

Oestrogen receptor beta (ERβ) ligand treatment is neuroprotective in experimental autoimmune encephalomyelitis. Kim et al. provide insights into the underlying mechanisms by showing that ERβ-ligand treatment has direct effects on ERβ in bone marrow-derived CD11c+ myeloid dendritic cells and macrophages, resulting in modulation of the inflammatory CNS microenvironment.
Keywords: macrophage, oestrogen receptor beta, experimental autoimmune encephalomyelitis, multiple sclerosis, neuroprotection
Abstract
Oestrogen treatments are neuroprotective in a variety of neurodegenerative disease models. Selective oestrogen receptor modifiers are needed to optimize beneficial effects while minimizing adverse effects to achieve neuroprotection in chronic diseases. Oestrogen receptor beta (ERβ) ligands are potential candidates. In the multiple sclerosis model chronic experimental autoimmune encephalomyelitis, ERβ-ligand treatment is neuroprotective, but mechanisms underlying this neuroprotection remain unclear. Specifically, whether there are direct effects of ERβ-ligand on CD11c+ microglia, myeloid dendritic cells or macrophages in vivo during disease is unknown. Here, we generated mice with ERβ deleted from CD11c+ cells to show direct effects of ERβ-ligand treatment in vivo on these cells to mediate neuroprotection during experimental autoimmune encephalomyelitis. Further, we use bone marrow chimeras to show that ERβ in peripherally derived myeloid cells, not resident microglia, are the CD11c+ cells mediating this protection. CD11c+ dendritic cell and macrophages isolated from the central nervous system of wild-type experimental autoimmune encephalomyelitis mice treated with ERβ-ligand expressed less iNOS and T-bet, but more IL-10, and this treatment effect was lost in mice with specific deletion of ERβ in CD11c+ cells. Also, we extend previous reports of ERβ-ligand’s ability to enhance remyelination through a direct effect on oligodendrocytes by showing that the immunomodulatory effect of ERβ-ligand acting on CD11c+ cells is necessary to permit the maturation of oligodendrocytes. Together these results demonstrate that targeting ERβ signalling pathways in CD11c+ myeloid cells is a novel strategy for regulation of the innate immune system in neurodegenerative diseases. To our knowledge, this is the first report showing how direct effects of a candidate neuroprotective treatment on two distinct cell lineages (bone marrow derived myeloid cells and oligodendrocytes) can have complementary neuroprotective effects in vivo.
Introduction
Multiple sclerosis is an autoimmune, neurodegenerative disease of the CNS characterized by immune cell infiltration, glial activation, demyelination and axonal loss (Steinman, 1996). Experimental autoimmune encephalomyelitis (EAE) is the most widely used animal model to understand disease mechanisms and develop treatments for multiple sclerosis (Gold et al., 2006). Current multiple sclerosis treatments reduce relapse rates, but have modest effects on slowing disability worsening. Thus, there is a need to develop new therapeutic strategies that are neuroprotective in multiple sclerosis.
Oestrogens are known to be neuroprotective in several models of neurodegenerative diseases including multiple sclerosis, Alzheimer’s disease, Parkinson’s disease, ischaemic stroke and spinal cord injury (Suzuki et al., 2009; Samantaray et al., 2010; Spence and Voskuhl, 2012; McFarland et al., 2013; Uchoa et al., 2016). Oestradiol signals through binding to oestrogen receptors (ER)s, alpha (ERα) and/or beta (ERβ). ERα-ligand treatment mediates protection in vivo during EAE by acting on T lymphocytes (Morales et al., 2006; Lelu et al., 2011), and astrocytes (Spence et al., 2011), but not neurons (Spence et al., 2011). Unfortunately, treatments using oestradiol or ERα ligands for years in humans may cause adverse effects on breast and uterus, so targeted strategies are needed to achieve neuroprotection while minimizing off-target toxicity. One approach is to use ERα ligands designed to act in a tissue-specific manner in the CNS, but not in the periphery (Prokai et al., 2015). Another is to use oestrogen compounds or selective oestrogen receptor modifiers (SERMs) that are neuroprotective through ERβ activation with minimum ERα activation. Oestriol, a naturally occurring oestrogen that is expressed in high levels during the third trimester of pregnancy (Tulchinsky et al., 1972; Lindberg et al., 1974), binds ERs weakly compared to oestradiol, and has a higher affinity for ERβ than ERα (β/α, 2:1) (Kuiper et al., 1997). Oestriol treatment is neuroprotective in EAE (Palaszynski et al., 2004; Ziehn et al., 2012) and is safe when administered chronically to humans (Lauritzen, 1987; Takahashi et al., 2000). In a pilot clinical trial, multiple sclerosis patients treated with oestriol had reduced inflammatory lesions in brain (Sicotte et al., 2002). In a phase 2b double-blinded, placebo-controlled trial, the oestriol-treated group had lower relapse rates and less fatigue (Voskuhl et al., 2016). Also, higher blood levels of oestriol correlated with improved cognition, which was associated with decreased cerebral cortical atrophy (Voskuhl et al., 2016). Another phase 2b trial of oestriol treatment with cognitive improvement as the primary outcome measure is ongoing (NCT01466114). These three clinical trials of oestriol treatment in multiple sclerosis highlight the importance of understanding mechanisms underlying the neuroprotective effects of ERβ activation, since this may lead to new treatment strategies for multiple sclerosis and other neurodegenerative diseases (Nilsson et al., 2011; Paterni et al., 2014).
Regarding a tailored oestrogen receptor ligand approach, treatment with the classic ERβ-ligand diarylpropionitrile (DPN) is neuroprotective in the later phase of chronic EAE, sparing axons and myelin, while reducing grey matter atrophy by MRI (Tiwari-Woodruff et al., 2007; Mackenzie-Graham et al., 2012; Itoh et al., 2017). This occurs without decreasing peripheral adaptive immune responses or reducing levels of white matter inflammation in the CNS (Tiwari-Woodruff et al., 2007), albeit with some qualitative effects on dendritic cells in the CNS (Du et al., 2011). Also, two high-throughput in vitro screens to discover molecules to enhance maturation of oligodendrocytes and promote remyelination have identified SERMs (Mei et al., 2014; Lariosa-Willingham et al., 2016) as top candidates and suggested a role for ERβ specifically (Lariosa-Willingham et al., 2016). That said, an effect on remyelination is not mutually exclusive of a direct effect of ERβ on the innate immune system during multiple sclerosis and other neurodegenerative diseases.
