

Tau proteins form diverse conformers known as strains in Alzheimer's disease and allied disorders. Ono et al. report differential selectivity of two tau PET radioligands, [11C]PBB3 and [18F]AV-1451, for these fibril subspecies, supporting the utility of these imaging agents for pursuing roles of tau strains in the pathogenesis of tauopathies.
Keywords: tau PET ligand, PBB3/AV-1451, Alzheimer’s disease, N279K/G272V FTDP-17-MAPT mutation, progressive supranuclear palsy/corticobasal degeneration/Pick’s disease
Abstract
Diverse neurodegenerative disorders are characterized by deposition of tau fibrils composed of conformers (i.e. strains) unique to each illness. The development of tau imaging agents has enabled visualization of tau lesions in tauopathy patients, but the modes of their binding to different tau strains remain elusive. Here we compared binding of tau positron emission tomography ligands, PBB3 and AV-1451, by fluorescence, autoradiography and homogenate binding assays with homologous and heterologous blockades using tauopathy brain samples. Fluorescence microscopy demonstrated intense labelling of non-ghost and ghost tangles with PBB3 and AV-1451, while dystrophic neurites were more clearly detected by PBB3 in brains of Alzheimer’s disease and diffuse neurofibrillary tangles with calcification, characterized by accumulation of all six tau isoforms. Correspondingly, partially distinct distributions of autoradiographic labelling of Alzheimer’s disease slices with 11C-PBB3 and 18F-AV-1451 were noted. Neuronal and glial tau lesions comprised of 4-repeat isoforms in brains of progressive supranuclear palsy, corticobasal degeneration and familial tauopathy due to N279K tau mutation and 3-repeat isoforms in brains of Pick’s disease and familial tauopathy due to G272V tau mutation were sensitively detected by PBB3 fluorescence in contrast to very weak AV-1451 signals. This was in line with moderate 11C-PBB3 versus faint 18F-AV-1451 autoradiographic labelling of these tissues. Radioligand binding to brain homogenates revealed multiple binding components with differential affinities for 11C-PBB3 and 18F-AV-1451, and higher availability of binding sites on progressive supranuclear palsy tau deposits for 11C-PBB3 than 18F-AV-1451. Our data indicate distinct selectivity of PBB3 compared to AV-1451 for diverse tau fibril strains. This highlights the more robust ability of PBB3 to capture wide-range tau pathologies.
Introduction
Accumulation of filamentous tau protein aggregates in the brain is characteristic of Alzheimer’s disease and allied neurodegenerative disorders collectively referred to as tauopathies (Lee et al., 2001). The discovery of tau gene mutations in familial tauopathy termed frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) and investigations of various tau transgenic mouse lines provided compelling evidence for the mechanistic implication of tau abnormalities in neurotoxic insults (Lee et al., 2001). Composition of tau isoforms may give rise to conformational diversity of tau fibrils, which are ultrastructurally identified as paired helical filaments and straight filaments or ribbons (Bibow et al., 2011; Murray et al., 2014). These and other minor conformational variants, dubbed ‘strains’, determine subcellular, cellular and regional localizations of tau aggregates in close association with pathological and clinical phenotypes of each tauopathy (Feany and Dickson, 1995; Murray et al., 2014). To date, converging experimental evidence supports the view that misfolding of tau can propagate intercellularly similar to prion proteins, with an original tau strain being preserved during the transmission process (Goedert et al., 2014; Sanders et al., 2014).
As there has been growing demand for diagnostic and therapeutic approaches to tau pathologies, small-molecule agents for PET have been developed to visualize tau deposits in the brains of living subjects. These tracers are in principle ligands that bind to a β-pleated sheet secondary structure forming in tau filaments, and are likely to dock a binding pocket on the β-sheet of tau, which may differ structurally from binding pockets in amyloid-β fibrils (Berriman et al., 2003). Similarly, there may be conformational and structural differences among tracer binding components on distinct tau fibril strains, particularly in with respect to tau isoform compositions (Hasegawa et al., 2014; Taniguchi-Watanabe et al., 2016), since an alternative splicing domain, exon 10, is a constituent of the β-sheet domain of fibrillary tau assemblies (Li et al., 2002; von Bergen et al., 2006; Daebel et al., 2012).
Among three classes of tau PET tracers, 11C-pyridinyl-butadienyl-benzothiazole 3 (11C-PBB3) was the first reported to detect a broad range of tau inclusions, and in vivo PET data supported its utility for detecting tau lesions not only in patients with Alzheimer’s disease but also in subjects with non-Alzheimer’s disease dementias as exemplified by corticobasal degeneration (CBD) (Maruyama et al., 2013). Alzheimer’s disease tau aggregates composed of all six tau isoforms are dominated by paired helical filaments (Goedert et al., 1992), while straight filaments are present as the major morphology of tau filaments in a large subset of tau-positive frontotemporal lobar degenerations (FTLDs) characterized by deposition of tau isoforms (FTLD-Tau) with 4-repeat domains, such as progressive supranuclear palsy (PSP) and CBD (Flament et al., 1991; Ksiezak-Reding et al., 1994), and FTLD-Tau disorders with 3-repeat domains, such as Pick’s disease (Kato and Nakamura, 1990). It has also been documented that other groups of tau radioligands, including 18F-AV-1451 (also known as 18F-T807), THK-5117 and THK-5351, react with Alzheimer’s disease tau tangles as well as a some FTLD-Tau non-Alzheimer’s disease tau deposits (Chien et al., 2013; Okamura et al., 2013; Harada et al., 2014, 2015, 2016; Rabinovici et al., 2015; Vettermann et al., 2016). Meanwhile, a recent study has raised the possibility that 18F-AV-1451 selectively binds to Alzheimer’s disease paired helical filaments but binds less avidly to other tau fibril types (Marquié et al., 2015; Lowe et al., 2016; Sander et al., 2016). Identification of tau fibril strains accessible to each tau PET ligand would be of critical significance for the use of such imaging compounds as conformational probes, serving for early and differential diagnosis of dementing tauopathies and therapeutic application of a strain- and/or conformation-specific tau antibody immune therapy (Yanamandra et al., 2013). Towards this goal, a head-to-head comparison of different tau tracers using the same brain samples would provide crucial information for the selection of imaging ligands capable of binding to one or more tau fibril strains.
Here, we generated non-labelled and 18F-labelled AV-1451, and performed fluorescence microscopic and autoradiographic analyses along with binding assays using human neurodegenerative brain tissues, in order to comparatively examine binding properties of PBB3 and AV-1451. The results reported here suggest that there is a differential selectivity of these two compounds for subspecies of tau deposits in Alzheimer’s disease and non-Alzheimer’s disease tauopathies, and sensitive detection of non-Alzheimer’s disease tau lesions by PBB3 relative to AV-1451.
Materials and methods
Post-mortem brain tissues
Post-mortem human brains were obtained from autopsies carried out at the Department of Neuroscience of the Mayo Clinic on patients with PSP, CBD and FTDP-17 harbouring the N279K mutation in the MAPT gene (Tsuboi et al., 2002; Arvanitakis et al., 2007); at the Center for Neurodegenerative Disease Research of the University of Pennsylvania Perelman School of Medicine on patients with Alzheimer’s and Pick’s diseases; at the Department of Neuropathology of the Aichi Medical University on patients with diffuse neurofibrillary tangles with calcification (DNTC) (Iwasaki et al., 2009); at the Department of Neurology at the Chiba-East National Hospital on patients with PSP; and at the Department of Neurology of the Erasmus Medical Center on patients with FTDP-17 harbouring the G272V mutation in the MAPT gene (Hutton et al., 1998; Spillantini et al., 1998). Tissues for homogenate binding assays were frozen, and tissues for histochemical, immunohistochemical and autoradiographic labelling were frozen or fixed in 10% neutral buffered formalin followed by embedding in paraffin blocks.
Compounds and antibodies
PBB3 [2-((1E,3E)-4-(6-(methylamino)pyridine-3-yl)buta-1,3-dienyl)benzo[d]thiazol-6-ol] and desmethyl precursor of 11C-PBB3 protected with a silyl group {5-[4-(6-tert-butyl-dimrthylsilyloxy-benzothiazol-2-yl)buta-1,3-dienyl]pyridine-2-amine} were custom-synthesized (Nard Institute) (Maruyama et al., 2013). AV-1451 [7-(6-fluoropyridine-3-yl)-5H-pyrido[4,3-b]indole] and nitro precursor of 18F-AV-1451 [7-(6-nitropyridin-3-yl)-5H-pyrido[4,3-b]indole] were in-house synthesized according to a previous report (US 2012/0302755 A1). Excitation and emission wavelengths of AV-1451 in methanol determined with Spectra Max M5 (Molecular Devise) were 305 nm and 375 nm, and were not changed when the compound bound to a sarkosyl-insoluble tau fraction extracted from the temporal cortex of a patient with Alzheimer’s disease and motor cortex of a patient with PSP. An analogue of PBB3, PBB5 (Maruyama et al., 2013), is commercially available (Sigma-Aldrich). Anti-tau monoclonal antibodies including AT8 against tau phosphorylated at Ser202 and Thr205 (Endogen), and RD4 against 4-repeat domains (Millipore), and a polyclonal antibody pS199/202, which is specific for tau phosphorylated at Ser199 and Ser202 (Invitrogen), are also commercially available. A polyclonal antibody against the unmodified N-terminus of amyloid-β, N1D, was provided by Dr T. C. Saido (RIKEN Brain Science Institute, Japan) (Saido et al., 1995).
