

Abstract
Background. Vitamin D deficiency and excess of circulating fibroblast growth factor 23 (FGF23) contribute to cardiovascular mortality in patients with chronic kidney disease (CKD). FGF23 activates FGF receptor 4 and (FGFR4) calcineurin/nuclear factor of activated T cells (NFAT) signaling in cardiac myocytes, thereby causing left ventricular hypertrophy (LVH). Here, we determined if 1,25-dihydroxyvitamin D (calcitriol) inhibits FGF23-induced cardiac signaling and LVH.
Methods. 5/6 nephrectomized (5/6 Nx) rats were treated with different doses of calcitriol for 4 or 10 weeks and cardiac expression of FGF23/FGFR4 and activation of calcineurin/NFAT as well as LVH were analyzed. FGFR4 activation and hypertrophic cell growth were studied in cultured cardiac myocytes that were co-treated with FGF23 and calcitriol.
Results. In 5/6Nx rats with LVH, we detected elevated FGF23 expression in bone and myocardium, increased cardiac expression of FGFR4 and elevated cardiac activation of calcineurin/NFAT signaling. Cardiac expression levels of FGF23 and FGFR4 significantly correlated with the presence of LVH in uremic rats. Treatment with calcitriol reduced LVH as well as cardiac FGFR4 expression and calcineurin/NFAT activation. Bone and cardiac FGF23 expression were further stimulated by calcitriol in a dose-dependent manner, but levels of intact cardiac FGF23 protein were suppressed by high-dose calcitriol. In cultured cardiac myocytes, co-treatment with calcitriol blocked FGF23-induced activation of FGFR4 and hypertrophic cell growth.
Conclusions. Our data suggest that in CKD, cardioprotective effects of calcitriol stem from its inhibitory actions on the cardiac FGF23/FGFR4 system, and based on their counterbalancing effects on cardiac myocytes, high FGF23 and low calcitriol synergistically contribute to cardiac hypertrophy.
Keywords: cardiac hypertrophy, chronic kidney disease, fibroblast growth factor 23, fibroblast growth factor receptor 4, vitamin D
INTRODUCTION
Chronic kidney disease (CKD) is a global public health epidemic that increases the risks of cardiovascular disease (CVD) and death [1, 2]. As an early and common metabolic complication of CKD, elevated levels of circulating fibroblast growth factor 23 (FGF23) reduce serum levels of 1,25-dihydroxyvitamin D (calcitriol) [3] and both are strongly associated with a greater risk of CKD progression, CVD events and mortality [4, 5]. Clinical studies suggest that elevated FGF23 and calcitriol deficiency causally contribute to adverse outcomes in CKD, but underlying molecular mechanisms are incompletely understood.
FGF23 is a hormone produced by osteocytes that regulates mineral metabolism [6, 7]. In the kidney, FGF23 binds FGF receptor (FGFR)–klotho co-receptor complexes [8, 9] to stimulate phosphaturia, inhibit parathyroid hormone (PTH) secretion and calcitriol levels are decreased because of/due to the inhibition of 1a-hydroxylase and the stimulation of 24-hydroxylase [10–12]. Beginning in CKD stage 2, FGF23 levels rise progressively as renal function declines in order to maintain normal phosphate levels [13]. Elevated FGF23 serum levels are associated with left ventricular hypertrophy (LVH) [4, 14, 15], which promotes diastolic dysfunction, congestive heart failure, arrhythmia and sudden death [16, 17].
In previous experimental studies we have shown that circulating FGF23 can directly contribute to LVH [18]. FGF23 induces hypertrophic growth of cardiac myocytes via FGFR-dependent signaling, but independent of klotho, which is not expressed in the heart [8, 18]. In contrast to klotho-expressing target cells that respond to FGF23 by activating the Ras/mitogen-activated protein kinase (MAPK) cascade [9], FGF23 activates phospholipase Cγ (PLCγ)/calcineurin/nuclear factor of activated T cells (NFAT) signaling in cardiac myocytes [18], which is a potent inducer of LVH in established animal models of pathologic cardiac remodeling [19]. We demonstrated that FGF23 specifically activates FGFR4 in cardiac myocytes and that administration of an FGFR4-specific blocking antibody protects 5/6 nephrectomized (5/6Nx) rats from developing LVH [20]. Moreover, expression levels of FGFR4, calcineurin and NFAT are elevated in the myocardial tissue of patients with CKD and positively correlate with the presence of LVH [21], suggesting that this FGF23-driven pro-hypertrophic signaling pathway also exists in humans.
Vitamin D is a hormone with a variety of tissue-protective functions [22]. Calcitriol initiates biological responses by binding the cytoplasmic vitamin D receptor (VDR), which modulates transcription of a large number of genes [23]. Deficiency of calcitriol is an important consequence of elevated FGF23 in CKD [24] and is associated with death and CVD, including LVH [25]. Clinical studies suggest a survival benefit of active vitamin D therapy in CKD [26, 27], and several observations demonstrate that calcitriol can directly target the heart and mediate cardioprotection. Cardiac myocytes express VDR and the enzymatic system for vitamin D activation [28], and gene polymorphisms in both are associated with cardiac hypertrophy [29]. Experimental studies have shown that vitamin D metabolites reduce the expression of genes that are involved in the development of cardiac hypertrophy and that administration of calcitriol blocks cardiac hypertrophy in rodents and isolated cardiac myocytes [30, 31]. Interestingly, cardiac myocyte-specific VDR deletion results in LVH in the absence of hypertension [31], indicating that a loss of vitamin D signaling in the heart directly causes LVH.
It has been shown that calcitriol prevents isoproterenol-induced cardiac hypertrophy by blocking calcineurin/NFAT signaling in cardiac myocytes [31]. Since FGF23 induces LVH via FGFR4-dependent activation of PLCγ/calcineurin/NFAT signaling in cardiac myocytes [20], we wanted to determine whether calcitriol treatment affects cardiac expression of FGF23 and/or of the downstream FGFR4/calcineurin/NFAT signaling system in uremic rats and inhibits the FGF23-mediated hypertrophic growth of cardiac myocytes.
MATERIALS AND METHODS
Detailed methodology is provided in the online-only Supplementary Data.