Here, we hypothesized that CD11c+ immune cells could be a direct target of ERβ-ligand treatment. CD11c is a marker for dendritic cells in the peripheral immune system and is expressed in dendritic cells in perivascular areas in EAE (Bulloch et al., 2008; Prodinger et al., 2011). These cells release cytokines and chemokines to propagate an immunological cascade leading to demyelination and axonal degeneration (Karman et al., 2004; Greter et al., 2005). CD11c+ resident microglia, as well as CD11c+ peripheral myeloid dendritic cells and macrophages that have infiltrated into the CNS, are known to be critical in EAE pathogenesis (Bailey et al., 2007; King et al., 2009; Wlodarczyk et al., 2014; Clarkson et al., 2015). Involvement of CD11c+ myeloid dendritic cells in progression of later phases of EAE (Miller et al., 2007) was consistent with the timing of the ERβ-ligand treatment effect (Tiwari-Woodruff et al., 2007). That said, CD11c+ resident microglia and CD11c+ peripheral myeloid dendritic cells and macrophages have each demonstrated pro-inflammatory and anti-inflammatory properties (Wlodarczyk et al., 2015), and either could be a target of ERβ-ligand treatment. CD11c+ cells express both ERα and ERβ during disease (Paharkova-Vatchkova et al., 2004), but effects of ERβ signalling on these cells in vivo during neurodegenerative diseases has remained unknown. Finally, cells of this lineage have been implicated in human diseases, namely multiple sclerosis (Mishra and Yong, 2016) and Alzheimer’s disease (Srinivasan et al., 2016), and cell-type specific similarities in gene expression changes have been observed across these distinct diseases (Itoh and Voskuhl, 2017). Here we determine the functional significance of ERβ in CD11c+ cells in vivo during EAE using cell-specific conditional knockouts (CKO) of ERβ in CD11c+ cells, and then use bone marrow chimeras to discern whether ERβ-ligand treatment effects are directly mediated though ERβ in CD11c+ myeloid dendritic cells and macrophages versus CD11c+ resident microglia.
Materials and methods
Animals
Mice with ERβ selective deletion in CD11c+ and Olig1+ cells were generated by crossing transgenic mice that express Cre under the regulation of the CD11c (Itgax) promoter and Olig1 promotor, respectively. C57BL/6J-Tg(Itgax-cre,EGFP)4097Ach/J mice (CD11c-cre) and B6;129S4-Olig1tm1(cre)Rth/J (Olig1-cre) were purchased from Jackson Laboratory (Lu et al., 2002; Stranges et al., 2007), and crossed with C57BL/6J-ERβfloxed/floxed mice carrying the ERβ (Esr2) gene flanked by LoxP sites, a gift from Dr Pierre Chambon, France (Antal et al., 2008). For the bone marrow chimera study, we used a C57BL/6J congenic strain B6.SJL-PtprcaPepcb/BoyJ (CD45.1) mice (Shen et al., 1985). Animals were maintained under standard conditions in a 12-h dark/12-h light cycle with access to food and water ad libitum. All procedures were done in accordance to the guidelines of the National Institutes of Health and the Chancellor's Animal Research Committee of the University of California, Los Angeles Office for the Protection of Research Subjects.
Ovariectomy
To eliminate the effects of circulating endogenous oestrogens during the experiments, ovariectomy was performed in female mice. Detailed methods are described in the Supplementary material.
Experimental autoimmune encephalomyelitis induction and ERβ-ligand treatment
Female mice were immunized subcutaneously with MOG, amino acids 35–55 (200 μg per mouse, Mimotopes) emulsified in Complete Freund’s Adjuvant, supplemented with Mycobacterium tuberculosis H37Ra (200 μg per mouse, Difco Laboratories), over two sites drained by left inguinal and auxiliary lymph nodes in a total volume of 0.1 ml per mouse (Day 0). Pertussis toxin (500 ng per mouse, List Biological Laboratories) was injected intraperitoneally on Day 0 and Day 2. On Day 7, a booster immunization was delivered over contralateral lymph nodes. Ages of female mice for EAE induction were 8 to 12 weeks. The animals were monitored daily for EAE signs based on a standard EAE 0–5 scale scoring system: 0, healthy; 1, complete loss of tail tonicity; 2, loss of righting reflex; 3, partial paralysis; 4, complete paralysis of one or both hind limbs; and 5, moribund (Kim et al., 2014). For ERβ-ligand treatment we used DPN. Treatment was initiated 1 week prior to EAE induction. Briefly, DPN was dissolved in 100% ethanol and then mixed into 100% Miglyol®812 (CREMER Oleo) at 1:9 ratio making the final ethanol concentration 10%, and given subcutaneously every other day at a dose of 8 mg/kg per day until the end of each experiment. Selectivity of DPN at this dose was shown using the sensitive off-target outcome of uterine weight. Specifically, ERα-ligand treatment increased uterine weight (Harris et al., 2002), while ERβ-ligand treatment alone did not affect uterine weight, but blocked ERα-ligand mediated increases in uterine weight when used in combination (Frasor et al., 2003). Selectivity for the 8 mg/kg DPN dose was shown by two groups (Meyers et al., 2001; Tiwari-Woodruff et al., 2007). In addition, this dose of DPN was efficacious for EAE in wild-type mice, but was not efficacious in global ERα knock outs (Tiwari-Woodruff et al., 2007). Mechanistic studies herein used this previously optimized dose. All assessments were done in a blinded fashion with regards to knowledge of treatment randomization. For randomization of mice, after ovariectomy and before EAE induction, recovered healthy mice from each genotype were randomly assigned to treatment with vehicle or ERβ-ligand. For blinding of drug treatment, third party concealment with colour-coded labelling of vehicle or ERβ-ligand treatment syringes was done before treatment started for each experiment.
Histological preparation
Mice were exposed to a lethal dose of isoflurane and perfused transcardially with ice-cold PBS for 8 min, followed by 10% buffered formalin for 8 min. Spinal cords were dissected and stored in 10% buffered formalin for 12 h overnight, then submerged in 30% sucrose, 0.1% sodium azide, PBS for 24 h at 4°C. Thoracic and lumbar portion of the spinal cords were cut and embedded in optical cutting temperature compound (O.C.T., Tissue Tek) and stored at −80°C after flash freezing in an isopropanol bath chilled with liquid nitrogen. Tissues were cyrosectioned at −25°C into 40-μm thick sections using a cryostat (Leica CM1860) and stored in 0.1% sodium azide, PBS at 4°C before used for immunofluorescence staining.
Immunofluorescence staining
Spinal cord sections were washed thoroughly using PBS with 0.1% Triton™ X-100 (PBSt) to remove residual sodium azide. In the case of anti-MBP staining, tissue sections were incubated with 5% glacial acetic acid, 95% ethanol solution for 30 min at room temperature and then washed with PBSt before blocking. For blocking, tissues were treated with 10% normal goat serum in PBSt for 1 h at room temperature. Primary antibody and secondary antibody staining was done at appropriate concentrations with 2% normal goat serum, PBSt for overnight at 4°C and 1 h at room temperature, respectively. Antibodies are listed in the Supplementary material. Nuclei were stained with DAPI at 1:5000 concentrations in PBS. After serial washes sections were mounted onto slides (Superfrost®Plus, VWR) allowed to semi-dry, and cover slipped (24 × 60, Fisher Scientific) with Fluoromount-G® (SouthernBiotech) for confocal microscopy.