Radiosynthesis
11C-PBB3 was radiosynthesized using its desmethyl precursor as described previously (Maruyama et al., 2013; Hashimoto et al., 2014). Specific activity of 11C-PBB3 at the end of synthesis was 28.9–93.1 GBq/µmol, and 11C-PBB3 maintained its radioactive purity exceeding 90% for over 3 h after formulation. Radiolabelling of 18F-AV-1451 was accomplished by reacting 18F-fluoride with a nitro precursor in the presence of dimethylsulphoxide, K2CO3, and K222 at 130°C for 15 min. After fluorination, the crude reaction mixture was transferred into a reservoir for semi-preparative high performance liquid chromatography (HPLC) using a Wakopak Fluofix 120sN column (10 × 250 mm, Wako Pure Chemical Industries) with a mobile phase consisting of methanol/50 mM ammonium acetate (1/1) at a flow rate of 4 ml/min. The fraction containing 18F-AV-1451 was collected in a flask, and was evaporated to complete dryness under a vacuum. The residue was dissolved in HPLC solvent consisting of methanol/50 mM ammonium acetate (1/1), and transferred into a reservoir for preparative HPLC as for the abovementioned semi-preparative HPLC. The fraction corresponding to 18F-AV-1451 was collected in a flask containing 100 µl of 25% ascorbic acid solution and 75 µl of Tween 80 in 300 µl of ethanol, and was evaporated to dryness under a vacuum. The residue was dissolved in 2 ml of saline (pH 7.4) to obtain 18F-AV-1451. The final formulated product was chemically and radiochemically pure (≥95%) as detected by analytical HPLC using a Wakopak Fluofix 120N column (4.6 × 250 mm) with a mobile phase consisting of methanol/50 mM ammonium acetate (1/1) at a flow rate of 1 ml/min. Specific activity of 18F-AV-1451 at the end of synthesis was 36–105.1 GBq/µmol, and 18F-AV-1451 maintained its radioactive purity exceeding 90% for over 3 h after formulation.
In vitro autoradiography
In vitro autoradiography was performed using 6-µm thick deparaffinized sections derived from Alzheimer’s disease, DNTC, CBD, and N279K and G272V mutant FTDP-17 brains and 20-µm thick fresh frozen sections post-fixed in 4% paraformaldehyde solution derived from PSP brains. For labelling with 11C-PBB3, sections were preincubated in 50 mM Tris-HCl buffer, pH 7.4, containing 20% ethanol at room temperature for 30 min, and incubated in 50 mM Tris-HCl buffer, pH 7.4, containing 20% ethanol and a radioligand (5 nM for Alzheimer’s disease, DNTC and N279K mutant FTDP-17 samples, and 10 nM for PSP, CBD and G272V mutant FTDP-17 samples) at room temperature for 60 min. The samples were then rinsed with ice-cold Tris-HCl buffer containing 20% ethanol twice for 2 min, and dipped into ice-cold water for 10 s. For labelling with 18F-AV-1451, dried sections were incubated in phosphate-buffered saline (PBS) containing a radioligand (5 nM for Alzheimer’s disease, DNTC and N279K mutant FTDP-17 samples, and 10 nM for PSP, CBD and G272V mutant FTDP-17 samples) at room temperature for 60 min, and then serially washed for 1 min in PBS, for 2 min in 70% ethanol/PBS, for 1 min in 30% ethanol/PBS, and for 1 min in PBS (Xia et al., 2013). The sections labelled with 11C-PBB3 and 18F-AV-1451 were subsequently dried by treating with warm air, and exposed to an imaging plate (BAS-MS2025; Fuji Film). The imaging plate was scanned with a BAS-5000 system (Fuji Film) to acquire autoradiograms. Intensities of autoradiographic signals were quantified using Multi Gauge software (Fuji Film). Excess concentration (100 µM) of PBB5 was added to the reaction to determine non-specific radioligand binding. All autoradiography procedures with 11C-PBB3 were performed without exposure to fluorescence lights to circumvent photo-isomerization of this compound (Hashimoto et al., 2014).
Histological examination
For fluorescence labelling with PBB3 (Maruyama et al., 2013), PBB5 (Maruyama et al., 2013) and AV-1451, deparaffinized sections were incubated in 50% ethanol containing 56.5 µM of PBB3 or AV-1451 or 2 µM of PBB5 at room temperature for 30 min. The samples were rinsed with 50% ethanol for 5 min, dipped into distilled water twice for 3 min, and mounted in non-fluorescent mounting media (VECTASHIELD; Vector Laboratories). Fluorescence images were captured using a DM4000 microscope (Leica) equipped with Filter cube A for AV-1451 (excitation band-pass at 340–380 nm and suppression low-pass with 425 nm cut-off) and custom filter cube for PBB3 (excitation band-pass at 391–437 nm and suppression low-pass with 458 nm cut-off), and using an FV-1000 confocal laser scanning microscope (Olympus) for PBB5 (excitation at 635 nm and emission at 645–720 nm) (Maruyama et al., 2013). The filter set for AV-1451 was selected to acquire intense fluorescence labelling of ghost tangles in Alzheimer’s disease brains, and intensity of labelling of other tau lesions (e.g. non-ghost tangles and neuropil threads) was semiquantitatively evaluated by comparison with ghost tangles. We did not use cyano-AV-1451 and other AV-1451 analogues producing brighter fluorescence than AV-1451, because structural diversities of these chemicals may result in differential affinity and selectivity for various tau conformers.
Following autoradiography and fluorescence microscopy, all sections labelled with radiolabelled or non-labelled tau ligands were autoclaved for antigen retrieval, immunostained with AT8. Alzheimer’s disease sections were also stained with N1D. Immunolabelling was then examined using DM4000. Finally, the tested samples were used for Gallyas-Braak silver stain (GB) with Nuclear Fast Red (Sigma-Aldrich) counter-staining after pretreatment with 0.25% KMnO4 followed by 2% oxalic acid.
In vitro binding assay
Frozen tissues derived from the temporal cortex of a patient with Alzheimer’s disease and the motor cortex of a PSP patient were homogenized in 50 mM Tris-HCl buffer, pH 7.4, containing protease inhibitor cocktail (cOmpleteTM, EDTA-free; Roche), and stored at −80°C pending analyses. To assay radioligand binding with homologous or heterologous blockade, these homogenates (100 µg tissue) were incubated with 5 nM 11C-PBB3 (specific radioactivity: 78–93.1 GBq/µmol) or 1 nM 18F-AV-1451 (specific radioactivity: 36–105.1 GBq/µmol) in the absence or presence of unlabelled PBB3 or AV-1451 at varying concentrations ranging from 10−11 to 10−6 M in Tris-HCl buffer containing 10% ethanol, pH 7.4, for 30 min at room temperature. These radioligand concentrations were determined in consideration of radioactive half-lives of 11C (∼20 min) and 18F (∼110 min), to equalize signal-to-noise ratios in assays with 11C-PBB3 and 18F-AV-1451. Non-specific binding of 11C-PBB3 and 18F-AV-1451 was determined in the presence of 5 × 10−7 M PBB3 and 5 × 10−7 M AV-1451, respectively. Samples were run in duplicates and specific radioligand binding was determined as pmol/g tissue. Inhibition constant (Ki) and percentage of displacement were determined by using non-linear regression to fit a concentration-binding plot to one-site and two-site binding models derived from the Cheng-Prusoff equation with GraphPad Prism version 5.0 (GraphPad Software), followed by F-test for model selection. In a one-site homologous blockade model, dissociation constant (Kd) and maximum number of binding sites (Bmax) were calculated from homologous competitive binding using this function:
| (1) |
| (2) |
where IC50 and [Radioligand] are concentration of the competitor inducing 50% inhibition and radioligand concentration, respectively, and Top and Bottom are upper and lower plateaus of the plot curve, respectively. Experiments with 11C-PBB3 and unlabelled PBB3 were performed in a dimly lit condition to avoid photoconversion of the compounds.
Results
Fluorescence labelling of Alzheimer’s disease tau pathologies with PBB3 and AV-1451
Nearly adjacent sections of the hippocampal formation derived from an Alzheimer’s disease patient were stained with non-labelled PBB3 and AV-1451, which are both fluorogenic, followed by AT8 immunostaining and GB. GB-positive, AT8-negative ghost tangles and GB-positive, AT8-positive non-ghost tangles were strongly labelled with PBB3 and AV-1451 (Fig. 1A and B). Fluorescence signals of PBB3 and AV-1451 were barely detected in GB-negative, AT8-positive pretangles (Fig. 1A and B). PBB3 labelled neuropil threads and plaque neurites more intensely than AV-1451 (Fig. 1A and B). Quadruple labelling demonstrated the staining of dense-core plaques with PBB3 and AV-1451, and that fluorescence signals of PBB3 bound to primitive plaques, which is accumulation of abnormally swollen neurites without cores of extracellular amyloid (Ikeda et al., 1990), were more clearly detectable than those of AV-1451 (Fig. 1C and D). Semiquantitative scores of PBB3 versus AV-1451 fluorescence labelling were summarized in Fig. 1E.
Figure 1.

Fluorescence labelling of Alzheimer’s disease pathologies with PBB3 and AV-1451. (A and B) Triple staining of tau lesions with 56.5 µM of PBB3 (A) or AV-1451 (B), Gallyas-Braak silver stain (GB) and AT8. From the top, labelling of ghost tangle (GT), non-ghost tangle (T), pre-tangle (P-T), neuropil threads and plaque neurites are displayed. (C and D) Quadruple staining of senile plaques with 56.5 µM of PBB3 (C) or AV-1451 (D), GB, AT8 and N1D. Top and bottom panels demonstrate labelling of dense-core plaque in the hippocampus and primitive plaque in the parahippocampal gyrus. (E) The overall intensity and amount of PBB3 and AV-1451 fluorescence signals in fibrillary tau lesions and plaques graded and labelled none (−), faint/sparse (±), moderate (+) and intense/abundant (++). Scale bars = 20 µm (A–D).
Autoradiographic labelling of Alzheimer’s disease hippocampal formation with 11C-PBB3 and 18F-AV-1451
To compare sub-regional distributions of autoradiographic labelling with 11C-PBB3 and 18F-AV-1451 at a tracer concentration, nearly adjacent sections of the hippocampal formation derived from a patient with Alzheimer’s disease were reacted with these radioligands, followed by double immunostaining with AT8 and N1D and subsequent GB. Total binding of 11C-PBB3 and 18F-AV-1451 was markedly abolished by the addition of excessive non-radioactive PBB5 (Fig. 2A), indicating the saturability of radioligand binding. Both radioligands abundantly labelled CA1 (area 1) and entorhinal cortex (area 5) enriched with densely packed tau aggregates forming neurofibrillary tangles (Fig. 2A and C).
Figure 2.

Autoradiographic labelling of Alzheimer’s disease hippocampal formation sections with 11C-PBB3 and 18F-AV-1451, followed by histochemical and immunohistochemical microscopic assays. (A) Autoradiographic labelling of closely adjacent brain sections of a patient with Alzheimer’s disease with 5 nM of 11C-PBB3 (left) and 18F-AV-1451 (right) in the absence (top) and presence (bottom) of 100 µM of non-radioactive PBB5. Slices contain the hippocampal CA1 sector (1), subiculum (2), presubiculum sectors close to the subiculum (3) and parasubiculum (4), and entorhinal cortex (5). (B) Ratio of 11C-PBB3 (filled bars) and 18F-AV-1451 (open bars) autoradiographic signal intensity in a target area to a white matter value estimated in sections shown in A. (C) Triple labelling of the section used for 11C-PBB3 autoradiography (shown in A) with GB, AT8 and N1D. Areas correspond to locations indicated by squares in A. The hippocampal CA1 sector (1) contained abundant ghost tangles, non-ghost tangles and neuritic plaques. The subiculum (2) harboured abundant ghost tangles and a fewer number of non-ghost tangles and neuritic plaques. The presubiculum adjacent to the subiculum (3) showed a small number of ghost tangles, non-ghost tangles and neuropil threads. The presubiculum in proximity to the parasubiculum (4) was enriched with highly diffuse amyloid-β deposits with few tau lesions. The entorhinal cortex (5) contained abundant ghost tangles. Immunolabelling of the section used for 18F-AV-1451 autoradiography also revealed tau and amyloid-β lesions similar to the above-mentioned findings (data not shown). (D) Low-power photomicrographs of closely adjacent sections of a presubicular sector flanking the subiculum stained with 56.5 µM of PBB3 (left) and AV-1451 (right). Scale bars = 20 µm (A, C and D).
The subiculum (area 2) contained ghost tangles with lower packing density according to the appearance in GB and massive neuropil threads (Fig. 2C), and 11C-PBB3 modestly bound to this sub-region distinct from the very weak binding of 18F-AV-1451 (Fig. 2A). The presubiculum sector adjacent to the subiculum (area 3) was relatively mildly involved in Alzheimer’s disease tau pathologies (Fig. 2C), but strong binding of 18F-AV-1451 to this area was in sharp contrast with the minimum 11C-PBB3 labelling (Fig. 2A). The abundance of PBB3- and AV-1451-positive tangles, threads and plaque neurites in this area was similarly low relative to CA1 and subiculum (Fig. 2D), which did not account for the distinct autoradiographic labelling with these two ligands. A portion of the presubiculum close to the parasubiculum (area 4) was labelled intensely with 11C-PBB3 but only minimally with 18F-AV-1451 (Fig. 2A), and this sub-region harboured highly diffuse amyloid-β deposits (Fig. 2C) as reported previously (Ji et al., 2015). The intensity of autoradiographic signals was quantified as the target to white matter ratio (Fig. 2B), indicating profound differences between the rather uniform binding of 11C-PBB3 across the hippocampal formation and the subregion-specific labelling with 18F-AV-1451.
Binding of PBB3 and AV-1451 to DNTC tau lesions consisting of all six tau isoforms
DNTC is a rare presenile dementia characterized by deposition of all six tau isoforms without amyloid-β deposition. Autoradiographic labelling of nearly adjacent sections of the temporal cortex derived from a patient with DNTC revealed strong binding of 11C-PBB3 and 18F-AV-1451 to grey matter in differential laminar patterns (Fig. 3A). Indeed, bilaminar labelling with 18F-AV-1451 was well contrasted with rather diffuse trans-laminar labelling of grey matter with 11C-PBB3 (Fig. 3A). Subsequent GB of the same sections and double-staining of closely adjacent sections with AT8 and PBB3 and singly with AV-1451 demonstrated abundant 11C-PBB3 and 18F-AV-1451 radiosignals in layer V (area 1) and layer II–III containing numerous GB-positive, AT8-negative ghost tangles (Fig. 3B). Meanwhile, neuropil threads diffusely present across grey matter layers were densely labelled by PBB3 using fluorescence microscopy as compared with weak AV-1451 staining (Fig. 3B). This was in agreement with 11C-PBB3 autoradiograms showing intense labelling in all layers including area 2, which was only weakly labelled with 18F-AV-1451 (Fig. 3A and B). These findings suggest that Alzheimer’s disease paired helical filament-type tau fibrils composed of all six isoforms can be clearly captured by 11C-PBB3 and 18F-AV-1451 in Alzheimer’s disease and DNTC sections, while 11C-PBB3 may yield more intense signals originating from neuropil threads than 18F-AV-1451.
Figure 3.

Autoradiographic labelling of DNTC temporal cortex sections with 11C-PBB3 and 18F-AV-1451, followed by histochemical and immunohistochemical microscopic assays. (A) Autoradiographic labelling of subadjacent sections with 5 nM of 11C-PBB3 (left) and 18F-AV-1451 (right) in the absence (top) and presence (bottom) of 100 µM of non-radioactive PBB5. Bi-layer distribution of 18F-AV-1451 radiosignals in grey matter are indicated by arrows. (B) GB staining of sections used for autoradiography, along with double labelling of a subadjacent section with AT8 and PBB3 and single labelling of another subadjacent section with AV-1451. GB and AT8 staining in grey matter at low power is compared with corresponding autoradiography with 11C-PBB3 and 18F-AV-1451 in top panels. Middle and bottom panels depict high-power photomicrographs in middle (1) and superficial (2) grey matter layers, respectively, indicated by the squares in the top panel. Area 1 was enriched with ghost tangles, which were GB-positive, AT8-negative, and were intensely labelled with PBB3 and AV-1451. Area 2 was heavily loaded with neuropil threads, and were clearly stained with PBB3, but only faintly labelled with AV-1451. Scale bars = 200 µm (top in B), 20 µm (middle and bottom in B).
Ligand binding to 4-repeat lesions in corticobasal degeneration, progressive supranuclear palsy and N279K mutant FTDP-17
Triple staining of brain sections with the self-fluorescent ligands as well as GB and AT8 illustrated strong binding of PBB3 to tufted astrocytes in PSP, astrocytic plaques in CBD, and coiled bodies and argyrophilic grains and threads in both diseases (Fig. 4A and C), supporting the reactivity of PBB3 with non-Alzheimer’s disease 4-repeat aggregates. By contrast, AV-1451 fluorescent staining of these lesions was very faint and fewer compared to PBB3 in closely adjacent sections stained with PBB3 (Fig. 4B and D). As PBB5 is a near-infrared fluorescence compound, it was usable for double fluorescence staining in combination with AV-1451 without overlaps of emission wavelengths. Similar to PBB3, PBB5 labelled neuronal and glial tau inclusions in PSP and CBD brains, while only a small subset of the same lesions was weakly labelled with AV-1451 (Fig. 4B and D).
Figure 4.

Binding of non-labelled and radiolabelled PBB3 and AV-1451 to PSP and CBD tau lesions. (A and B) Triple staining of tau lesions in the motor cortex of a patient with PSP with 56.5 µM of PBB3, AT8 and GB (A), and quadruple staining with 56.5 µM of AV-1451, 2 µM of PBB5, AT8 and GB (B). From the top, tufted astrocyte (TA), coiled body (CB) and argyrophilic threads (ArT) are displayed. (C and D) Triple staining of astrocytic plaques (AP) in the caudate/putamen of a patient with CBD with 56.5 µM of PBB3, AT8 and GB (C), and quadruple staining with 56.5 µM of AV-1451, 2 µM of PBB5, AT8 and GB (D). Arrowheads in A–D indicate tau inclusions. (E) Autoradiographic labelling of closely adjacent fresh frozen sections of the motor cortex derived from a PSP patient with 10 nM of 11C-PBB3 (left) and 18F-AV-1451 (right) in the absence (top) and presence (bottom) of 100 µM of non-radioactive PBB5. (F) High-power photomicrographs of double staining with GB and AT8 in areas indicated by squares in E. Strong GB staining primarily in astrocytes and oligodendrocytes was abundantly observed in a transition zone between grey matter and white matter (area 1), consistent with the laminar pattern of autoradiographic signals of 11C-PBB3 in E. Such strongly GB-positive glial inclusions were slightly less abundant in the middle layers of grey matter (areas 2 and 3), and were nearly absent in a superficial portion of grey matter (area 4), as reflected by moderate and minimal autoradiographic labelling with 11C-PBB3, respectively, in E. (G) Autoradiographic labelling of adjacent sections of the caudate/putamen derived from a CBD patient with 10 nM of 11C-PBB3 (left) and 18F-AV-1451 (right) in the absence (top) and presence (bottom) of 100 µM of non-radioactive PBB5. Radioligand binding was highest in the internal capsule (area 5), while weak radiosignals were present in a grey matter portion of the striatum (area 6). (H) High-power photomicrographs of double staining with GB and AT8 in areas indicated by squares in G. In line with 11C-PBB3 autoradiography, argyrophilic tau inclusions in neurites and coiled bodies were present with high density in the internal capsule (5), but were not abundant in a striatal grey matter area (6). Scale bars = 20 µm (A–D, F and H).
Autoradiography clearly demonstrated specific binding of 11C-PBB3 in grey and white matter of PSP motor cortex sections, and the distribution of radiosignals corresponded exactly to the abundance of tau inclusions stained by AT8 and GB (Fig. 4E and F). The highest 11C-PBB3 binding was noted around the border between grey and white matter (area 1), which was involved in massive GB-positive, AT8-positive tau pathologies. By contrast, superficial grey matter layers (area 4) were devoid of specific 11C-PBB3 labelling, which showed infrequent tau immunostained aggregates. Autoradiographic labelling of a closely adjacent section with 18F-AV-1451 was much weaker than 11C-PBB3 autoradiograms despite similarities between localizations of labelling with these two ligands (Fig. 4E). Likewise, 11C-PBB3 yielded autoradiographic signals in the internal capsule neighbouring the caudate/putamen of a CBD patient (area 5) in contrast with minimum radioligand binding to a grey matter portion of the striatum (area 6) (Fig. 4G). The signal distribution was consistent with the abundance of GB-positive, AT8-positive tau inclusions mostly in oligodendrocytes and neurites as assessed in the same section (Fig. 4H). Unlike 11C-PBB3, there was almost no specific binding of 18F-AV-1451 to an adjacent section (Fig. 4G).
In brain section from an FTDP-17 patient carrying the N279K MAPT mutation, 4-repeat aggregates were abundantly found in white matter axons and oligodendrocytes, with neuropathological resemblance to PSP (Delisle et al., 1999; de Silva et al., 2006; Arvanitakis et al., 2007; Slowinski et al., 2007; Spector et al., 2011). These lesions were fluorescently labelled with PBB3 and were GB-positive, while they were partially immunostained with AT8 (Fig. 5A). Fluorescent PBB3 signals were locally intensified in the alveus. Consistently, autoradiography of a section sub-adjacent to fluorescently stained slices depicted localization of 11C-PBB3 binding to white matter maximizing along the alveus (area 2) (Fig. 5C), and the intensities of radiosignals reflected the abundance of GB-positive neuritic and oligodendrocytic aggregates partly immunoreactive with AT8 (Fig. 5D). Quadruple labelling of sections demonstrated very weak AV-1451 fluorescence from these GB-positive, partially AT8-positive pathologies, which were clearly detected by PBB5 (Fig. 5B). Moreover, faint autoradiographic labelling with 18F-AV-1451 was observed only in the alveus (Fig. 5C).
Figure 5.
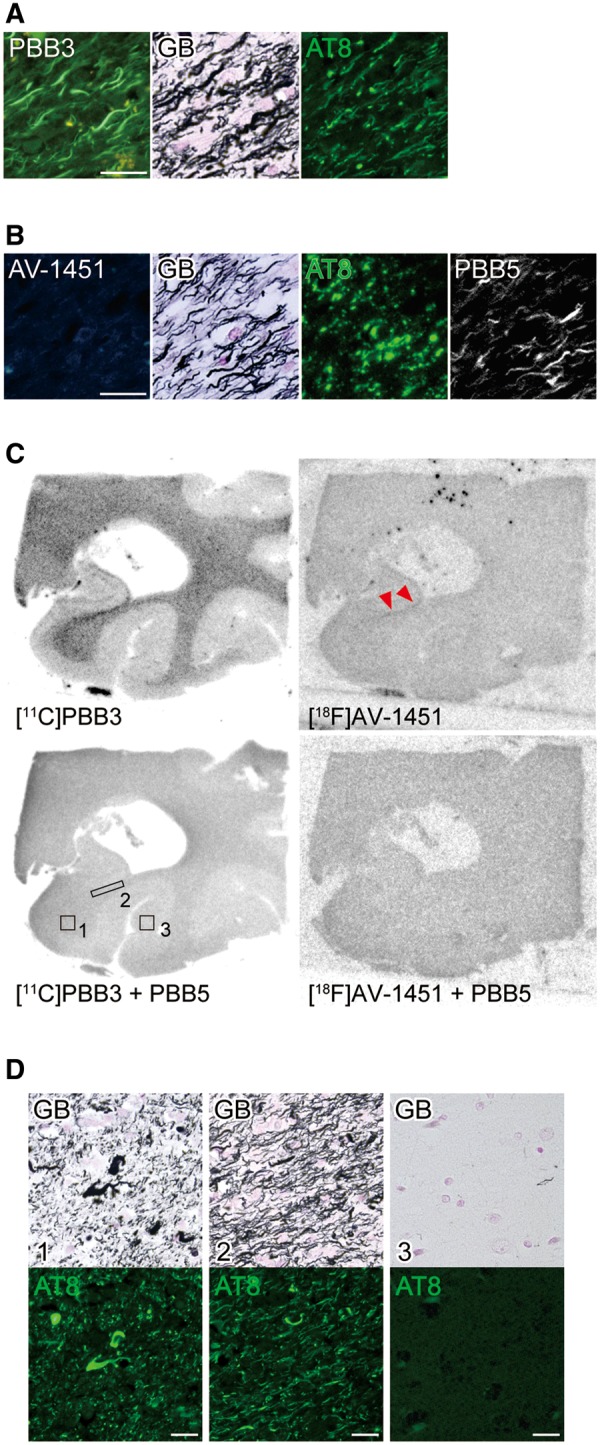
Binding of non-labelled and radiolabelled PBB3 and AV-1451 to tau lesions in the hippocampal formation of an FTDP-17 patient with the N279K MAPT mutation. (A and B) High-power photomicrographs of the alvear pathway triple stained with 56.5 µM of PBB3, AT8 and GB (A) and quadruple stained with 56.5 µM of AV-1451, 2 µM of PBB5, AT8 and GB (B). (C) Autoradiographic labelling of closely adjacent sections including the posterior hippocampus and lateral geniculate nucleus with 5 nM of 11C-PBB3 (left) or 18F-AV-1451 (right) in the absence (top) and presence (bottom) of 100 µM of non-radioactive PBB5. Specific 11C-PBB3 radiosignals were mostly present in white matter (1) with maximal intensity localized in the alveus (2), and were negligible in grey matter (3). Very faint labelling with 18F-AV-1451 was observed in the alveus only (arrowheads). (D) Photomicrographs showing double staining of the section used for 11C-PBB3 autoradiography with GB and AT8. Areas correspond to those indicated by boxes in C. White matter areas were enriched with GB-positive tau lesions in neurites and oligodendrocytes, while grey matter was almost devoid of these pathologies. Scale bars = 20 µm (A, B and D).
These findings have collectively provided evidence for the differential reactivity of PBB3 and AV-1451 with 4-repeat fibril strains in non-Alzheimer’s disease 4-repeat tauopathies.
Methodological assessment of autoradiographic techniques to examine radioligand binding to 4-repeat pathologies
It should be noted that off-target binding of 18F-AV-1451 in the basal ganglia and several other brain areas (Marquié et al., 2015; Lowe et al., 2016) was in general agreement with in vivo PET findings (Shcherbinin et al., 2016). Similarly, there is a possibility of cross-reactivity of 11C-PBB3 with a non-tau component in certain brain regions, including the basal ganglia and midbrain, possibly affecting quantification of tau accumulations in these areas of PSP brains. To address this concern, binding of 11C-PBB3 in normal control brain tissues was assessed by a supplemental autoradiographic analysis. Our data demonstrated that off-target binding displaceable by non-radioactive PBB3 and PBB5 was minimal in grey matter portions of both basal ganglia and frontal cortex sections, and that low-level non-specific radioligand binding in white matter portions did not differ between these two regions (Supplementary Fig. 1 and Supplementary material). This finding rules out the possibility that in vitro off-target binding components for 11C-PBB3 exist in the basal ganglia with higher abundance than other areas, notwithstanding further needs for investigating correlations between in vivo PET and post-mortem autoradiographic data.
In the present autoradiographic experiments, choice of protocols for sample preparation, reaction with a radioligand and washing might lead to results biased toward either 11C-PBB3 or 18F-AV-1451. In this consideration, we conducted a supplemental assay to compare the present procedure for post-fixation of brain slices (4% paraformaldehyde overnight) with a method for 18F-AV-1451 autoradiography (100% methanol for 20 min) (Marquié et al., 2015). As shown in Supplementary Fig. 2 (see also Supplementary material), post-fixation with methanol resulted in higher contrasts for labelling of 4-repeat lesions with 11C-PBB3 in PSP motor cortex than paraformaldehyde fixation, whereas labelling of these pathologies with 18F-AV-1451 was not overtly affected by post-fixation conditions.
We also examined influences of protocols for washing of sections, which were separately optimized for 11C-PBB3 according to our previous work (Maruyama et al., 2013) and for 18F-AV-1451 following a commonly employed method (Xia et al., 2013; Marquié et al., 2015; Loew et al., 2016). As displayed in Supplementary Fig. 3 (experimental procedures are provided in the Supplementary material), labelling of PSP motor cortex slices with 18F-AV-1451 in washing conditions identical to those for 11C-PBB3 autoradiography yielded unaltered or slightly reduced specific binding of 18F-AV-1451 to 4-repeat tau aggregates, along with a notable increase of non-specific radioligand binding in white matter, in comparison with the original settings (c.f.Supplementary Fig. 3 and Fig. 4E). These data indicate that the present autoradiographic comparisons of the two radioligands in 4-repeat pathologies such as PSP motor cortex (Fig. 4E) and CBD (Fig. 4F) were done under conditions optimized for each radioligand.
Ligand binding to 3-repeat lesions in Pick’s disease and G272V mutant FTDP-17
In brain sections from a patient with FTDP-17 carrying the G272V MAPT mutation, 3-repeat aggregates were abundantly found in neurons, with neuropathological resemblance to Pick’s disease (Hutton et al., 1998; Spillantini et al., 1998). Triple staining of brain sections with the self-fluorescent ligands, GB and AT8 illustrated strong binding of PBB3 to Pick bodies in brain slices derived from a patient with Pick’s disease (Fig. 6A). Likewise, intense PBB3 fluorescence labelling of 3-repeat inclusions, which were pS199/202-positive and RD4-negative, was observed in the frontal cortex of an FTDP-17 patient with the G272V MAPT mutation (Fig. 6C). These results support the reactivity of PBB3 with non-Alzheimer’s disease 3-repeat aggregates. By contrast, AV-1451 fluorescence on these lesions was faint and sparse compared to PBB3 in sections closely to those stained with PBB3 (Fig. 6B and D).
Figure 6.

Binding of non-labelled and radiolabelled PBB3 and AV-1451 to tau lesions in the frontal cortex of patients with Pick’s disease and FTDP-17 due to the G272V MAPT mutation. (A and B) Staining of Pick bodies in the frontal cortex of a Pick’s disease patient with 56.5 µM of PBB3, AT8 and GB (A), and with 56.5 µM of AV-1451, AT8 and GB (B). (C and D) Quadruple staining of tau lesions in the frontal cortex of an FTDP-17 patient due to the G272V MAPT mutation with 56.5 µM of PBB3, pS199/pS202, RD4 and GB (C), and with 56.5 µM of AV-1451, pS199/pS202, RD4 and GB (D). (E) Autoradiographic labelling of closely adjacent sections of the frontal cortex derived from an FTDP-17 patient due to the G272V MAPT mutation with 10 nM of 11C-PBB3 (left) and 18F-AV-1451 (right) in the absence (top) and presence (bottom) of 100 µM of non-radioactive PBB5. (F) High-power photomicrographs of triple staining with GB, pS199/pS202 and RD4 in areas indicated by squares in E. Intense GB staining was abundantly observed in grey matter (area 1), consistent with the pattern of autoradiographic signals of 11C-PBB3 in E. These strongly GB-positive inclusions were almost absent in white matter (areas 2), as reflected by minimal autoradiographic labelling with 11C-PBB3 in E. Arrowheads in A and B indicate Pick bodies and those in C, D and F indicate tau inclusions comprised of 3-repeat isoforms. Scale bars = 20 µm (A–D and F).
Autoradiography clearly demonstrated specific binding of 11C-PBB3 in grey matter of G272V mutant FTDP-17 frontal cortex sections, and the distribution of radiosignals corresponded to the abundance of tau inclusions stained by pS199/202 and GB (Fig. 6E and F). Intense 11C-PBB3 radiolabelling was noted in grey matter (area 1), which was involved in massive GB-positive, pS199/202-positive, RD4-negative tau pathologies. By contrast, white matter (area 2) were devoid of specific 11C-PBB3 labelling and exhibited infrequent tau immunoreactivities. Autoradiographic labelling of a closely adjacent section with 18F-AV-1451 was much weaker than 11C-PBB3 autoradiograms, despite similarities between localizations of labelling with these two ligands (Fig. 6E).
These findings have collectively provided evidence for the differential reactivity of PBB3 and AV-1451 with 3-repeat fibril strains in non-Alzheimer’s disease 3-repeat tauopathies.
Quantification of radioligand binding sites and affinities by homologous and heterologous blocking assays
Binding properties of 11C-PBB3 and 18F-AV-1451 were compared by conducting homologous and heterologous blocking of radioligand binding in human brain homogenates. Total (specific + non-specific) radioligand binding and its blockade are depicted in Fig. 7 and Table 1, and error bars in the plots indicate that qualities of data in assays with 11C-PBB3 and 18F-AV-1451 are comparable. Specific 11C-PBB3 binding in Alzheimer’s disease temporal cortex tissue was homologously inhibited by non-labelled PBB3 in a concentration-dependent fashion, and the blocking curve was demonstrated better by a two-site model (Fig. 7A), indicating the presence of high-affinity binding sites for 11C-PBB3 with 1.3 nM of Ki (= Kd). Non-labelled AV-1451 induced heterologous blocking of 11C-PBB3 binding with relatively large Ki (97.2 nM), and ∼60% of specific 11C-PBB3 remained unblocked by AV-1451 (Fig. 7A). High-affinity, one-site binding of 18F-AV-1451 in the same homogenate was also observed with 0.3 nM of Ki (= Kd) in homologous blocking studies (Fig. 7B). Unlabelled PBB3 inhibited the binding of 18F-AV-1451 with high affinity for the targets (Ki = 5.2 nM), and ∼70% of radioligand binding was blocked by PBB3 (Fig. 7B). Hence, Alzheimer’s disease brains may contain tau fibril strains harbouring common binding sites for 11C-PBB3 and 18F-AV-1451 and selective binding sites for 11C-PBB3.
Figure 7.

Binding of 11C-PBB3 and 18F-AV-1451 in cortical homogenates derived from Alzheimer’s disease and PSP cases. (A) Total (specific + non-specific) binding of 5 nM of 11C-PBB3 in an Alzheimer’s disease (AD) temporal cortex sample blocked homologously by non-labelled PBB3 (filled squares) and heterologously by non-labelled AV-1451 (grey triangles). Inhibition of radioligand binding by PBB3 and AV-1451 was described by 2-site and 1-site models, respectively, and parameters resulting from curve fits are indicated. (B) Total binding of 1 nM of 18F-AV-1451 in the same Alzheimer’s disease sample blocked homologously by non-labelled AV-1451 (grey triangles) and heterologously by non-labelled PBB3 (filled squares). Inhibition of radioligand binding by AV-1451 and PBB3 was described by a 1-site model, and parameters resulting from curve fits are indicated. (C) Total binding of 5 nM of 11C-PBB3 in a PSP motor cortex sample blocked homologously by non-labelled PBB3 (filled squares) and heterologously by non-labelled AV-1451 (grey triangles). Inhibition of radioligand binding by PBB3 was described by a 1-site model. (D) Total binding of 1 nM of 18F-AV-1451 in the same PSP sample blocked homologously by non-labelled AV-1451 (grey triangles) and heterologously by non-labelled PBB3 (black squares). Inhibition of radioligand binding by AV-1451 was described by a 1-site model. See Table 1 for binding parameters for 11C-PBB3 and 18F-AV-1451 determined by non-linear fitting of a one-site homologous blockade model to data shown in A–D. Data are presented as mean ± standard error.
Table 1.
Binding parameters for 11C-PBB3 and 18F-AV-1451 determined by non-linear fitting of a one-site homologous blockade model to data shown in Fig. 7A–D
| Kd (nM) | Bmax (pmol/g) | BP | ||
|---|---|---|---|---|
| AD | PBB3 | 6.3 | 4303 | 687.4 |
| Temporal coretx | AV-1451 | 0.7 | 94.3 | 135 |
| PSP | PBB3 | 4.8 | 589.7 | 122.5 |
| Motor cortex | AV-1451 | 3.3 | 62.8 | 18 |
AD = Alzheimer's disease; BP = binding potential.
11C-PBB3 presented specific one-site binding in PSP motor cortex homogenates with 5.9 nM of Ki (= Kd) in homologous blocking experiments (Fig. 7C). Notably, this radioligand binding was not inhibited by non-labelled AV-1451 (Fig. 7C). Conversely, specific binding of 18F-AV-1451 was detected in the same sample with 3.3 nM of Ki (= Kd) in homologous blocking experiments, which was not inhibited by non-radioactive PBB3 (Fig. 7D). Kd and Bmax were then calculated by a one-site homologous blockade model, as summarized in Table 1. 11C-PBB3 and 18F-AV-1451 displayed high-affinity binding in Alzheimer’s disease homogenates, while Bmax for 11C-PBB3 was much higher than that for 18F-AV-1451, resulting in a higher binding potential (= Bmax / Kd) for 11C-PBB3. High-affinity binding components for 11C-PBB3 and 18F-AV-1451 also existed in PSP homogenates with lower abundance (Bmax) than those in Alzheimer’s disease tissues, and binding potential for 18F-AV-1451 was only 15% of the value for 11C-PBB3 (Table 1). Accordingly, tau fibril strains in PSP brains appear to harbour distinctly different binding sites for 11C-PBB3 and 18F-AV-1451, inducing moderate and weak radioligand binding, respectively.
Discussion
The present work has revealed distinct binding modes of two tau PET radioligands, 11C-PBB3 and 18F-AV-1451, in Alzheimer’s disease as well as in non-Alzheimer’s disease tauopathy brains. Autoradiography and homogenate binding assays demonstrated that tau fibrils in Alzheimer’s disease and DNTC patients consisting of all six isoforms yielded heterogeneous binding components with differential selectivity for 11C-PBB3 and 18F-AV-1451. This observation was in line with intense fluorescence labelling of ghost and non-ghost tangles by both PBB3 and AV-1451 while modest AV-1451 fluorescence contrasted with strong PBB3 signals in neuropil threads and plaque neurites. The differential binding properties of 11C-PBB3 and 18F-AV-1451 were even more prominent in 4-repeat tauopathies, including sporadic PSP and CBD as well as FTDP-17 due to the N279K MAPT mutation, since PBB3 yielded fluorescent and autoradiographic labelling much more intensely than AV-1451. This was also in agreement with the abundant binding components for 11C-PBB3 relative to 18F-AV-1451 binding sites in PSP tissue homogenates, and these binding pockets appear to be distinct and exclusive for either of the two radioligands. Moreover, PBB3 yielded fluorescent and autoradiographic labelling of 3-repeat pathologies much more intensely than AV-1451 in the brains of sporadic Pick’s disease and FTDP-17 due to the G272V MAPT mutation. Taken together, the current results provide evidence for the conformational diversity of tau fibrils from different tauopathy brains with different tau isoform compositions (all six tau isoforms versus 4-repeat or 3-repeat isoforms) and possibly reflecting conformational variant or strains that may be attributable to different post-translational modifications, interactions with other cellular constituents and fibril structures.
The detection of extracellular ghost tangles indicates that long and densely packed tau filaments are associated with neuronal death, and both 11C-PBB3 and 18F-AV-1451 may allow sensitive in vivo imaging of these tau aggregates following their externalization or release from dying and dead neurons. As documented previously (Maruyama et al., 2013), ghost tangles were labelled by fluorescent PBB3 but not by the anti-phospho-tau antibodies (e.g. AT8), which are widely used for detecting diverse forms of tangles, although they do not stain ghost tangles very well (Dickson et al., 1992). Factors hampering the access of these antibodies to ghost tangles are not clear, but coverage and/or engulfment of tau filaments by extracellular components, ranging from small peptides to astrocytic processes, is likely to alter the reactivity and accessibility of tangles (Braak and Braak, 1991; Schmidt et al., 1998). On the other hand, there could exist subtle conformational differences between pathological tau assemblies in neurites and cell bodies, and it might be presumed that the former contains binding components preferring 11C-PBB3 to 18F-AV-1451. Because no Stokes shift of fluorescence wavelength of AV-1451 occurred upon its binding to sarkosyl-insoluble tau aggregates, weaker signals in neuropil threads, plaque neurites and ghost tangles than those in non-ghost tangles could not be attributed to out-of-range fluorescence, but were likely due to the lower affinity for these pathologies. In light of the notion that the emergence of neuropil threads in the transentorhinal pre-α layer is one of the initial events in the emergence of tau pathology in Alzheimer’s disease followed by intense accumulations of these lesions in the stratum radiatum and in the stratum oriens of the hippocampal CA1 sector (Braak and Braak, 1991), differential binding of the two ligands to neuritic tau inclusions might affect their ability to visualize early pathological processes heralding the onset of Alzheimer’s disease and other tauopathies. This possibility would need to be further examined by a head-to-head comparison of 11C-PBB3 and 18F-AV-1451 in autoradiographic and tau PET imaging of brains of patients in transition from normal ageing to prodromal tauopathy disorders. Finally, as neuropil thread pathology is more abundant than tau tangle pathology (Mitchell et al., 2000), it is important to identify tau imaging ligands that can detect neuropil thread pathology in addition to tangles and plaque neurite tau pathology.
A two-site model described specific binding of 11C-PBB3 in Alzheimer’s disease tissues better than a one-site model, implying the presence of high-affinity and moderate-affinity components in Alzheimer’s disease-type tau fibrils (Fig. 7A). In light of Ki values (97.2 nM) of 18F-AV-1451 for blocking of 11C-PBB3 binding, none of these two components can be a high-affinity binding site for 18F-AV-1451. Accordingly, there might be at least three distinct binding elements in Alzheimer’s disease tau aggregates, and one of them with high reactivity with 11C-PBB3 and low reactivity with 18F-AV-1451 may abundantly exist in neuropil threads and plaque neurites. Composition of such binding components in tangles and threads is also likely to alter along the evolution of tau strains from normal ageing to Alzheimer’s disease, and this will be clarified by further PET and histopathological investigations.
It should also be noted that both 11C-PBB3 and 18F-AV-1451 exhibit off-target binding to non-tau components in Alzheimer’s disease brains. The two radioligands labelled dense cores of classic plaques, but the contribution of cored plaques to total radiosignals was relatively low, as determined in a previous work (Maruyama et al., 2013). Moreover, differential distributions of 11C-PBB3 and 18F-AV-1451 were not attributed to their binding to cored plaques, since the difference was also observed in DNTC tissues in the absence of amyloid-β aggregates. 11C-PBB3 additionally bound to highly diffuse amyloid-β deposits in a presubicular subregion connected to the parasubiculum (Ji et al., 2015), while intense 18F-AV-1451 signals were present in a different sub-portion of the presubiculum close to the subiculum, which lacked abundant tau accumulations. Molecules with binding sites for 18F-AV-1451 to this area remain unclear, and previous findings raised the probability that 18F-AV-1451 cross-reacts with monoamine oxidase A (MAOA) (Vermeiren et al., 2015), neuromelanin- and melanin-bearing cells (Marquié et al., 2015; Lowe et al., 2016) and acute and subacute haemorrhagic lesions (Marquié et al., 2015). Accordingly, we investigated the immunohistochemical distribution of MAOA, but immunolabelling of MAOA, which was primarily localized in neurons, was not very intense in the hippocampal subiculum (Ono et al., unpublished data). The non-specific binding of 11C-PBB3 and 18F-AV-1451, however, was confined to rather small anatomical structures in the hippocampal formation, and may not elicit strong in vivo PET signals. It is noteworthy that 11C-PiB displayed tight binding to presubicular diffuse amyloid-β pathologies (Ji et al., 2015), but it’s in vivo PET signals in the hippocampal formation are known to be minimal in patients with Alzheimer’s disease (Maruyama et al., 2013). Additionally, cross-reactivity of these radioligands with non-tau amyloid fibrils needs to be taken into account in a subset of neurodegenerative disorders. A previous study demonstrated no and minimal binding of AV-1451 to α-synuclein and TAR DNA-binding protein of 43 kDa (TDP-43) inclusions, respectively (Lowe et al., 2016), and our preliminary analyses also imply reactivity of PBB3 with α-synuclein inclusions in dementia with Lewy bodies (DLB) and multiple system atrophy and TDP-43 aggregates in FTLD-TDP to some extent (Ono et al., unpublished data). As our pilot binding assays using DLB brain homogenates have demonstrated that Kd value for binding of 11C-PBB3 to α-synuclein deposits is 10–50 times larger than the Kd for binding to tau deposits in Alzheimer’s disease and PSP brain samples (Ono et al., unpublished data), 11C-PBB3-PET should not sensitively capture α-synuclein pathologies, unless α-synuclein burden is extremely high.
Previous reports also implied the presence of off-target binding sites for tau radioligands in several subcortical structures (Shcherbinin et al., 2016). Although these off-target molecules are yet to be identified, binding of 18F-AV-1451 in the basal ganglia and midbrain was found to increase with normal ageing. Our observations similarly minimal binding of 11C-PBB3 in the putamen and frontal cortex of normal controls (Supplementary Fig. 1), suggesting that off-target binding of 11C-PBB3 does not overtly influence in vitro autoradiographic labelling in these areas. However, it is likely that binding of 11C-PBB3 to non-tau components increases in other brain regions including the midbrain, and off-target radiosignals might be noticeable in samples derived from even older normal subjects than those who were examined here (47 and 61 years of age).
The current analyses delineated pronounced distinctions between Alzheimer’s disease-type tau fibrils containing all six isoforms and PSP-type 4-repeat or Pick’s disease-type 3-repeat fibril strains. Characteristic morphologies known as paired helical filaments versus straight filaments (Bibow et al., 2011; Murray et al., 2014) reflect the different structures of these tau fibril assemblies which presumably reflects different conformational tau units in these filaments. Binding of 11C-PBB3 to 4-repeat or 3-repeat lesions was much higher than that of 18F-AV-1451, and this difference was greater than what could be seen in Alzheimer’s disease and DNTC samples. In general, this observation was consistent with an earlier report showing negative (Marquié et al., 2015) or minimal (Lowe et al., 2016) autoradiographic labelling of 4- and 3-repeat pathologies with 18F-AV-1451, although the low-grade binding of 18F-AV-1451 in PSP, CBD and FTDP-17 due to the N279K and G272V MAPT mutation tissues shown here leaves open the possibility that tau pathologies in these FTLD-Tau syndromes could be detected by tau PET with this radioligand.
In autoradiographic comparisons of different radioligands, care needs to be taken in consideration of experimental conditions possibly modifying radiolabelling contrasts. Our supplemental assays implied that distinct binding of 11C-PBB3 and 18F-AV-1451 to PSP brain sections shown here were not confounded by differences in methods of post-fixation and buffer preparation. Accordingly, the negative autoragiographic labelling of 4-repeat lesions with 18F-AV-1451 in a previous report (Marquié et al., 2015) versus weak specific binding of this radioligand to PSP and CBD tau pathologies in this study may be attributed to variability of tau burdens, because we could not detect labelling of 4-repeat inclusions with 18F-AV-1451 in brain slices containing tau deposits with relatively low abundance (data not shown). However, more conclusive insights should be provided by comprehensive across-ligand comparisons covering additional assay conditions and diverse pathologies other than PSP.
It is of great significance that 11C-PBB3 labelled 4-repeat inclusions in white matter, which corresponded to the aggregation of abnormal tau in axons, astrocytes and oligodendrocytes, and is in sharp contrast to Alzheimer’s disease tau tangles and threads localized in grey matter. These data may indicate that 4-repeat strains of pathological tau are prone to propagate intracellularly from neuronal somas to distal axons and intercellularly from neurons to neighbouring glia cells, and imply the capability of 11C-PBB3 to capture transmittable tau species in living brains. Furthermore, the heterogeneity of tau fibril strains can exist among 4-repeat tauopathies, as shown here for tau inclusions in proximal astrocytic processes (i.e. tufted astrocytes) in PSP versus distal processes (i.e. astrocytic plaques) in CBD. Such conformational diversity might also result in modest 18F-AV-1451 labelling of tau pathology in PSP and CBD brain sections versus its very weak binding in N279K mutant FTDP-17 samples. Our pilot in vitro fluorescence binding assays have also demonstrated that PBB3 tightly bound to recombinant 4-repeat assemblies, while no binding of AV-1451 to these fibrils was observed (Jang et al. unpublished data), suggesting affinity of PBB3 for a wide range of 4-repeat strains as compared with AV-1451.
Our results support the utility of 11C-PBB3 in capturing a wide range of tau conformers in the brains of living subjects by PET, although care should be taken in translating in vitro observations to in vivo settings. There remains a possibility that other in vivo properties of 11C-PBB3, including rapid generation of a brain-entering radiometabolite, photoisomerization, spill-over of radioactivity from venous sinuses, and short half-life of 11C (Maruyama et al., 2013; Kimura et al., 2015), may impede high-contrast detection of target pathologies relative to 18F-AV-1451. This notion raises the need for a head-to-head comparison of PET with 11C-PBB3 and 18F-AV-1451 in the same individuals with different species of tau deposits. Likewise, in vivo comparisons of these radioligads with 18F-THK-5351, a PET probe reported to bind to tau aggregates in Alzheimer’s disease (Harada et al., 2016) and PSP (Vettermann et al., 2016), will be required for acquiring a comprehensive picture of relationships between chemical structures of radioligands and activities on a variety of tau fibril strains in living brains. Toward this aim, it will be of significance to conduct in vitro and in vivo comparisons of multiple radioligands covering a broader range of tauopathies, such as FTDP-17 due to other MAPT mutations, argyrophilic grain disease and chronic traumatic encephalopathy.
Taken together, the data presented here support distinct roles played by different chemical classes of tau imaging agents in visualizing a diversity of tau fibril strains. Accordingly, the use of multiple tau probes would allow us to clarify the contributions of each tau strain to the regional detection of tau pathology by PET imaging, localized neuronal loss and focal clinical symptoms. Since a subset of tau strains may propagate more rapidly than others, serial tau PET imaging could determine the rate of disease progression. Further, comparative multi-tracer tau PET imaging assays could enable selection of a suitable therapy targeting a specific tau conformer (Yanamandra et al., 2013) on an individual basis.
Supplementary Material
Acknowledgements
We are indebted to all the subjects who donated their brains. We thank Dr Hiroyasu Akatsu and Norihiro Ogawa from Choju Medical Institute of Fukushimura Hospital, for kindly sharing the post-mortem brain sections from healthy subject, Dr Takaomi C Saido, from the Brain Science Institute, RIKEN, for kindly sharing the N1D antibody, and Ms Hiromi Kondo and Mr Kyohei Mikami, from the Center for Basic Technology Research, Tokyo Metropolitan Institute of Medical Science, for guidance of Gallyas-Braak silver stain.
Funding
This work is supported by grants from the Takeda Science Foundation to M.O., Grants-in-Aid for Core Research for Evolutional Science and Technology to T.S. and M.H. (14533254), the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS; 15653129) and Research and Development Grants for Dementia (16768966) to M.H. from the Japan Agency for Medical Research and Development, and the Japan Advanced Molecular Imaging Program and Scientific Research on Innovative Areas (“Brain Environment” 23111009 to M.H., and “Brain Protein Aging” 26117001 to N.S.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Support for this work was also provided by grants from the National Institute on Aging of the National Institutes of Health (AG-10124, and AG-17586 to V.M.Y.L. and J.Q.T.).
Glossary
Abbreviations
- CBD
corticobasal degeneration
- DNTC
diffuse neurofibrillary tangles with calcification
- FTDP-17
frontotemporal dementia and parkinsonism linked to chromosome 17
- FTLD
frontotemporal lobar degeneration
- GB
Gallyas-Braak silver stain
- PSP
progressive supranuclear palsy
Supplementary material
Supplementary material is available at Brain online.
References
- Arvanitakis Z, Witte RJ, Dickson DW, Tsuboi Y, Uitti RJ, Slowinski J. et al. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation). Parkinsonism Relat Disord 2007; 13: 230–9. [DOI] [PubMed] [Google Scholar]
- Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-beta structure. Proc Natl Acad Sci USA 2003; 100: 9034–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibow S, Mukrasch MD, Chinnathambi S, Biernat J, Griesinger C, Mandelkow E. et al. The dynamic structure of filamentous tau. Angew Chem Int Ed Engl 2011; 50: 11520–4. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY. et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis 2013; 34:457–68. [DOI] [PubMed] [Google Scholar]
- Daebel V, Chinnathambi S, Biernat J, Schwalbe M, Habenstein B, Loguet A. et al. β-Sheet core of tau paired helical filaments revealed by solid-state NMR. J Am Chem Soc 2012; 134: 13982–9. [DOI] [PubMed] [Google Scholar]
- Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, Soulages X. et al. A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol 1999; 98: 62–77. [DOI] [PubMed] [Google Scholar]
- de Silva R, Lashley T, Strand C, Shiarli AM, Shi J, Tian J. et al. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol 2006; 111: 329–40. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Ksiezak-Reding H, Liu WK, Davies P, Crowe A, Yen SH. Immunocytochemistry of neurofibrillary tangles with antibodies to subregions of tau protein: identification of hidden and cleaved tau epitopes and a new phosphorylation site. Acta Neuropathol 1992; 84: 596–605. [DOI] [PubMed] [Google Scholar]
- Feany MB, Dickson DW. Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 1995; 146: 1388–96. [PMC free article] [PubMed] [Google Scholar]
- Flament S, Delacourte A, Verny M, Hauw JJ, Javoy-Agid F. Abnormal Tau proteins in progressive supranuclear palsy. Similarties and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol 1991; 81: 591–6. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brains isoforms. Neuron 1992; 8: 159–68. [DOI] [PubMed] [Google Scholar]
- Goedert M, Falcon B, Clavaguera F, Tolnay M. Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies. Curr Neurol Neurosci Rep 2014; 14: 495. [DOI] [PubMed] [Google Scholar]
- Harada R, Okamura N, Furumoto S, Tago T, Yoshikawa T, Akatsu H. et al. Binding characterization of tau PET tracer 18F-THK5117 in non-alzheimer’s neurodegenerative diseases. Alzheimers Dement 2014; 10: 307–8. [Google Scholar]
- Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N. et al. [18F]THK-5117 PET for assessing neurofibrillary pathology in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 2015; 42: 1052–61. [DOI] [PubMed] [Google Scholar]
- Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N. et al. 18F-THK5351: A novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J Nucl Med 2016; 57: 208–14. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Watanabe S, Kondo H, Akiyama H, Mann DM, Saito Y. et al. 3R and 4R tau isoforms in paired helical filaments in Alzheimer’s disease. Acta Neuropathol 2014; 127: 303–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Kawamura K, Igarashi N, Takei M, Fujishiro T, Aihara Y. et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J Nucl Med 2014; 55: 1532–8. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H. et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998; 393: 702–5. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Haga C, Kosaka K. Light and electron microscopic examination of amyloid-rich primitive plaques: comparison with diffuse plaques. J Neurol 1990; 237: 88–93. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Ito M, Mori K, Deguchi A, Nagaoka M, Yoshida M. et al. An autopsy case of diffuse neurofibrillary tangles with calcification: early stage pathologic findings. Neuropathology 2009; 29: 697–703. [DOI] [PubMed] [Google Scholar]
- Ji B, Chen CJ, Bando K, Ashino H, Shiraishi H, Sano H. et al. Distinct binding of amyloid imaging ligands to unique amyloid-β deposited in the presubiculum of Alzheimer’s disease. J Neurochem 2015; 135: 859–66. [DOI] [PubMed] [Google Scholar]
- Kato S, Nakamura H. Presence of two different fibril subtypes in the Pick body: an immunoelectron microscopic study. Acta Neuropathol 1990; 81: 125–9. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Ichise M, Ito H, Shimada H, Ikoma Y, Seki C. et al. PET quantification of tau pathology in human brain with 11C-PBB3. J Nucl Med 2015; 56: 1359–65. [DOI] [PubMed] [Google Scholar]
- Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH. et al. Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol 1994; 145: 1496–508. [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ Neurodegenerative tauopathies [Review]. Annu Rev Neurosci 2001; 24: 1121–59. [DOI] [PubMed] [Google Scholar]
- Li L, von Bergen M, Mandelkow EM, Mandelkow E. Structure, stability, and aggregation of paired helical filaments from tau protein and FTDP-17 mutants probed by tryptophan scanning mutagenesis. J Biol Chem 2002; 277: 41390–400. [DOI] [PubMed] [Google Scholar]
- Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE. et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun 2016; 4: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG. et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 2015; 78: 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J. et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013; 79: 1094–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TW, Nissanov J, Han LY, Mufson EJ, Schneider JA, Cochran EJ. et al. Novel method to quantify neuropil threads in brains from elders with or without cognitive impairment. J Histochem Cytochem 2000; 48: 1627–38. [DOI] [PubMed] [Google Scholar]
- Murray ME, Kouri N, Lin WL, Jack CR, Jr, Dickson DW, Vemuri P. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimers Res Ther 2014; 6: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N, Furumoto S, Harada R, Tago T, Yoshikawa T, Fodero-Tavoletti M. et al. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med 2013; 54: 1420–7. [DOI] [PubMed] [Google Scholar]
- Rabinovici G, Schonhaut D, Ossenkoppele R, Baker S, Lazaris A, Lockhart S. et al. Initial Experience with [18F]AV1451 PET in AD and non-AD Tauopahies. Neurology 2015; 84: P5.005. [Google Scholar]
- Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 1995; 14: 457–66. [DOI] [PubMed] [Google Scholar]
- Sander K, Lashley T, Gami P, Gendron T, Lythgoe MF, Rohrer JD. et al. Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and dementias. Alzheimers Dement 2016; 12: 1116–24. [DOI] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A. et al. Distinct tau prion strains propagate in dells and mice and define different tauopathies. Neuron 2014; 82: 1271–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, Gur RE, Gur RC, Trojanowski JQ. Intraneuronal and extracellular neurofibrillary tangles exhibit mutually exclusive cytoskeletal antigens. Ann Neurol 1998; 23: 184–9. [DOI] [PubMed] [Google Scholar]
- Shcherbinin S, Schwarz AJ, Joshi AD, Navitsky M, Flitter M, Shankle WR. et al. Kinetics of the tau PET tracer 18F-AV-1451 (T807) in subjects with normal cognitive function, mild cognitive impairment and Alzheimer’s disease. J Nucl Med 2016; 57: 1535–42. [DOI] [PubMed] [Google Scholar]
- Slowinski J, Dominik J, Uitti RJ, Ahmed Z, Dickson DD, Wszolek ZK. Frontotemporal dementia and Parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology 2007; 27: 73–80. [DOI] [PubMed] [Google Scholar]
- Spector AR, Dugger BN, Wszolek ZK, Uitti RJ, Fredrickson O, Kaplan J. et al. Anatomy of disturbed sleep in pallido-ponto-nigral degeneration. Ann Neurol 2011; 69: 1014–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol 1998; 153: 1359–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi-Watanabe S, Arai T, Kametani F, Nonaka T, Masuda-Suzukake M, Tarutani A. et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol 2016; 131: 267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi Y, Uitti RJ, Delisle MB, Ferreira JJ, Brefel-Courbon C, Rascol O. et al. Clinical features and disease haplotypes of individuals with the N279K tau gene mutation: a comparison of the pallidopontonigral degeneration kindred and a French family. Arch Neurol 2002; 59: 943–50. [DOI] [PubMed] [Google Scholar]
- Vermeiren C, Mercier J, Viot D, Mairet-Coello G, Hannestad J, Courade JP. et al. T807, a reported selective Tau tracer, binds with nanomolar affinity to monoamine oxidase A. Alzheimers Dement 2015; 11: P283. [Google Scholar]
- Vettermann F, Brendel M, Schönecker S, Höglinger G, Denek A, Levin J. et al. [18F]THK-5351 PET in patients with clinically diagnosed progressive supranuclear palsy. J Nucl Med 2016; 57: 457. [Google Scholar]
- von Bergen M, Barghorn S, Müller SA, Pickhardt M, Biernat J, Mandelkow EM. et al. The core of tau-paired helical filaments studied by scanning transmission electron microscopy and limited proteolysis. Biochemistry 2006; 45: 6446–57. [DOI] [PubMed] [Google Scholar]
- Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D. et al. [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement 2013; 9: 666–76. [DOI] [PubMed] [Google Scholar]
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE. et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013; 80: 402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.