Animal experiments
Kidney disease was induced in male Sprague Dawley rats using the 5/6Nx method, as described previously [32]. Two weeks after surgery, 5/6Nx rats were randomized into eight groups and treated once a day by mouth with 30, 100 or 300 ng/kg body weight of calcitriol or vehicle for 4 weeks and 10 weeks, respectively. At the end, animals were euthanized, blood was collected for serological analyses and organs were isolated and prepared for further analyses. All experimental procedures were performed in accordance with national animal protection guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Isolation and culture of neonatal rat ventricular myocytes (NRVMs)
NRVMs were isolated using a standard isolation system as described previously [18]. For immunofluorescence analysis, 3 × 105 cells were seeded per well on glass coverslips in 24-well plates. For co-immunoprecipitation studies, 5 × 106 cells were seeded in 10-cm culture dishes. After 4 days, isolated cardiac myocytes were cultured in the presence of recombinant murine FGF23 or FGF2 (both at 25 ng/ml) for different time points.
Histomorphometry
Cardiac mid-chamber (MC) sections were incubated with wheat germ agglutinin (WGA) to visualize cellular borders of individual cardiac myocytes and immunofluorescence images were taken on a Zeiss AxioObserver Z1 microscope with a Plan-Apo 63×/N.A. 1.4 oil objective followed by quantification of myocardial cross-sectional area (in µm2) of 100 cardiac myocytes on average.
Immunostaining for quantification of protein localization and cell size
For immunohistochemistry, MC sections were incubated with primary antibodies against NFAT. Appropriate biotin-conjugated secondary antibodies were used, followed by the avidin-biotin complex technique and antigen labeling with 3,3′-diaminobenzidine substrate. Images were taken on a Keyence BZ-9000 microscope with 20× objective.
Immunocytochemistry and morphometry of NRVMs
To assess the effects of calcitriol on FGF23- and FGF2-mediated hypertrophic growth of cardiac myocytes [18], isolated NRVMs were pretreated with 100 nM calcitriol for 60 min prior to addition of the FGFs. After 48 h, NRVMs were incubated with primary mouse monoclonal antibody against sarcomeric α-actinin and Cy3-conjugated goat anti-mouse secondary antibody. Immunofluorescence images were taken on a Leica TCS-SP5 confocal microscope with a 40× air objective. Myocyte cross-sectional area was measured using Leica AF6000 fluorescence software.
RNA isolation and quantitative real-time polymerase chain reaction analysis
For RNA isolation of snap-frozen rat myocardial tissue, the RNeasy Mini Kit was used. For RNA isolation from long bone, the entire femur was crunched without bone marrow and incubated with QIAzol lysis reagent following lysis using TissueLyser. RNA isolation was performed using the RNeasy Lipid Tissue Kit, total RNA was transcribed into cDNA using the QuantiTect Reverse Transcription Kit and real-time polymerase chain reaction (RT-PCR) was performed in triplicate with appropriate primers (Supplementary data, Table S1) using the QuantiFAST SYBR Green PCR Kit including ROX dye. Relative gene expression values, adjusted for the same Ct-threshold and baseline settings, were calculated with the 2−ΔΔCt method using Gapdh as a housekeeping gene.
Protein isolation, immunoblotting and immunoprecipitation
For protein extraction from snap-frozen myocardial specimens of the left ventricle, tissue was homogenized in RIPA extraction buffer and total protein was analyzed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting. Antibodies to FGF23, calcineurin A, NFATc4 and GAPDH were used as primary antibodies, and IRDye conjugated goat anti-mouse and goat anti-rabbit were used as secondary antibodies. For endogenous immunoprecipitation studies from NRVMs [20], cells were pretreated with vehicle, 100 nM calcitriol or with human monoclonal FGFR4 blocking antibody at 10 μg/ml for 1 h followed by treatment with growth factors for 20 min. Cells were scraped in RIPA buffer and protein lysates were incubated with anti-PLCγ or anti-GFP antibody as negative controls. A mix of Protein A and Protein G beads was added, followed by incubation, protein elution in sample buffer and SDS-PAGE and Western blot analyses. Antibodies to FGFR4 and PLCγ were used as primary antibodies and horseradish peroxidase–conjugated goat anti-rabbit was used as a secondary antibody.
Statistics
Data are presented as mean ± SEM, if not indicated otherwise. Comparisons between groups were done by unpaired t-tests in case of normally distributed data or Mann–Whitney U test in case of non-Gaussian distributions for two independent samples. Different groups of NRVMs were compared by one-way analysis of variance and Bonferroni’s multiple comparison test. Correlation analyses were done by a two-sided Pearson’s or Spearman’s rank correlation after testing for Gaussian distribution with the Kolmogorov–Smirnov test. Two-tailed P-values <0.05 were considered statistically significant.
RESULTS
Administration of calcitriol attenuates LVH in uremic rats
Here we analyzed cardiac effects of calcitriol treatment in 5/6Nx rats in a time- and dose-dependent manner. Calcitriol was given at 30, 100 or 300 ng/kg body weight per day beginning 2 weeks after nephrectomy and then daily for an additional 4 or 10 weeks. Renal function was significantly impaired in rats that underwent 5/6Nx versus sham nephrectomy (Table 1). There were no significant differences in animals with short-term versus long-term uremia, and calcitriol did not improve kidney function. As expected [33, 34], high PTH serum levels in 5/6Nx rats were reduced by calcitriol in a dose-dependent manner, and high-dose calcitriol increased serum calcium levels after 4 weeks of treatment (Table 1).
Table 1.
Serum biochemistry
| Group | Calcitriol |
Creatinine (mg/dL) | BUN (mg/dL) | Calcium++ (mmol/L) | Phosphate (mmol/L) | PTH (pg/mL) | 1,25(OH)2D3 (pg/mL) | |
|---|---|---|---|---|---|---|---|---|
| Dosage (ng/kg BW/day) | Duration (weeks) | |||||||
| Sham | 0 | 4 | 0.25 ± 0.05 | 15 ± 2 | 2.48 ± 0.07 | 2.64 ± 0.44 | 187 ± 89 | n.a. |
| 5/6Nx | 0 | 4 | 0.59 ± 0.12a | 127 ± 21a | 2.54 ± 0.12 | 2.95 ± 0.60 | 262 ± 123 | 79.2 ± 22.6 |
| 5/6Nx | 30 | 4 | 0.75 ± 0.31 | 132 ± 26 | 2.24 ± 0.26 | 2.46 ± 0.34 | 119 ± 66b | 57.7 ± 27.4 |
| 5/6Nx | 100 | 4 | 0.84 ± 0.29b | 156 ± 67 | 2.62 ± 0.40 | 2.84 ± 0.60 | 33 ± 65b | 65.2 ± 30.4 |
| 5/6Nx | 300 | 4 | 0.65 ± 0.27 | 156 ± 35b | 2.87 ± 0.40b | 2.73 ± 0.60 | 2 ± 0 b | 73.3 ± 38.9 |
| Sham | 0 | 10 | 0.33 ± 0.08 | 17 ± 2 | 2.60 ± 0.20 | 2.79 ± 0.20 | 149 ± 84 | n.a. |
| 5/6Nx | 0 | 10 | 0.62 ± 0.09a | 119 ± 26a | 2.60 ± 0.15 | 2.75 ± 0.19 | 330 ± 236 | 46.2 ± 43.6 |
| 5/6Nx | 30 | 10 | 0.78 ± 0.22 | 134 ± 37 | 2.07 ± 0.50 | 2.34 ± 0.60 | 121 ± 67b | 58.7 ± 32.3 |
| 5/6Nx | 100 | 10 | 1.43 ± 1.42 | 177 ± 66 | 2.50 ± 0.70 | 2.91 ± 0.70 | 10 ± 12b | 53.7 ± 41.9 |
| 5/6Nx | 300 | 10 | 0.76 ± 0.25 | 132 ± 29 | 2.68 ± 0.45 | 2.80 ± 0.49 | 23 ± 57b | 77.7 ± 54.9 |
Values are presented as mean ± SD. BW, body weight; BUN, blood urea nitrogen.
Significant versus sham.
Significant versus untreated 5/6Nx.
5/6Nx induced LVH, as demonstrated by a significant increase in ratios of heart weight to body weight (Figure 1A) and heart weight to tibia length (Figure 1B) and in cross-sectional area of individual cardiac myocytes (Figure 1C and D) when compared with controls. We did not detect significant differences in cardiac changes observed in rats that were not treated and studied 6 versus 12 weeks post-surgery, suggesting that between these two time points LVH did not further progress. Compared with vehicle-treated 5/6Nx rats, treatment with calcitriol for 4 weeks attenuated LVH in a dose-dependent manner, as demonstrated by decreases in the ratios of heart weight to body weight and heart weight to tibia length, as well as myocyte cross-sectional area (Figure 1A–D). Changes became significant in animals that received the highest dose of calcitriol. In 5/6Nx rats that were treated with calcitriol for 10 weeks, the increase in cardiac myocyte area but not in relative heart weight was significantly reduced (Figure 1A–D). We next investigated the expression of pro-hypertrophic genes, known to be involved in pathologic cardiac remodeling [35]. Beta myosin heavy chain (βMHC) and brain natriuretic peptide (BNP) mRNA levels were increased in 5/6Nx rats (Figure 1E and F), suggesting an induction of pathologic cardiac hypertrophy. Calcitriol treatment significantly reduced expression levels of both markers in a time-dependent manner. These data indicate that treatment of 6/5Nx rats with a VDR agonist attenuates the development of LVH in a dose- and time-dependent manner.
FIGURE 1.

Calcitriol ameliorates LVH in uremic rats. (A) The heart weight:body weight ratio and (B) the heart weight:tibia length ratio of sham-operated and 5/6 Nx rats were determined 6 and 12 weeks after surgery. Compared with sham, 5/6Nx significantly increases relative heart weight. Treatment of uremic rats with calcitriol for 4 and 10 weeks reduces this effect in a dose-dependent manner. (C) Cardiac mid-chamber sections of sham and 5/6Nx rats were stained with WGA (red; original magnification ×63; scale bar 50 µm). (D) Cardiac myocyte cross-sectional area of vehicle-treated 5/6Nx rats is significantly larger when compared with sham. In uremic rats, the increase in cardiac myocyte area is reduced after 4 and 10 weeks of calcitriol treatment in a dose-dependent manner when compared with vehicle-treated 5/6Nx rats (N = 100 cells/section). Quantitative RT-PCR analysis of expression levels of (E) βMHC and (F) BNP demonstrate a significant increase in both pro-hypertrophic genes in 5/6Nx rats compared with sham, indicating pathologic remodeling in myocardial tissue. Calcitriol treatment reduces expression levels of both markers in a time- and dose-dependent manner. N = 4–13 animals in each group; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus sham; †P < 0.05, ††P < 0.01, †††P < 0.001 versus vehicle-treated 5/6Nx.
Cardiac expression of FGF23 and FGFR4 is elevated in uremic rats and reduced by calcitriol treatment
We investigated whether anti-hypertrophic effects of vitamin D treatment in 5/6Nx rats correlated with changes in expression levels of the cardiac FGF23/FGFR4 system. Compared with sham-operated animals, Fgf23 expression in bone was increased in vehicle-treated 5/6Nx rats and ∼100-fold elevated by calcitriol dosages of 100 and 300 ng/kg in a time-dependent manner (Figure 2A). Cardiac Fgf23 mRNA levels were elevated in vehicle-treated 5/6Nx rats after 4 and 10 weeks and further enhanced by treatment with calcitriol at 30 ng/kg for 4 weeks (Figure 2B). However, in contrast to Fgf23 expression in bone, long-term treatment or higher dosages of calcitriol did not further increase cardiac Fgf23 levels.
FIGURE 2.

Cardiac Fgf23/Fgfr4 expression is elevated in uremic rats and reduced by calcitriol treatment. (A) Quantitative RT-PCR analysis of Fgf23 mRNA expression in bone reveals significantly increased levels in vehicle-treated 5/6Nx rats compared with sham. Bone Fgf23 mRNA levels in uremic rats are elevated by calcitriol dosages of 100 and 300 ng/kg in a time-dependent manner. (B) Cardiac Fgf23 mRNA levels significantly increase in vehicle-treated 5/6Nx rats 6 and 12 weeks after surgery compared with sham. The expression of cardiac Fgf23 in uremic rats does not further increase by long-term treatment or higher dosages of calcitriol. Cardiac levels of the Fgf23 full-length protein are significantly induced in uremic rats compared with sham demonstrated by (C) immunoblot analysis of total heart extracts followed by (D) densitometric quantification. Cardiac full-length Fgf23 protein levels in 5/6Nx rats decrease upon calcitriol treatment at higher dosages for 10 weeks compared with vehicle-treated uremic rats. Gapdh serves as a loading control. In uremic rats, the amount of full-length cardiac Fgf23 protein positively correlates with (E) the heart weight:body weight ratio and with the enhanced (F) βMHC and (G) BNP mRNA levels. (H) Fgfr4 expression levels are significantly elevated in heart tissue of 5/6Nx rats when compared with sham animals as determined by quantitative RT-PCR analysis. Higher calcitriol dosages reduce cardiac Fgfr4 expression in a dose-dependent manner. (I) In uremic rats, the expression of Fgfr4 correlates positively with the cardiac Fgf23 mRNA levels. N = 4–13 animals in each group; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus sham; †P < 0.05, ††P < 0.01, †††P < 0.001 versus vehicle-treated 5/6Nx.
As determined by immunoblot analysis of total heart extracts followed by densitometric quantification, cardiac levels of the Fgf23 full-length protein were elevated in uremic rats and further increased by a low dose of calcitriol (30 ng/kg) at 4 weeks (Figure 2C and D). Cardiac Fgf23 protein levels in 5/6Nx rats tended to decrease at higher calcitriol dosages and were significantly reduced by calcitriol treatment at 300 ng/kg for 10 weeks compared with vehicle-treated animals. In vehicle- and calcitriol-treated 5/6Nx rats, the amount of full-length cardiac Fgf23 protein positively correlated with the heart weight:body weight ratio (Figure 2E) and with enhanced βMHC and BNP expression levels (Figure 2F and G).
Expression of Fgfr4 was significantly elevated in heart tissue from 5/6Nx rats compared with controls (Figure 2H). Low-dose treatment with calcitriol for 4 weeks further elevated Fgfr4 expression. However, higher calcitriol dosages reduced cardiac Fgfr4 mRNA levels in a dose-dependent manner, irrespective of the duration of treatment (Figure 2H). In uremic animals, expression levels of Fgfr4 correlated positively with cardiac Fgf23 levels (Figure 2I). Combined, our data indicate that cardiac expression levels of FGF23 and FGFR4 are elevated in uremic rats. Calcitriol treatment reduces cardiac expression of FGF23 and FGFR4, which might contribute to the anti-hypertrophic effects of calcitriol treatment in uremic rats.
Uremic rats exhibit activated calcineurin/NFAT signaling in the heart, which is abrogated by calcitriol treatment
To determine the activity of cardiac calcineurin/NFAT signaling in 5/6Nx rats, we investigated protein levels of total calcineurin and phosphorylated NFAT by immunoblot analysis of heart extracts. Activated calcineurin dephosphorylates NFAT in the cytoplasm, inducing the translocation of NFAT into the nucleus and thereby the expression of NFAT target genes involved in LVH [36]. Induction of uremia significantly increased calcineurin protein levels in the heart (Figure 3A and B), accompanied by a significant reduction in the levels of phosphorylated NFAT (Figure 3C and D). Calcitriol significantly reduced calcineurin protein levels, irrespective of the dose and duration of treatment (Figure 3A and B), and caused the rephosphorylation of NFAT in a dose-dependent manner (Figure 3C and D). Moreover, nuclear NFAT protein expression was increased in 5/6Nx rats when compared with controls (Figure 3E). Calcitriol treatment caused a shift in NFAT localization from a nuclear to a cytoplasmic pattern. Finally, expression levels of regulator of calcineurin 1 (Rcan1) and transient receptor potential channel 6 (TRPC6), two established NFAT target genes [37, 38], were increased in 5/6Nx rats compared with controls, and reduced by calcitriol treatment in a dose-dependent manner (Figure 3F and G).
FIGURE 3.

Calcineurin/NFAT expression and signaling is elevated in uremic rats and reduced by calcitriol treatment. (A, B) Calcineurin protein levels are significantly increased in the heart of 5/6Nx rats compared with sham-operated animals, demonstrated by immunoblot analysis of total heart extracts followed by densitometric quantification. (C, D) In parallel, levels of phosphorylated NFAT (pNFAT) are significantly reduced in uremic rats. In 5/6Nx rats, calcineurin protein levels are significantly reduced by calcitriol treatment in a dose-dependent manner, thereby causing re-phosphorylation of NFAT. Gapdh serves as a loading control. (E) Immunohistochemical analysis of myocardial sections stained for NFAT (brown) with hematoxylin counterstain (blue) reveals an increase in nuclear localization of NFAT in 5/6Nx rats when compared with sham. Treatment of calcitriol in 5/6Nx induces a shift in localization of NFAT protein from the nucleus to the cytoplams (original magnification ×63; scale bar 50 µm). In 5/6Nx rat, (F) Rcan1 and (G) TRPC6 mRNA expression levels are increased compared with sham-operated rats and calcitriol treatment reduces both Rcan1 and TRPC6 levels in a dose-dependent manner. N = 4–13 animals in each group; *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; †P < 0.05, ††P < 0.01, †††P < 0.001, ††††P < 0.0001 versus vehicle-treated 5/6Nx.
In vehicle- and calcitriol-treated uremic rats, the cardiac phosphorylation state of NFAT negatively correlated with the cross-sectional area of cardiac myocytes (Figure 4A) and with Fgfr4 mRNA levels (Figure 4B). Furthermore, phospho-NFAT levels (Figure 4C) and expression levels of Rcan1 and TRPC6 (Figure 4D and E) positively correlated with full-length cardiac Fgf23 protein. The expression of NFAT target genes negatively correlated with NFAT phosphorylation (Figure 4F and G). Our data indicate that calcineurin protein levels and NFAT activity are elevated in the myocardium of uremic rats. Calcitriol treatment reduces cardiac expression of calcineurin and activation of NFAT, which might contribute to the anti-hypertrophic effects of vitamin D in uremic rats.
FIGURE 4.

Cardiac expression of FGF23 and FGFR4 correlates with cardiac hypertrophy in uremic rats. In the myocardium of vehicle- and calcitriol-treated 5/6Nx rats, phosphorylation of NFAT (pNFAT) negatively correlates with (A) the cross-sectional area of myocytes and (B) Fgfr4 mRNA levels. (C) The levels of pNFAT as well as mRNA levels of (D) Rcan1 and (E) TRPC6 correlate with cardiac protein levels of full-length Fgf23. The expression of NFAT target genes (F) Rcan1 and (G) TRPC6 negatively correlates with NFAT phosphorylation. N = 59–61 animals.
Calcitriol inhibits FGF23-mediated FGFR4 activation and hypertrophic growth in cultured cardiac myocytes
To determine if calcitriol could directly modify the pro-hypertrophic effects of FGF23 on cardiac myocytes, we co-treated NRVMs with FGF23 and calcitriol for 48 h. FGF23 significantly increased NRVM cross-sectional area, which did not occur in the presence of calcitriol (Figure 5A and B). In contrast, when NRVMs were stimulated with FGF2, a paracrine FGF family member that induces hypertrophic growth of cardiac myocytes by activating Ras/MAPK signaling [18], co-treatment with calcitriol had no effect.
FIGURE 5.
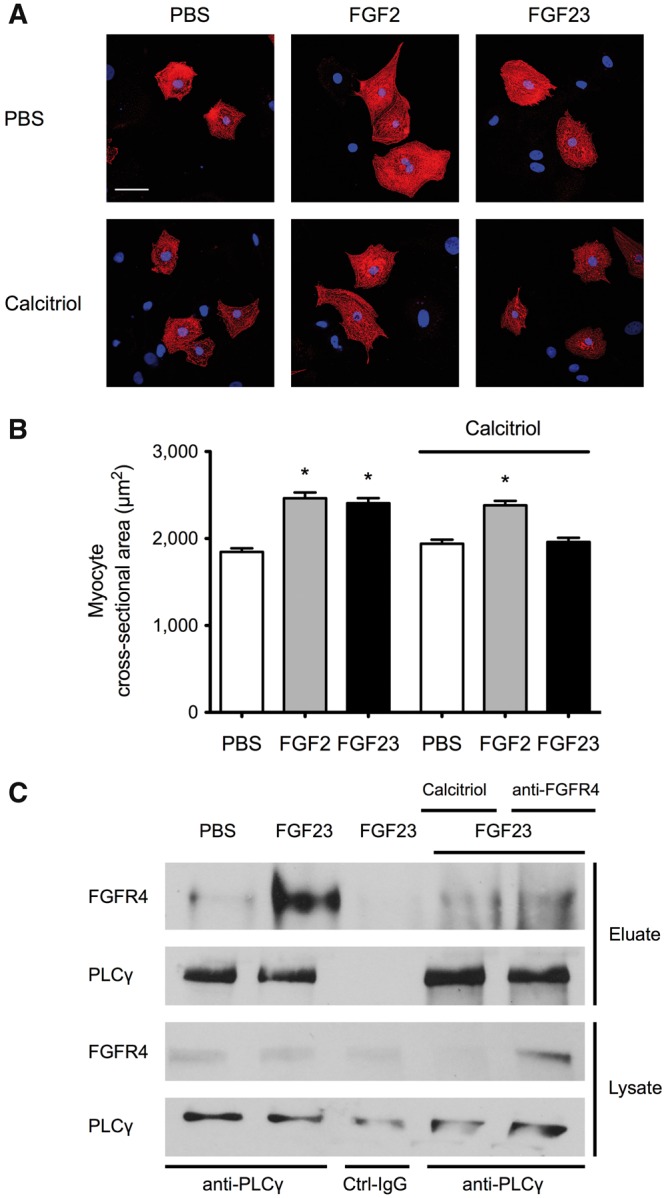
Calcitriol inhibits FGF23-mediated hypertrophic growth and FGFR4 signaling in NRVMs. (A) Immunofluorescence confocal images of isolated NRVMs that were co-treated with FGF2 or FGF23 and calcitriol for 48 h. Myocytes are labeled with anti-α-actinin (red), and DAPI (blue) identifies nuclei (original magnification ×40; scale bar 50 μm). NRVMs treated with FGF23 or FGF2 appear larger than PBS-treated control cells. Calcitriol blocks the effect of FGF23 but not FGF2. (B) Compared with PBS-treated control cells, 48 h of treatment with FGF23 or FGF2 significantly increases the cross-sectional area of isolated NRVMs (mean ± SEM). Co-treatment with calcitriol prevents FGF23- but not FGF2-induced hypertrophy (150 cells/condition; n = 3 independent isolations of NRVMs; *P < 0.0001 compared with PBS). (C) NRVMs were co-treated with FGF23 or FGF2 and calcitriol or an FGFR4 blocking antibody (anti-FGFR4) for 30 min followed by anti-PLCγ immunoprecipitation from total protein extracts. The amount of co-precipitated FGFR4 is increased in cells that were treated with FGF23 compared with PBS-treated control cells. In the presence of anti-FGFR4 or calcitriol, levels of FGFR4 in anti-PLCγ eluates are reduced. Immunoprecipitation with control antibody (Ctrl-IgG) does not result in purification of PLCγ or FGFR4.
Next we determined if calcitriol’s anti-hypertrophic effect towards FGF23 involved an inhibition of FGFR4/PLCγ signaling. Co-immunoprecipitation studies in NRVMs revealed that FGF23 treatment induced an interaction between FGFR4 and PLCγ. Pretreatment with an FGFR4-specific blocking antibody inhibited the interaction between FGFR4 and PLCγ. Similarly co-treatment with calcitriol reduced the amount of FGFR4 co-purified with PLCγ. Our in vitro data indicate that by blocking the interaction between FGFR4 and PLCγ and PLCγ activation, calcitriol inhibits FGF23-mediated hypertrophic growth of cardiac myocytes.
DISCUSSION
Here we show that calcitriol protects 5/6Nx rats, an established animal model of CKD with associated pathologic cardiac remodeling, from developing LVH in a dose-dependent manner. Calcitriol’s cardioprotective effect is accompanied by a significant reduction of FGF23 production in the heart and by reduced cardiac expression levels of FGFR4 and calcineurin and cardiac activity of NFAT. In the heart, FGF23 expression levels positively correlate with FGFR4 expression and NFAT activity as well as hypertrophy. Our findings indicate the existence of a paracrine FGF23/FGFR4/calcineurin/NFAT signaling axis in the heart that contributes to LVH under uremic conditions. This outcome is in line with our recent study in CKD patients showing similar positive associations between myocardial expression levels of these signal mediators and LVH [21] and with our mechanistic study using loss- and gain-of-function approaches in cell culture and animal models that revealed a direct causative role of FGF23-induced FGFR4 activation and subsequent calcineurin/NFAT signaling in the development of LVH [20].
Although it has been shown before that VDR agonists have anti-hypertrophic effects in 5/6Nx rats [39, 40], the current study is the first to indicate that this cardioprotective effect involves an inhibition of the cardiac FGF23/FGFR4/calcineurin/NFAT signaling pathway. Most likely the anti-hypertrophic effects of VDR agonists are multifactorial, including the reduction of blood pressure [39, 40] and the blockade of myocardial FGF23 effects. Although in the current model we cannot distinguish between the respective contributions of both mechanisms, our previous animal studies have shown that inhibition of cardiac FGF23/FGFR4/calcineurin/NFAT signaling by pharmacologic blockade of FGFR4 or calcineurin, protects from LVH in a blood pressure–independent manner [20, 41]. Therefore, we postulate that under uremic conditions, in addition to its established anti-hypertensive effects [42], calcitriol protects the heart by directly blocking FGF23 effects on the heart. This hypothesis is supported by our cell culture studies, showing that co-treatment with calcitriol inhibits FGF23-induced hypertrophic growth of isolated cardiac myocytes.
It appears that by directly inhibiting calcineurin/NFAT signaling in the heart, calcitriol mediates beneficial anti-hypertrophic effects in other scenarios of primary cardiac injury, e.g. when induced by β-adrenergic stimulation of cardiac myocytes [31]. Therefore, VDR and NFAT may be two counterbalancing factors that modulate cardiac remodeling under physiological conditions when levels or activation states of their respective upstream regulators undergo modest and short-term alterations. This balance might shift to NFAT overactivation and pathological changes when VDR activity is reduced, and even more so if the inducer of NFAT activity is elevated. Since we have demonstrated that FGF23 causes LVH via FGFR4-dependent activation of NFAT signaling in the myocardium [20] and calcitriol can block FGF23-induced hypertrophy of isolated cardiac myocytes, and since CKD is a stage of high FGF23 and low calcitriol [3], CKD might be such a pathological situation.
Currently the underlying molecular mechanism for a potential cardiac crosstalk between FGF23 and calcitriol is not understood. VDR and NFAT function as transcription factors [23, 36] that might communicate with each other on a genomic level by acting on the same promoter elements with opposite effects on the respective gene expression. Since VDR can induce and repress expression of target genes by recruiting co-repressors and restructuring the chromatin architecture of gene promoters [23], it is possible that calcitriol represses pro-hypertrophic NFAT target genes in cardiac myocytes following FGF23 stimulation. Such a scenario is likely, since it has been shown that VDR activation reduces atrial natriuretic peptide (ANP) and BNP expression in the heart by interacting with their promoter elements [28], and ANP and BNP are known NFAT target genes [43] whose expression in cardiac myocytes is increased by FGF23 [18]. Further evidence supporting such direct transcriptional repressive effects of VDR on NFAT target genes comes from T cell biology where NFAT binding to NFAT-responsive elements in target genes, such as interleukin-2—a key step in T cell activation—is directly inhibited by calcitriol VDR [44]. Future studies in cardiac myocytes should test if liganded VDR can bind to promoter regions of pro-hypertrophic genes and inhibit formation of the NFAT/DNA complex.
Calcitriol VDR can also act on a non-genomic level, which involves the regulation of calcium-dependent signal transduction [45]. Interestingly, it has been shown that calcitriol VDR can directly interact with PLCγ and thereby regulate PLCγ activity in keratinocytes and T cells [46, 47]. PLCγ is the enzyme largely responsible for maintaining increased levels of intracellular calcium through its effects on calcium channels at the cell surface and the production of inositol 1,4,5-triphosphate (IP3) to stimulate calcium release from intracellular stores [48]. Since PLCγ inhibition can block FGF23-induced cardiac hypertrophy [18], and PLCγ is an upstream activator of calcineurin/NFAT signaling [36], VDR may interfere with NFAT signaling by reducing PLCγ activity in cardiac myocytes. Such a mechanism is supported by our in vitro data indicating that following calcitriol treatment, VDR can interfere with PLCγ binding to FGFR4 and subsequent PLCγ activation.
It has been shown that cardiac myocytes can produce FGF23 [49] and that myocardial FGF23 levels are elevated in patients with dilated cardiomyopathy and ischemic heart disease as well as in mice with inflammatory heart failure [49] and myocardial infarct [50]. Cardiac FGF23 expression is increased in CKD patients and correlated with the development of LVH [21]. Overall, it seems that myocardial and circulating levels of FGF23 associate with cardiac hypertrophy in CKD [14, 18] and non-CKD [4, 51–53]. Here we show that although FGF23 production in bone and heart is elevated in 5/6Nx rats, only myocardial FGF23 levels are reduced by calcitriol treatment that correlates with a reduction in LVH, indicating pro-hypertrophic effects of cardiac-derived FGF23. Future animal studies should aim to determine if paracrine and endocrine FGF23 differ in their ability to target the heart and to contribute to cardiac hypertrophy.
Observational studies suggest a survival benefit of calcitriol therapy and its analogues in CKD [26, 27], but it raises circulating FGF23 serum levels, which has harmful effects [4, 54]. Our study suggests that calcitriol attenuates FGF23 toxicity in the heart downstream of FGFR4 by interfering with pathological NFAT signaling. This counterbalancing mechanism would reconcile the clinical paradox of how calcitriol treatment can be associated with improved survival despite raising total FGF23 levels, which are associated with mortality. However, in line with previous studies, mild hypercalcemia was observed in uremic animals treated with high-dose 1,25D [55] and high-dose calcitriol treatment was shown to be associated with an increased risk of vascular calcifications in CKD patients [55, 56]. Our finding suggests that even low doses of calcitriol ameliorate FGF23-driven LVH, hence recommending the use of low-dose calcitriol instead of pharmacological doses to avoid toxic side effects.
Our study indicates that calcitriol has anti-hypertrophic effects that at least partially derive from VDR activation in the myocardium. Although the randomized PRIMO study of CKD stage 3–4 patients failed to reach its primary anatomical endpoint—paricalcitol failed to reduce left ventricular mass versus placebo—it decreased left atrial volume and blunted the increase in BNP [57], suggesting a functional benefit. Indeed, paricalcitol reduced CVD hospitalization rates compared with placebo in PRIMO participants during 18 months of follow-up [58, 59]. Furthermore, PRIMO did not measure changes in serum FGF23 levels, which may have modified the effects of paricalcitol. Mechanistic animal studies are needed to understand FGF23/calcitriol interactions in the heart.
Although some studies have indicated that FGF23 might not be capable of directly targeting the heart [60–62] and that serum FGF23 levels are elevated in response to cardiac injury but by itself might not have cardiotoxic effects [63], other translational studies have provided compelling evidence showing that in CKD, circulating levels of FGF23 are elevated early in disease progression [14, 28] and that FGF23 can directly activate specific signaling pathways in cardiac myocytes, thereby inducing cardiac hypertrophy [14, 19, 64]. A limitation of this study is that all results are based on experimental rat data and must be confirmed in the human setting. Nevertheless, our findings indicate that under uremic conditions, high FGF23 and low calcitriol synergistically contribute to LVH, suggesting that a combination of pharmacological interventions activating VDR and blocking FGF23/FGFR4 signaling might have synergistic cardio-protective effects in patients with CKD.
SUPPLEMENTARY DATA
Supplementary data are available online at http://ndt.oxfordjournals.org.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Anja Ziolek for expert technical assistance, as well as Anja Rahn and Birgit Salewski for skillful assistance during 5/6 nephrectomy of rats and dedicated care of the animals throughout the experiments. This study was supported by the American Heart Association (A.G., C.F.), the American Diabetes Association (C.F.), and grant R01HL128714 to C.F. from the National Institutes of Health.
CONFLICT OF INTEREST STATEMENT
C.fF. has served as a consultant for Ultragenyx and has received research support from U3 Pharma and Hoffmann-La Roche. All other authors have declared that no conflict of interest exists.
REFERENCES
- 1. Saran R, Li Y, Robinson B. et al. US Renal Data System 2015 Annual Data Report: epidemiology of kidney disease in the United States. Am J Kidney Dis 2016; 67(3 Suppl 1): Svii, S1–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Go AS, Chertow GM, Fan D. et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351: 1296–1305 [DOI] [PubMed] [Google Scholar]
- 3. Gutierrez O, Isakova T, Rhee E. et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 2005; 16: 2205–2215 [DOI] [PubMed] [Google Scholar]
- 4. Isakova T, Xie H, Yang W. et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011; 305: 2432–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ravani P, Malberti F, Tripepi G. et al. Vitamin D levels and patient outcome in chronic kidney disease. Kidney Int 2009; 75: 88–95 [DOI] [PubMed] [Google Scholar]
- 6. Benet-Pages A, Lorenz-Depiereux B, Zischka H. et al. FGF23 is processed by proprotein convertases but not by PHEX. Bone 2004; 35: 455–462 [DOI] [PubMed] [Google Scholar]
- 7. Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest 2008; 118: 3820–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuro-o M, Matsumura Y, Aizawa H. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997; 390: 45–51 [DOI] [PubMed] [Google Scholar]
- 9. Urakawa I, Yamazaki Y, Shimada T. et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006; 444: 770–774 [DOI] [PubMed] [Google Scholar]
- 10. Shimada T, Urakawa I, Yamazaki Y. et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 2004; 314: 409–414 [DOI] [PubMed] [Google Scholar]
- 11. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V. et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest 2007; 117: 4003–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saito H, Maeda A, Ohtomo S. et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 2005; 280: 2543–2549 [DOI] [PubMed] [Google Scholar]
- 13. Isakova T, Gutierrez OM, Wolf M.. A blueprint for randomized trials targeting phosphorus metabolism in chronic kidney disease. Kidney Int 2009; 76: 705–716 [DOI] [PubMed] [Google Scholar]
- 14. Gutierrez OM, Januzzi JL, Isakova T. et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009; 119: 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kirkpantur A, Balci M, Gurbuz OA. et al. Serum fibroblast growth factor-23 (FGF-23) levels are independently associated with left ventricular mass and myocardial performance index in maintenance haemodialysis patients. Nephrol Dial Transplant 2011; 26: 1346–1354 [DOI] [PubMed] [Google Scholar]
- 16. Glassock RJ, Pecoits-Filho R, Barberato SH.. Left ventricular mass in chronic kidney disease and ESRD. Clin J Am Soc Nephrol 2009; 4(Suppl 1): S79–S91 [DOI] [PubMed] [Google Scholar]
- 17. Towbin JA, Bowles NE.. The failing heart. Nature 2002; 415: 227–233 [DOI] [PubMed] [Google Scholar]
- 18. Faul C, Amaral AP, Oskouei B. et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121: 4393–4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilkins BJ, Dai YS, Bueno OF. et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res 2004; 94: 110–118 [DOI] [PubMed] [Google Scholar]
- 20. Grabner A, Amaral AP, Schramm K. et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab 2015; 22: 1020–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leifheit-Nestler M, Grosse Siemer R, Flasbart K. et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant 2016; 31: 1088–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lavie CJ, Lee JH, Milani RV.. Vitamin D and cardiovascular disease will it live up to its hype? J Am Coll Cardiol 2011; 58: 1547–1556 [DOI] [PubMed] [Google Scholar]
- 23. Haussler MR, Whitfield GK, Kaneko I. et al. Molecular mechanisms of vitamin D action. Calcif Tissue Int 2013; 92: 77–98 [DOI] [PubMed] [Google Scholar]
- 24. Hasegawa H, Nagano N, Urakawa I. et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int 2010; 78: 975–980 [DOI] [PubMed] [Google Scholar]
- 25. Gunta SS, Thadhani RI, Mak RH.. The effect of vitamin D status on risk factors for cardiovascular disease. Nat Rev Nephrol 2013; 9: 337–347 [DOI] [PubMed] [Google Scholar]
- 26. Shoben AB, Rudser KD, de Boer IH. et al. Association of oral calcitriol with improved survival in nondialyzed CKD. J Am Soc Nephrol 2008; 19: 1613–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teng M, Wolf M, Ofsthun MN. et al. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol 2005; 16: 1115–1125 [DOI] [PubMed] [Google Scholar]
- 28. Chen S, Glenn DJ, Ni W. et al. Expression of the vitamin D receptor is increased in the hypertrophic heart. Hypertension 2008; 52: 1106–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Testa A, Mallamaci F, Benedetto FA. et al. Vitamin D receptor (VDR) gene polymorphism is associated with left ventricular (LV) mass and predicts left ventricular hypertrophy (LVH) progression in end-stage renal disease (ESRD) patients. J Bone Miner Res 2010; 25: 313–319 [DOI] [PubMed] [Google Scholar]
- 30. Bodyak N, Ayus JC, Achinger S. et al. Activated vitamin D attenuates left ventricular abnormalities induced by dietary sodium in Dahl salt-sensitive animals. Proc Natl Acad Sci USA 2007; 104: 16810–16815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen S, Law CS, Grigsby CL. et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation 2011; 124: 1838–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haffner D, Hocher B, Muller D. et al. Systemic cardiovascular disease in uremic rats induced by 1,25(OH)2D3. J Hypertens 2005; 23: 1067–1075 [DOI] [PubMed] [Google Scholar]
- 33. Silver J, Naveh-Many T, Mayer H. et al. Regulation by vitamin D metabolites of parathyroid hormone gene transcription in vivo in the rat. J Clin Invest 1986; 78: 1296–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ritter CS, Armbrecht HJ, Slatopolsky E. et al. 25-Hydroxyvitamin D3 suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int 2006; 70: 654–659 [DOI] [PubMed] [Google Scholar]
- 35. Komuro I, Yazaki Y.. Control of cardiac gene expression by mechanical stress. Annu Rev Physiol 1993; 55: 55–75 [DOI] [PubMed] [Google Scholar]
- 36. Crabtree GR, Olson EN.. NFAT signaling: choreographing the social lives of cells. Cell 2002; 109: S67–S79 [DOI] [PubMed] [Google Scholar]
- 37. Yang J, Rothermel B, Vega RB. et al. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res 2000; 87: E61–E68 [DOI] [PubMed] [Google Scholar]
- 38. Kuwahara K, Wang Y, McAnally J. et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest 2006; 116: 3114–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Freundlich M, Li YC, Quiroz Y. et al. Paricalcitol downregulates myocardial renin-angiotensin and fibroblast growth factor expression and attenuates cardiac hypertrophy in uremic rats. Am J Hypertens 2014; 27: 720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu-Wong JR, Chen YW, Wessale JL.. Vitamin D receptor agonist VS-105 improves cardiac function in the presence of enalapril in 5/6 nephrectomized rats. Am J Physiol Renal Physiol 2015; 308: F309–F319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Di Marco GS, Reuter S, Kentrup D. et al. Cardioprotective effect of calcineurin inhibition in an animal model of renal disease. Eur Heart J 2011; 32: 1935–1945 [DOI] [PubMed] [Google Scholar]
- 42. Li YC, Kong J, Wei M. et al. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110: 229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Akazawa H, Komuro I.. Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res 2003; 92: 1079–1088 [DOI] [PubMed] [Google Scholar]
- 44. Takeuchi A, Reddy GS, Kobayashi T. et al. Nuclear factor of activated T cells (NFAT) as a molecular target for 1α,25-dihydroxyvitamin D3-mediated effects. J Immunol 1998; 160: 209–218 [PubMed] [Google Scholar]
- 45. Bikle DD. Vitamin D: an ancient hormone. Exp Dermatol 2011; 20: 7–13 [DOI] [PubMed] [Google Scholar]
- 46. von Essen MR, Kongsbak M, Schjerling P. et al. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol 2010; 11: 344–349 [DOI] [PubMed] [Google Scholar]
- 47. Xie Z, Bikle DD.. The role of phospholipase C-gamma1 in 1α,25-dihydroxyvitamin D3 regulated keratinocyte differentiation. Steroids 2001; 66: 339–345 [DOI] [PubMed] [Google Scholar]
- 48. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000; 103: 211–225 [DOI] [PubMed] [Google Scholar]
- 49. Richter M, Lautze HJ, Walther T. et al. The failing heart is a major source of circulating FGF23 via oncostatin M receptor activation. J Heart Lung Transplant 2015; 34: 1211–1214 [DOI] [PubMed] [Google Scholar]
- 50. Andrukhova O, Slavic S, Odorfer KI. et al. Experimental myocardial infarction upregulates circulating fibroblast growth factor-23. J Bone Miner Res 2015; 30: 1831–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seiler S, Cremers B, Rebling NM. et al. The phosphatonin fibroblast growth factor 23 links calcium-phosphate metabolism with left-ventricular dysfunction and atrial fibrillation. Eur Heart J 2011; 32: 2688–2696 [DOI] [PubMed] [Google Scholar]
- 52. Wohlfahrt P, Melenovsky V, Kotrc M. et al. Association of fibroblast growth factor-23 levels and angiotensin-converting enzyme inhibition in chronic systolic heart failure. JACC Heart Fail 2015; 3: 829–839 [DOI] [PubMed] [Google Scholar]
- 53. Andersen IA, Huntley BK, Sandberg SS. et al. Elevation of circulating but not myocardial FGF23 in human acute decompensated heart failure. Nephrol Dial Transplant 2016; 31: 767–772 [DOI] [PubMed] [Google Scholar]
- 54. Gutierrez OM, Mannstadt M, Isakova T. et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359: 584–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zebger-Gong H, Muller D, Diercke M. et al. 1,25-Dihydroxyvitamin D3-induced aortic calcifications in experimental uremia: up-regulation of osteoblast markers, calcium-transporting proteins and osterix. J Hypertens 2011; 29: 339–348 [DOI] [PubMed] [Google Scholar]
- 56. Briese S, Wiesner S, Will JC. et al. Arterial and cardiac disease in young adults with childhood-onset end-stage renal disease-impact of calcium and vitamin D therapy. Nephrol Dial Transplant 2006; 21: 1906–1914 [DOI] [PubMed] [Google Scholar]
- 57. Thadhani R, Appelbaum E, Pritchett Y. et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: the PRIMO randomized controlled trial. JAMA 2012; 307: 674–684 [DOI] [PubMed] [Google Scholar]
- 58. Tamez H, Packham D, Wenger J. et al. Long-term outcomes for vitamin D treatment on patients with chronic kidney disease. J Am Soc Nephrol 2012; 23: P318A [Google Scholar]
- 59. Tamez H, Zoccali C, Packham D. et al. Vitamin D reduces left atrial volume in patients with left ventricular hypertrophy and chronic kidney disease. Am Heart J 2012; 164: 902–909 [DOI] [PubMed] [Google Scholar]
- 60. Agarwal I, Ide N, Ix JH. et al. Fibroblast growth factor-23 and cardiac structure and function. J Am Heart Assoc 2014; 3: e000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shalhoub V, Shatzen EM, Ward SC. et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest 2012; 122: 2543–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xie J, Yoon J, An SW. et al. Soluble klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol 2015; 26: 1150–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fuernau G, Poss J, Denks D. et al. Fibroblast growth factor 23 in acute myocardial infarction complicated by cardiogenic shock: a biomarker substudy of the Intraaortic Balloon Pump in Cardiogenic Shock II (IABP-SHOCK II) trial. Crit Care 2014; 18: 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Touchberry CD, Green TM, Tchikrizov V. et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am J Physiol Endocrinol Metab 2013; 304: E863–E873 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.