Confocal microscopy and image analysis
Stained sections were examined and imaged using Olympus BX51 fluorescence microscope with a DP50 digital camera. Images were taken in stacks, ×10 images were taken at 10-μm thickness with 2 μm stacks and ×40 images were taken at 5-μm thickness with 1 μm stacks. All images were taken and processed using the integrated software program Slidebook4.2 (Intelligent Imaging Innovations). ImageJ (NIH) was used to preform integration and analysis of images. All analyses were done in a blinded fashion with regards to knowledge of treatment randomization.
Electron microscopy
Mice were exposed to a lethal dose of isoflurane and perfused transcardially with ice-cold PBS for 8 min, followed by 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M phosphate buffer, 0.9% sodium chloride (PBS) for 8 min. Thoracic portions of the spinal cords were dissected and post-fixed for 2 h at room temperature in the same fixative and stored at 4°C until processing. Tissues were washed with PBS, post-fixed in 1% OsO4 in double-distilled H2O for 90 min on ice, dehydrated in a graded series of ethanol, treated with propylene oxide and infiltrated with Eponate 12™ (Ted Pella) overnight. Tissues were embedded in fresh Eponate, and polymerized at 60°C for 48 h. Approximately 50–60-nm thick sections were cut on a RMC MT-X ultramicrotome and picked up on Formvar®-coated copper grids. The sections were stained with uranyl acetate and lead citrate and examined on a JEOL 100CX electron microscope at 60 kV. Images were collected on type 4489 EM film and the negatives scanned to create digital files. Films (Kodak) were developed and scanned at high resolution. Analysis was done with ImageJ and g-ratio was used for extent of remyelination on axons. g-ratio = (axon diameter) / (axon + outer myelin diameter).
CNS immune cell isolation and flow cytometry and cell sorting
Animals were overdosed with isoflurane and perfused transcardially with 10 ml PBS within 2 min. Brain and spinal cords were collected into a 15 ml tube with 2 ml of 2% foetal bovine serum in PBS (FACS buffer). Collected CNS tissues were passed through 70 μm followed by 40 μm cell strainers to obtain single cell suspensions. Cells were centrifuged at 1500 rpm for 5 min at 4°C and resuspended in 30% Percoll solution (GE Healthcare Biosciences). The resuspended solution was then layered onto 70% Percoll solution, carefully so that the two solutions do not mix, and centrifuged for 1500 rpm for 20 min at 4°C. After centrifugation, mononuclear CNS immune cells were collected at the 70%:30% interface of Percoll solution and washed. Cells were stained with premixed combination of antibodies: APC-anti-CD11c (N418, BioLegend), PE-anti-CD45 (30-F11, Biolegend), PerCP/Cy5.5-anti-CD11b (M1/70, Biolegend), incubated for 30 min at 4°C and washed thoroughly. For FACS sorting, cells were stained with premixed combination of antibodies: APC-anti-CD11c and PE-anti-CD45, and incubated for 30 min at 4°C. Flow cytometry was performed using BD LSRFortessa cytometer and FACS sorting was performed using BD FACSAriaIII High-Speed Cell Sorter at the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards P30 CA016042 and 5P30 AI028697, and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, the UCLA Chancellor's Office, and the UCLA Vice Chancellor's Office of Research. Analysis was performed with FlowJo software (Tree Star).
RNA isolation and quantitative PCR of sorted CD11c+ cells from the CNS
FACS sorted cells were centrifuged at 2000 rpm for 5 min at 4°C and resuspended in TRI Reagent® (Zymo Research). RNA was isolated using the Direct-zol™ RNA MiniPrep Plus kit (Zymo Research) according to manufacturer’s instructions. cDNA was synthesized using the Tetro cDNA Synthesis kit (Bioline) according to manufacturer’s instructions. Quantitative PCR was performed using PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific) on the Bio-Rad Opticon 2 qPCR / Peltier Thermal Cycler according to manufacturer’s instruction. Primers are listed in the Supplementary material. The efficiency of each set of primers was assessed by quantitative PCR on serial dilution of cDNA from spleen immune cells and was confirmed to be above 90%. All gene expression levels were normalized to levels of β-actin by using the ΔΔCt and expressed as fold change relative to CD11c+ microglia cells sorted from vehicle treated wild-type EAE mice.
Bone marrow chimera
Bone marrow cell collection from donor mice
Cells were isolated from femurs and tibias of age-matched female donors. Bone marrow cells were subjected to T and B cell depletion by AutoMACS Magnetic Cell Separator (Miltenyi Biotec). Briefly, cells were incubated with anti-CD90.2 and anti-CD19 Miltenyi Biotec microbeads at 1:9 dilutions in AutoMACS buffer solution for 15 min at 4°C. Cells were then washed with AutoMACS buffer, resuspended at concentration of 108 cells per 500 μl, and passed through AutoMACS Magnetic Cell Separator columns using the ‘Deplete’ program. Collected cells were washed and resuspended at the concentration of 1.5 × 107 cells per 0.2 ml per mouse in lactated ringer’s solution (Abbott Laboratories).
Preparation and irradiation of recipient mice
Female mice were first ovariectomized at 4 to 5 weeks old and rested 2 weeks for recovery, as described earlier. Mice used for bone marrow chimera analyses received sulfamethoxazole and trimethoprim (TMS, 0.5 mg/ml water) for antibiotics. The mice were then subjected to irradiation once with a dose of 8.5 Gy using 137Cs source irradiator (Model MKI-68A, JL Shepherd) provided by the UCLA Center for AIDS Research Humanized Mouse Core. Within 1 to 2 h, irradiated mice received bone marrow cells collected from the donors via tail vein injections. After transplantation, animals were given TMS mixed in autoclaved water for 7 weeks.
Experimental autoimmune encephalomyelitis induction and drug treatment
Female mice were immunized subcutaneously with MOG peptide 35–55 emulsified in Freund’s Complete Adjuvant, supplemented with M. tuberculosis H37Ra, over four sites drained by inguinal and auxiliary lymph nodes on both sides in a total volume of 0.1 ml per mouse and received one dose of pertussis toxin (500 ng per mouse, List Biological Laboratories) intraperitoneally on Day 0. Ages of female mice for EAE induction were 14 to 16 weeks due to the time needed for recovery from transplantation and reconstitution. ERβ-ligand was treated 1 week prior to induction, where healthy mice from each genotype were randomly assigned to treatment with vehicle or ERβ-ligand. Treatment was given subcutaneously every other day at a dose of 8 mg/kg per day until the end of each experiment.
Reconstitution rate
Mouse blood immune cells collected by retro-orbital puncture were used to investigate reconstitution rates. Detailed methods are described in the Supplementary material.
