

Abstract
Background
An ELISA to analyse uromodulin in human serum (sUmod) was developed, validated and tested for clinical applications.
Methods
We assessed sUmod, a very stable antigen, in controls, patients with chronic kidney disease (CKD) stages 1–5, persons with autoimmune kidney diseases and recipients of a renal allograft by ELISA.
Results
Median sUmod in 190 blood donors was 207 ng/mL (women: men, median 230 versus 188 ng/mL, P = 0.006). sUmod levels in 443 children were 193 ng/mL (median). sUmod was correlated with cystatin C (rs = −0.862), creatinine (rs = −0.802), blood urea nitrogen (BUN) (rs = −0.645) and estimated glomerular filtration rate (eGFR)–cystatin C (rs = 0.862). sUmod was lower in systemic lupus erythematosus-nephritis (median 101 ng/mL), phospholipase-A2 receptor- positive glomerulonephritis (median 83 ng/mL) and anti-glomerular basement membrane positive pulmorenal syndromes (median 37 ng/mL). Declining sUmod concentrations paralleled the loss of kidney function in 165 patients with CKD stages 1–5 with prominent changes in sUmod within the ‘creatinine blind range’ (71–106 µmol/L). Receiver-operating characteristic analysis between non-CKD and CKD-1 was superior for sUmod (AUC 0.90) compared with eGFR (AUC 0.39), cystatin C (AUC 0.39) and creatinine (AUC 0.27). sUmod rapidly recovered from 0 to 62 ng/mL (median) after renal transplantation in cases with immediate graft function and remained low in delayed graft function (21 ng/mL, median; day 5–9: relative risk 1.5–2.9, odds ratio 1.5–6.4). Immunogold labelling disclosed that Umod is transferred within cytoplasmic vesicles to both the apical and basolateral plasma membrane. Umod revealed a disturbed intracellular location in kidney injury.
Conclusions
We conclude that sUmod is a novel sensitive kidney-specific biomarker linked to the structural integrity of the distal nephron and to renal function.
Keywords: kidney failure, renal biomarker, serum assay, thick ascending limb, uromodulin
INTRODUCTION
Uromodulin (Umod) is a glycoprotein exclusively synthesized in the human kidney by epithelial cells of the thick ascending limb (TAL) of the loop of Henle and the early distal convoluted tubule [1–4]. This glycosylphosphatidylinositol (GPI)-anchored protein is predominantly localized to the apical plasma membrane, from which it is released into the tubular lumen by proteolytic cleavage [5]. Umod of urinary origin (uUmod) is a high molecular weight polymer and the most abundant urinary protein, detectable at a concentration of approximately 75 mg/24 h [6–8]. As a renal defensin, Umod protects against urinary tract infection by clotting fibrillary type-1 and type-S adhesins of bacteria [6–8]. Furthermore, uUmod prevents renal stone formation [9], is involved in handling tubular Na+/K+/Cl− transport and is suspected to contribute to arterial hypertension [10–12]. Umod exhibits immunomodulatory properties, activates granulocytes, mediates monocyte cytotoxicity and inhibits T-cell proliferation [13–15]. In vitro studies showed that Umod can be either pro- or anti-inflammatory [16, 17]. Epithelia of the TAL are specifically susceptible to oxidative stress, hypoxia and nephrotoxins. There, vulnerable, sublethally damaged cells may undergo apoptosis and necrosis, finally resulting in acute kidney injury (AKI) as was shown in animal models [18–23].
Up to now very little attention has been paid to the fact that uromodulin is also present in serum (sUmod; [24–26]). This might originate from a frequently overlooked basolateral release of Umod into the interstitium that was shown in a mouse model [27].
Various studies have focused on the role of uUmod in chronic kidney diseases (CKD) of different a aetiologies [28–31]. Excretion of uUmod was reduced in patients with CKD or chronic graft failure and paralleled the estimated glomerular filtration rate (eGFR) [32–34]. In contrast, a case–control study reported that increased uUmod concentrations precede the development of CKD [35]. UMOD was confirmed as one of the most important loci associated with end-stage renal failure [36–42].
Preanalytic data showed that the urinary form impairs precision and reliability of the measurements [2, 31, 43]. It is speculated that the polymeric structure and varying degrees of polymerization hamper antibody-based detection.
In the present work, we describe the development and application of an ELISA for the determination of sUmod, based on monoclonal antibodies (moAb). We assessed sUmod levels in healthy persons, patients with different stages of CKD and patients after kidney transplantation (KTX). Our immunohistochemical findings support the view that newly biosynthesized Umod is translocated and released bidirectionally by TAL cells, such that the basolateral pathway might contribute to sUmod levels. The observed progressive decrease of sUmod levels related to CKD stages suggests altered cell viability in the TAL segment, which mirrors a gradual and more global renal parenchymal injury.
MATERIALS AND METHODS
Study populations
Physiological levels of sUmod were assessed in 190 blood donors (18–60 years, Table 1) and in 443 children and adolescents (1–17 years). Thirty-three patients without kidney disease (non-CKD) and 132 patients with CKD stages 1–5 were enrolled in a nonrandomized manner. This cohort was used to determine the correlation of sUmod with cystatin C, creatinine and eGFR (Table 2). sUmod was studied in patients with autoimmune diseases classified by clinical symptoms, CKD stages and autoantibody tests (Euroimmun, Lübeck, Germany), i.e. systemic lupus erythematosus (SLE; n = 53), anti-glomerular basement membrane (GBM)-positive pulmorenal syndromes (n = 10), phospholipase-A2 receptor (PLA2-R) antibody (Ab)-positive glomerulonephritis (n = 20). Forty-four kidney transplant patients [17 with delayed graft function (DGF) and 27 with immediate graft function (IGF)] were analysed for sUmod levels (Table 3). Details of immunosuppressive therapy in transplant patients have been published elsewhere [44]. All individuals underwent physical examination and completed a detailed questionnaire. Ethics approval was obtained from two ethics committees and all participants provided informed consent, in adherence to the declarations of Helsinki and Istanbul.
Table 1.
Demographic characteristics and serum uromodulin concentration of blood donors; data are presented as mean ± SD and median, with IQR in brackets
| Parameter | Total (n = 190) | Male (n = 89) | Female (n = 101) | P-value |
|---|---|---|---|---|
| Age (years) | 38.7 ± 11.9, 40.0 (28.0, 49.0) | 37.4 ± 11.8, 38.0 (25.5, 47.5) | 39.8 ± 11.9, 42.0 (29.8, 50) | 0.165 |
| Uromodulin (ng/mL) | 221.5 ± 95.2, 207.4 (148.5, 275.1) | 199.3 ± 78.5, 188.2 (144.4, 250.7) | 241.1 ± 103.5, 229.9 (164.7, 307.6) | 0.006 |
Table 2.
