

Abstract
Arrhythmogenic cardiomyopathy (AC) is a progressive disease with high risk of life-threatening ventricular arrhythmias. A genetic mutation is found in up to 50–60% of probands, mostly affecting desmosomal genes. Diagnosis of AC is made by a combination of data from different modalities including imaging, electrocardiogram, Holter monitoring, family history, genetic testing, and tissue properties. Being a progressive cardiomyopathy, repeated cardiac imaging is needed in AC patients. Repeated imaging is important also for risk assessment of ventricular arrhythmias. This expert consensus document gives clinical recommendations for how to use multi-modality imaging in the different aspects of AC disease, including diagnosis, family screening, follow-up, risk assessment, and differential diagnosis.
Keywords: arrhythmogenic cardiomyopathy, echocardiography, speckle tracking echocardiography, multi-modality imaging, cardiac magnetic resonance, late gadolinium enhancement, multidetector computed tomography
Current knowledge
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is considered as an inherited cardiomyopathy predisposing to ventricular arrhythmias, sudden cardiac death (SCD), and more rarely ventricular dysfunction and heart failure. Due to the frequent involvement of the left ventricle (LV) the term arrhythmogenic cardiomyopathy (AC) has recently been proposed to encompass both LV and RV disease and this expert consensus group acknowledges and recommends replacing ARVC with AC.1
The 2010 Task Force Criteria (TFC 2010) have improved the sensitivity of AC diagnosis and combine data from different categories including imaging, electrical parameters from resting electrocardiogram (ECG) and Holter monitoring, family history, genetic testing, and tissue properties.2 Imaging modalities include LV angiogram, echocardiography, and cardiac magnetic resonance (CMR), with qualitative assessment of RV wall motion abnormalities. In addition, a few quantitative measures are included in diagnosis with cut-off values for the RV outflow tract (RVOT) diameter and RV fractional area change (RV-FAC) by echocardiography and RV volumes and ejection fraction (RV-EF) by CMR.
Being a progressive cardiomyopathy, repeated cardiac imaging is needed in AC patients to follow disease progression and importantly, for risk assessment of life-threatening ventricular arrhythmias. This expert consensus document will give clinical recommendations for how to use multi-modality imaging in the different aspects of AC disease, including diagnosis, family screening, follow-up, risk assessment, and differential diagnosis.
Definition and pathogenesis of AC
Anatomy and morphology
Arrhythmogenic cardiomyopathy is a cardiomyopathy characterized by an acquired and progressive replacement of the ventricular myocardium by fibrous and fatty tissue.3,4 The process starts from the epicardium or mid-myocardium and then extends to become transmural in the RV, leading to wall thinning and aneurysms, typically located at the inferior, apical, and infundibular walls (triangle of dysplasia). The RV involvement can be either segmental or diffuse. The LV involvement is present in more than half of the cases, typically located in the subepicardium or mid-myocardium, and often confined to the posterolateral region;3 a circumferential distribution is also observed. Also, hearts with isolated or predominantly LV involvement have been reported.1,5 The septum is affected in a minority of cases, typically on the right side. Fatty infiltration is not sufficient to achieve a clear-cut diagnosis and replacement-type fibrosis and myocyte degenerative changes are essential.6 Inflammatory cell infiltrates, together with myocyte injury, are often observed in the early stages of disease and during the process of evolution, mimicking myocarditis.3,5,7
Pathogenesis, genetic background, and inheritance
Dysontogenetic (dysplasia), inflammatory (myocarditis), and degenerative (myocardial dystrophy) theories have been proposed as the mechanisms of AC.3 A familial background consistent with autosomal-dominant inheritance is present in most AC patients.8,9 Recessive families with cardiocutaneous manifestations (palmoplantar keratoderma and woolly hair) have been reported and have provided the substrate for the initial recognition of disease-causing genes (Naxos disease, Carvajal syndrome).10,11 Up to 50–60% of probands harbour a disease-causing gene mutation, leading to the current belief of a genetically determined ‘cardiomyopathy’. Disease-causing genes mostly encode major components of the cardiac desmosomes and include junction plakoglobin, desmoplakin, plakophillin-2, desmoglein-2, and desmocollin-2.5 Compound/digenic heterozygosity was identified in up to 25% of AC-causing desmosomal gene mutation carriers, in part explaining the phenotypic variability. Non-desmosomal genes, including transforming growth factor beta-3, desmin, catenin-aT, phospholamban, Lamin A/C, and titin, have been reported causing AC mimicking disease. However, these phenotypes often have features untypical for ‘classical’ AC, e.g. including skeletal muscle symptoms, etc.12
How the mechanical and/or functional disruption of cell junctions by mutant desmosomal proteins leads to cardiomyocyte death and subsequent repair with fibrous and adipose tissue is under investigation. Abnormal cell–cell adhesion as well as intracellular signalling pathways (Wnt and Hippo/YAP) have been implicated in adipogenesis, fibrogenesis, and apoptosis. Finally, gap junction and ion channel remodelling have also been demonstrated, suggesting that impaired mechanical coupling might account for abnormal electrical coupling.12,13
Key points:
Arrhythmogenic cardiomyopathy is a progressive disease. Morphological changes start from the epi- or mid-myocardium and usually progress through all layers as a transmural myocardial disease.
The left ventricle is affected in >50% of cases and the term ‘arrhythmogenic cardiomyopathy’ has been proposed to include biventricular disease.
A genetic mutation is found in up to 50–60% of probands, mostly affecting desmosomal genes.
Inheritance is most frequently autosomal dominant.
Clinical characteristics
Symptoms and ECG
Family studies suggest that most individuals carrying potentially disease-causing variants go undiagnosed.14 Data on clinical presentation will depend on the sources, ranging from the primary care physician to the coroner’s pathologist. The classic clinical presentation is with exercise-related ventricular tachycardia (VT) associated with awareness of rapid heartbeat, lightheadedness, and/or syncope. Palpitation, when described, is of sudden onset and offset, rapid and different from the awareness of a ‘heavy heart beat’ typical of ventricular ectopic beats.7 Atypical chest pain which is precordial, but neither exertional nor relieved by rest, is reported in publications from referral centres, and may initially focus investigations on the exclusion of coronary artery disease. The mechanism is uncertain, but may relate to inflammation associated with a potential arrhythmic or hot phase of the condition.
The earliest disease manifestations are usually seen on the ECG, whereas the structural abnormalities which are best identified with imaging occur later in the natural history of the disease.15 Twelve-lead ECG changes reflect myocardial areas of disease involvement. AC involving the RV free wall will typically manifest with inverted T waves in V1–V3 (Figure 1), and where there is LV involvement the T-wave inversion may extend to lateral chest leads (V4–V6). T-wave changes in inferior leads (II, III, aVF) often reflect RV infero-posterior wall involvement. Most patients present in sinus rhythm with a normal QRS axis, normal PR and QRS intervals, though a delayed upstroke of the S wave or localized QRS widening may be a subtle early disease manifestation, and is considered minor AC diagnostic criterion.2,16 A normal ECG may occur in a mutation carrier, but in these individuals the risk of a life-threatening event as the initial clinical manifestation is very low. Similarly and importantly, a diagnosis based on imaging criteria with completely normal ECG should be considered suspicious, and warrants careful review. AC with morphological changes limited to or predominately affecting the LV is increasingly recognized.1,5,17,18 Such patients may have inferior and/or lateral T-wave inversion and ventricular arrhythmias with a configuration suggestive of an LV origin, often from the posterior–lateral LV wall. In contrast, ventricular ectopy with left bundle branch block (LBBB) morphology, particularly with a superior axis, is typical of ARVC. LBBB morphology ectopy/ventricular arrhythmia with an inferior axis (R wave positive in leads II and III and negative in lead aVL) requires careful evaluation and differentiation from RVOT tachycardia, a condition which is not familial and rarely life-threatening (see ‘Right ventricular outflow tract-VT’ section). Ambulatory ECG monitoring during normal daily activity, ideally with 12-lead recordings, is an important diagnostic evaluation and often reveals ventricular arrhythmia in the absence of symptoms in affected individuals. The signal-averaged ECG provides a marker of slow conduction and arrhythmic risk, but is neither a sensitive nor specific diagnostic test.19
Key points:
The classical clinical presentation is the occurrence of ventricular arrhythmias, typically with LBBB morphology and superior axis.
LBBB morphology ventricular arrhythmias with an inferior axis requires careful evaluation and differentiation from the more benign RVOT tachycardia.
ECG changes are often the first manifestation.
An AC diagnosis based on imaging criteria with completely normal ECG should be considered suspicious.
Figure 1.
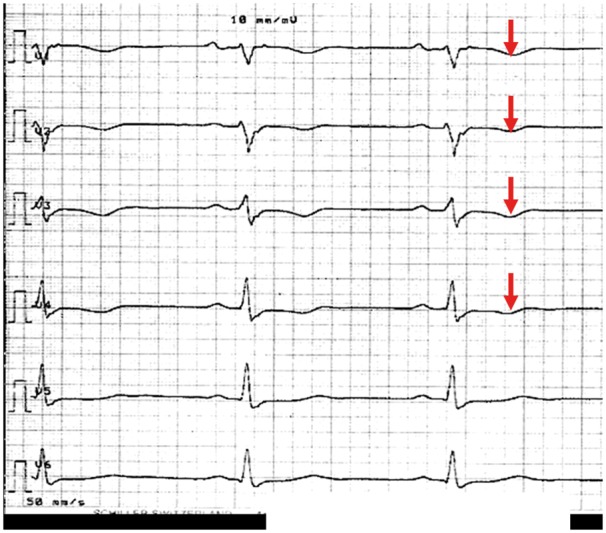
Precordial ECG leads from an AC patient at rest with typical T-wave inversions in V1–V4 (red arrows).
Structural remodelling in AC—RV/LV dominant types
Echocardiography and CMR are pivotal to detect structural and functional abnormalities in patients with suspected or genetically proven AC. LV angiogram is also included in the TFC 2010, but it is an invasive procedure, now rarely performed. The importance of high-sensitivity non-invasive tools for diagnosing early disease is emphasized by the fact that SCD may occur in individuals with no or subtle evidence of structural heart disease. Accordingly, it is important to distinguish between individuals with early, pre-symptomatic disease, where no or minor and subtle structural changes can be detected in a single-specific region of the RV (inflow tract, outflow tract, or apex, the ‘triangle of dysplasia’) and symptomatic individuals with advanced disease, where severe RV involvement is evident, with or without LV involvement, typically affecting the posterior–lateral wall. On the other hand, specificity is also important and in subjects with non-specific symptoms and/or subtle abnormalities, over-diagnosing, especially by non-experienced operators, must be avoided.
According to the TFC 20102 the cardiac imaging diagnosis of AC is based on RV global or regional dysfunction and structural changes, with or without associated LV dysfunction (Table 1). Compared with previously published diagnostic criteria,20 TFC 2010 imaging criteria increased sensitivity to around 70%, keeping specificity high (around 95%). According to the TFC 2010,2 regional RV wall motion abnormalities are the main imaging criteria to diagnose AC, and the level of RV dilatation and RV global dysfunction determines whether or not a major or minor AC criterion is present. Despite the undisputed usefulness of the TFC 2010 criteria as a diagnostic tool for AC, concerns have been raised about its practical applicability as a screening tool. These include the complexity of the diagnostic workup, the necessity of memorizing several different imaging cut-off values both for major and minor criteria, the non-inclusion of RV inflow tract data, and the low sensitivity of conventional echocardiographic criteria for the detection of subtle regional RV wall abnormalities in early disease stages.
Table 1.
Imaging criteria for AC from the Modified Task Force Criteria from 2010
| Global or regional dysfunction and structural alterations |
|---|
| Major |
| 2D echo criteria |
| Regional RV akinaesia, dyskinaesia or aneurysm and one of the following measured at end diastole |
| PLAX RVOT ≥ 32 mm |
| PSAX RVOT ≥ 36 mm |
| Fractional area change ≤ 33% |
| CMR criteria |
| Regional RV akinaesia or dyskinaesia or dyssynchronous RV contraction and one of the following |
| Ratio of RV end-diastolic volume to BSA ≥110 mL/m2 (male) or ≥ 100 mL/m2 (female) |
| RV ejection fraction ≤ 40% |
| RV angiography criteria |
| Regional RV akinaesia, dyskinaesia, or aneurysm |
| Minor |
| 2D echo criteria |
| Regional RV akinaesia or dyskinaesia and one of the following measured at end diastole |
| PLAX RVOT ≥29 to < 32 mm |
| PSAX RVOT ≥32 to < 36 |
| Fractional area change >33% to ≤ 40% |
| CMR criteria |
| Regional RV akinaesia or dyskinaesia or dyssynchronous RV contraction and one of the following |
| Ratio of RV end-diastolic volume to BSA ≥100 to < 110 mL/m2 (male) or ≥ 90 to < 100 mL/m2 |
| RV ejection fraction >40 to ≤ 45% |
Diagnostic imaging criteria in AC modified from Marcus et al.2
AC, arrhythmogenic cardiomyopathy; BSA, body surface area; PLAX RVOT, parasternal long-axis right ventricular outflow tract; PSAX RVOT, parasternal short-axis right ventricular outflow tract; RV, Right ventricle.
Echocardiography
Conventional echocardiographic methods
Echocardiography is the first-line imaging modality in AC, and the most commonly used imaging tool for follow-up of AC patients. Echocardiography is inexpensive, widely available, and well tolerated. However, the echocardiographic diagnosis of AC is challenging and needs specific expertise. The echocardiographic protocol requires more views than those usually included in a routine study. Segmental evaluation of the RV is difficult due to the complex shape of the chamber and the position of the RVOT immediately under the sternum, narrowing the acoustic window for proximal structures. Moreover, the quantitative analysis of RV function is also difficult due to the complex RV anatomy and to its load dependency. A systematic qualitative assessment of RV function and dimensions should be performed initially, followed by quantitative measurements using conventional and advanced echocardiography.21,22
The typical morphological features in AC patients are regional wall motion abnormalities or global RV dysfunction and RV dilatation. The TFC 20102 only includes 2D echocardiography as imaging criteria to diagnose AC: presence of RV akinaesia, dyskinaesia, or aneurysms together with measurements of RVOT diameter and RV-FAC (Figures 2 and 3, Supplementary data online, Video S1).
Figure 2.

Proximal RV outflow diameters (RVOT PLAX and PSAX) and RV basal diameter (RVD) (diastole). Courtesy: Dr J. Saberniak.
Figure 3.

RV fractional area change (RV-FAC) calculated from (A) RV end-diastolic area and (B) EV end-systolic area as (RVEDA − RVESA)/RVEDA × 100. (C) Left dominant type of AC with reduced LV function, LV dilatation, CMR showed epicardial fibrosis, ECG showed T-inversion V1–V6 and in inferior leads. The patient suffered from ventricular arrhythmia and was implanted with an ICD. Courtesy: Dr J. Saberniak.
The RVOT diameter2,21,22 can be measured from parasternal long axis (PLAX) (Figure 2) or from parasternal short axis (PSAX) views. When possible, the proximal RVOT diameter should be measured from PSAX as a robust and reproducible measure (Figure 2B).
In addition, we recommend the routine and systematic assessment of RV basal diameter23 (Figure 2C), as well as the inclusion of additional quantitative echo-strain data (Table 2). The new parameters are reported in a few studies24–27 and are not validated in larger or multicentre studies. However, careful assessment of RV morphology and function will add to the accuracy of AC diagnosis. The use of contrast agents has not been investigated.
Table 2.
Quantitative standard and advanced echocardiographic parameters (with respective cut-off points) recommended in patients with suspected or ascertained AC
| Parameter | Abnormal if: |
|---|---|
| PLAX RVOT (mm)a | ≥29 |
| PLAX RVOT index (mm/m2)a | ≥16 |
| PSAX RVOT (mm)a | ≥32 |
| PSAX RVOT index (mm2)a | ≥18 |
| RV basal diameter (mm) | >41 |
| RV fractional area change (%)a | ≤40 |
| TAPSE (mm) | <17 |
| RV longitudinal strain of lateral RV free wall (%) | Worse than −23 |
| RV mechanical dispersion (SD of time-to-peak strain) (ms) | >25–30b |
| 3D RV-EF (%) | ≤40 |
| LV GLS (%) | Worse than −18 |
aMinor criteria from the TFC 2010.
bThree-segment model and six-segment model.
EF, ejection fraction; GLS, global longitudinal strain; LV, left ventricular; PLAX RVOT, parasternal long-axis right ventricular outflow tract; PSAX RVOT, parasternal short-axis right ventricular outflow tract; RV, right ventricle; TAPSE, tricuspid annular plane systolic excursion.
Advanced 2D echocardiographic methods
New echocardiographic techniques may increase the performance of conventional echocardiography. Despite its intrinsic limitations, such as angle dependency, Doppler tissue imaging is useful in the assessment of RV longitudinal systolic function [peak velocity of the systolic velocity of lateral tricuspid annulus in apical four-chamber view, s′] (Figure 4). RV function can be further assessed by speckle tracking echocardiography (STE), traced from the four-chamber view with focus on the RV. RV peak systolic longitudinal strain values from six RV segments are averaged to calculate RV global longitudinal strain (RV GLS) (Figure 5). Alternatively, peak systolic strains from three RV free wall segments are averaged as a measure of RV free wall strain which will result in higher absolute values compared with RV GLS.28 Both RV GLS and RV free wall strain have been reported to be reduced in the early phases of AC.24–27 In addition to amplitude parameters, temporal parameters such as the time-to-peak strain (time from onset R on ECG to maximum RV longitudinal shortening by STE) could also be assessed. RV mechanical dispersion can be calculated as the standard deviation of time-to-peak strain in a six-RV-segment model25 (Figure 5) or in a three-segment model24 (Figures 5B and 6). The six-RV-segment model is more robust for calculation of standard deviation (Table 2).
Figure 4.

Reduced longitudinal function in a patient with AC (TAPSE: 17 mm, s′ tricuspid annulus: 9.5 cm/s).
Figure 5.

Reduced RV strain and increased mechanical dispersion in an AC patient. Vertical white markers indicate peak longitudinal strain and horizontal white arrows indicate the time from R on QRS to peak longitudinal strain. (A) Six RV segments including the interventricular septum. Right ventricular global strain is calculated as average peak strain rom six RV segments. Mechanical dispersion is calculated as standard deviation of time-to-peak longitudinal strain in six RV segments. (B) RV free wall strain is calculated as average peak strain from three RV free wall segments. Courtesy: Dr Ø.H. Lie.
Figure 6.

Mechanical dispersion defined as standard deviation of time to LV peak longitudinal strain in AC. Compared with the healthy volunteer (left panel), the asymptomatic mutation carrier (mid panel) shows increased mechanical dispersion and reduced RV free wall strain. The AC patient with overt disease (right panel) has most pronounced mechanical dispersion and worse RV free wall strain (modified from Sarvari et al.25).
Figure 8.

3D TTE images showing the potential of this technique to diagnose AC: multiplanar display of the subtricuspid aneurism. Left upper: 4CH, right upper: 2CH; left lower: long axis; right lower: short axis at the level of the aneurysm (blue-dotted line on longitudinal views). Yellow arrows indicate the location of the aneurysm. Courtesy: Dr D. Muraru.
3D echocardiography
3D echocardiography (3DE) allows measurements of RV volumes overcoming the limitations of conventional 2D views with respect to orientation and reference points. RV and LV volume measurements will be of interest in patients with overt AC, whereas increased volumes are rare in early phases of AC. 3DE can obtain a reasonably accurate estimate of RV volumes and provide RV-EF, but expertise is needed and practical recommendations regarding RV acquisition and analysis by 3DE have to be followed. Technical challenges include particularly patients with imperfect image quality or advanced AC patients with severely enlarged RV (Figures 7–11). Normal 3DE values of RV volumes are available.22,29 However, no data currently exist for AC patients. Although 3DE tends to underestimate RV volumes, excellent correlations with CMR have been described.30 According to current guidelines, in laboratories with appropriate 3D platforms and experience, 3DE-derived RV-EF should be considered as a method of quantifying RV systolic function. Roughly, an RV-EF lower than 40–45% usually reflects abnormal RV systolic function.21,22
Figure 7.
3D TTE images showing the potential of this technique to diagnose AC: subtricuspid valve aneurysm. Courtesy: Dr D. Muraru.
Figure 11.

Longitudinal (upper panels) and transversal (lower panels) cut planes showing the localized aneurysm (asterisk). Courtesy: Dr D. Muraru.
Figure 9.

3D TTE images showing the potential of this technique to diagnose AC: 12 slice display of the subtricuspidal aneurysm. Yellow arrow indicates the location of the aneurysm. Courtesy: Dr D Muraru.
Figure 10.

3D TTE images showing the potential of this technique to diagnose AC: localized aneurysm. Left panel: RV focused apical 4CH view = normal; central panel: cropped longitudinal apical view showing the localized aneurysm (yellow arrows); right panel: en face view of the entry orifice of the aneurysm. Courtesy: Dr D. Muraru.
In summary, we recommend a broader approach of imaging methods than the current TFC, considering the inclusion of specific indications for imaging methods in probands (index patients, presenting with overt disease), follow- up of probands, and in family screening and in follow-up of mutation positive family members (see ‘Imaging follow-up in AC’ section). Abnormal RV or LV function by other parameters will probably strengthen the suspicion of AC in unclear cases. In addition to parameters used in the TFC, we propose to evaluate additional parameters in patients with suspected or established AC:
Conventional echo parameters: tricuspid annular plane systolic excursion (TAPSE) and RV basal diameter.23
Advanced echo parameters: RV GLS, LV GLS, and RV and LV mechanical dispersion by 2D-STE,25,31 in family screening and follow-up of early ARVC in which sensitive echocardiographic parameters are needed.
3DE parameters: However, the value of 3DE and cut-off values for AC diagnosis and risk stratification need to be established.
These parameters are not yet proved to increase diagnostic sensitivity but may be evaluated to prospectively gain new data in AC. New cut-off limits need to be considered and should be based on data from multiple large cohorts.
Key points:
RV wall motion abnormalities in addition to quantitative measurements of RVOT diameter and RV-FAC are the echo parameters included in the TFC 2010.
We recommend a broader, comprehensive, and systematic assessment of RV geometry and function using both conventional and advanced echocardiographic techniques (Tables 1 and 2).
Cardiac magnetic resonance
The accuracy of CMR to detect subtle RV regional functional and structural wall abnormalities has been shown to be higher than conventional 2D echocardiography, due to the higher spatial resolution of the former.32 The tomographic, high-spatial resolution, non-invasive tissue characterization nature of CMR is particularly suited for the assessment of cardiomyopathies, including AC. CMR is erroneously considered the ‘gold standard’ test to diagnose AC. As the TFC 20102 emphasizes, the diagnosis of this disease is a composite of familiar, ECG, arrhythmic, histological, functional, and structural features, in which CMR may play a role only to the latter two aspects. Great caution must be employed when the only abnormality in a presumed AC patient is found at the level of the RV on CMR, as it is uncommon for AC patients to have a normal ECG and Holter monitoring but an abnormal CMR exam.33
The CMR parameters from the TFC 2010 include RV regional dysfunction, reduced RV-EF and enlarged indexed RV end-diastole volume, as well as localized RV wall thinning and aneurysmal formations (Table 1, Figures 12 and 13, Supplementary data online, Video S2). Despite the ability of CMR to detect myocardial fibro-fatty replacement in current routine clinical practice with the late gadolinium enhancement (LGE) and gradient echo with fat saturation technique, this aspect is not included in the TFC 2010 as a diagnostic criterion. In fact, at the time of compiling the 2010 TFC, fibro-fatty replacement by CMR was not considered a robust parameter that could be consistently reproduced in different laboratories and lack of a control population was considered a major drawback. CMR plays also an important role in detecting AC phenocopies: unrecognized disease that could mimic AC such as RV volume overload, and myocardial scarring.34
Figure 12.

CMR RVOT in and out view, cine image in end diastole (A) and end systole (B). Micro-aneurysms of the RVOT and RV diaphragmatic wall are present (white arrows).
Figure 13.

CMR four-chamber view, LGE image (A) and corresponding image in cine end-diastolic frame (B). The white arrow shows myocardial LGE (fibrosis) of the RV free wall.
Of note, the TFC 2010 lacks specific diagnostic criteria for the non-classical variant of AC, which includes the dominant or isolated LV disease. LGE is typically located in a subepicardial/mid-wall distribution confined to the LV.1 However, LV dominant disease can be under-diagnosed and the abnormalities can be attributed to other disorders, such as myocarditis, dilated or hypertrophic cardiomyopathy.
Key points:
RV wall motion abnormalities in addition to RV volumes and RV-EF are the CMR diagnostic criteria included in the TFC 2010.
LGE on CMR is an important sign of disease and can be the only sign of LV involvement.
CMR alterations alone, without ECG and Holter abnormalities, are uncommon in AC disease except for the LV variant.
CMR protocol in the Supplementary data online, Supplementary data
Computed tomography
Cardiac computed tomography (CT) is not included in the diagnostic algorithm and thus usually is not part of the initial screening of patients with suspected AC. Nevertheless, multidetector computed tomography (MDCT) has an excellent spatial resolution and allows accurate quantification of RV and LV volumes/function and, similar to CMR, detection of fatty tissue in the myocardium.35 Currently, low-radiation-dose scan can be performed (1–2 mSv), while maintaining good temporal and spatial resolution.
According to the 2010 ACC/AHA appropriate use criteria,36 MDCT is appropriate for the evaluation of structural RV and/or LV remodelling in suspected AC, in particular for those patients with inadequate echocardiographic images and contraindications for CMR.35 Relevant MDCT parameters include RV dilatation, reduction of RV-EF, severe segmental dilatation, and regional hypokinesis which are part of the major or minor criteria for the diagnosis of AC.2 In addition the visualization of epicardial and intramyocardial fat helps to assess biventricular involvement.
CT protocol in the Supplementary data online, Supplementary data
Radionuclide angiography/single-photon emission computed tomography/positron emission tomography
Nuclear imaging is not included in the diagnostic algorithm of patients with suspected AC. Radionuclide angiography may provide measurements of RV volumes, RV ejection fraction, and the standard deviation of regional times of end systole which can help to recognize diffuse or localized forms of AC37 in patients with ventricular arrhythmias when the acoustic window is inadequate for echocardiography and the patient has contraindications to CMR.38
Abnormal presynaptic myocardial sympathetic function, demonstrated using 123I-MIBG single-photon emission computed tomography, is associated with a markedly higher risk of future recurrent life-threatening ventricular tachyarrhythmias both in patients with heart failure and in patients with AC.39,40 These findings are associated with downregulation of LV myocardial b-adrenergic receptor density, demonstrated by 11C-CGP-12177 positron emission tomography (PET).41 Nuclear imaging of myocardial sympathetic function may therefore be an additional individualized risk stratification tool in AC independently from other common structural features.
Key points:
MDCT is appropriate for the evaluation of structural remodelling in patients with suspected AC who have inadequate echocardiographic images and contraindications for CMR.
Nuclear imaging may be a potential risk stratification tool, but still is a research tool and needs further investigation.
Role of imaging in early disease
Early signs
The overt stage of AC is preceded by a concealed stage with no or only minor signs of disease. However, life-threatening arrhythmias can occur with only discrete myocardial structural changes.27,42–45 On the other side, presence of structural abnormalities highly increases the risk of ventricular arrhythmias. The echocardiographic diagnosis is challenging at the early stage of AC when only mild RV hypokinaesia and dilatation are present.43,46,47 In this situation, additional information can be obtained from CMR, strain echocardiography,25,48 and potentially from 3DE,49 although data are still limited or missing in the latter two imaging techniques. Teske et al.27 focused on early detection of AC and found abnormal RV strain in 71% of asymptomatic carriers. A dilated RV basal diameter and pronounced RV mechanical dispersion may be other early echocardiographic signs of AC disease (Table 2).24,25,50
The role of CMR is important and it is the preferred imaging modality in the early diagnosis of AC, providing assessment of function and tissue characterization of both ventricles. LGE is used to assess myocardial fibro-fatty replacement which may help to diagnose AC, although LGE is not specific for AC and may be present also in other cardiomyopathies43 (Table 3).
Table 3.
Advantages and limitations of imaging techniques
| Echocardiography | CMR | CT | Nuclear | |
|---|---|---|---|---|
| Advantages | Large availability | Assessment of structure and function | If inadequate echocardiographic images and contraindications for CMR | For research purposes |
| New techniques (strain) | Assess myocardial fibro-fatty replacement | |||
| Limitations | Need of specific expertise | Radiation | Limited documentation | |
| Need of specific expertise | Frequent ectopic beats interfere with acquisition | Limited documentation | ||
| Complex anatomy of the RV | Complex anatomy of the RV | Complex anatomy of the RV | ||
| Load dependency of the RV | Load dependency of the RV | Load dependency of the RV | ||
| Quantitative evaluation difficult | Limited availability | |||
| Limited value at the early stage | Difficult at the early stage | |||
| Risk of under diagnosis | Risk of over diagnosis |
CMR, cardiac magnetic resonance; CT, computed tomography; RV, right ventricle.
Role of imaging and genetic testing
First symptom of AC is most often VT or syncope. However, imaging findings like a dilated RV with wall motion abnormalities can sometimes be the first finding. To decide whether a patient with echocardiographic RV/LV pathology should undergo genetic testing for AC can be challenging. Importantly, due to high genetic variability an AC phenotype should always be present when referring a patient for AC genetic testing to reduce the risk of over-interpretation of genetic variants of uncertain significance. Patients with RV pathology and no previous arrhythmias should be carefully examined for other causes of RV pathology, e.g. pulmonary hypertension, ASD, ischaemia, etc. Furthermore, family history, occurrence of syncopes or arrhythmias, and history of sports participation increase the probability for AC correct genetic diagnosis. Finally, obvious RV dilation and motion abnormalities by visual assessment in combination with a normal ECG are rare in AC and genetic testing for AC is rarely indicated.
Key points:
Advanced echo modalities and CMR are the preferred imaging techniques in the early diagnosis of AC, allowing to detect subtle changes in biventricular function and tissue characterization.
Genetic testing can be considered in patients with accidentally diagnosed AC-resembling RV pathology in presence of ECG markers or other markers. Imaging pathology in asymptomatic patients with a normal ECG should not lead to genetic testing for AC.
Imaging in risk stratification of ventricular arrhythmias
Severely reduced RV function and LV involvement are important risk factors for ventricular arrhythmias in AC.51,52 RV dilatation, reduced TAPSE, and low FAC have been associated with arrhythmic risk in two studies.25,47 Also right atrial dilation and tricuspid regurgitation have been related to arrhythmic events in one study on AC.47 Based on these and other findings, AC patients with abnormal RV function (Table 2) should be followed closely and be continuously evaluated for implantable cardioverter defibrillator (ICD) implantation.44 The role of right chamber volume modifications for risk stratification of arrhythmias is controversial. In some cohorts, RV-EF and RV end-diastolic volume did not change significantly during long-term follow-up.8,52,53 Importantly, involvement of LV function is a strong marker for risk of ventricular arrhythmias.52
A single study has indicated that an impairment of adrenergic innervation (obtainable by 123I-MIBG) was associated with a higher incidence for future recurrences of VT in AC patients40 as discussed above (see ‘Radionuclide angiography/single-photon emission computed tomography/positron emission tomography’ section). This finding needs to be further investigated. To date, cardiac CT has not shown power in predicting VT in AC patients.
Key points:
Presence of any morphological or functional abnormalities in combination with electrical changes is the basis of AC diagnosis and indicates increased risk of ventricular arrhythmias.
Risk stratification is commonly performed on an individual basis and definite evidence-based parameters are lacking.
Imaging follow-up in AC
Patients with AC and their family members should be followed clinically and undergo imaging testing on a regular basis, preferably at tertiary referral hospitals with specific experience in managing patients with AC.54 It is essential that all diagnostic imaging options are used in a rational and comprehensive way, in order to establish:
AC diagnosis;
AC disease staging and progression;
risk assessment of ventricular arrhythmias;
assessment of heart failure and evaluation for cardiac transplant.
Imaging modality and follow-up intervals differ between AC probands, family members and in AC patients with ICD. In probands (index cases of AC, presenting with overt disease), RV structural changes are usually present and the diagnosis is often made with echocardiography alone. CMR may also be performed at the initial diagnosis, if available, for a better morphological and functional characterization, particularly if ICD is planned. CMR is mandatory in selected cases where the diagnosis depends on the presence or absence of imaging criteria and to assess the LV involvement by LGE CMR. AC probands often have an ICD due to ventricular arrhythmias. Newer ICDs are CMR conditional, i.e. patients can be safely scanned following a specific protocol. During follow-up, echocardiography is repeated at regular intervals (Table 4) and CMR should be repeated if there are changes in clinical status and in questions not answered by echo. Family screening and follow-up of mutation positive family members usually include individuals with no or only subtle morphological and functional findings. Because of the low sensitivity of echocardiography, both echo and CMR must be used to detect early phenotypes of the disease and repeated at regular time intervals.32,54
Table 4.
Proposed imaging methods and follow-up intervals in patients with definite AC diagnosis and in family members (mutation-positive or first-degree relatives from families without identified mutations)
| Echocardiography | CMR | CT | |
|---|---|---|---|
| AC patients with ICD | When clinically indicated by heart failure symptoms | Not indicated or contraindicateda | |
| AC patients without ICD | Every year or when clinically indicatedb | First visit. Every 3–5 years or when changes in clinical status and/or ECG occur | Patients who are difficult to assess with echo and unsuitable for CMR |
| In patients who are difficult to explore at echo | |||
| Family members from approximately 10 years of age with borderline findings | Every year in subjects <40 years of age or when indicated.b Every 2 years in subjects >40 years of age or when indicatedb | First visit. Every 1–2 years in subjects <40 years of age or when indicatedb | Patients who are difficult to assess with echo and unsuitable for CMR |
| Every 3–5 years or when indicated in subjects >40 yearsb | |||
| Family members from approximately 10 years of age without any morphological findings | Every 1–2 years in subjects <40 years of age or when indicated.b Every 3–5 years or when indicated in subjects >40 years of ageb | First visit. Every 1–2 years in subjects <40 years of age or when indicatedb | Patients who are difficult to assess with echo and unsuitable for CMR |
| Every 3–5 years or when indicated in subjects >40 years of ageb |
aIn patients with CMR incompatible devices.
bClinical or ECG changes suggestive of disease progression.
AC, arrhythmogenic cardiomyopathy; CMR, cardiac magnetic resonance; CT, multidetector computed tomography; ICD, implantable cardioverter defibrillator.
Importantly, there is no current evidence to guide the use of imaging during follow-up of patients with definite or possible diagnosis or in patients at-risk of AC. The following recommendations are based on cost-effectiveness considerations and consensus among members of the writing committee.
Patients with AC, implanted with ICD
Arrhythmogenic cardiomyopathy patients implanted with ICD are often probands and imaging is used to establish AC diagnosis and to follow disease progression. These patients are normally followed with ICD checks every 6–12 months. Echocardiography should be performed to detect progressive LV heart failure and help evaluation for potential start of heart failure treatment. Furthermore, in AC patients with severe RV or biventricular failure, echocardiographic follow-up may be needed for timing of heart transplantation. CMR is usually contraindicated (except in those with CMR conditional devices) and is not useful for clinical/management purposes if the diagnosis is already established.
Patients with definite diagnosis of AC and no ICD
In patients with a definite diagnosis of AC who are not implanted with an ICD, close follow-up is necessary. The potential need for ICD implantation must be discussed with the patient and considered at each visit. Careful evaluation of ECG, signal averaged ECG, Holter, CMR, and echocardiographic changes are warranted. Patients must be informed to contact their centre if they experience palpitations, syncope, or chest pain and report of these symptoms should lead to timely investigation of the patient and often ICD implantation. We propose at least yearly follow-up visits including ECG and Holter and with a low threshold of new visits if the patient reports any new symptoms. Exercise stress test can be used to assess exercise capacity and to provoke ventricular arrhythmias. Echocardiography should be performed in all patients with definite diagnosis of AC who report changes in clinical status or in whom new ECG changes are detected. Echocardiography studies should include quantitative assessment of RV and LV size and function, and RVOT diameter (Table 2). Risk of ventricular arrhythmias increases along with severity of structural and functional changes in RV and LV. In clinically stable AC patients without ICD, it seems reasonable to routinely repeat echocardiography every year. For CMR measures, no cut-off values are established indicating when primary prophylaxis ICD implantation is indicated. Therefore, the repeated use of CMR is currently not indicated. However, LGE LV involvement and progressive RV dysfunction indicate progression of disease and thereby higher risk of arrhythmias. CMR may therefore be appropriate to perform at first visit and to repeat routinely every 3–5 years or on an individual basis.54
Mutation-positive family members, early diagnosis
The follow-up strategy for mutation-positive family members of patients with AC or first-degree relatives from families without identified mutations remains to be defined. Penetrance of disease in family members is approximately 35%55 and efforts should aim at identifying these individuals. ECG changes often precede overt morphofunctional abnormalities of the RV, and only small changes in conventional imaging parameters are expected during follow-up of family members.56,57 Importantly, the finding of morphofunctional abnormalities in family members should lead to close follow-up due to the increased risk of ventricular arrhythmias.44 However, tissue alterations, forming the substrate for ventricular arrhythmias, may be present although no wall motion abnormalities or RV dilatation are detected by conventional imaging parameters and technique. The tissue substrate may potentially be detected by LGE CMR and strain echocardiography.58 The delay in myocardial activation and contraction inhomogeneity due to subtle tissue alterations could be detected by measuring RV mechanical dispersion by STE.25,59 We recommend echocardiographic follow-up every year and CMR at baseline and for follow-up at every 1–2 years in younger mutation-positive family members with borderline findings. In young AC mutation positive without any symptoms or signs of the disease, echocardiography should be performed yearly while CMR intervals can be expanded. Furthermore, in asymptomatic mutation-positive family members >40 years of age, CMR could be repeated at longer intervals. Detection of CMR abnormalities during follow-up may further lead to closer follow-up with ECG and Holter monitoring and evaluation of prophylactic ICD implantation. Follow-up of family members should start from age 10, as events before this age are extremely rare.
Key points:
Imaging follow-up strategies differ between AC patients with and without ICD and in family members.
Imaging in AC patients with ICD should focus on disease progression in order to start heart failure treatment or initiate evaluation for transplantation.
AC patients with definite or possible diagnosis without ICD should be closely monitored due to high risk of arrhythmias. Worsening of imaging findings should tend to close arrhythmia monitoring or ICD implantation.
AC family members should undergo full non-invasive testing at first visit and be followed with ECG and Holter.
Echocardiography should be repeated if changes in clinical status or ECG changes occur and be routinely repeated every year in mutation carriers with borderline ARVC diagnosis and every 2–3 years in patients without any clinical, ECG, or morphological findings.
CMR should be repeated at first visit and be routinely repeated every 1–2 years in mutation carriers with borderline ARVC diagnosis and every 3–5 years in patients without any clinical, ECG, or morphological findings.
Other diagnostic modalities in AC and shortcomings of TFC 2010
Updated diagnostic criteria were published in 2010 to improve sensitivity, but with the important prerequisite of maintaining diagnostic specificity. Quantitative imaging cut-off points RV measures, morphofunctional abnormalities, tissue characterization by endomyocardial biopsy, ECG, and signal-averaged ECG have been introduced. Genetic data have been also incorporated. However, although the TFC 2010 criteria acknowledge the existence of a broad disease spectrum, that includes also LV and biventricular subtypes, the revised criteria are addressing only the classical RV variant. No specific diagnostic guidelines do exist for the LV involvement, with the exception of: a moderate-to-severe LV dysfunction on imaging, lateral or inferolateral T-wave inversion (leads V5, V6, Lead I, and aVL) and low voltage QRS complex on standard limb leads on ECG, and right bundle branch block/polymorphic ventricular arrhythmias.
Endomyocardial biopsy is proposed for AC diagnosis in cases who remain undetermined after extensive non-invasive testing and in patients with suspected AC phenocopies. Biopsy may reveal myocyte atrophy and replacement by fibrous and fatty tissue. An ARVC major criterion is fulfilled when residual myocytes constitute <60% by morphometric analysis with fibrous replacement of the right ventricle free wall myocardium, with or without fatty replacement of tissue. Residual myocytes between 60% and 75% by morphometric analysis serve as a minor criterion.2
Electro anatomical mapping is another diagnostic tool used for diagnosis and to guide catheter ablation in AC patients with recurrent VT. This technique can be used to identify the abnormal low-voltage areas, which have been demonstrated to correspond to the loss of electrically active myocardium caused by fibro-fatty tissue, and is particularly useful for differential diagnosis with idiopathic RVOT tachycardia.60
Finally, the new AC criteria include the identification of a pathogenic mutation categorized as associated or probably associated with AC in the patient under evaluation. However, it has been emphasized that a pathogenic mutation is a DNA alteration associated with AC that alters, or is expected to alter, the encoded protein, and which is unobserved or rare in a large non-AC control population. Furthermore, a pathogenic mutation either alters or is predicted to alter the structure or function of the protein or has demonstrated linkage to the disease phenotype in a conclusive pedigree (so-called co-segregation). Strict adherence to these criteria is essential, particularly at present with the advent of next-generation sequencing.
Key point:
Genetic data, endomyocardial biopsy, and electro anatomical mapping are additional diagnostic techniques helpful in the evaluation of AC.
Imaging pre- and post-RF ablation (see Supplementary data online, Supplementary data)
How to differentiate between AC from other arrhythmic diseases and acquired conditions
Right ventricular outflow tract-VT
The RVOT is the most common site of origin for idiopathic VT and frequent premature ventricular complexes in patients with structurally normal hearts, known as RVOT-VT. In contrast to AC, RVOT-VT is usually a relatively benign condition, with generally well-tolerated ventricular arrhythmias.7,60 However, the RVOT area may also be origin of VT in patients with AC and in early stages of AC the distinction to RVOT-VT may be challenging. The treatment and prognosis differ substantially and an incorrect diagnosis may be devastating. Patients with RVOT-VT commonly have structurally normal ventricles, but frequent premature ventricular contractions may cause myocardial remodelling with subsequent reduced function and dilatation, which further complicates the discrimination to AC. By cardiac imaging, fibro-fatty replacement is not present in patients with RVOT-VT, whereas this is a hallmark of AC. Any findings of regional RV hypokinaesia and dyskinaesia by echo or CMR in addition to RVOT dilatation make the diagnosis of AC more probable and prognosis more severe.61
Other diagnostic tests include family history, which is normally negative in RVOT-VT patients, genetic testing for AC related mutations, and Holter monitoring. ECG markers for AC as T-wave inversions in the precordial leads are absent in RVOT-VT patients. High numbers of VPC (>9000 per 24 h) in the absence of severe structural alterations increase the probability that the diagnosis is RVOT-VT and not AC.24 In selected cases, electrovoltage anatomic mapping and endomyocardial biopsy can be determinants for differential diagnosis.60
Key point:
RVOT VT patients have more frequent premature ventricular contractions than AC patients and usually no structural or mechanical alterations.
Sarcoidosis and myocarditis
Patients with cardiac sarcoidosis or myocarditis have a few significantly different cardiac imaging characteristics when compared with patients with AC. The cardiac volume, in addition to the degree and location of cardiac involvement, can be used to distinguish between these disease entities. For instance, the presence of mediastinal lymphadenopathy, LV septal scar (LGE), significant LV dysfunction, and segmental areas of decreased uptake (201Thallium, 99-mTc Sestamibi, etc.) in the ventricular myocardium that disappears or decrease in size during stress, and intense PET-FDG (fludeoxyglucose) uptake in the myocardium should raise the suspicion for cardiac sarcoidosis.62 Consideration of cardiac sarcoidosis should be given if these imaging findings are observed during the evaluation for possible AC, particularly in presence of conduction disturbances or systemic disease and in the absence of family history.
Inflammatory rheumatic diseases, particularly systemic sclerosis can mimic AC and cardiac involvement is a major cause of death in these patients. Regional wall motion abnormalities can occur irrespective of pulmonary artery hypertension in systemic sclerosis. However, the diagnosis of systemic sclerosis differentiates from AC by multiorgan affection.
In myocarditis the LV is most often involved and the RV is rarely affected selectively. Areas of segmental perfusion defects coupled with FDG uptake, local oedema (T2-weighted images), epicardial/mid-wall LGE on CMR, and the presence of severe global or regional hypokinaesia without a specific coronary territory distribution are in favour of myocarditis, when clinically suspected.63 Of note, although endomyocardial biopsy is still considered the gold standard, it can be inconclusive since these diseases tend to be focal.64 Higher sensitivity is achieved through imaging-guided endomyocardial biopsy.65,66
Key points:
Sarcoidosis and systemic sclerosis can mimic AC. PET-FDG could be reasonably recommended and a positive test raise the suspicion for cardiac sarcoidosis.
Myocarditis may also mimic AC, and endomyocardial biopsy—although showing a low sensitivity—remains the gold standard for diagnosis
Dilated cardiomyopathies
Dilated cardiomyopathy may be particularly difficult to distinguish from non-classic (biventricular or left-dominant) forms of AC when there is early affection of the LV. Echocardiography has a limited role in this regard since LV dilation and/or systolic impairment are non-specific features. In many cases of left-dominant AC, LV structural abnormalities are localized in the posterolateral region.67,68 CMR may further aid to diagnosis by providing tissue characterization and identification of intra-myocardial fat and fibrosis in addition to assessment of LV morphology and function. Although findings like mid-wall LGE may be observed in both left dominant AC and dilated cardiomyopathy, the subepicardial distribution favours AC.69 Moreover, LV fatty infiltration was shown to be a prevalent finding in AC, often involving the subepicardial LV lateral wall and resulting in myocardial wall thinning.33,70
Key point:
Left dominant AC can mimic dilated cardiomyopathy. Frequent arrhythmias and subepicardial fibro-fatty replacement on CMR favour AC diagnosis.
Congenital heart diseases
Congenital heart diseases with RV overload including anomalous venous drainage, ASD, can be misdiagnosed as AC. ECG changes may help correct diagnosis in addition to careful imaging including transoesophageal echocardiography. Right heart catheterization may be necessary to detect potential shunts.
Athlete’s heart
Right ventricular and right atrium dilation are not specific to AC and are commonly found in athletes performing high-intensity exercise. Other important findings in athletes can be mild functional tricuspid regurgitation and dilation of the inferior vena cava.71,72 These findings partly resemble echocardiographic findings in AC and can make it challenging to distinguish an athlete heart from AC by traditional cardiac imaging methods. Furthermore, athletic activity aggravates structural disease in AC patients which further complicate discrimination from athlete’s heart.50,73 RV cavity dilatation in athletes involves particularly RV inflow tract and is almost always associated with LV enlargement (balanced enlargement).72 Typical findings in AC, like RV thinning, bulging, and aneurysms will normally not be found in athletes. Importantly, TAPSE is typically normal and no convincing differences in strain measurements are found even in dilated RV in athletes.74–76 AC or another type of RV cardiomyopathy should be suspected if RV strain values or TAPSE suggests a decreased RV function.25,77 Furthermore, LV GLS is most often normal in athletes.72 CMR must always be requested if the diagnosis of AC is suspected in athletes. However, CMR RV alterations alone, without abnormalities on ECG and Holter, are uncommon in AC disease.
Key points:
AC is aggravated by athletic activity.
Abnormal measures of deformation strain imaging favour AC diagnosis.
CMR should be performed in unclear cases. CMR alterations alone, without abnormalities on ECG and Holter monitoring, should favour athlete-induced changes and not AC diagnosis.
Brugada syndrome
Although Brugada syndrome (BrS) is not associated with overt RV and LV structural abnormalities, endomyocardial biopsy, MDCT, and CMR have identified mild RV abnormalities.78–82 In particular, RV wall motion abnormalities of the inferior wall have been detected by 2D echocardiography. A recent study has also shown in BrS mild reduction of speckle tracking-derived RV GLS and of regional basal or mid RV free wall longitudinal strain which are, however, less pronounced than in AC.83,84 Based on these findings, advanced cardiac imaging could be useful to distinguish BrS from AC, in overlapping syndromes and when the clinical picture and ECG signs do not provide decisive elements for diagnosis.
Key point:
Overlap exists between AC and BrS phenotypes including RV wall motion abnormalities. The clinical implication is unclear.
Potential future role of different imaging modalities in AC (see Supplementary data online, Supplementary data)
Summary
In this expert consensus document, we have provided an overview of currently available imaging modalities and parameters to be used in diagnosis and follow-up of patients evaluated for AC. We propose a wider spectrum of echocardiographic parameters which can increase the sensitivity of the modality for AC. Furthermore, we have provided the recommendations of imaging use during follow-up in AC patients and their family members. Finally, we have presented the most important differential diagnoses and how to distinguish these from AC.
Supplementary data
Supplementary data are available at European Heart Journal – Cardiovascular Imaging online.
Conflict of interest: None declared.
Acknowledgements
CBD is supported by the Bristol NIHR Cardiovascular BRU. The views expressed are those of the authors and not necessarily those of the UK National Health Service, National Institute for Health Research or Department of Health.
Supplementary Material
Contributor Information
EACVI Scientific Documents Committee, EACVI Board members and external reviewers:
Victoria Delgado, Bernard Cosyns, Erwan Donal, Massimo Lombardi, Denisa Muraru, Philipp Kauffmann, Ruxandra Jurcut, Jutta Bergler Klein, and Leyla Elif Sade
References
- 1. Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D. et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 2008;52:2175–87. [DOI] [PubMed] [Google Scholar]
- 2. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA. et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010;31:806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996;94:983–91. [DOI] [PubMed] [Google Scholar]
- 4. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988;318:129–33. [DOI] [PubMed] [Google Scholar]
- 5. Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol 2012;9:223–33. [DOI] [PubMed] [Google Scholar]
- 6. Basso C, Thiene G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat? Cardiovasc Pathol 2005;14:37–41. [DOI] [PubMed] [Google Scholar]
- 7. Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009;373:1289–300. [DOI] [PubMed] [Google Scholar]
- 8. Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C. et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2000;36:2226–33. [DOI] [PubMed] [Google Scholar]
- 9. Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G. et al. Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol 1988;12:1222–8. [DOI] [PubMed] [Google Scholar]
- 10. McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A. et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000;355:2119–24. [DOI] [PubMed] [Google Scholar]
- 11. Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE. et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 2000;9:2761–6. [DOI] [PubMed] [Google Scholar]
- 12. Pilichou K, Thiene G, Bauce B, Rigato I, Lazzarini E, Migliore F. et al. Arrhythmogenic cardiomyopathy. Orphanet J Rare Dis 2016;11:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paul M, Meyborg M, Boknik P, Gergs U, Gerss J, Schmitz W. et al. Autonomic dysfunction in patients with arrhythmogenic right ventricular cardiomyopathy: biochemical evidence of altered signaling pathways. Pacing Clin Electrophysiol 2014;37:173–8. [DOI] [PubMed] [Google Scholar]
- 14. Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR. et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol 2002;40:1445–50. [DOI] [PubMed] [Google Scholar]
- 15. Nasir K, Bomma C, Tandri H, Roguin A, Dalal D, Prakasa K. et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity: a need to broaden diagnostic criteria. Circulation 2004;110:1527–34. [DOI] [PubMed] [Google Scholar]
- 16. Cox MG, Nelen MR, Wilde AA, Wiesfeld AC, van der Smagt JJ, Loh P. et al. Activation delay and VT parameters in arrhythmogenic right ventricular dysplasia/cardiomyopathy: toward improvement of diagnostic ECG criteria. J Cardiovasc Electrophysiol 2008;19:775–81. [DOI] [PubMed] [Google Scholar]
- 17. Norman M, Simpson M, Mogensen J, Shaw A, Hughes S, Syrris P. et al. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 2005;112:636–42. [DOI] [PubMed] [Google Scholar]
- 18. Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G. et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J 2005;26:1666–75. [DOI] [PubMed] [Google Scholar]
- 19. Kamath GS, Zareba W, Delaney J, Koneru JN, McKenna W, Gear K. et al. Value of the signal-averaged electrocardiogram in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 2011;8:256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G. et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J 1994;71:215–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, Chandrasekaran K. et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr 2010;23:685–713; quiz 86-8. [DOI] [PubMed] [Google Scholar]
- 22. Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L. et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging 2015;16:233–70. [DOI] [PubMed] [Google Scholar]
- 23. Leren IS, Saberniak J, Haland TF, Edvardsen T, Haugaa KH. Echocardiography combined with ECGs improve identification of arrhythmic events in early ARVC. JACC Cardiovasc Imaging 2016;pii:S1936-878X(16)30619-2. [DOI] [PubMed] [Google Scholar]
- 24. Saberniak J, Leren IS, Haland TF, Beitnes JO, Hopp E, Borgquist R. et al . Comparison of patients with early-phase arrhythmogenic right ventricular cardiomyopathy and right ventricular outflow tract ventricular tachycardia. Eur Heart J Cardiovasc Imaging 2016pii: jew014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sarvari SI, Haugaa KH, Anfinsen OG, Leren TP, Smiseth OA, Kongsgaard E. et al. Right ventricular mechanical dispersion is related to malignant arrhythmias: a study of patients with arrhythmogenic right ventricular cardiomyopathy and subclinical right ventricular dysfunction. Eur Heart J 2011;32:1089–96. [DOI] [PubMed] [Google Scholar]
- 26. Teske AJ, Cox MG, De Boeck BW, Doevendans PA, Hauer RN, Cramer MJ. Echocardiographic tissue deformation imaging quantifies abnormal regional right ventricular function in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Soc Echocardiogr 2009;22:920–7. [DOI] [PubMed] [Google Scholar]
- 27. Teske AJ, Cox MG, Te Riele AS, De Boeck BW, Doevendans PA, Hauer RN. et al. Early detection of regional functional abnormalities in asymptomatic ARVD/C gene carriers. J Am Soc Echocardiogr 2012;25:997–1006. [DOI] [PubMed] [Google Scholar]
- 28. Muraru D, Onciul S, Peluso D, Soriani N, Cucchini U, Aruta P. et al. Sex- and method-specific reference values for right ventricular strain by 2-dimensional speckle-tracking echocardiography. Circ Cardiovasc Imaging 2016;9:e003866. [DOI] [PubMed] [Google Scholar]
- 29. Maffessanti F, Muraru D, Esposito R, Gripari P, Ermacora D, Santoro C. et al. Age-, body size-, and sex-specific reference values for right ventricular volumes and ejection fraction by three-dimensional echocardiography: a multicenter echocardiographic study in 507 healthy volunteers. Circ Cardiovasc Imaging 2013;6:700–10. [DOI] [PubMed] [Google Scholar]
- 30. Muraru D, Spadotto V, Cecchetto A, Romeo G, Aruta P, Ermacora D. et al. New speckle-tracking algorithm for right ventricular volume analysis from three-dimensional echocardiographic data sets: validation with cardiac magnetic resonance and comparison with the previous analysis tool. Eur Heart J Cardiovasc Imaging 2015;pii:jev309. [DOI] [PubMed] [Google Scholar]
- 31. Kjaergaard J, Hastrup Svendsen J, Sogaard P, Chen X, Bay Nielsen H, Kober L. et al. Advanced quantitative echocardiography in arrhythmogenic right ventricular cardiomyopathy. J Am Soc Echocardiogr 2007;20:27–35. [DOI] [PubMed] [Google Scholar]
- 32. Borgquist R, Haugaa KH, Gilljam T, Bundgaard H, Hansen J, Eschen O. et al. The diagnostic performance of imaging methods in ARVC using the 2010 Task Force criteria. Eur Heart J Cardiovasc Imaging 2014;15:1219–25. [DOI] [PubMed] [Google Scholar]
- 33. te Riele AS, Tandri H, Bluemke DA. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update. J Cardiovasc Magn Reson 2014;16:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quarta G, Husain SI, Flett AS, Sado DM, Chao CY, Tome Esteban MT. et al. Arrhythmogenic right ventricular cardiomyopathy mimics: role of cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2013;15:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Te Riele AS, Tandri H, Sanborn DM, Bluemke DA. Noninvasive multimodality imaging in ARVD/C. JACC Cardiovasc Imaging 2015;8:597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taylor AJ, Cerqueira M, Hodgson JM, Mark D, Min J, O'Gara P. et al. ACCF/SCCT/ACR/AHA/ASE/ASNC/NASCI/SCAI/SCMR 2010 appropriate use criteria for cardiac computed tomography. A report of the American College of Cardiology Foundation Appropriate Use Criteria Task Force, the Society of Cardiovascular Computed Tomography, the American College of Radiology, the American Heart Association, the American Society of Echocardiography, the American Society of Nuclear Cardiology, the North American Society for Cardiovascular Imaging, the Society for Cardiovascular Angiography and Interventions, and the Society for Cardiovascular Magnetic Resonance. J Am Coll Cardiol 2010;56:1864–94. [DOI] [PubMed] [Google Scholar]
- 37. Mariano-Goulart D, Dechaux L, Rouzet F, Barbotte E, Caderas de Kerleau C, Rossi M. et al. Diagnosis of diffuse and localized arrhythmogenic right ventricular dysplasia by gated blood-pool SPECT. J Nucl Med 2007;48:1416–23. [DOI] [PubMed] [Google Scholar]
- 38. Hendel RC, Berman DS, Di, Carli MF, Heidenreich PA, Henkin RE, Pellikka PA. et al. ACCF/ASNC/ACR/AHA/ASE/SCCT/SCMR/SNM 2009 appropriate use criteria for cardiac radionuclide imaging: a report of the American College of Cardiology Foundation Appropriate Use Criteria Task Force, the American Society of Nuclear Cardiology, the American College of Radiology, the American Heart Association, the American Society of Echocardiography, the Society of Cardiovascular Computed Tomography, the Society for Cardiovascular Magnetic Resonance, and the Society of Nuclear Medicine. Circulation 2009;119:e561–87. [DOI] [PubMed] [Google Scholar]
- 39. Jacobson AF, Senior R, Cerqueira MD, Wong ND, Thomas GS, Lopez VA. et al. Myocardial iodine-123 meta-iodobenzylguanidine imaging and cardiac events in heart failure. Results of the prospective ADMIRE-HF (AdreView Myocardial Imaging for Risk Evaluation in Heart Failure) study. J Am Coll Cardiol 2010;55:2212–21. [DOI] [PubMed] [Google Scholar]
- 40. Paul M, Wichter T, Kies P, Gerss J, Wollmann C, Rahbar K. et al. Cardiac sympathetic dysfunction in genotyped patients with arrhythmogenic right ventricular cardiomyopathy and risk of recurrent ventricular tachyarrhythmias. J Nucl Med 2011;52:1559–65. [DOI] [PubMed] [Google Scholar]
- 41. Wichter T, Schafers M, Rhodes CG, Borggrefe M, Lerch H, Lammertsma AA. et al. Abnormalities of cardiac sympathetic innervation in arrhythmogenic right ventricular cardiomyopathy: quantitative assessment of presynaptic norepinephrine reuptake and postsynaptic beta-adrenergic receptor density with positron emission tomography. Circulation 2000;101:1552–8. [DOI] [PubMed] [Google Scholar]
- 42. Saffitz JE. Arrhythmogenic cardiomyopathy: advances in diagnosis and disease pathogenesis. Circulation 2011;124:e390–2. [DOI] [PubMed] [Google Scholar]
- 43. Haugaa KH, Haland TF, Leren IS, Saberniak J, Edvardsen T. Arrhythmogenic right ventricular cardiomyopathy, clinical manifestations, and diagnosis. Europace 2015; 18: 965–72. [DOI] [PubMed] [Google Scholar]
- 44. Te Riele AS, James CA, Groeneweg JA, Sawant AC, Kammers K, Murray B. et al. Approach to family screening in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur Heart J 2016;37:755–63. [DOI] [PubMed] [Google Scholar]
- 45. Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol 2013;61:1945–8. [DOI] [PubMed] [Google Scholar]
- 46. Yoerger DM, Marcus F, Sherrill D, Calkins H, Towbin JA, Zareba W. et al. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the multidisciplinary study of right ventricular dysplasia. J Am Coll Cardiol 2005;45:860–5. [DOI] [PubMed] [Google Scholar]
- 47. Saguner AM, Vecchiati A, Baldinger SH, Rueger S, Medeiros-Domingo A, Mueller-Burri AS. et al. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circul Cardiovasc Imaging 2014;7:230–9. [DOI] [PubMed] [Google Scholar]
- 48. Prakasa KR, Wang J, Tandri H, Dalal D, Bomma C, Chojnowski R. et al. Utility of tissue Doppler and strain echocardiography in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol 2007;100:507–12. [DOI] [PubMed] [Google Scholar]
- 49. Prakasa KR, Dalal D, Wang J, Bomma C, Tandri H, Dong J. et al. Feasibility and variability of three dimensional echocardiography in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol 2006;97:703–9. [DOI] [PubMed] [Google Scholar]
- 50. Saberniak J, Hasselberg NE, Borgquist R, Platonov PG, Sarvari SI, Smith HJ. et al. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur J Heart Fail 2014;16:1337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A. et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an International Task Force Consensus Statement. Circulation 2015;132:441–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pinamonti B, Dragos AM, Pyxaras SA, Merlo M, Pivetta A, Barbati G. et al. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: results from a 10-year registry. Eur Heart J 2011;32:1105–13. [DOI] [PubMed] [Google Scholar]
- 53. Folino AF, Bauce B, Frigo G, Nava A. Long-term follow-up of the signal-averaged ECG in arrhythmogenic right ventricular cardiomyopathy: correlation with arrhythmic events and echocardiographic findings. Europace 2006;8:423–9. [DOI] [PubMed] [Google Scholar]
- 54. Haugaa KH, Bundgaard H, Edvardsen T, Eschen O, Gilljam T, Hansen J. et al. Management of patients with Arrhythmogenic Right Ventricular Cardiomyopathy in the Nordic countries. Scand Cardiovasc J 2015;49:299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C. et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circul Cardiovasc Genet 2015;8:437–46. [DOI] [PubMed] [Google Scholar]
- 56. te Riele AS, Bhonsale A, James CA, Rastegar N, Murray B, Burt JR. et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1761–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. te Riele AS, James CA, Rastegar N, Bhonsale A, Murray B, Tichnell C. et al. Yield of serial evaluation in at-risk family members of patients with ARVD/C. J Am Coll Cardiol 2014;64:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Perazzolo Marra M, Rizzo S, Bauce B, De Lazzari M, Pilichou K, Corrado D. et al. Arrhythmogenic right ventricular cardiomyopathy. Contribution of cardiac magnetic resonance imaging to the diagnosis. Herz 2015;40:600–6. [DOI] [PubMed] [Google Scholar]
- 59. Tops LF, Prakasa K, Tandri H, Dalal D, Jain R, Dimaano VL. et al. Prevalence and pathophysiologic attributes of ventricular dyssynchrony in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2009;54:445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Corrado D, Basso C, Leoni L, Tokajuk B, Turrini P, Bauce B. et al. Three-dimensional electroanatomical voltage mapping and histologic evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J Am Coll Cardiol 2008;51:731–9. [DOI] [PubMed] [Google Scholar]
- 61. Aquaro GD, Pingitore A, Strata E, Di Bella G, Molinaro S, Lombardi M. Cardiac magnetic resonance predicts outcome in patients with premature ventricular complexes of left bundle branch block morphology. J Am Coll Cardiol 2010;56:1235–43. [DOI] [PubMed] [Google Scholar]
- 62. Steckman DA, Schneider PM, Schuller JL, Aleong RG, Nguyen DT, Sinagra G. et al. Utility of cardiac magnetic resonance imaging to differentiate cardiac sarcoidosis from arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 2012;110: 575–9. [DOI] [PubMed] [Google Scholar]
- 63. Blankstein R, Osborne M, Naya M, Waller A, Kim CK, Murthy VL. et al. Cardiac positron emission tomography enhances prognostic assessments of patients with suspected cardiac sarcoidosis. J Am Coll Cardiol 2014;63:329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pieroni M, Dello Russo A, Marzo F, Pelargonio G, Casella M, Bellocci F. et al. High prevalence of myocarditis mimicking arrhythmogenic right ventricular cardiomyopathy differential diagnosis by electroanatomic mapping-guided endomyocardial biopsy. J Am Coll Cardiol 2009;53:681–9. [DOI] [PubMed] [Google Scholar]
- 65. Corrado D, Basso C, Leoni L, Tokajuk B, Bauce B, Frigo G. et al. Three-dimensional electroanatomic voltage mapping increases accuracy of diagnosing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2005;111:3042–50. [DOI] [PubMed] [Google Scholar]
- 66. Vasaiwala SC, Finn C, Delpriore J, Leya F, Gagermeier J, Akar JG. et al. Prospective study of cardiac sarcoid mimicking arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol 2009;20:473–6. [DOI] [PubMed] [Google Scholar]
- 67. Pinamonti B, Pagnan L, Bussani R, Ricci C, Silvestri F, Camerini F. Right ventricular dysplasia with biventricular involvement. Circulation 1998;98:1943–5. [DOI] [PubMed] [Google Scholar]
- 68. Lindstrom L, Nylander E, Larsson H, Wranne B. Left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy—a scintigraphic and echocardiographic study. Clin Physiol Funct Imaging 2005;25:171–7. [DOI] [PubMed] [Google Scholar]
- 69. Raman SV, Basso C, Tandri H, Taylor MR. Imaging phenotype vs genotype in nonhypertrophic heritable cardiomyopathies: dilated cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy. Circul Cardiovasc Imaging 2010;3:753–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Te Riele AS, James CA, Philips B, Rastegar N, Bhonsale A, Groeneweg JA. et al. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol 2013;24:1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. La Gerche A, Claessen G, Van de Bruaene A, Pattyn N, Van Cleemput J, Gewillig M. et al. Cardiac MRI: a new gold standard for ventricular volume quantification during high-intensity exercise. Circul Cardiovasc Imaging 2013;6:329–38. [DOI] [PubMed] [Google Scholar]
- 72. Galderisi M, Cardim N, D’Andrea A, Bruder O, Cosyns B, Davin L. et al. The multi-modality cardiac imaging approach to the Athlete's heart: an expert consensus of the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging 2015;16:353. [DOI] [PubMed] [Google Scholar]
- 73. James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H. et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Oxborough D, Sharma S, Shave R, Whyte G, Birch K, Artis N. et al. The right ventricle of the endurance athlete: the relationship between morphology and deformation. J Am Soc Echocardiogr 2012;25:263–71. [DOI] [PubMed] [Google Scholar]
- 75. D'Andrea A, Caso P, Bossone E, Scarafile R, Riegler L, Di Salvo G. et al. Right ventricular myocardial involvement in either physiological or pathological left ventricular hypertrophy: an ultrasound speckle-tracking two-dimensional strain analysis. Eur J Echocardiogr 2010;11:492–500. [DOI] [PubMed] [Google Scholar]
- 76. Pagourelias ED, Kouidi E, Efthimiadis GK, Deligiannis A, Geleris P, Vassilikos V. Right atrial and ventricular adaptations to training in male Caucasian athletes: an echocardiographic study. J Am Soc Echocardiogr 2013;26: 1344–52. [DOI] [PubMed] [Google Scholar]
- 77. La Gerche A, Connelly KA, Mooney DJ, MacIsaac AI, Prior DL. Biochemical and functional abnormalities of left and right ventricular function after ultra-endurance exercise. Heart (British Cardiac Society) 2008;94:860–6. [DOI] [PubMed] [Google Scholar]
- 78. Frustaci A, Priori SG, Pieroni M, Chimenti C, Napolitano C, Rivolta I. et al. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005;112:3680–7. [DOI] [PubMed] [Google Scholar]
- 79. Corrado D, Nava A, Buja G, Martini B, Fasoli G, Oselladore L. et al. Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J Am Coll Cardiol 1996;27:443–8. [DOI] [PubMed] [Google Scholar]
- 80. Takagi M, Aihara N, Kuribayashi S, Taguchi A, Shimizu W, Kurita T. et al. Localized right ventricular morphological abnormalities detected by electron-beam computed tomography represent arrhythmogenic substrates in patients with the Brugada syndrome. Eur Heart J 2001;22:1032–41. [DOI] [PubMed] [Google Scholar]
- 81. Catalano O, Antonaci S, Moro G, Mussida M, Frascaroli M, Baldi M. et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur Heart J 2009;30:2241–8. [DOI] [PubMed] [Google Scholar]
- 82. Tessa C, Del Meglio J, Ghidini Ottonelli A, Diciotti S, Salvatori L, Magnacca M. et al. Evaluation of Brugada syndrome by cardiac magnetic resonance. Int J Cardiovasc Imaging 2012;28:1961–70. [DOI] [PubMed] [Google Scholar]
- 83. Murata K, Ueyama T, Tanaka T, Nose Y, Wada Y, Matsuzaki M. Right ventricular dysfunction in patients with Brugada-like electrocardiography: a two dimensional strain imaging study. Cardiovasc Ultrasound 2011;9:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Iacoviello M, Forleo C, Puzzovivo A, Nalin I, Guida P, Anaclerio M. et al. Altered two-dimensional strain measures of the right ventricle in patients with Brugada syndrome and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur J Echocardiogr 2011;12:773–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.