Statistical analyses
Statistical analyses of EAE experiments were evaluated using two-way ANOVA with Bonferroni’s multiple comparison tests. This test was performed due to the existence of two variables, conditional knockout and drug treatment. In addition, repeated measures were used to observe the treatment effects over time during EAE. Statistical analyses of neuropathological experiments were evaluated using one-way ANOVA with Bonferroni’s multiple comparison tests, comparing treatment effects in two different transgenic groups. Data are presented as means ± standard error of the mean (SEM), with error bars representing biological variability between mice within each group. Power calculations for EAE experiments were determined for sample size to reach P < 0.05 by Dr Myung S. Sim, M.S. Dr PH, in the Department of Medicine Statistic Core at UCLA. A minimum of five animals per EAE group was stated to provide statistical significance at a level of P < 0.05 using 95% power analysis, consistent with numbers for EAE experiments in the field. Mice that never exhibited signs of EAE or died during an experiment were excluded, based on previously established criteria in the lab. Specifically, mice with EAE induced, but which never exhibit signs of EAE, would be excluded from the study. In addition, mice that died during experiments would be excluded, because of inability to carry forward previous scores over time during repeated measures. This criterion was applied equally to all groups. Data distribution was assumed to be normal. All statistical analyses were performed using Prizm 6 (version 6.01) software (GraphPad, CA).
Results
Neuroprotective effects mediated by CD11c+ myeloid cells
To determine whether ERβ-ligand treatment effects in vivo during EAE are driven by ERβ in CNS resident microglia and peripherally derived myeloid dendritic cells and macrophages, we focused on CD11c+ cells. Since CD11c expression is low in the healthy CNS, we first verified which population of cells expresses CD11c in the CNS during disease using flow cytometry of CNS mononuclear immune cells isolated from brains and spinal cords of EAE mice. CNS CD45+ cells were divided into two distinct populations; CD45int CNS resident microglia and CD45hi peripherally derived immune cells. The populations were then further divided into: CD45intCD11c+CD11b+ or CD45intCD11c−CD11b+ microglia, CD45hiCD11c+CD11b+ myeloid dendritic cells and macrophages, CD45hiCD11c−CD11b+ macrophages and CD45hiCD11c−CD11b− lymphocytes (Fig. 1A). These data demonstrate CD11c expression in microglia and myeloid dendritic cells and macrophages in the CNS during EAE.
Figure 1.

ERβ-specific deletion in CD11c+ cells during EAE. (A) Flow cytometry dot plots of CNS mononuclear immune cells isolated from brains and spinal cords of EAE mice. CNS mononuclear immune cells were gated based on FSC and CD45 expression. CNS CD45+ gated cells included two separate populations, CD45hi and CD45int, within the R1 gate. CD45+ cells were further gated based on CD11c expression into R2: CD45+CD11c− and R3: CD45+CD11c+. CD11b staining revealed that R2 included CD45hiCD11c−CD11b− lymphocytes, CD45hiCD11c−CD11b+ macrophages, and CD45intCD11c−CD11b+ resident microglia, and R3 included CD45hiCD11c+CD11b+ peripherally derived myeloid dendritic cells and macrophages (DC/Mφ) and CD45intCD11c+CD11b+ resident microglia. FSC = forward scatter. (B) Immunofluorescence images of spinal cord tissues stained with CD11c-GFP (green), ERβ (red) and nuclear stain DAPI (blue) with merged images for co-localization (yellow) on right. Top row: CD11c-cre-GFP+ control mice showed co-localization (white arrows) of CD11c-GFP and ERβ. Bottom row: CD11c-cre-GFP+;ERβfl/fl CKO mice did not show co-localization (white arrows). Orange arrows represent other cells in the CNS expressing ERβ. Scale bar = 20 μm. (C) Quantitative analysis of CD11c-GFP and ERβ co-localization in immunofluorescence images of CD11c-cre-GFP+ (Control) and CD11c ERβ CKO (CKO) mice with EAE. (D) Representative flow cytometry plots of isolated CNS mononuclear immune cells from a pool of three individual mice. CNS mononuclear immune cells were gated based on SSC and FSC (left), and subpopulations were identified using CD11c and CD45 staining (right). Cell populations labelled as CD11c+ microglia, CD11c+ myeloid dendritic cells and macrophages, and CD11c− cells were FACS sorted for mRNA isolation and quantitative PCR analysis. (E) Quantitative analysis of ERβ (Esr2) mRNA expression in sorted CNS CD11c+ microglia, CD11c+ myeloid dendritic cells and macrophages cells, and CD11c− cells from mice with ERβ deleted in CD11c+ cells (CKO) and wild-type mice, each with EAE.
To investigate whether neuroprotective effects of ERβ-ligand treatment are mediated through CD11c+ cells in vivo during EAE, we created mice with specific deletion of ERβ in CD11c+ cells using the Cre-LoxP system, by crossing CD11c-cre-GFP+ mice with ERβfloxed/floxed (ERβfl/fl) mice to obtain CD11c-cre-GFP+;ERβfl/fl mice (CD11c ERβ CKO). We then confirmed the specific deletion of ERβ in CD11c+ cells in the CNS in CKO mice by inducing EAE and collecting spinal cord tissues for immunofluorescence. ERβ expression in CD11c+ cells was shown by co-localization (merge) of ERβ (red) and CD11c-EGFP (green) in control EAE spinal cord tissues, while CKO EAE spinal cords did not show co-localization (Fig. 1B). Quantification of co-localization confirmed the efficiency of ERβ deletion in CD11c+ cells (Fig. 1C). FACS sorted mononuclear immune cells from the CNS of EAE mice with analysis of ERβ (Esr2) mRNA expression by quantitative PCR showed deletion of ERβ from CD11c+ microglia and CD11c+ myeloid dendritic cells and macrophages, but not from CD11c− cells (Fig. 1D and E). Together, these data validated the deletion of ERβ from CD11c+ microglia and CD11c+ myeloid dendritic cells and macrophages in the CNS of mice with EAE.
To determine the functional significance of ERβ expression in CD11c+ cells, we assessed effects of ERβ-ligand treatment during EAE in CKO and CD11c-cre-GFP−;ERβfl/fl (wild-type) littermates (Fig. 2A). For ERβ-ligand treatment we used DPN. Vehicle treated wild-type EAE mice exhibited the typical EAE disease course in C57BL/6J mice, whereas ERβ-ligand treated wild-type EAE mice showed amelioration of EAE during the chronic phase, consistent with previous reports (Tiwari-Woodruff et al., 2007; Spence et al., 2013). In contrast, ERβ-ligand treated CKO EAE mice did not exhibit clinical disease protection, with EAE scores identical to those in vehicle treated CKO EAE mice (Fig. 2A, Supplementary Tables 1 and 2). Next, we performed neuropathological studies on the CKO and wild-type EAE mice.
Figure 2.

ERβ expression in CD11c+ cells is necessary for neuroprotection in EAE. (A) Breeding scheme for creating wild-type (WT) and CKO mice of ERβ in CD11c+ cells. Briefly, CD11c-Cre-GFP+ mice were crossed with mice carrying an ERβ (Esr2) gene flanked by LoxP sites (ERβfl/fl). Homozygous ERβfl/fl mice without (WT) or with (CKO) Cre were generated. Each genotype was separated into two groups and received either vehicle or ERβ-ligand, DPN, treatment (tx). EAE was induced and animals were monitored daily and scored using the standard EAE 0–5 scale. ERβ-ligand treated wild-type EAE mice (WT-ERβ, blue solid) had significantly better clinical scores compared to vehicle treated wild-type EAE mice (WT-V, blue clear), ***P (WT-V versus WT-ERβ) = 0.0002, after Day 20 of EAE. In contrast, ERβ-ligand mediated protection did not occur in ERβ-ligand treated CKO EAE mice (CKO-ERβ, black solid) when compared to vehicle treated CKO EAE mice (CKO-V, black clear), P (CKO-V versus CKO-ERβ) > 0.9999. Detailed EAE statistics are in Supplementary Tables 1 and 2. (B) Representative images and quantitative analyses of NF200+, SMI32+, and βAPP+ axons in dorsal white matter of the spinal cord. Images were taken at 40× magnification. Scale bar = 20 μm. (C) Representative images and quantitative analyses of MBP+ and CNPase+ mean intensity in dorsal white matter of the spinal cord. Scale bar = 100 μm. (D) Representative electron microscopy images of ultraresolution of axons and myelin thickness in the dorsal white matter of spinal cord. Myelin thickness in the wild-type and CKO EAE mice treated with vehicle and ERβ-ligand was measured using the g-ratio (axon diameter) / (axon + outer myelin diameter) and shown in comparison with normal. Quantitative analysis showed that ERβ-ligand treated wild-type EAE mice had a decrease in g-ratio due to increased outer myelin diameter, while CD11c ERβ CKO mice with EAE did not. Scale bar = 1 μm. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Data are representative of three repeated experiments.
Healthy axons were evaluated by counting the number of phosphorylated neurofilament 200 (NF200)-positive intact axons, and injured axons by the number of non-phosphorylated NF200 (SMI32) and beta amyloid precursor protein (βAPP)-positive axons in spinal cord white matter. We observed a reduction of NF200+ healthy axons and an increase in SMI32+ and βAPP+ injured axons in vehicle treated wild-type EAE mice compared to healthy controls. Axons were protected in ERβ-ligand treated wild-type EAE mice as shown by an increase in NF200+ axons and a decrease in SMI32+ and βAPP+ axons. In contrast, ERβ-ligand treatment of CKO EAE mice failed to protect axons during EAE (Fig. 2B). Myelin pathology was evaluated by measuring the intensity of MBP and 2,3-cyclic-nucleotide 3-phosphodiesterase (CNPase). We observed a reduction of myelin intensity in vehicle treated wild-type EAE mice compared to healthy controls. Myelin was protected in ERβ-ligand treated wild-type EAE mice, but not in ERβ-ligand treated CKO EAE mice (Fig. 2C). Lastly, ultrastructural analysis of myelination using electron microscopy showed that vehicle treated wild-type EAE mice had an increased g-ratio [axon diameter / (axon + outer myelin diameter)] compared to healthy controls. ERβ-ligand treatment of EAE wild-type mice decreased the g-ratio compared to vehicle treated, consistent with increased myelin staining by immunofluorescence during ERβ-ligand treatment. In contrast, ERβ-ligand treated CKO EAE mice did not show this protective effect on myelin (Fig. 2D). Together, these results show that ERβ expression in CD11c+ cells is necessary for ERβ-ligand treatment to provide neuroprotection during EAE.
ERβ-ligand treatment reduces the pro-inflammatory phenotype of myeloid cells in the CNS
Despite the beneficial effects of ERβ-ligand treatment on axons and myelin during EAE, there was no effect on quantitative levels of CD3+ T cells, Iba1+ globoid macrophages, or Iba1+ ramified microglia in spinal cord white matter (Supplementary Fig. 1), as described (Tiwari-Woodruff et al., 2007; Spence et al., 2013). However, there were qualitative effects of ERβ-ligand treatment on Iba1+ myeloid cells. During EAE, vehicle treated wild-type EAE mice showed an increase in MHCII expression on Iba1+ myeloid cells compared to healthy controls, and ERβ-ligand treatment abrogated this increase in MHCII. In contrast, this effect of ERβ-ligand treatment on Iba1+ myeloid cells was lost in CKO EAE mice (Fig. 3A and D). We then measured expression levels of pro-inflammatory inducible nitrogen oxide synthase (iNOS) and anti-inflammatory arginase-1 (ARG1), on Iba1+ myeloid cells. We observed that iNOS expression on Iba1+ myeloid cells was reduced in ERβ-ligand treated wild-type EAE mice compared to vehicle treated wild-type EAE mice, but this treatment effect did not occur in ERβ-ligand treated CKO EAE mice (Fig. 3B and E). There was no effect on ARG1 expression (Fig. 3C and F). Together, these results show that ERβ-ligand treatment reduced the pro-inflammatory phenotype of myeloid cells in the CNS during EAE, and this was mediated through direct effects of ERβ-ligand treatment on CD11c+ cells.
Figure 3.

Qualitative effects on inflammatory markers on CNS resident and infiltrated Iba1+ myeloid cells. (A) Representative images of spinal cord tissues stained with MHCII (red) and Iba1 (green), (B) iNOS (red) and Iba1 (green), and (C) ARG1 (red) and Iba1 (green). Scale bar = 50 μm and 10 μm (inset). Inset: white arrows indicate co-localization. (D) MHCII+Iba1+ myeloid cells were increased in vehicle treated wild-type EAE mice (WT-V) compared to healthy controls (N), while ERβ-ligand treated wild-type EAE mice (WT-ERβ) showed a reduction in per cent MHCII+Iba1+ myeloid cells. In contrast, ERβ-ligand treated CKO EAE mice (CKO-ERβ) were no different from vehicle treated CKO EAE mice (CKO-V). (E) iNOS+Iba1+ myeloid cells were also reduced in ERβ-ligand treated wild-type EAE, but not in CKO EAE mice. (F) ARG1+Iba1+ myeloid cells were no different between groups. *P < 0.05; **P < 0.01; ***P < 0.001. Data are representative of two repeated experiments.
The relationship between neuroprotective effects on CD11c+ cells and oligodendrocyte lineage cells
Previous reports showed that ERβ in Olig2+ oligodendrocyte lineage cells (OLCs) (Khalaj et al., 2013), but not GFAP+ astrocytes (Spence et al., 2013) or NSE+ neurons (Spence et al., 2013), mediated neuroprotective effects of ERβ-ligand treatment during EAE. This neuroprotection entailed increased oligodendrocyte maturation and remyelination (Khalaj et al., 2013; Kumar et al., 2013). To understand these reports in the context of our new finding of a direct effect of ERβ-ligand treatment on CD11c+ cells, we determined levels of Olig2+ OLC subpopulations using an oligodendrocyte precursor cell (OPC) marker chondroitin sulphate proteoglycan (NG2) and more mature oligodendrocyte markers, adenomatous polyposis coli (CC1) and glutathione S-transferase-pi (GSTπ) during EAE in wild type and CD11c+ ERβ CKO mice (Fig 4A). The percentages of Olig2+GSTπ+ mature and Olig2+CC1+ immature/mature oligodendrocytes were reduced in wild-type EAE mice compared to normal controls, and ERβ-ligand treatment increased these cells in wild-type EAE mice. In contrast, ERβ-ligand treatment of EAE in CD11c ERβ CKO mice did not induce an increase in these OLC subpopulations (Fig 4B and C) showing that ERβ-ligand acting on CD11c+ cells was necessary for this effect. Olig2+NG2+ OPCs did not show changes with disease or with ERβ-ligand treatment (Fig. 4D). Together, these results demonstrate that ERβ-ligand treatment increases mature OLCs during EAE, but when the immunomodulatory effects of ERβ-ligand treatment on CD11c+ cells is removed, this effect no longer occurs.
Figure 4.

ERβ-ligand treatment acts on CD11c+ cells to permit increases in mature oligodendrocytes during EAE. (A) Immunofluorescence images of spinal cord tissues stained with Olig2 (red), and co-stained with GSTπ (green), CC1 (green), and NG2 (green). On the left is a representative image of Olig2+ oligodendrocyte lineage cells (OLC) in dorsal white matter of the spinal cord. On the right are representative images of each co-stain; Olig2-GSTπ (top), Olig2-CC1 (middle) and Olig2-NG2 (bottom). Scale bar = 50 μm (left) and 20 μm (right). White box indicates the area where the co-stains were imaged. Quantitative analyses of (B) Olig2+GSTπ+ mature OLCs, (C) Olig2+CC1+ immature/mature OLCs, and (D) Olig2+NG2+ oligodendrocyte precursor cells, each in dorsal white matter of the spinal cord. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Data are representative of two repeated experiments.
To further examine effects of ERβ-ligand treatment on OLCs, we used mice with a specific deletion of ERβ specifically in Olig1+ OLCs. We crossed Olig1-cre mice with ERβfl/fl mice to generate Olig1-cre+;ERβfl/fl mice (Olig1 ERβ CKO) and littermate controls Olig1-cre−;ERβfl/fl (Olig1-WT), then induced EAE and treated with ERβ-ligand or vehicle (Fig. 5A). ERβ-ligand treated Olig1-WT EAE mice exhibited protection from EAE compared to vehicle treated Olig1-WT EAE mice. In contrast, Olig1 ERβ CKO EAE mice did not show clinical disease protection with ERβ-ligand treatment, thereby showing direct effects of ERβ-ligand treatment on Olig1+ OLCs (Fig. 5B, Supplementary Tables 1 and 2). Neuropathology of axons and myelin showed that the ERβ-ligand treatment mediated neuroprotective effects were lost in Olig1 ERβ CKO EAE mice (Fig. 5B and D). In contrast, the immunomodulatory effect of ERβ-ligand treatment on MHCII expression on Iba1+ myeloid cells was not lost in Olig1 ERβ CKO mice with EAE (Fig. 5D), differing from how treatment effects on MHCII were lost in CD11c ERβ CKO EAE mice (Fig 3D). Furthermore, ERβ-ligand treatment increased both the percentage of Olig2+GSTπ+ mature oligodendrocytes and Olig2+CC1+ immature/mature oligodendrocytes during EAE in Olig1-WT mice, but both of these effects were lost in Olig1 ERβ CKO mice with EAE (Fig. 5E). Together, these data show that direct effects of ERβ-ligand treatment on CD11c+ cells and on Olig1+ cells are both necessary for neuroprotection during EAE, for one without the other is not sufficient.
Figure 5.

ERβ expression on Olig1+ cells is necessary for neuroprotection in EAE. (A) Breeding scheme for creating wild-type (WT) and CKO mice of ERβ in Olig1+ cells. Briefly, Olig1-Cre + mice were crossed with mice carrying an ERβ (Esr2) gene flanked by LoxP sites (ERβfl/fl). Homozygous ERβfl/fl mice without (Olig1-WT) or with (Olig1-CKO) Cre were generated. Each genotype was separated into two groups and received either vehicle or ERβ-ligand treatment (tx), EAE was induced and mice were scored for EAE severity as above. ERβ-ligand treated Olig1-WT EAE mice (Olig1-WT-ERβ, blue solid) had significantly better clinical scores compared to vehicle treated Olig1-WT EAE mice (Olig1-WT-V, blue clear), **P (Olig1-WT-V versus Olig1-WT-ERβ) = 0.0095, after Day 20 of EAE. In contrast, ERβ-ligand mediated protection did not occur in ERβ-ligand treated Olig1-CKO EAE mice (Olig1-CKO-ERβ, black solid) when compared to vehicle treated Olig1-CKO EAE mice (Olig1-CKO-V, black clear), P (Olig1-CKO-V versus Olig1-CKO-ERβ) = 0.7967. Detailed EAE statistics are in Supplementary Tables 1 and 2. Quantitative analyses of (B) NF200+, SMI32+, and βAPP+ axonal counts, (C) MBP+ and CNPase+ myelin intensity, and (D) MHCII+Iba1+ myeloid cells in dorsal white matter of the spinal cord. (E) Quantitative analyses of total Olig2+ OLCs, and the percentage of subpopulations: Olig2+NG2+ OPCs, Olig2+CC1+ immature/mature oligodendrocytes, and Olig2+GSTπ+ mature oligodendrocytes in dorsal white matter of the spinal cord. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Data are representative of three repeated experiments.
CD11c+ myeloid dendritic cells and macrophages, not microglia, mediate neuroprotection
Next, we determined whether direct effects of ERβ-ligand treatment on CD11c+ cells during EAE were due to effects on resident microglia or myeloid dendritic cells and macrophages. Studying these cell populations independently has been difficult since they share the same lineage (Ginhoux et al., 2010). The recent identification of microglia-specific markers is now permitting microglia-specific studies in vitro without contamination of peripherally derived myeloid cells; however, in vivo models are not yet available (Bennett et al., 2016). Therefore, to investigate which CD11c+ population mediates the protective effects of ERβ-ligand treatment in vivo, we created CKO mice with ERβ deleted in one population, but not the other, using bone marrow chimeras. Bone marrow chimera experiments used C57BL/6J CD45.1 wild-type mice as donors to determine the reconstitution efficiency of CD45.2 CKO or wild-type recipient mice using flow cytometry with FITC-anti-CD45.2 and PE-anti-CD45.1 labelling of blood leucocytes. The reconstitution efficiency was ∼95% (Supplementary Fig. 2). Bone marrow chimera mice with the CKO of ERβ in CD11c+ cells originating from the CNS (CD11c+ microglia-CKO) were created by injecting wild-type donor bone marrow cells into irradiated wild-type versus CKO recipient mice (Fig. 6A). We observed that chimeric mice with deletion of ERβ in CNS resident CD11c+ microglia (wild-type→CKO) exhibited protection from clinical EAE with ERβ-ligand treatment, identical to that in chimeric wild-type (wild-type→wild-type) EAE mice (Fig. 6B and Supplementary Tables 1 and 2). Neuropathology confirmed clinical EAE data, with ERβ-ligand treatment sparing NF200+ axons and MBP+ myelin and reducing βAPP+ axons in both groups regardless of selective ERβ deletion in CD11c+ microglia of recipients (Fig. 6C−E). These results demonstrate that ERβ expression on CNS resident CD11c+ microglia is not necessary for neuroprotective effects of ERβ-ligand treatment during EAE.
Figure 6.

CD11c+ microglial ERβ expression is not necessary for neuroprotection in EAE. (A) Diagram for creating CD11c+ microglia-CKO using BMC. Briefly, CD45.1 wild-type mice were used as donors for bone marrow cells, and CD45.2 wild-type (WT→WT) or CKO (WT→CKO) were used as irradiated recipients. Each genotype was separated into two groups and received either vehicle or ERβ-ligand treatment (tx), EAE was induced and mice were scored for EAE severity. (B) ERβ-ligand treated wild-type (WT→WT-ERβ, blue solid) EAE mice had significantly better scores compared to vehicle treated wildtype (WT→WT-V, blue clear) EAE mice, *P (WT→WT-V versus WT→WT-ERβ) = 0.0188. Similarly, ERβ-ligand treated CKO (WT→CKO-ERβ, black solid) EAE mice also had significantly better scores compared to vehicle treated conditional knockout (WT→CKO-V, black clear) EAE mice, **P (WT→CKO-V versus WT→CKO-ERβ) = 0.0077. Detailed EAE statistics are in Supplementary Tables 1 and 2. Quantitative analysis of (C) NF200+ axonal count, (D) βAPP+ axonal count, (E) MBP+ myelin intensity, and (F) MHCII expression on Iba1+ myeloid derived cells (percentage) in dorsal white matter of the spinal cord. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Data are representative of two repeated experiments.
Since CD11c+ microglia were not the target of ERβ-ligand treatment, we next created CKO mice with ERβ deleted from CD11c+ cells originating from the periphery (CD11c+ myeloid dendritic cells and macrophages CKO) by collecting donor bone marrow cells from CD45.2 wild-type or CKO mice and injecting them into irradiated CD45.1 wild-type recipient mice (Fig. 7A). In contrast to the protection observed with ERβ-ligand treatment of chimeric wild-type (wild-type→wild-type) EAE mice, ERβ-ligand treatment was not protective in EAE mice with deletion of ERβ in CD11c+ myeloid dendritic cells and macrophages (CKO→wild-type) (Fig. 7B, Supplementary Tables 1 and 2). Neuropathological analyses of NF200+ axons, βAPP+ axons, and MBP+ myelin confirmed that ERβ-ligand treatment mediated neuroprotective effects were lost in CD11c+ myeloid dendritic cells and macrophages ERβ CKO mice (Fig. 7C–E). There was no effect on quantitative levels of immune cells in the CNS (Supplementary Fig. 3). Together, these results show that ERβ expression on CD11c+ myeloid dendritic cells and macrophages mediates neuroprotective effects of ERβ-ligand treatment during EAE.
Figure 7.

ERβ expression on CD11c+ myeloid dendritic cells and macrophages is necessary for neuroprotection in EAE. (A) Diagram for creating CD11c+ myeloid dendritic cells and macrophages (DC/MΦ) CKO using bone marrow chimeras. Briefly, CD45.2 wild-type (WT→WT) versus CKO (CKO→WT) mice were used as donors for bone marrow cells, and CD45.1 wild-type mice were used as irradiated recipients. Each genotype was separated into two groups and received either vehicle or ERβ-ligand treatment (tx), EAE was induced and mice were scored for EAE severity. (B) ERβ-ligand treated wild-type (WT→WT-ERβ, blue solid) EAE mice had significantly better EAE scores compared to vehicle treated wild-type (WT→WT-V, blue open) EAE mice, ***P (WT→WT-V versus WT→WT-ERβ) = 0.0005, after Day 20 of EAE, whereas ERβ-ligand mediated protection did not occur in ERβ-ligand treated conditional knockout (CKO→WT-ERβ, black solid) compared to vehicle treated CKO (CKO→WT-V, black clear) EAE mice, P (CKO→WT-V versus CKO→WT-ERβ) > 0.9999. Detailed EAE statistics are in Supplementary Tables 1 and 2. Quantitative analysis of (C) NF200+ axonal count, (D) βAPP+ axonal count, (E) MBP+ myelin intensity, and (F) MHCII expression on Iba1+ myeloid derived cells (percentage) in dorsal white matter of the spinal cord. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Finally, to characterize the effect of ERβ-ligand treatment on CD11c+ microglia and CD11c+ myeloid dendritic cells and macrophages in the CNS during EAE, we determined expression levels of iNOS, T-bet, CCR2, IL-10, ARG1 and YM-1 in FACS sorted CD11c+ microglia and CD11c+ myeloid dendritic cells and macrophages (Fig. 8A) from the CNS of CD11c ERβ CKO and wild-type EAE mice treated with vehicle or ERβ-ligand. Pro-inflammatory iNOS and T-bet were reduced, while anti-inflammatory IL-10 was increased, in CD11c+ myeloid dendritic cells and macrophages from wild-type EAE mice treated with ERβ-ligand. In contrast, this ERβ-ligand treatment effect did not occur in CD11c ERβ CKO mice (Fig. 8B). ARG1 and YM-1 did not show significant changes. Interestingly, the ERβ-ligand treatment effect on iNOS, T-bet and IL-10 observed in CD11c+ myeloid dendritic cells and macrophages was not observed in CD11c+ microglia. This was consistent with bone marrow chimera results showing that CD11c+ myeloid dendritic cells and macrophages play a role mediating neuroprotective effects of ERβ-ligand treatment during EAE.
Figure 8.
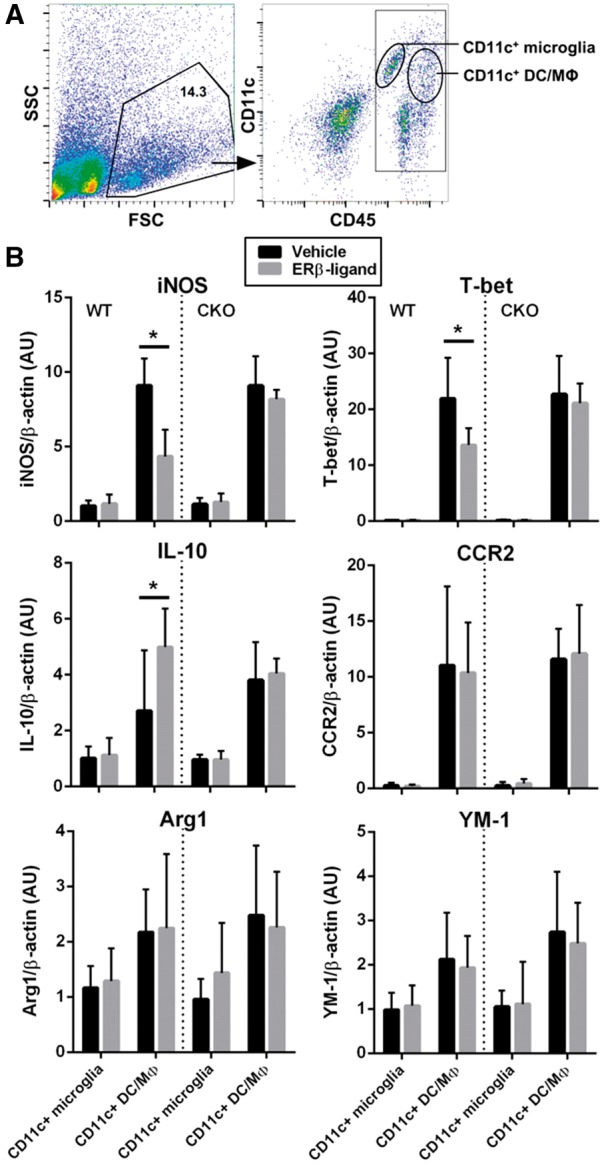
Gene expression profiles of CD11c+ microglia and CD11c+ dendritic cells and macrophages cells from the CNS of ERβ-ligand or vehicle treated mice with EAE. (A) Representative flow cytometry plots of isolated CNS mononuclear immune cells from a pool of two to four individual mice. CNS mononuclear immune cells were gated based on SSC and FSC (left), and subpopulations were identified using CD11c and CD45 staining (right). Cell populations labelled as CD11c+ microglia and CD11c+ myeloid dendritic cells and macrophages were FACS sorted for mRNA isolation and quantitative PCR analysis. (B) Quantitative analysis of iNOS, T-bet, IL-10, CCR2, ARG1, and YM-1 mRNA expression levels of sorted CD11c+ microglia and CD11c+ myeloid dendritic cells and macrophages from wild type (WT) (left) and CD11c ERβ CKO (right) mice with EAE that were treated with vehicle or ERβ-ligand. Data are from three separate experiments, with error bar representing variation between experiments. *P < 0.05.
Discussion
Here we show for the first time that ERβ expressed in CD11c+ cells plays an important role in mediating neuroprotection during EAE. When ERβ was specifically deleted from CD11c+ cells, the protective effect of ERβ-ligand treatment was lost. We then used bone marrow chimeras to show that ERβ in peripherally derived CD11c+ myeloid dendritic cells and macrophages was responsible, while ERβ in CD11c+ microglia was not. We also found that ERβ in Olig1+ OLCs can mediate direct neuroprotective effects during EAE. When effects of ERβ-ligand treatment on either myeloid dendritic cells and macrophages or OLCs were removed, treatment was no longer neuroprotective. Together these in vivo mechanistic studies show that effects on these two cells types are complementary. Direct effects on OLCs increase numbers of mature OLCs during EAE, but direct effects on CD11c+ myeloid dendritic cells and macrophages are also needed to modulate the inflammatory microenvironment in the CNS and avoid an arrest in OLC maturation. These findings underscore the need for combination treatment strategies in EAE that both modulate the inflammatory microenvironment of myeloid dendritic cells and macrophages in the CNS and promote the maturation of oligodendrocytes.
Distinguishing between the effects of ERβ-ligand treatment on CNS resident microglia versus peripheral myeloid dendritic cells and macrophages has therapeutic implications, suggesting that ERβ signalling pathways in myeloid dendritic cells and macrophages are candidate targets. The majority of myeloid dendritic cells and macrophages in the CNS during EAE release pro-inflammatory cytokines and cause myelin and axonal damage. During EAE, the infiltrates are classified as CD45hiCD11c+CD11b+ myeloid dendritic cells and macrophages with elevated levels of MHCII, IL-6, iNOS and other pro-inflammatory molecules (King et al., 2009; Vainchtein et al., 2014; Wlodarczyk et al., 2014). Recruitment of these cells through the CCL2-CCR2 axis has been shown to be critical for further recruitment of Th1/Th17 encephalitogenic cells and progression of chronic EAE (Dogan et al., 2008). When MHCII was ablated from these cells, this recruitment was prevented and EAE was ameliorated (Greter et al., 2005; Clarkson et al., 2015). These reports are consistent with our findings that protection from EAE with ERβ-ligand treatment coincided with reductions in MHCII, iNOS and T-bet, with an increase in IL-10, in CD11c+ myeloid dendritic cells and macrophages in the CNS. That said, the infiltration of myeloid dendritic cells and macrophages is not always detrimental in EAE since they can perform phagocytosis to clear debris and secrete neurotrophic factors for recruitment, differentiation and maturation of OPCs to facilitate remyelination (Miron et al., 2013; Rawji and Yong, 2013). However, direct ERβ activation on CD11c+ myeloid dendritic cells and macrophages did not increase expression of ARG1 and YM-1 markers of this protective phenotype.
A limitation of our study is that bone marrow chimeras are imperfect and can be associated with the microglial pool being a mixed population of resident microglia and those which repopulate from the periphery after irradiation. This could confound interpretation of our negative findings in Fig. 6 where deletion of ERβ in the CD11c+ microglial population did not prevent ERβ-ligand induced disease protection. The protective effect of ERβ-ligand treatment may have remained in wild-type→CKO EAE mice if not all of the CD11c+ microglia were devoid of ERβ due to repopulation. However, data in Fig. 8, where effects of ERβ-ligand treatment on iNOS, T-bet, and IL-10 were found on the CD11c+ myeloid dendritic cell and macrophage population, but not on the CD11c+ microglial population, make an effect of ERβ-ligand treatment acting on repopulating CD11c+ microglial less likely. That said, our findings focusing on CD11c+ cells do not rule out additional actions of ERβ in CD11c− microglia.
When ERβ ligands were first shown to provide neuroprotection during late, but not early, EAE (Tiwari-Woodruff et al., 2007), it suggested that ERβ-ligand conferred protective effects that were unique from other previous EAE treatments, which reduced early EAE as a prelude to reducing late EAE. Translational studies then showed efficacy when ERβ-ligand treatment was started after EAE onset (Kim et al., 1999; Tiwari-Woodruff et al., 2007; Wisdom et al., 2013), together prompting investigations using cell-specific knock outs to assess neuroprotective mechanisms in vivo and laying the foundation for development of next generation oestrogen treatments to maximize ERβ-mediated neuroprotective efficacy, while minimizing ERα-mediated adverse effects, for treatment of neurodegenerative diseases (Nilsson et al., 2011; