care and Use of Laboratory Animals that have been approved by the University of Cincinnati Institutional Animal Care and Use Committee.
Ahrfx/fxNkx2.5+/+mice with a floxed Ahr allele were a generous gift from Dr Christopher Bradfield (University of Wisconsin-Madison) (Walisser et al., 2005). Ahr+/+Nkx2.5+/cre mice, bearing a cre recombinase transgene knocked-in in the Nkx2.5 promoter by homologous recombination were a generous gift of Dr Jeffery Molkentin (Cincinnati Children’s Hospital) and Dr R.J. Schwartz (Baylor College of Medicine, Houston, Texas) (Moses et al., 2001). Ahrfx/fx Nkx2.5+/+ mice were initially bred into Ahr+/+Nkx2.5+/cre mice to generate Ahr+/fxNkx2.5+/cre mice. Female Ahr+/fxNkx2.5+/cre mice were crossed to male Ahrfx/fxNkx2.5+/+ mice to generate Ahrfx/fxNkx2.5+/+(Ahr pseudo-wild type, Nkx2.5 wild type), Ahrfx/+Nkx2.5+/+ (Ahr pseudo-wild type, Nkx2.5 wild type), Ahrfx/fxNkx2.5+/cre (Ahr floxed, Nkx2.5 haploinsufficient), and Ahrfx/+Nkx2.5+/cre(Ahr pseudo-wild type, Nkx2.5 haploinsufficient). The Ahr-wild type genotypes Ahr+/+Nkx2.5+/+ and Ahr+/+Nkx2.5+/cre replaced the pseudo-Ahr wild type genotypes Ahrfx/+Nkx2.5+/+and Ahrfx/+Nkx2.5+/cre genotypes in the experiments shown in Figures 2 and 3. All mice used in the experiments reported here were backcrossed for at least 6 generations into a C57BL/6J background. To avoid litter effects, we used 1–2 males and the same number of females from each of several independent litters. The total number of mice in each experiment is indicated in the figure legends. When mice reached 3 months of age, groups of 5–8 mice were treated with corn oil or 1 μg TCDD/kg/week by oral gavage for 2 additional months, measuring their body weight once per week throughout the duration of the TCDD treatment. After discontinuation of TCDD treatment, mice were examined for echocardiographic parameters at 5, 7, and 12 months of age. Some mice were sacrificed at 9 months to determine organ weights. To genotype the cre transgene and the Ahr floxed, excised, or wild-type alleles, we used PCR analysis of DNA isolated from snipped ear tissue. Analysis of Ahrfx/fx excision was carried out by multiplex PCR of DNA isolated from several tissues and organs including heart ventricles using 2 forward primers and 1 reverse primer. The genotypes of all the mice used in these analyses were verified by PCR amplification. Information of primers used for genotyping and excision are shown in Table 1.
Figure 2.
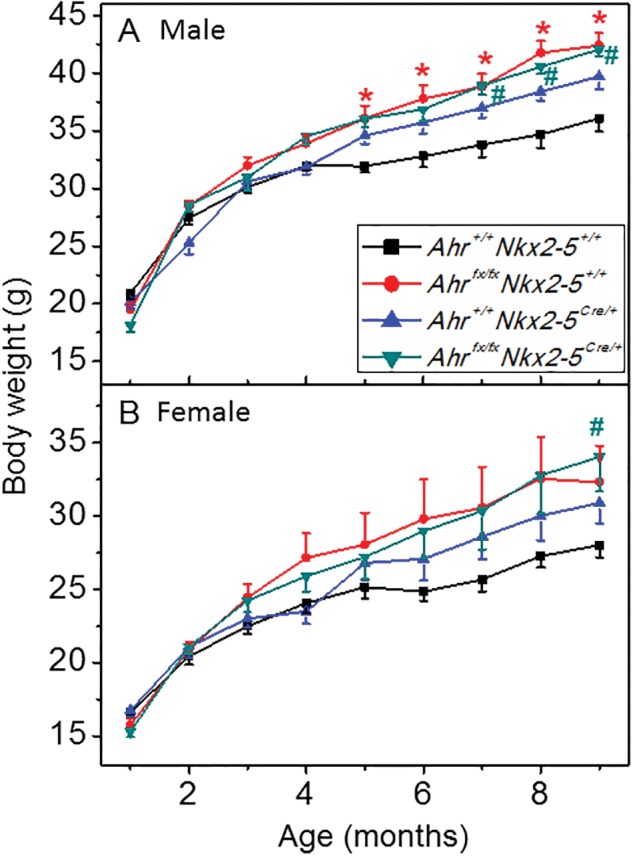
Body weight changes in cardiomyocyte-specific Ahr knockout mice. Five to eight mice from each sex, male (A) and female (B), were used for body weight measurements. Data are expressed as the mean ± SEM. * indicates significance in the comparison between Ahr+/+Nkx2.5+/+ and Ahrfx/fxNkx2.5+/+ and # indicates significance in the comparison between Ahr+/+Nkx2.5+/+ and Ahrfx/fxNkx2.5Cre/+(*#P < 0.05).
Figure 3.
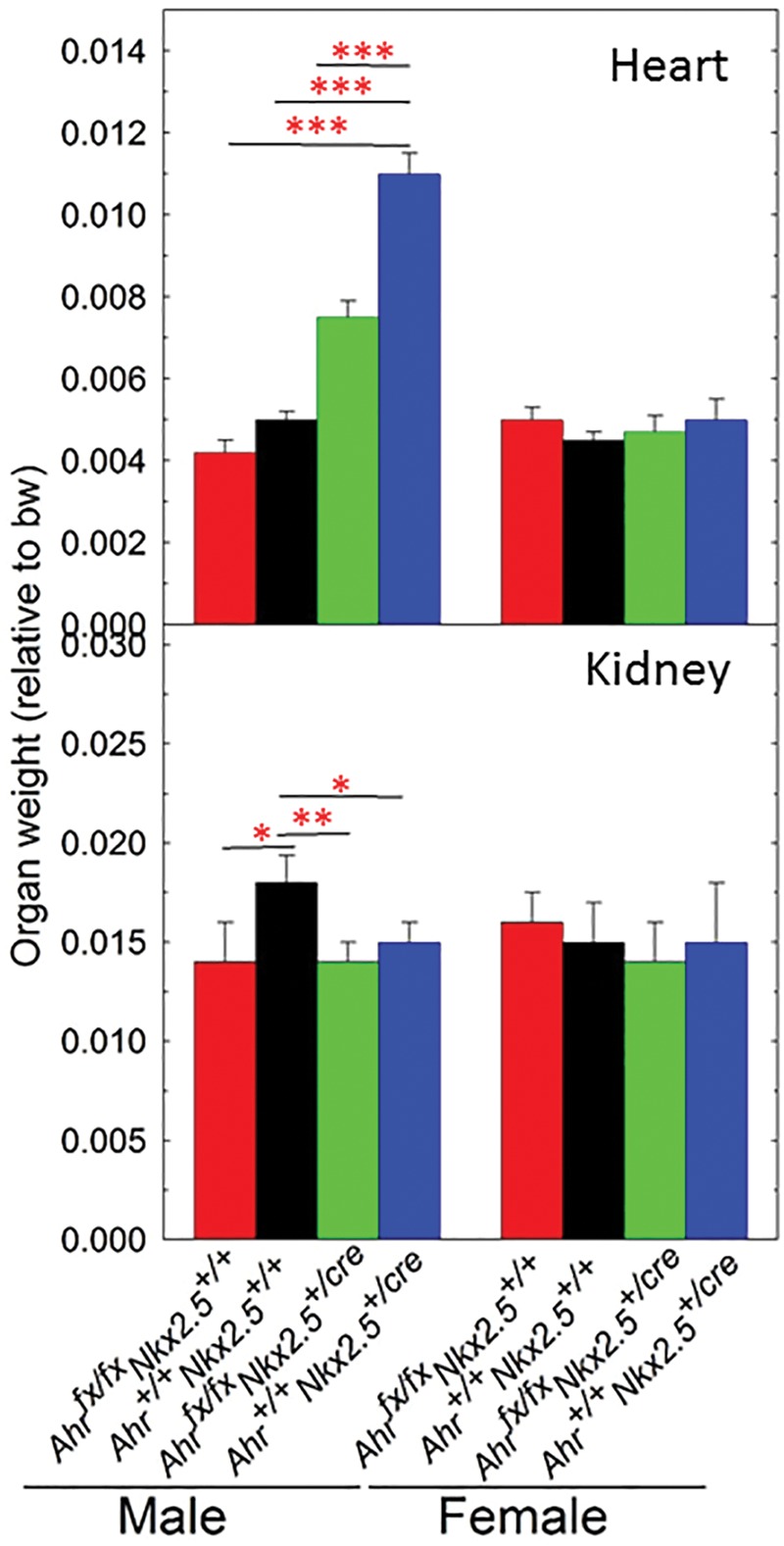
Organ weight (normalized to body weight) of heart and kidney at 9 months of age. Five to eight mice per group were used for weight determinations of male and female mice from 4 genotypes, Ahr+/+Nkx2.5+/+, Ahrfx/fxNkx2.5+/+, Ahr+/+Nkx2.5+/cre, and Ahrfx/fxNkx2.5+/cre. Data are expressed as the mean ± SEM. (*P < 0.05, **P < 0.01, and ***P < 0.001).
Table 1.
Primer List for Genotyping
| Forward (5′ > 3′) | Reverse (5′ > 3′) | Amplicon size (bp) | Target |
|---|---|---|---|
| GGCGTTTTCTGAGCATACCT | CTACACCAGAGACGGAAATCC | 587 | Nkx2.5cre allele |
| CAGTGGGAATAAGGCAAGAGTGA | GGTACAAGTGCACATGCCTGC | Ahr wt allele: 106 | Ahr+ or Ahrfx allele |
| Ahr fx allele: 140 | |||
| CAGTGGGAATAAGGCAAGAGTGA | GTCACTCAGCATTACACTTTCTA | 180 | Excised Ahrfx allele |
Echocardiography
A full echocardiographic study was carried out to determine functional abnormalities, as previously described (Rubinstein et al., 2014) at the times indicated. Briefly, mice were anesthetized with isoflurane (1.5%–2%), and images were obtained from a parasternal long axis view between 2 and 10 mm in depth in both M-mode and B-mode using the Vevo 2100 Ultrasound system equipped with a MS400 probe (30-MHz centerline frequency). As previously described, data were postprocessed using the Vevostain software (Visualsonic, Vevo 2100, v1.1.1 B1455) by an investigator blinded to the experiment working at a separate workstation. The B-mode, color, and Doppler images were analyzed for valvular function and structural abnormalities, while the M-mode images were postprocessed for cardiac functional analysis including ventricular size, ejection fraction, and cardiac output.
Collection of tissue samples
Following euthanasia, tissues were harvested, rinsed in PBS, gently blotted to remove excess fluids, and weighed using a high definition scale. Hearts were longitudinally bisected with one-half used for morphological studies (ie, histological processing) and the other one-half processed for molecular studies (real time RT-qPCR) as detailed later. When so indicated for reporting, organ weights were normalized to body weights.
Statistical analyses
All results are expressed as the mean ± SEM of biological replicates. Statistical analysis was performed using IBM SPSS Statistics ver. 19.0. A 1- or 2-way ANOVA followed by Bonferroni’s post hoc test was performed to compare means of body and tissue weights, and echocardiography. A P value of < 0.05 was considered statistically significant.
RESULTS
Cardiomyocyte-Specific Excision of the Ahr Gene
To investigate the potential interactions between AHR and the master cardiac transcription factor NKX2.5 in the developing heart, we used the cre-loxP system to generate cardiomyocyte-specific Ahr knockout mice. We crossed Nkx2.5+/cre, which express the cre recombinase gene driven by the cardiomyocyte-specific Nkx2.5 promoter, with Ahrfx/fxNkx2.5+/+ mice that have 2 floxed Ahr alleles. Further mating gave rise to mice of the genotypes described in the Materials and Methods section. To verify the excision of the floxed Ahr allele, we tested 15 different tissues from Ahrfx/fxNkx2.5+/cre mice for the loss of the Ahrfx allele. Only heart tissue showed significant excision of the floxed Ahr allele with no detectable excision in other tissues, strongly supporting the cardiomyocyte-specificity of the excision (Figure 1). Cardiomyocytes constitute < 50% of the heart cells (Ali et al., 2014) and noncardiomyocyte cells, such as fibroblasts, do not express NKX2.5. Most likely, the unexcised Ahrfx alleles that can still be detected in the heart (Figure 1) are present in these other cell types.
Figure 1.

Determination of tissue-specific Ahr excision in cardiomyocyte-specific Ahr knockout mice. A, PCR genotyping and gel electrophoresis analysis of Ahrfx/fxNkx2.5+/+; Ahr+/fxNkx2.5+/+; Ahrfx/fxNkx2.5+/cre; and Ahr+/fxNkx2.5+/cre in heart ventricles of 2-day postnatal (PND2) old mice. The panel shows the cre allele in Nkx2.5creand wild-type, excised, and unexcised Ahr floxed alleles. B, Genotyping of several organs in 30-day-old Ahrfx/fxNkx2.5+/cre mice. H, heart; L, liver; K, kidney; SP, spleen; TH, thymus; P, pancreas; ST, stomach; CO, colon; IN, intestine; M, muscle; LU, lung; B, brain; E, eye; and BW, bone marrow.
Ahr Ablation in Males Protects From Cardiac Hypertrophy Induced by Nkx2.5 Haploinsufficiency
Starting at 7 months of age, NKX2.5 haploidy in Nkx2.5+/cre mice caused an increased in the body weight of males compared with wild type (Figure 2A). Knocking out Ahr in NKX2.5 haploinsufficient mice significantly increased the body weight of male Ahrfx/fxNkx2.5+/cre even at an earlier age (5 months) (Figure 2A). In contrast, a significant increase of body weight in female mice of the same genotype was observed only at 9 months of age (Figure 2B), suggestive of a possible protective effect of sex. The bearing of 2 unexcised floxed Ahr alleles in mice of Ahrfx/fxNkx2.5+/+ genotype was sufficient to cause an increase of body weight in male mice from 5 to 9 months of age (Figure 2A) but not in females (Figure 2B). To determine whether genotype also had an effect on the organ weights of these mice, we measured organ weights in a subset of the mice when they reached 9 months of age. Compared with wild type, Ahr+/+Nkx2.5+/cre haploinsufficient males showed a significant increase in mean heart weight relative to body weight, consistent with the cardiac hypertrophy observed by others in these mice (Lyons et al., 1995; Tanaka et al., 2002). Knockout of the Ah receptor blunted the hypertrophy of the haploinsufficient Ahrfx/fxNkx2.5+/cre male mice and caused their mean heart weight to drop by 30% and be more similar to the weight of the fully wild-type Ahr+/+Nkx2.5+/+ mice (Figure 3-Heart). In contrast, Nkx2.5 haploinsufficiency showed no effect on the relative heart weights of females (Figure 3-Heart). All male mice of Ahrfx/fxNkx2.5+/+, Ahr+/+Nkx2.5+/cre, and Ahrfx/fxNkx2.5+/cre genotypes showed significantly lower kidney weights than wild-type males, although, surprisingly the presence of the loxP site in the unexcised floxed Ahr alleles in Ahrfx/fxNkx2.5+/+ was sufficient to cause a reduction in the kidney weight of these mice relative to the wild type (Figure 2-Kidney). We have no explanation for this phenomenon beyond the possibility that the insertion of the loxP site might somehow influence the expression levels of the Ahr gene in kidney. As in the heart, genotype had no other effect on the relative kidney weight of females (Figure 3-Kidney). In both males and females, genotype caused no changes in the relative weights of liver, spleen, or fat tissues (data not shown).
Ahr Ablation in the Heart Perturbs Cardiac Function
To determine whether Ahr ablation in the heart would hinder adult heart function and aggravate potential responses to TCDD, we sought to determine the extent to which echocardiographic parameters of heart physiology were altered by genetic disruption of the Ahr in cardiomyocytes. Resting (conscious) heart rates were determined in male and female mice at 5, 7, and 12 months of age. Echocardiographic assessment revealed no genotype-dependent difference in heart rate of males or females at 5 or 7 months (data not shown) but significant differences at the later age of 12 months. At this time phenotypic differences were evident in both males and females, leading to a significant heart rate depression in haploinsufficient Ahrfx/fxNkx2.5+/cre mice, with 2 deleted cardiac Ahr alleles, relative to their Ahr+/fxNkx2.5+/cre counterparts, with just one deleted Ahr allele, or to the genotypes with wild-type Nkx2.5+/+. This effect of Ahr genotype on heart rate was evident in both sexes (Figure 4A).
Figure 4.

Echosonographic determination of cardiac functions in mice with a cardiomyocyte-specific Ahr deletion. Five to eight mice of each sex and genotype, as shown in the figure, were used for echocardiographic determination of heart rate (A), volume in systole (B), and diastole (C); cardiac output (D), ejection fraction (E), and stroke volume (F) at 12 months of age. Data are expressed as the mean ± SEM. Asterisks indicate significance (*P < 0.05, **P < 0.01, and ***P < 0.001). Only the indicated comparisons reached statistical significance.
Loss of AHR in the heart also caused significant genotype- and sex-dependent changes in ventricular volume of Nkx2.5+/cre haploinsufficient mice. In males, significant differences due to genotype were observed both in systolic and diastolic volumes. Deletion of both Ahr alleles in these mice caused a significant difference in both systolic and diastolic volumes relative to wild-type males and a significant increase relative to the pseudo-wild type Ahr+/fxNkx2.5+/cre with only one Ahr allele excised (Figs. 4B and 4C). The effect of genotype on these parameters in females was either strongly blunted (Figs. 4B and 4D–F) or mildly significant (Figure 4C).
We also found significant changes due to genotype in cardiac output, ejection fraction, and stroke volume, but only in the male experimental groups, not in the female. Haploinsufficient mice of Ahr+/fxNkx2.5+/cre had significantly reduced cardiac output, ejection fraction, and stroke volume but ablation of both Ahrfx alleles in Ahrfx/fxNkx2.5+/cre males restored these functions to wild type-like levels (Figs. 4D–F).
TCDD Exposure Restores Cardiac Physiology in Aging Males
To determine whether TCDD exposure affected the outcome of gene interactions between Ahr and Nkx2.5, we focused on mice of 2 Nkx2.5+/cre genotypes, bearing either a wild-type Ahr+/fx allele combination—Ahr+/fxNkx2.5+/cre—or the fully ablated Ahrfx/fx complement—Ahrfx/fxNkx2.5+/cre. Echocardiographic parameters in these mice were determined at the end of a 2-month exposure to 1 μg TCDD/kg/week, from 3 to 5 months of age, and subsequently after 2 and 7 additional months unexposed to TCDD. No obvious differences of cardiac function in response to TCDD exposure were observed at 5 and 7 months but significant differences from control mice were found in aging 12-month-old males. No change in heart rate was evident in exposed mice relative to controls (Figure 5A) but exposure, terminated 7 months earlier, caused a significant reduction in ventricular volumes in systole and in diastole for both genotypes in 12-month-old males (Figs. 5B and 5C). Accordingly, we found a major TCDD exposure-dependent difference between 2 genotypes in cardiac output, ejection fraction, and stroke volume. As shown earlier in Figure 4, cardiac function was significantly depressed in male Ahr+/fxNkx2.5+/cremice and restored to wild-type levels by full ablation of 2 Ahrfx alleles. TCDD exposure caused no change in the parameters in Ahrfx/fxNkx2.5+/cre mice but increased them to wild-type levels in Ahr+/fxNkx2.5+/cremales (Figs. 5D–F).
Figure 5.

Echocardiographic analyses of heart function in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposed and control mice with a cardiomyocyte-specific Ahr deletion. Five to eight 3-month-old mice of each sex and genotype were exposed for 2 months to 1 µg/kg/week of TCDD or corn oil vehicle by gavage. At 5 months of age, exposure was terminated and the mice maintained until they reach 12 months, at which time they were used for echocardiographic determination of the same parameters analyzed in Figure 4. C, corn oil control and T, TCDD. Data are expressed as the mean ± SEM. Asterisks indicate significance (*P < 0.05, **P < 0.01, and ***P < 0.001).
DISCUSSION
In this study, we have investigated the role of the interactions between NKX2.5, AHR, and TCDD on cardiac function and structure. Previous studies on whole-body Ahr knockout mice have shown a phenotype of heart hypertrophy and fibrosis, suggesting that the AHR plays an endogenous role in cardiac function and in TCDD-induced cardiotoxicity (Fernandez-Salguero et al., 1997). Similar studies on whole-body Ahr knockout mice (Kopf et al., 2008; Kopf and Walker, 2009) and in mice transgenic for a constitutively activated AHR (Brunnberg et al., 2006) demonstrated that adult TCDD exposure induces an increase in blood pressure and heart hypertrophy, supporting the conclusion that endogenous cardiac AHR functions are responsible for heart toxicity in response to TCDD exposure. Recently, to determine the role of cardiomyocyte-specific Ahr in heart function and its disruption by TCDD, we generated another cardiomyocyte-specific Ahr knockout mouse in which loxP-targeted Ahrfx/fx alleles were deleted by expression of cre recombinase driven by the promoter of the βMhc (myosin heavy chain-beta) gene (Kurita et al., 2016). In these mice, AHR expression protected adult female mice from TCDD-exposure induced heart dysfunction, including a significant increase in body weight, blood pressure, and cardiac hypertrophy and a decrease in cardiac ejection fraction (Kurita et al., 2016). Because expression of myosin heavy chain-beta, which starts at E10.5 in mice, is a later event in cardiomyocyte development than expression of NKX2.5 (Ng et al., 1991), we argued that an earlier Ahr deletion might have more dire consequences in the development of a heart disease phenotype. Hence, we chose to drive expression of cre recombinase with the promoter of the Nkx2.5 gene, the earliest known cardiogenesis marker, expressed as early as E7.5 in mouse embryos (Schwartz and Olson, 1999). In addition to the earlier expression time, a second advantage of this choice was that the cre gene is integrated into the Nkx2.5 locus, such that “knock-in” Nkx2.5+/cre mice bear a single functional Nkx2.5 allele and thus are NKX2.5 haploinsufficient (Lyons et al., 1995; Tanaka et al., 1999). Using these mice as the genetic background of the Ahr ablation would help determine whether AHR cooperates with or antagonizes NKX2.5 haploinsufficiency.
Nkx2.5 haploinsufficient mice had significantly increased left ventricular systolic and diastolic volume in both Ahr+/fxNkx2.5+/cre and Ahrfx/fxNkx2.5+/cre males, with no effect of Ahr genotype detected in Nkx2.5+/cre females. Cardiac deletion of both Ahr alleles in Ahrfx/fxNkx2.5+/cre males restored the loss of cardiac function of Ahrfx/+Nkx2.5+/cre. Females, however, showed no loss of cardiac function, providing strong evidence of sexual dimorphism. For the most part, AHR-mediated responses were absent in females and present in males, albeit at times with a deleterious effect. Ahr ablation provided males with significant protection against body weight and heart weight increase, and increase of left ventricle volume, presumably the result of Nkx2.5 haploinsufficiency. Paradoxically, the interactions between Ahr and Nkx2.5 had in some occasions a detrimental effect on male heart function. Conversely, females showed AHR-independent responses or no response at all to Nkx2.5 haploinsufficiency in most of the parameters tested. Cardiomyocytes constitute < 50% of the cells in the heart (Ali et al., 2014); for which reason, it is unfeasible to determine whether cardiomyocyte AHR-independent responses in females are truly cardiomyocyte AHR independent or are dependent on the mesenchymal or epicardial AHR, which are not ablated in the Ahrfx/fxNkx2.5Cre/+ mice. In contrast, the AHR-dependent responses in the males are unambiguously dependent on the cardiomyocyte AHR.
Interestingly, a majority of the findings driven by whole-body AHR disruption that we have observed in the past were generally more pronounced in females than in males (Carreira et al., 2015a). Although our current results do not address sexual dimorphism directly, it is reasonable to surmise that a network of interacting signals regulated by Ahr and other genes might be responsible for sex-specific responses that may also be expressed in a tissue-specific manner. Our findings suggest that the compensatory mechanisms at play in the heart may be sufficient in one sex but not the other to cope with environmental stressors. Preeminent among these are dimorphic hormonal responses, such as the complexes formed between the AHR and the estrogen receptor, which may modify estrogenic responses (Carlson and Perdew, 2002).
In this work, we set out to test whether interactions between Ahr and Nkx2.5, in combination with TCDD exposure might aggravate the cardiac insufficiency resulting from Nkx2.5 hemizygosity. Our data indicate that in this context insufficiency is in fact a function of at least 3 components: Ahr genetics, TCDD exposure, and cell lineage identity. The large increase in ventricular volume resulting from Nkx2.5 hemizygosity is increased even further by Ahr ablation, but regardless of Ahr deletion in cardiomyocytes, the increase is suppressed by TCDD, suggesting that the effect is either AHR-independent or -dependent on the unexcised Ahr gene present on noncardiomyocyte cardiac cells. In contrast, all 3, cardiac output, stroke volume, and ejection fraction, are largely reduced by Nkx2.5 hemizygosity, and all 3 are rescued by deletion of the Ahr gene, suggesting a detrimental cooperation between NKX2.5 and AHR. It is somewhat surprising that in this case TCDD promotes a beneficial outcome, rescuing the mice from the detrimental effects of the AHR-NKX2.5 interaction.
It is intriguing that the response to TCDD is so protracted, to the extent that at 12 months of age, 7 months after termination of exposure, TCDD- and AHR-dependent disruption of echocardiographic parameters still persists in males. Continued presence of TCDD in circulation is unlikely to be the reason of this persistence because the biological half-life of TCDD in mice is only a few days (DeVito and Birnbaum, 1995). It is striking that deletion of the Ahr gene on embryonic day E7.5, the time of Nkx2.5 expression, has such protective consequences in adult heart dysfunction and handling of TCDD. Ironically, rather than being detrimental, adult TCDD exposure had no cardiac dysfunction effects on the Ahrfx/fxNkx2.5+/cremice but instead brought these functions back to normal in mice of Ahr+/fxNkx2.5+/cre genotype, likely reflecting multiple interconnected compensatory responses in cardiac function and structure. Possibly, the ones that we observe are just the more evident subset of complex molecular departures from homeostasis that translate into a more severe adult phenotype (Carreira et al., 2015a,b). To identify the sustainable functional signals and long-term biological processes triggered by Ahr ablation would be fundamental to understand the central role that the AHR signaling network plays in cardiovascular function and dysfunction.
ACKNOWLEDGMENTS
The authors thank Dr Ying Xia, Dr Chia-I Ko, Dr Hongxia Zhang, Andrew VonHandorf, and Matthew de Gannes for a critical reading of the article.
FUNDING
This research was supported by National Institute of Environmental Health Sciences (NIEHS) grants (R01 ES06273, R01 ES024744, and R01 ES10807).
REFERENCES
- Ali S. R., Hippenmeyer S., Saadat L. V., Luo L., Weissman I. L., Ardehali R. (2014). Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc. Natl. Acad. Sci. U.S.A. 111, 8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biben C., Weber R., Kesteven S., Stanley E., McDonald L., Elliott D. A., Barnett L., Koentgen F., Robb L., Feneley M., et al. (2000). Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ. Res. 87, 888–895. [DOI] [PubMed] [Google Scholar]
- Brunnberg S., Andersson P., Lindstam M., Paulson I., Poellinger L., Hanberg A. (2006). The constitutively active Ah receptor (CA-Ahr) mouse as a potential model for dioxin exposure–effects in vital organs. Toxicology 224, 191–201. [DOI] [PubMed] [Google Scholar]
- Carlson D. B., Perdew G. H. (2002). A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J. Biochem. Mol. Toxicol. 16, 317–325. [DOI] [PubMed] [Google Scholar]
- Carreira V. S., Fan Y., Kurita H., Wang Q., Ko C. I., Naticchioni M., Jiang M., Koch S., Zhang X., Biesiada J., et al. (2015a). Disruption of Ah receptor signaling during mouse development leads to abnormal cardiac structure and function in the adult. PLoS One 10, e0142440.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira V. S., Fan Y., Wang Q., Zhang X., Kurita H., Ko C. I., Naticchioni M., Jiang M., Koch S., Medvedovic M., et al. (2015b). Ah receptor signaling controls the expression of cardiac development and homeostasis genes. Toxicol. Sci. 147, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito M. J., Birnbaum L. S. (1995). The importance of pharmacokinetics in determining the relative potency of 2,3,7,8-tetrachlorodibenzo-p-dioxin and 2,3,7,8-tetrachlorodibenzofuran. Fundam. Appl. Toxicol. 24, 145–148. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P., Hilbert D. M., Rudikoff S., Ward J. M., Gonzalez F. J. (1996). Aryl hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 140, 173–179. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P., Pineau T., Hilbert D. M., McPhail T., Lee S. S., Kimura S., Nebert D. W., Rudikoff S., Ward J. M., Gonzalez F. J. (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P. M., Ward J. M., Sundberg J. P., Gonzalez F. J. (1997). Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 34, 605–614. [DOI] [PubMed] [Google Scholar]
- Flesch-Janys D., Berger J., Gurn P., Manz A., Nagel S., Waltsgott H., Dwyer J. H. (1995). Exposure to polychlorinated dioxins and furans (PCDD/F) and mortality in a cohort of workers from a herbicide-producing plant in Hamburg, Federal Republic of Germany. Am. J. Epidemiol. 142, 1165–1175. [DOI] [PubMed] [Google Scholar]
- Gu Y. Z., Hogenesch J. B., Bradfield C. A. (2000). The PAS superfamily: Sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 40, 519–561. [DOI] [PubMed] [Google Scholar]
- Harvey R. P. (1996). NK-2 homeobox genes and heart development. Dev. Biol. 178, 203–216. [DOI] [PubMed] [Google Scholar]
- Humblet O., Birnbaum L., Rimm E., Mittleman M. A., Hauser R. (2008). Dioxins and cardiovascular disease mortality. Environ. Health Perspect. 116, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivnitski-Steele I. D., Friggens M., Chavez M., Walker M. K. (2005). 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibition of coronary vasculogenesis is mediated, in part, by reduced responsiveness to endogenous angiogenic stimuli, including vascular endothelial growth factor A (VEGF-A). Birth Defects Res. A Clin. Mol. Teratol. 73, 440–446. [DOI] [PubMed] [Google Scholar]
- Jay P. Y., Harris B. S., Maguire C. T., Buerger A., Wakimoto H., Tanaka M., Kupershmidt S., Roden D. M., Schultheiss T. M., O’Brien T. X., et al. (2004). Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J. Clin. Invest. 113, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H. K., Dalager N. A., Needham L. L., Patterson D. G. Jr, Lees P. S., Yates K., Matanoski G. M. (2006). Health status of Army Chemical Corps Vietnam veterans who sprayed defoliant in Vietnam. Am. J. Ind. Med. 49, 875–884. [DOI] [PubMed] [Google Scholar]
- Kim J. S., Lim H. S., Cho S. I., Cheong H. K., Lim M. K. (2003). Impact of Agent Orange exposure among Korean Vietnam veterans. Ind. Health 41, 149–157. [DOI] [PubMed] [Google Scholar]
- Kopf P. G., Huwe J. K., Walker M. K. (2008). Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are associated with increased superoxide. Cardiovasc. Toxicol. 8, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf P. G., Walker M. K. (2009). Overview of developmental heart defects by dioxins, PCBs, and pesticides. J. Environ. Sci. Health C. Environ. Carcinog. Ecotoxicol. Rev. 27, 276–285. [DOI] [PubMed] [Google Scholar]
- Kurita H., Carreira V. S., Fan Y., Jiang M., Naticchioni M., Koch S., Rubinstein J., Puga A. (2016). Ah receptor expression in cardiomyocytes protects adult female mice from heart dysfunction induced by TCDD exposure. Toxicology 355–356, 9–20. [DOI] [PubMed] [Google Scholar]
- Lyons I., Parsons L. M., Hartley L., Li R., Andrews J. E., Robb L., Harvey R. P. (1995). Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 9, 1654–1666. [DOI] [PubMed] [Google Scholar]
- Ma C., Marlowe J. L., Puga A. (2009). The aryl hydrocarbon receptor at the crossroads of multiple signaling pathways. EXS 99, 231–257. [DOI] [PubMed] [Google Scholar]
- Moses K. A., DeMayo F., Braun R. M., Reecy J. L., Schwartz R. J. (2001). Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 31, 176–180. [DOI] [PubMed] [Google Scholar]
- Nebert D. W., Dalton T. P., Okey A. B., Gonzalez F. J. (2004). Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 279, 23847–23850. [DOI] [PubMed] [Google Scholar]
- Ng W. A., Grupp I. L., Subramaniam A., Robbins J. (1991). Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ. Res. 68, 1742–1750. [DOI] [PubMed] [Google Scholar]
- Pesatori A. C., Zocchetti C., Guercilena S., Consonni D., Turrini D., Bertazzi P. A. (1998). Dioxin exposure and non-malignant health effects: A mortality study. Occup. Environ. Med. 55, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana M. S., Christoffels V. M., Moorman A. F. (2013). A molecular and genetic outline of cardiac morphogenesis. Acta Physiol. 207, 588–615. [DOI] [PubMed] [Google Scholar]
- Reyes H., Reiz-Porszasz S., Hankinson O. (1992). Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256, 1193–1195. [DOI] [PubMed] [Google Scholar]
- Rubinstein J., Lasko V. M., Koch S. E., Singh V. P., Carreira V., Robbins N., Patel A. R., Jiang M., Bidwell P., Kranias E. G., et al. (2014). Novel role of transient receptor potential vanilloid 2 in the regulation of cardiac performance. Am. J. Physiol. Heart Circ. Physiol. 306, H574–H584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor M. A., Schnekenburger M., Marlowe J. L., Reichard J. F., Wang Y., Fan Y., Ma C., Karyala S., Halbleib D., Liu X., et al. (2009). Genomewide analysis of aryl hydrocarbon receptor binding targets reveals an extensive array of gene clusters that control morphogenetic and developmental programs. Environ. Health Perspect. 117, 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnekenburger M., Peng L., Puga A. (2007). HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation. Biochim. Biophys. Acta 1769, 569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott J. J., Benson D. W., Basson C. T., Pease W., Silberbach G. M., Moak J. P., Maron B. J., Seidman C. E., Seidman J. G. (1998). Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281, 108–111. [DOI] [PubMed] [Google Scholar]
- Schwartz R. J., Olson E. N. (1999). Building the heart piece by piece: Modularity of cis-elements regulating Nkx2-5 transcription. Development 126, 4187–4192. [DOI] [PubMed] [Google Scholar]
- Srivastava D., Olson E. N. (2000). A genetic blueprint for cardiac development. Nature 407, 221–226. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Berul C. I., Ishii M., Jay P. Y., Wakimoto H., Douglas P., Yamasaki N., Kawamoto T., Gehrmann J., Maguire C. T., et al. (2002). A mouse model of congenital heart disease: Cardiac arrhythmias and atrial septal defect caused by haploinsufficiency of the cardiac transcription factor Csx/Nkx2.5. Cold Spring Harb. Symp. Quant. Biol. 67, 317–325. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Chen Z., Bartunkova S., Yamasaki N., Izumo S. (1999). The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 126, 1269–1280. [DOI] [PubMed] [Google Scholar]
- Van Vliet P., Wu S. M., Zaffran S., Puceat M. (2012). Early cardiac development: A view from stem cells to embryos. Cardiovasc. Res. 96, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walisser J. A., Glover E., Pande K., Liss A. L., Bradfield C. A. (2005). Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. U.S.A. 102, 17858–17863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Chen J., Ko C. I., Fan Y., Carreira V., Chen Y., Xia Y., Medvedovic M., Puga A. (2013). Disruption of aryl hydrocarbon receptor homeostatic levels during embryonic stem cell differentiation alters expression of homeobox transcription factors that control cardiomyogenesis. Environ. Health Perspect. 121, 1334–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Fan Y., Puga A. (2010). Dioxin exposure disrupts the differentiation of mouse embryonic stem cells into cardiomyocytes. Toxicol. Sci. 115, 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]