Demographic characteristics and laboratory blood parameters; data are presented as mean ± SD and median, with IQR in brackets, or as absolute numbers (n) with percentage (% of the referring group) in brackets
| Total (n = 165) | Non-CKD (n = 33, 20.0%) | CKD 1 (n = 21, 12.7%) | CKD 2 (n = 27, 16.4%) | CKD 3 (n = 43, 26.1%) | CKD 4 (n = 38, 23.0%) | CKD 5 (n = 3, 1.8%) | P-value | |
|---|---|---|---|---|---|---|---|---|
| Age (years) | 61.9 ± 14.4, 65.0 (51.0, 73.0) | 60.8 ± 14.2, 66.0 (62.0, 67.0) | 56.0± 11.3, 55.0 (45.0, 63.0) | 60.5 ± 12.4, 62.0 (48.0, 72.0) | 64.0 ± 17.6, 71.0 (52.5, 77.0) | 63.9 ± 12.1, 66.0 (56.3, 73.0) | 63.3 ± 16.0, 53.0 (52.0, 69.5) | 0.159 |
| Gender (m/f in %) | 110/55 (66.7/33.3) | 20/13 (60.6/39.4) | 15/6 (71.4/28.6) | 18/9 (66.7/33.3) | 28/15 (65.1/34.9) | 26/12 (68.4/31.6) | 3/0 (100/0) | 0.888 |
| Hypertension | 74 (44, 8%) | 1 (3%) | 12 (57%) | 17 (60, 7%) | 19 (44%) | 23 (60, 5%) | 3 | |
| NIDDM | 27 | 0 | 6 (28, 5%) | 5 (17, 8%) | 8 (18, 6%) | 7 (18, 4%) | 1 | |
| IDDM | 13 | 0 | 0 | 0 | 5 (11, 6%) | 8 (21%) | 0 | |
| GN, others | 1 former GN in complete remission | mGN (1) SLE (1) ANCA (1) | GN 7 (25%), pHPT (2), sarkoidosis (3), myeloma (2) | GN 7 (16, 2%), PN 2 (4, 6%), myeloma 6 (13, 9%) | GN 8 (21%), toxN 6 (15, 7%), obstrN 3 (7, 9%) | mGN (1) | ||
| Proteinuria <1 g/g creat | 1 | 9 (42%) | 13 (46%) | 12 (28%) | 15 (39%) | 0 | ||
| Proteinuria >1 g/g creat | 0 | 1 (4, 7%) | 2 (7, 1%) | 8 (18, 6%) | 15 (31, 8) | 3 | ||
| Uric acid (µmol/L) | 375.3 ± 93.9, 369.0 (315.5, 424.1) | 316.0 ± 79.7, 315.5 (267.9, 381.0) | 353.4 ± 81.0, 330.4 (293.2, 431.5) | 332.5 ± 76.6, 333.3 (273.8, 378.0) | 391.9 ± 64.2, 372.0 (345.2, 422.6) | 469.0 ± 92.8, 422.6 (401.8, 523.8) | 333.3 ± 0.0, 333.3 (333.3, 333.3) | <0.001 |
| C-reactive protein (mg/dL) | 1.3 ± 1.9, 0.9 (0.4, 1.5) | 0.6 ± 0.2, 0.5 (0.4, 0.6) | 0.8 ± 0.5, 0.7 (0.6, 0.9) | 1.4 ± 0.2, 1.4 (1.3, 1.5) | 0.7 ± 0.4, 0.7 (0.4, 1.1) | 2.5 ± 3.2, 1.7 (1.2, 2.0) | 1.4 ± 0.0, 1.4 (1.4, 1.4) | <0.001 |
| Urea (mmol/L) | 11.9 ± 8.0, 8.4 (5.9, 16.7) | 5.9 ± 1.6, 5.7 (4.9, 6.8) | 5.1 ± 1.2, 4.9 (4.6, 5.9) | 7.3 ± 3.4, 6.2 (6.0, 7.5) | 11.9 ± 4.4, 11.8 (7.9, 15.5) | 21.8 ± 6.5, 20.2 (16.7, 26.7) | 28.4 ± 4.3, 29.5 (26.1, 31.2) | <0.001 |
| Sodium (mmol/L) | 138.0 ± 12.1, 139.0 (138.0, 141.0) | 135.1 ± 19.9, 139.0 (138.0, 140.0) | 138.2 ± 3.8, 138.0 (137.0, 141.0) | 139.4 ± 2.9, 139.0 (139.0, 140.0) | 139.1 ± 4.0, 139.0 (137.5, 141.0) | 137.4 ± 17.0 140.0, (139.0, 142.8) | 141.7 ± 0.5, 142.0 (141.5, 142.0) | 0.088 |
| Potassium (mmol/L) | 4.4 ± 0.6, 4.3 (4.0, 4.7) | 4.2 ± 0.4, 4.1 (3.9, 4.5) | 4.2 ± 0.9, 4.0 (3.9, 4.3) | 4.3 ± 0.5, 4.3 (4.0, 4.6) | 4.5 ± 0.5, 4.4 (4.2, 4.8) | 4.7 ± 0.7, 4.7 (4.2, 5.1) | 4.7 ± 0.2, 4.7 (4.6, 4.9) | <0.001 |
| Calcium (mmol/L) | 2.3 ± 0.2, 2.4 (2.3, 2.4) | 2.4 ± 0.1, 2.4 (2.3, 2.5) | 2.4 ± 0.1, 2.4 (2.3, 2.4) | 2.4 ± 0.1, 2.4 (2.3, 2.4) | 2.3 ± 0.2, 2.3 (2.3, 2.4) | 2.36 ± 0.2, 2.3 (2.2, 2.4) | 2.3 ± 0.1, 2.3 (2.3, 2.4) | 0.096 |
| Phosphate (mmol/L) | 1.2 ± 0.3, 1.1 (1.0, 1.3) | 1.1 ± 0.2, 1.1 (1.0, 1.2) | 1.0 ± 0.2, 1.1 (0.9, 1.1) | 1.0 ± 0.2, 1.0 (0.9, 1.2) | 1.1 ± 0.2, 1.1 (0.9, 1.3) | 1.4 ± 0.5, 1.3 (1.1, 1.5) | 1.6 ± 0.6, 1.6 (1.3, 2.0) | <0.001 |
| Total protein (g/L) | 75 ± 28, 73 (67, 76) | 73 ± 5, 73 (69, 77) | 73 ± 6, 74 (67, 77) | 73 ± 6, 74 (69, 76) | 74 ± 10, 73 (70, 76) | 84 ± 63, 67 (65, 73) | 73 ± 0, 73 (73, 73) | 0.325 |
| Cholesterol (mmol/L) | 5.7 ± 1.6, 5.8 (4.6, 7.0) | 6.6 ± 1.0, 6.6 (5.8, 7.4) | 6.0 ± 1.3, 6.1 (5.3, 7.2) | 5.5 ± 1.3, 5.1 (4.8, 6.4) | 4.8 ± 1.8, 4.4 (3.7, 5.0) | 6.0 ± 1.3, 5.9 (5.1, 6.9) | 4.9 ± 0.0, 4.9 (4.9 , 4.9) | <0.001 |
| HDL (mmol/L) | 1.5 ± 0.4, 1.4 (1.2, 1.7) | 1.6 ± 0.3, 1.6 (1.4, 1.8) | 1.4 ± 0.5, 1.3 (1.0, 1.7) | 1.4 ± 0.5, 1.2 (1.1, 1.7) | 1.4 ± 0.4, 1.4 (1.2, 1.6) | 1.5 ± 0.6, 1.3 (1.2, 1.6) | 1.1 ± 0.0, 1.1 (1.1, 1.1) | 0.112 |
| LDL (mmol/L) | 4.0 ± 1.3, 4.0 (3.0, 5.0) | 4.6 ± 0.8, 4.6 (4.0, 5.1) | 4.4 ± 1.2, 4.4 (3.9, 5.0) | 3.7 ± 0.8, 3.7 (3.1, 4.5) | 3.2 ± 1.4, 2.7 (2.4, 3.2) | 4.0 ± 1.3, 4.1 (3.1, 4.9) | 3.6 ± 0.0, 3.6 (3.6, 3.6) | <0.001 |
| Triglyceride (mmol/L) | 1.9 ± 1.4, 1.5 (1.0, 2.2) | 1.5 ± 0.5, 1.5 (1.1, 1.9) | 1.8 ± 0.8, 1.7 (1.3, 1.9) | 2.4 ± 2.1, 1.7 (1.1, 2.6) | 1.7 ± 1.2, 1.2 (0.9, 2.2) | 2.4 ± 1.7, 2.1 (1.1, 3.0) | 1.9 ± 0.0, 1.9 (1.9, 1.9) | 0.256 |
| Haemoglobin (mmol/L) | 8.3 ± 1.6, 8.7 (7.9, 9.2) | 8.9 ± 0.5, 8.9 (8.4, 9.2) | 8.8 ± 1.5, 9.2 (8.7, 9.5) | 8.2 ± 1.8, 8.6 (7.9, 9.2) | 8.0 ± 2.1, 8.1 (7.0, 9.0) | 7.7 ± 1.4, 8.2 (6.8, 8.9) | 7.8 ± 0.0, 7.8 (7.8, 7.8) | 0.006 |
| Leucocyte count (10e9/L) | 8.0 ± 7.4, 6.9 (5.9, 8.2) | 8.1 ± 11.1, 6.4 (5.4, 7.1) | 10.0 ± 10.0, 6.6 (6.1, 9.5) | 7.6 ± 2.6, 8.0 (6.8, 8.6) | 7.5 ± 4.6, 6.9 (6.1, 7.8) | 7.2 ± 1.3, 7.4 (6.2, 8.4) | 9.2 ± 0.0, 9.2 (9.2, 9.2) | 0.057 |
| Kidney biomarker level | ||||||||
| Uromodulin (ng/mL) | 115.6 ± 101.0, 84.8 (37.0, 180.0) | 260.4 ± 101.6, 227.6 (194.4, 277.8) | 150.7 ± 54.8, 153.4 (121.5, 180.7) | 116.7 ± 44.5, 111.8 (80.9, 139.4) | 70.5 ± 47.1, 51.8 (36.1, 86.2) | 29.1 ± 13.2, 30.3 (19.2, 38.0) | 10.3 ± 7.0, 12.7 (6.7, 15.1) | <0.001 |
| Cystatin C (nmol/L) | 147.1 ± 93.8, 113.8 (72.7, 206.3) | 66.7 ± 10.1, 68.2 (62.9, 73.4) | 62.1 ± 10.9, 66.7 (55.4, 70.4) | 91.5 ± 9.5, 89.1 (85.0, 98.9) | 151.9 ± 27.4, 147.6 (127.0, 176.4) | 273.8 ± 39.3, 271.1 (247.9, 304.3) | 453.1 ± 70.5, 445.7 (408.2, 494.3) | <0.001 |
| Creatinine (µmol/L) | 176.9 ± 126.1, 127.0 (93.0, 223.3) | 96.7 ± 22.1, 94.0 (80.0, 117.0) | 82.4 ± 16.8, 76.9 (72.0, 93.0) | 107.0 ± 21.8, 105.0 (91.5, 119.2) | 169.3 ± 52.7, 160.9 (125.0, 206.0) | 333.5 ± 131.5, 316.5 (246.1, 383.1) | 669.5 ± 153.0, 670.0 (575.9, 763.3) | <0.001 |
| BUN (mmol/L) | 11.9 ± 8.0, 8.4 (5.9, 16.6) | 5.9 ± 1.6, 5.7 (4.9, 6.8) | 5.1 ± 1.2, 4.9 (4.6 , 5.9) | 7.3 ± 3.4, 6.2 (6.0 , 7.4) | 11.9 ± 4.4, 11.8 (7.9 , 15.5) | 21.8 ± 6.5, 20.2 (16.7 , 26.7) | 28.3 ± 4.3, 29.4 (26.0 , 31.2) | <0.001 |
| eGFRHoek (mL/min) | 64.2 ± 37.9, 57.7 (29.7, 92.9) | 104.6 ± 20.7, 99.3 (91.9, 108.0) | 114.0 ± 26.8, 101.0 (96.0, 123.2) | 73.6 ± 7.8, 74.9 (67.1, 78.8) | 43.7 ± 8.7, 43.6 (35.7, 51.1) | 22.0 ± 3.8, 21.8 (19.1, 24.3) | 11.8 ± 2.5, 12.0 (10.3, 13.4) | <0.001 |
| eGFRCKD-EPI (mL/min) | 49.4 ± 31.1, 44.0 (20.5, 71.5) | 78.8 ± 17.5, 76.5 (65.1, 86.2) | 92.8 ± 18.3, 88.9 (85.1, 97.0) | 60.1 ± 11.5, 61.6 (51.5, 69.5) | 32.5 ± 8.7, 31.4 (25.7, 39.5) | 14.5 ± 4.0, 14.2 (11.6, 16.9) | 6.7 ± 1.2, 7.0 (6.0, 7.5) | <0.001 |
NIDDM, non-insulin-dependent diabetes mellitus type II; IDDM, insulin-dependent diabetes mellitus type II; GN, glomerulonephritis: biopsy proven; pHPT, primary hyperparathyroidism; obstrN, obstructive nephropathy; toxN, CKD due to former nephrotoxic chemotherapy, HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Table 3.
Kidney allograft recipient’s demographics
| Parameter | Total (n = 44) | Immediate graft function (n = 27) | Delayed graft function (n = 17) | P-value |
|---|---|---|---|---|
| Age (years) | 42.0 ± 10.8, 41.0 (33.8, 51.3) | 43.1 ± 10.6, 44.0, (34.5, 52.0) | 43.0 ± 11.0, 39.0, (32.0, 48.0) | 0.421 |
| Gender | ||||
| Male | 27 (61.4) | 16 (59.3) | 11(64.7) | 0.761 |
| Female | 17 (38.6) | 11(40.7) | 6 (35.3) | |
| Kind of donation | ||||
| Deceased donor | 44 (100) | 27 (100) | 17 (100) | 1.000 |
| Cold ischaemia time (min) | 952.8 ± 455.3, 859.5 (609.8, 1227.0) | 803.1 ± 347.9, 720.0 (570.5, 1005.0) | 1190.6 ± 502.1, 1200.0 (794.0, 1500.0) | 0.007 |
| Maximum panel cytotoxicity (>5%) | 11 (25.0) | 7 (25.9) | 4 (23.0) | 1.000 |
| Immunosuppression—Induction therapy | ||||
| ATG Fresenius, 9 mg/kg body weight intraoperatively (i.op.) | 30 (68.2) | 19 (70.4) | 11 (64.5) | 0.124 |
| Lymphoglobulin Merieux, 30 mg/kg body weight i.op. | 11 (25.0) | 8 (29.6) | 3 (17.6) | |
| ATG Biotest, 1.5 mg/kg body weight i.op. | 1 (2.3) | 0 (0) | 1 (5.9) | |
| None | 2 (4.5) | 0 (0) | 2 (11.8) | |
| Replacement (or additional therapies) therapy after Tx | ||||
| None | 27 (61.4) | 27 (100) | 0 (0) | <0.001 |
| Haemodialysis | 16 (36.4) | 0 (0) | 16 (94.1) | |
| Peritoneal dialysis | 1 (2.3) | 0 (0) | 1 (5.9) | |
| Number of dialysis sessions | 9.1 ± 5.5 | 0 | 9.1 ± 5.5 | |
| Patient with rejection during the first year | 16 (36.4) | 8 (29.6) | 8 (47.1) | 0.337 |
| Patient with Cytomegalovirus infections during the first year | 8 (18.2) | 4 (14.8) | 4 (23.5) | 0.690 |
| Biomarker levels at day of discharge | ||||
| Day of discharge after Tx | 40.7 ± 21.3, 37.5 (23.8, 50.3) | 30.4 ± 13.2, 25.0 (20.0, 42.5) | 56.9 ± 21.5, 51.0 (38.0, 63.0) | 0.004 |
| Uromodulin (ng/mL) | 60.1 ± 43.1, 47.6 (29.3, 75.8) | 76.6 ± 45.7, 63.0 (45.0, 95.1) | 33.9 ± 19.1, 29.4 (21.2, 39.0) | <0.001 |
| Creatinine (µmol/L) | 146.0 ± 53.5, 140.0, (107.8, 170.3) | 124.7 ± 30.6, 118.0 (105.0, 145.5) | 179.8 ± 63.7, 180.0, (161.0, 191.0) | <0.001 |
| eGFR (MDRD) (mL/min) | 49.2 ± 17.0, 47.1 (36.7, 58.9) | 55.4 ± 15.5, 53.6, (44.6, 66.5) | 39.4 ± 14.5, 37.4 (32.3, 47.0) | 0.004 |
Data are presented as mean ± SD and median with IQR in brackets or as absolute numbers (n) with percentage (%) in brackets. Tx, transplantation; MDRD, Modification of Diet in Renal Disease.
Definition of CKD stages and outcome parameters
Based on clinical and eGFR data patients were assigned to one of five stages of CKD. The underlying kidney diseases were histologically characterized by routine stains [haematoxylin & eosin (HE), periodic acid-Schiff (PAS)], immunohistology and ultrastructure of renal biopsy sections. GFR was determined by eGFRCysC. Patients were assigned to CKD stages according to Kidney Disease: Improving Global Outcomes (KDIGO) guidelines: CKD 1 with eGFR >90 mL/min, CKD 2 61–90 mL/min, CKD 3 31–60 mL/min, CKD 4 16–30 mL/min and CKD 5 0–15 mL/min. We included 33 patients without kidney disease as the control group (non-CKD).
Forty-four patients had undergone KTX between 1990 and 1995 at the Berlin-Friedrichshain Kidney Centre, Germany. Follow-up was until 2002, unless death occurred. Clinical characteristics and definitions of IGF and DGF are summarized in Table 3.
Antibodies
Mouse moAb were generated against purified human uUmod. BALB/c mice were immunized with emulsified purified Umod in ABM-S adjuvant. After booster injections (ABM-N adjuvant) mice were anesthetized, splenectomized and spleen cells were PEG fused with mouse X63Ag8.653 myeloma cells. Hybridoma supernatants were screened for specific antibodies by ELISA with purified Umod as antigen. Positive hybridomas were subcloned by limiting dilution. Distinct hybridomas were expanded and purified. Specificity of moAb was assessed by immunocytochemistry using paraffin-embedded tissue sections from human kidney, liver, intestine, muscle and lymph nodes. Only human kidney showed a positive histochemical reaction with anti-Umod antibodies.
Uromodulin ELISA
Purified IgG1 of moAb T112A12 for coating the microtiter plates and moAb T5.G as detection antibody were used to establish an ELISA. All analytical data (sensitivity, precision, linearity, etc.) for CE labelling were determined and are available from the manufacturer (Euroimmun). All measurements were done with a 1:101 serum dilution in sample buffer. The linear measurement range was 2–400 ng/mL.
Immunohistological and cytological analyses
Normal renal tissue was obtained from kidney resection in renal carcinoma patients and fixed in paraformaldehyde. The tissue was cut into small blocks and processed in an automatic freeze substitution unit (Reichert-Jung AFS, Leica Microsystems, Wetzlar, Germany). Umod moAb clonotypes 109F9 (IgG1), 112A12 (IgG1) and 5A2 (IgG2b) were used for staining.
Immunocytochemistry
After primary antibody incubation deparaffinized sections were processed with secondary biotinylated antibody and avidin-peroxidase complex according to standard procedures. Biopsy material was obtained from patients (21 men, 11 women, mean age 54.7 ± 17 years) with kidney diseases including IgA nephropathy (n = 12), membranous glomerulonephritis (mGN) (n = 8), ANCA-positive RPGN (n = 12), where a minimum of two to three corticomedullary nephrons were noticed, displaying interstitial fibrosis and tubular atrophy of more than 10%. In addition, we assessed sections of non-functioning allografts of nine patients (one female, eight males). Staining included HE, Masson-Trichrom, Giemsa and PAS. Sections were examined for Umod location by two different pathologists in a blinded manner.
Immunoelectron microscopy
Sections were incubated with the primary antibody, followed by gold-conjugated second antibody. Stained sections were studied in a Philips/FEI CM100 transmission electron microscope. In controls, sections were incubated with secondary antibodies alone or with non-specific moAb. None of the controls showed any labelling.
Statistical analysis
All statistical analyses were performed with SigmaPlot 13 and the language R for statistical computing. Continuous data are expressed as mean with standard deviation (SD) and median with interquartile range (IQR). Categorical variables are reported in absolute numbers and percentages. Differences concerning continuous parameters and biomarker concentrations between two groups were analysed using non-parametric Mann–Whitney U-test. The Kruskal–Wallis test was used for multiple group comparison. For categorical variables, possible differences between groups were examined using non-parametric chi-square test (Fisher's exact test). The association between sUmod and creatinine, cystatin C and eGFR was determined by Spearman’s rank correlation analysis. Multiple linear regression modelling was conducted to evaluate the association of Umod (independent variable) and eGFRCysC (dependent variable) adjusted for gender and age. As part of the regression modelling sUmod and eGFR were logarithmically transformed. Wilcoxon-rank test and/or receiver-operating characteristic (ROC) analysis were used to evaluate diagnostic validity of sUmod, cystatin C, creatinine, BUN and eGFRCysC to differentiate between non-CKD and CKD stage 1. Relative risk (RR) and odds ratio (OR) for DGF after KTX were calculated on the basis of mean cut-off (day 7–25 post-transplant). Daily cut offs were determined according to Youden’s index. The probability of error (type 1) of a two tailed P-value <0.05 was considered to be statistically significant.
RESULTS
sUmod stability
sUmod stability was analysed over 4 weeks (Supplementary data, Table S1). Serum samples from eight individuals were stored at either −20°C, 4°C or 37°C before monitoring Umod levels. A high concordance in Umod concentrations was observed for all three storage conditions with mean deviations of 4% (−20 versus 4°C) and 10% (−20 versus 37°C). Even up to five freeze/thaw cycles had no impact on sUmod recoveries (Supplementary data, Table S1).
sUmod expected normal values
sUmod was assessed in 190 blood donors (89 males, 101 females, age 18–60 years, median age 40 years). Donors were checked to be healthy by clinical examination and laboratory tests (normal controls, Table 1). Females revealed higher sUmod than males [females: median 230 ng/mL (IQR: 165, 308); males: median 188 ng/mL (IQR: 44, 251), P = 0.006]. In adults, no correlation between age and sUmod was observed (rs = −0.0197, P = 0.787). sUmod in children and adolescents (n = 443, age 1–17 years, median age 6 years) was comparable to values found in adults [median 193 ng/mL (IQR: 143, 254)]. sUmod levels did not differ between boys and girls (P = 0.389).
sUmod in renal failure
sUmod levels and the filtration markers creatinine and cystatin C were analysed in 165 patients (110 males, 55 females, age 22–94 years, median age 65 years) with various diseases potentially affecting the kidney. sUmod ranged from 1 to 667 ng/mL with a median of 85 ng/mL (Table 2). In univariate analysis, sUmod was significantly correlated with cystatin C, creatinine and all eGFR calculations. Scatter plots demonstrate an inverse hyperbolic relationship of sUmod with cystatin C (rs = −0.862, P < 0.001), creatinine (rs = −0.802, P < 0.001) and blood urea nitrogen (BUN, rs = −0.645, P < 0.001) and, accordingly, a positive relationship with cystatin C-based eGFR (eGFRCysC, Hoek’s equation) (rs = 0.862, P < 0.001) and eGFR (CKD-EPI) (rs = 0.842, P < 0.001) (Figure 1). The creatinine–sUmod ratio was not superior compared with assessing sUmod levels alone. The data disclose that sUmod parallels the progressive deterioration of kidney function and behaves oppositely to the filtration markers. In a multiple linear regression model, sUmod was adjusted for age and gender, which did not significantly influence the regression model with eGFRCysC (log) as a dependent variable. The correlation between sUmod (log) and eGFRCysC (log) was r2 = 0.735 (P < 0.001).
FIGURE 1.

Relationship between serum uromodulin and creatinine (A), blood urea nitrogen (B), cystatin C (C) and eGFRcystC (D). rs: Spearman’s rank correlation coefficient.
To study the relationship between sUmod and GFR, sUmod levels were assigned to the stages of renal failure CKD 1–5, based on the eGFRCysC data (Figure 2). sUmod decreased with progressive CKD (non-CKD: median 228 ng/mL; stage 1: 153 ng/mL, stage 2: 107 ng/mL; stage 3: 52 ng/mL; stage 4: 30 ng/mL; stage 5: 13 ng/mL). Differences in sUmod were shown for all pairs of CKD stages (P < 0.001), except for stages 1 and 2 (P = 0.051). The most prominent changes, i.e. the steepest decline in sUmod, occurred within the ‘creatinine blind range’ (Figure 2).
FIGURE 2.

Serum uromodulin concentration of patients without kidney diseases (non-CKD) and patients with CKD (stage 1 to stage 5) based on eGFRcysC assignment.
All traditional kidney markers significantly differentiated between all CKD stages from 1 to 5, but none showed a conclusive statistical difference between non-CKD and CKD 1. Creatinine and cystatin C were even higher in non-CKD compared with CKD stage 1 [creatinine: median 94.0 µmol/L (IQR: 80.0, 117.0) versus 76.9 µmol/L (72.0, 93.0), P = 0.011; cystatin C: median 68.2 nmol/L (IQR: 62.9, 73.4) versus 66.7 nmol/L (IQR: 55.4, 70.4), P = 0.153].
ROC analysis of sUmod to differentiate between non-CKD and CKD 1 resulted in an AUC of 0.90 (P < 0.001, 95% CI 0.808 vs 0.990) at an optimal cut-off of 163 ng/mL (sensitivity 57.1%, specificity 100%). None of the other parameters including eGFR discriminated between stages non-CKD and CKD 1 (cystatin C: AUC 0.39; creatinine: AUC 0.29; BUN: AUC 0.37; eGFRCysC: AUC 0.39; eGFR (CKD-EPI): AUC 0.26) (Figure 3).
FIGURE 3.

ROC analysing the ability of different parameters to differentiate patients without kidney disease (non-CKD) and CKD stage 1; BUN = blood urea nitrogen, eGFR = estimated glomerular filtration rate, AUC = area under the curve.
sUmod in autoimmune diseases
Three patient cohorts with autoimmune diseases, including pulmorenal syndromes (n = 10), SLE (n = 53) and PLA2-R Ab-positive glomerulonephritis (mGN, n = 20) were assessed. Patients with acute anti-GBM nephritis of CKD 3–5 had much lower sUmod compared with blood donors (median 37 ng/mL, P < 0.001, Supplementary data, Figure S1). Patients with SLE-nephritis (CKD 2–4) had median sUmod of 101 ng/mL, P < 0.001, while those without kidney involvement had similar sUmod levels compared with blood donors (median: 259 ng/mL, P = 0.485). Patients with mGN had median sUmod of 81 ng/mL, P < 0.001.
sUmod after KTX
As shown in Figure 4 prior to surgery sUmod was not detectable in all patients due to end-stage renal disease (ESRD). Immediately after transplantation sUmod rapidly increased in recipients with IGF and DGF until post-KTX day 5, reaching 43 ng/mL (median) in IGF and 33 ng/mL (median) in DGF, respectively. Thereafter, levels diverged from day 7 (IGF: 59.5 ± 41.2 ng/mL, DGF: 33.1 ± 20 ng/mL; P = 0.033) to day 25. The cohort with DGF failed to reach sUmod levels above 40 ng/mL (cut off 39.26 ng/mL). From day 5 on, the RR and OR to develop DGF rose dramatically on the basis of sUmod (day 5: RR 1.3, OR 1.5; day 9: RR 2.9, OR 6.4, etc.; day 25: RR 8.2, OR 26; Supplementary data, Table S2). Sixteen of 17 patients with DGF needed 3–24 postoperative haemodialysis treatments. Patients with IGF were discharged from hospital after a median of 25 days, with a median creatinine of 118 µmol/L and a stable ‘high’ sUmod (76.6 ± 46.6 ng/mL). Patients with DGF and additional complications were discharged from hospital much later, at a median of 51 days, with a median creatinine of 180 µmol/L and lower sUmod concentration (33.9 ± 19.7 ng/mL, P < 0.001).
FIGURE 4.

Post-kidney transplant comparison of serum uromodulin and creatinine in patients with immediate and delayed graft function.
Umod distribution in normal human kidney
Labelling with anti-Umod antibodies was confined to the distal tubule (Figure 5). Umod was seen throughout the cytoplasm and in apical and basolateral membranes of the distal tubule cells. The images illustrate the transition from the unlabelled thin ascending limb to the intensely labelled TAL. In the macula densa, the cells were largely unlabelled or weakly labelled, whereas the other distal tubule cells were intensely labelled.
FIGURE 5.
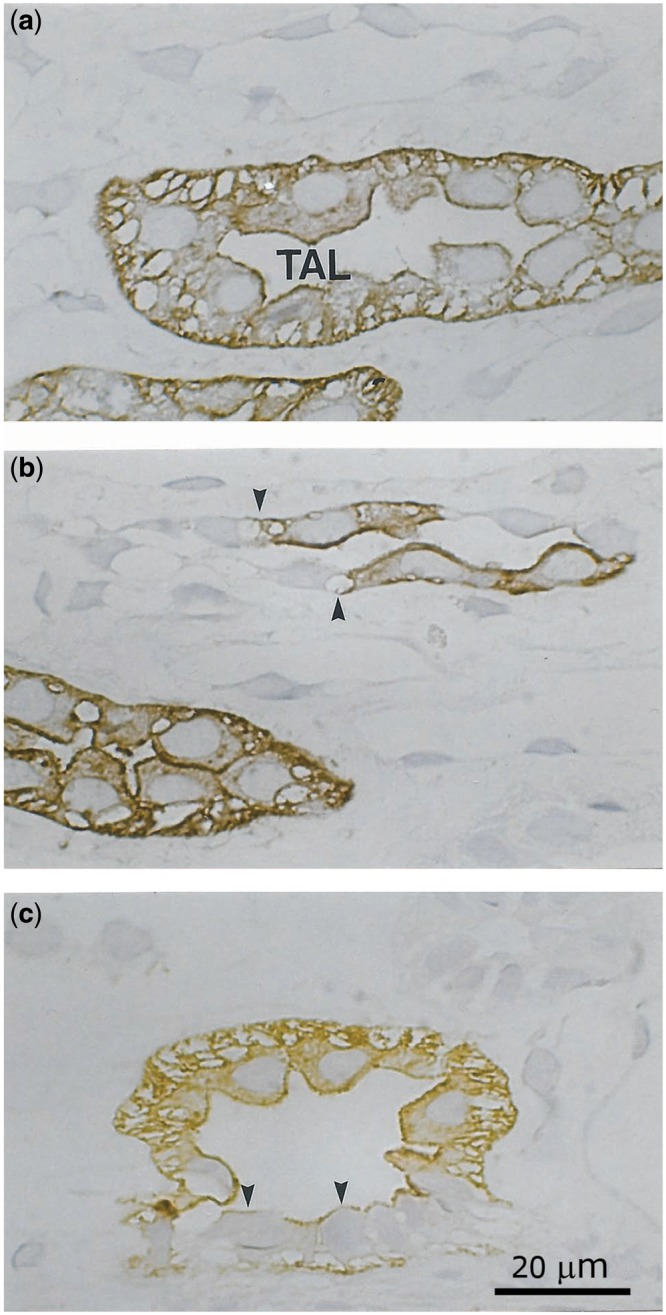
Immunohistochemical distribution of Umod in the normal human kidney (a–c); semithin cryosections. (a) Apical, basolateral and cytoplasmatic staining, (b) transition of the thin ascending limb (negative) to the TAL (positive, arrows), (c) macula densa reveals a faint staining pattern only (arrowheads).
Umod location in kidney diseases
Umod staining of biopsy material from patients with proven kidney diseases and of grafts that had to be explanted due to ESRD showed an irregular pattern and increased intracellular expression of the antigen compared with healthy controls (Supplementary data, Figure S2). In kidney biopsies, TAL segments were under-represented compared with other tubular segments and were not accessible to further morphometric analyses. In most Umod-positive cells, the antigen appeared to accumulate and condense around the nuclei, the endoplasmatic reticulum and the Golgi apparatus. The cytoplasma of TAL cells from explanted kidney grafts stained for Umod with a dramatically increased intensity; here potential changes in Umod expression of apical and basolateral membranes could not be assessed (Supplementary data, Figure S2).
Immunoelectron microscopy
Immunogold labelling of Umod was seen at the apical and basolateral membrane, in the cytoplasma, the granular endoplasmic reticulum, the Golgi apparatus and in vesicles located apically but also in vesicles close to the basement membrane (Figure 6). Labelling was largely confined to the external layer of the apical and external parts of the basolateral plasma membrane including their infoldings. No labelling was seen in the collecting duct either in principal or in intercalated cells.
FIGURE 6.

Immunogold labelling of Umod revealing the transport of the antigen within cytoplasmatic vesicles (small arrowheads) to the apical (a) as well as to the basolateral cell pole (b). Shuttle of uromodulin (c) from the endoplasmatic reticulum (RER) to the Golgi apparatus (G) (inset to C), through cytoplasmic vesicles (arrowheads). Basolateral membrane (BM) labelling (arrow). M, mitochondria.
DISCUSSION
The majority of studies on Umod to date focus on the urinary form of Umod. sUmod in contrast has rarely been investigated [25, 26] and its clinical significance, molecular structure and function in blood are poorly understood. Brezis et al. first emphasized that the renal medulla is a neglected tissue compartment, not adequately reached by renal biopsy. Umod is a major secreted protein produced by the renal medulla and experimental data support the pivotal role of medullary hypoxic injury in kidney diseases [12, 18–23, 27].
We assessed sUmod in patients with different CKD stages by ELISA. sUmod levels inversely correlated with creatinine and creatinine/cystatin C-based eGFR. The lowest sUmod was measured in patients with CKD stages 4 and 5. We speculate that low sUmod results from a disturbed biosynthesis in cells of the TAL. Patients with renal injury also show a decrease in urinary Umod levels [28, 32–34]. sUmod is not measurable in anephric patients [24]. In elderly patients, sUmod displayed inverse relationships with creatinine, cystatin C and urea, and, correspondingly, a positive relation with eGFR [26]. Contradicting all recent data, Prajczer et al. report that sUmod is very low in healthy controls and increased in patients with CKD 1–5 [25]. We can only speculate that these conflicting results depend on the design of their ELISA and potential additional preanalytic factors.
Addressing the limitations of our study we did not measure GFR by inulin or iohexol clearance. Instead we calculated eGFR on a creatinine and cystatin C basis, which is presently still the most accepted method under clinical routine conditions.
With the help of our sensitive assay, to the best of our knowledge, we first demonstrated Umod as a physiological and very stable antigen circulating in the blood of healthy adults and children without any known kidney problems.
Importantly, we defined conspicuous sUmod at values below the fifth percentile range of healthy individuals and found different thresholds for children and males (<80 ng/mL) and for females (<100 ng/mL). Higher sUmod levels in healthy female compared with men were also observed in a previous study [26] and parallel similar sex differences in the urinary excretion of Umod and other tubular markers [43–46]. Although gender-dependent endocrine, haemodynamic, metabolic and genetic factors, expression of receptors, as well as sex differences in the susceptibility of cell integrity may contribute to this observation, most questions are still unanswered [47, 48].
The clinical data presented here are limited to patients with CKD (apart from the cohorts after grafting) and need to be extended to cases suffering from AKI, which are now under study. In contrast to Risch et al. [26] we could not find a correlation between sUmod and age (age range from 18 to 60 years; r = −0.60, P < 0.001). A possible explanation is that Risch et al. focused on elderly participants (mean age 71 ± 7 years) and the physiological decrease of sUmod probably starts at the age of 60 years. Age-related changes in the healthy kidney assessed by multidetector CT scans showed diverged alterations (disequilibrium) of renal compartments, where cortical volume (and GFR in parallel) declines, whereas medullary parenchymal volume increases [49]. This suggests that synthesis and release of Umod by TAL into the blood might be maintained over a longer period.
According to as yet unpublished studies, we solely found monomeric sUmod and so far did not observe any aggregation processes in serum. As a key novel finding we noticed extraordinarily stable signals over weeks even at increased temperatures. In contrast, Umod from urine (uUmod) lacks stability depending on the storage conditions, centrifugation, vortexing, pH, electrolytes and osmolality [31, 43]. The changing conformation of uUmod and the varying degree of polymerization, possibly modulating antigenic binding sites, may impede the reproducibility of the results.
A major finding is the comparative analysis of sUmod with creatinine, cystatin C and eGFR indicating that lower sUmod reflects a decline in kidney function and may precede the onset of CKD, as was shown by us in another patient cohort [50].
Tubulointerstitial injury is closely linked to progression of renal disease [51, 52]. We assume that low sUmod parallels a structural derangement of the TAL. sUmod levels may drop due to altered intracellular processing and reduced basolateral exocytosis and mirror the reduced quantity of intact TAL cells. Acute and chronic renal injury characterized by epithelial atrophy and tubular loss of both cortical and medullary segments may also parallel changes of the vascular microarchitecture [53]. Interstitial vascular remodelling may influence the structural integrity of tubular cells, e.g. vulnerable TAL segments, and change glomerular morphology and haemodynamics, which would link sUmod levels with eGFR (CKD stages). In the present study, we could not assess the relation between histological alterations of Umod-expressing cells and sUmod. The amount of TAL segments varied greatly among the biopsies, and Umod-positive cells appear considerably under-represented in renal clinical pathology.
One known secretory pathway of Umod occurs at the apical membrane of the TAL cells. Umod concentrations in urine are approximately 1000-fold higher than in serum. Umod’s apical secretion is based on exocytosis and the formation of a GPI anchor, followed by its release from the membrane via proteolytic cleavage [3, 27]. However, besides the luminal secretion process of Umod an additional basolateral pathway was suggested [54, 55]. Our data confirm the view that Umod is synthesized in the endoplasmatic reticulum, transferred to the Golgi apparatus and transported to the apical membrane by vesicles. Finally it sheds into the luminal space. In addition, we clearly demonstrated a second, basolateral pathway of Umod secretion (Figure 6), which apparently contributes to sUmod levels (Figure 7).
FIGURE 7.
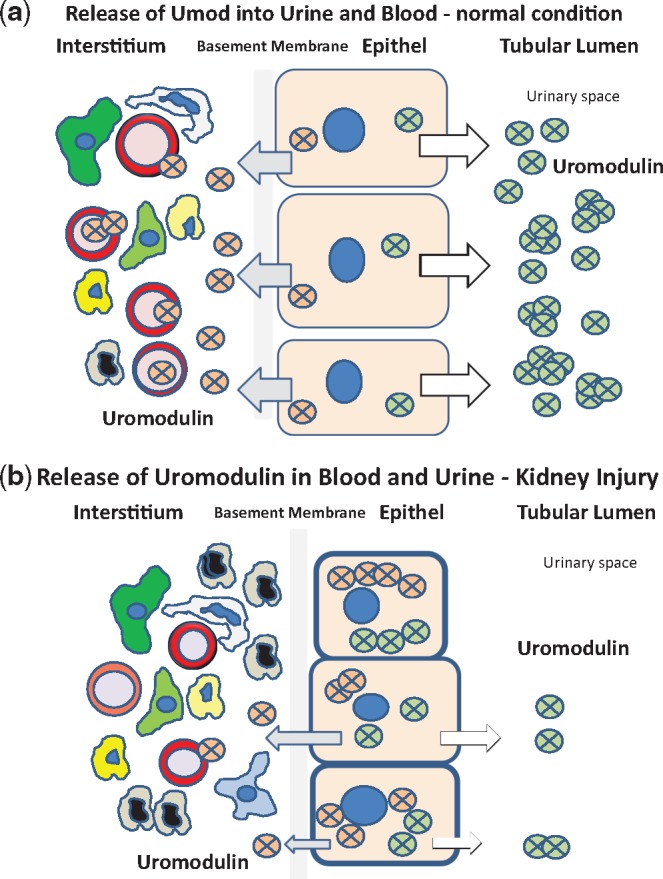
Cartoon demonstrating the proposed bidirectional physiological transfer of Umod to the urinary space and to the tubulointerstitial space. As shown in Figure 6b, Umod is physiologically released from basolateral plasma membranes and membrane infoldings into the interstitium and enters the blood compartment (a). Umod serum levels are correlated with eGFR. sUmod concentrations may depend on the cellular integrity of the TAL segment. Therefore, sUmod might be released into serum at a decreased rate in renal injury (b).
We interpret sUmod as an early indicator defining the integrity and the functional viability of the TAL. In contrast, the traditional endogenous filtration markers are not related to defined renal structures and need mathematical transformation. Established renal markers have limitations in detecting early or slight changes in kidney function, particularly in a range when approximately half of the filtration capacity is lost [56]. Following the analyses and conclusions from experimental animal data on TAL segments [18–23] we assume that sUmod levels may somehow reflect the amount of intact nephrons being linked to the ‘functioning kidney mass’.
Another important new finding is that, in accordance with eGFR, sUmod recovers to higher levels as shown in transplant patients, and may distinguish IGF from DGF. Worsening of graft function was always accompanied by long-lasting reduced sUmod levels and predictable after the seventh postoperative day. It is a subject of future research to clarify whether low sUmod levels in patients with DGF are predictive for later chronic graft dysfunction and serious patient outcome [57].
In conclusion, we have demonstrated that sUmod concentrations decrease in line with eGFR and with biopsy-classified stages of renal failure. Our data strengthen the link between sUmod and kidney disease and suggest that sUmod might be superior compared with the traditional filtration markers to detect early stages of CKD, independent of the underlying disease. sUmod should not only be interpreted as a complementary parameter describing global ‘kidney function’, but may shift our focus to potential structural and metabolic lesions of TAL cells. These encourage more extensive elucidation of the role of sUmod as a diagnostic tool in comparison with, and likely in concert with, other novel renal biomarkers [58–61].
SUPPLEMENTARY DATA
Supplementary data are available online at http://ndt.oxfordjournals.org.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Walter Nathrath, Institute of Pathology, Prof. Oliver Reich, Department of Urology, Klinikum Harlaching, LMU München, and Dr Florian Laenger, Institute of Pathology, Goethe-University Frankfurt am Main and Hannover Medical School, for supplying paraffin-embedded material and analysing tissue staining patterns. Parts of the work were presented in abstract form at the congress of the German Society of Nephrology, Berlin, September 2015, and the congress of Deutsche Transplantationsgesellschaft, Dresden, October 2015, and the 10th Intenational Congress on Autoimmunity, Leipzig, April 2016. We thank Prof. Seymour Rosen, Beth Israel Deaconess MC, Harvard Medical School, Boston, Prof. Anna Koettgen, Medizinische Klinik IV, Department of Nephrology, University of Freiburg, and Dr Ulrike Krummrei, Euroimmun, for critical proofreading of the manuscript, and Prof. Olivier Devuyst, University Zürich, Switzerland, and Prof. Hans-Joachim Anders, LMU Munich for collaboration and valuable discussions.
CONFLICT OF INTEREST STATEMENT
J.E.S. received travel support from Hoffmann-La Roche and Euroimmun, R.G. received travel support from Roche and ThermoScientific, V.H., M.B. and W.S. are full time employees of Euroimmun and J.K. is member of the supervisory board of Euroimmun. W.A.N., E.I.C. and H.S. declare no potential conflicts of interest.
REFERENCES
- 1. Tamm I, Horsfall FL.. Characterization and separation of an inhibitor of viral hemagglutination present in urine. Proc Soc Exp Biol Med 1990; 74: 106–108 [PubMed] [Google Scholar]
- 2. Devuyst O, Dahan K, Pirson Y.. Tamm–Horsfall protein or uromodulin: new ideas about an old molecule. Nephrol Dial Transplant 2005; 20: 1290–1294 [DOI] [PubMed] [Google Scholar]
- 3. Rampoldi L, Scolari F, Amoroso A. et al. The rediscovery of uromodulin (Tamm–Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int 2011; 80: 338–347 [DOI] [PubMed] [Google Scholar]
- 4. Mount DB. Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol 2014; 9: 1974–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cavallone D, Malagolini N, Serafini-Cessi F.. Mechanism of release of urinary Tamm–Horsfall glycoprotein from the kidney GPI-anchored counterpart. Biochem Biophys Res Commun 2001; 280: 110–114 [DOI] [PubMed] [Google Scholar]
- 6. Pak J, Pu X, Zhang ZT. et al. Tamm–Horsfall protein binds to type 1 fimbriated Escherichia coli and prevent E. coli from binding to uroplakin Ia and Ib receptors. J Biol Chem 2001; 276: 9924–9930 [DOI] [PubMed] [Google Scholar]
- 7. Raffi HS, Bates JM Jr, Laszik Z. et al. Tamm–Horsfall protein protects against urinary tract infection by Proteus mirabilis. J Urol 2009; 181: 2332–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Serafini-Cessa F, Monti A, Cavallone D.. N-glycans carried by Tamm–Horsfall glycoprotein have a crucial role in the defense against urinary tract diseases. Glycoconj J 2005; 22: 383–394 [DOI] [PubMed] [Google Scholar]
- 9. Mo L, Huang HY, Zhu XH. et al. Tamm–Horsfall protein is a critical renal defense factor protecting against calcium oxalate crystal formation. Kidney Int 2004; 66: 1159–1166 [DOI] [PubMed] [Google Scholar]
- 10. Graham LA, Padmanabhan S, Fraser NJ. et al. Validation of Uromodulin as a candidate gene for human essential hypertension. Hypertension 2014; 63: 551–558 [DOI] [PubMed] [Google Scholar]
- 11. Zacchia M, Capasso G.. The importance of uromodulin as regulator of salt reabsorption along the thick ascending limb. Nephrol Dial Transplant 2015; 30: 158–160 [DOI] [PubMed] [Google Scholar]
- 12. Trudu M, Janas S, Lanzani C. et al. Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med 2013; 19: 1655–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scolari F, Izzi C, Ghiggeri GM.. Uromodulin: from monogenic to multifactorial diseases. Nephrol Dial Transplant 2015; 30: 1250–1256 [DOI] [PubMed] [Google Scholar]
- 14. Kreft B, Jabs WJ, Laskay T. et al. Polarized expression of Tamm–Horsfall protein by renal tubular epithelial cells activates human granulocytes. Infect Immun 2002; 70: 2650–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iorember FM, Vehaskari VM.. Uromodulin: old friend with new roles in health and disease. Pediatr Nephrol 2014; 29: 1151–1158 [DOI] [PubMed] [Google Scholar]
- 16. Säemann MD, Weichhart T, Zeyda M. et al. Tamm–Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J Clin Invest 2005; 115: 468–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Darisipudi MN, Thomasova D, Mulay SR. et al. Uromodulin triggers IL-1ß-dependent innate immunity via the NLRP3 inflammasome. J Am Soc Nephrol 2012; 23: 1783–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schley G, Klanke B, Schödel J. et al. Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J Am Soc Nephrol 2011; 22: 2004–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brezis M, Rosen S.. Hypoxia of the renal medulla—its implications for disease. N Engl J Med 1995; 332: 647–655 [DOI] [PubMed] [Google Scholar]
- 20. Heyman SN, Rosenberger C, Rosen S.. Experimental ischemia—reperfusion: biases and myths—the proximal vs. distal hypoxic tubular injury debate revisited. Kidney Int 2010; 77: 9–16 [DOI] [PubMed] [Google Scholar]
- 21. Rosen S, Stillman IE.. Acute tubular necrosis is a syndrome of physiologic and pathologic dissociation. J Am Soc Nephrol 2008; 19: 871–875 [DOI] [PubMed] [Google Scholar]
- 22. Srichai MB, Hao C, Davis L. et al. Apoptosis of the thick ascending limb results in acute kidney injury. J Am Soc Nephrol 2008; 19: 1538–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharfuddin AA, Molitoris BA.. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 2011; 7: 189–200 [DOI] [PubMed] [Google Scholar]
- 24. Thornley C, Dawnay A, Cattell WR.. Human Tamm–Horsfall glycoprotein: urinary and plasma levels in normal subjects and patients with renal disease determined by a fully validated radioimmunoassay. Clin Sci (Lond) 1985; 68: 529–535 [DOI] [PubMed] [Google Scholar]
- 25. Prajczer S, Heidenreich U, Pfaller W. et al. Evidence for a role of uromodulin in chronic kidney disease progression. Nephrol Dial Transplant 2010; 25: 1896–1903 [DOI] [PubMed] [Google Scholar]
- 26. Risch L, Lhotta K, Meier D. et al. The serum uromodulin level is associated with kidney function. Clin Chem Lab Med 2014; 52: 1755–1761 [DOI] [PubMed] [Google Scholar]
- 27. El-Achkar TM, McCracken R, Liu Y. et al. Tamm–Horsfall protein translocates to the basolateral domain of thick ascending limbs, interstitium, and circulation during recovery from acute kidney injury. Am J Physiol Renal Physiol 2013; 304: F1066–F1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaden J, Groth J, May G. et al. Urinary Tamm–Horsfall protein as a marker of renal transplant function. Urol Res 1994; 22: 131–136 [DOI] [PubMed] [Google Scholar]
- 29. Kobayashi K, Fukuoka S.. Conditions for solubilization of Tamm–Horsfall protein/uromodulin in human urine and establishment of a sensitive and accurate enzyme-linked immunosorbent assay (ELISA) method. Arch Biochem Biophys 2001; 388: 113–120 [DOI] [PubMed] [Google Scholar]
- 30. Catalano C, Torffvit O.. Urinary excretion of Tamm–Horsfall protein in normotensive, normo-albuminuric type 1 diabetic patients. Nephron 1996; 72: 436–441 [DOI] [PubMed] [Google Scholar]
- 31. Youhanna S, Weber J, Beaujean V. et al. Determination of uromodulin in human urine: influence of storage and processing. Nephrol Dial Transplant 2014; 29: 136–145 [DOI] [PubMed] [Google Scholar]
- 32. Shlipak MG, Li Y, Fox C. et al. Uromodulin concentrations are not associated with incident CKD among persons with coronary artery disease. BMC Nephrol 2011; 12: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou J, Chen Y, Liu Y. et al. Urinary uromodulin excretion predicts progression of chronic kidney disease resulting from IgA nephropathy. PLoS One 2013; 8: e71023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garimella PS, Biggs ML, Katz R. et al. Urinary uromodulin, kidney function, and cardiovascular disease in elderly adults. Kidney Int 2015; 88: 1126–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Köttgen A, Hwang SJ, Larson MG. et al. Uromodulin levels associate with common UMOD variant and risk for incident CKD. J Am Soc Nephrol 2010; 21: 337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Olden M, Corre T, Hayward C. et al. Common variants in UMOD associate with urinary uromodulin levels: a meta-analysis. J Am Soc Nephrol 2014; 25: 1869–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kumar S. Mechanisms of injury in Uromodulin-associated kidney disease. J Am Soc Nephrol 2007; 18: 10–12 [DOI] [PubMed] [Google Scholar]
- 38. Bleyer AJ, Hart PS, Kmoch S.. Hereditary interstitial kidney disease. Semin Nephrol 2010; 30: 366–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ekici AB, Hackenbeck T, Morinière V. et al. Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin. Kidney Int 2014; 86: 589–599 [DOI] [PubMed] [Google Scholar]
- 40. Dahan K, Devuyst O, Smaers M. et al. A cluster of mutations in the UMOD gene causes familial juvenile hyperuricemic nephropathy with abnormal expression of uromodulin. J Am Soc Nephrol 2003; 14: 2883–2893 [DOI] [PubMed] [Google Scholar]
- 41. Hart TC, Gorry MC, Hart PS. et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet 2002; 39: 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reznichenko A, Böger CA, Snieder H. et al. UMOD as a susceptibility gene for end-stage renal disease. BMC Med Genet 2012; 13: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pruijm M, Ponte B, Ackermann D. et al. Association of urinary Uromodulin with clinical characteristics and markers of tubular function in the general population. Clin J Am Soc Nephrol 2016; 11: 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaden J. Optimal management of induction therapy with ATG in kidney allograft rejection. Int J Immunother 1999; 15: 115–124 [Google Scholar]
- 45. Glauser A, Hochreiter W, Jaeger P. et al. Determinants of urinary excretion of Tamm–Horsfall protein in non-selected kidney stone formers and healthy subjects. Nephrol Dial Transplant 2000; 15: 1580–1587 [DOI] [PubMed] [Google Scholar]
- 46. Trachtenberg F, Barregard L.. The effect of age, sex, and race on urinary markers of kidney damage in children. Am J Kidney Dis 2007; 50: 938–945 [DOI] [PubMed] [Google Scholar]
- 47. Neugarten J, Golestaneh L.. Gender and the prevalence and progression of renal disease. Adv Chronic Kidney Dis 2013; 20: 390–395 [DOI] [PubMed] [Google Scholar]
- 48. Silbiger SR. Raging hormones: gender and renal disease. Kidney Int 2011; 79: 382–384 [DOI] [PubMed] [Google Scholar]
- 49. Wang X, Vrtiska TJ, Avula RT. et al. Age, kidney function, and risk factors associate differentially with cortical and medullary volumes of the kidney. Kidney Int 2014; 85: 677–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Steubl D, Block M, Herbst V. et al. Plasma uromodulin correlates with kidney function and identifies early stages in chronic kidney disease patients. Medicine 2016; 95: e3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis 1992; 20: 1–17 [DOI] [PubMed] [Google Scholar]
- 52. Healy E, Brady HR.. Role of tubule epithelial cells in the pathogenesis of tubulointerstitial fibrosis induced by glomerular disease. Curr Opin Nephrol Hypertens 1998; 7: 525–530 [DOI] [PubMed] [Google Scholar]
- 53. Farris AB, Ellis CL, Rogers TE. et al. Renal medullary and cortical correlates in fibrosis, epithelial mass, microvascularity, and microanatomy using whole slide image analysis morphometry. PLoS One 11: e0161019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. El-Achkar TM, Wu XR.. Uromodulin in kidney injury: an instigator, bystander, or protector? Am J Kidney 2012; 59: 452–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jennings P, Aydin S, Kotanko P. et al. Membrane targeting and secretion of mutant uromodulin in familial juvenile hyperuricemic nephropathy. J Am Soc Nephrol 2007; 18: 264–273 [DOI] [PubMed] [Google Scholar]
- 56. Biesen van W, Nagler EV.. A Swiss army knife for estimating kidney function: why new equations will not solve the real problem. Nephrol Dial Transplant 2016; 31: 685–687 [DOI] [PubMed] [Google Scholar]
- 57. Gill J, Dong J, Rose C. et al. The risk of allograft failure and the survival benefit of kidney transplantation are complicated by delayed graft function. Kidney Int 2016; 89: 1331–1336 [DOI] [PubMed] [Google Scholar]
- 58. Scherberich JE. The king’s new cloths—the infinite history of kidney tissue proteinuria (histuria) as a renal biomarker. Nieren- und Hochdruckkrankh 2012; 41: 436–450 [Google Scholar]
- 59. Scherberich JE, Wolf G.. Disintegration and recovery of kidney membrane proteins: consequence of acute and chronic renal failure. Kidney Int Suppl 1994; 47: S52–S57 [PubMed] [Google Scholar]
- 60. Johnson KR. Strengths and weaknesses of renal markers as risk factors and surrogate markers. Kidney Int 2011; 79: 1272–1274 [DOI] [PubMed] [Google Scholar]
- 61. Filler G, Huang SH, Lindsay RM.. The search for more reliable estimated GFR biomarkers. Am J Kidney Dis 2016; 67: 5–8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.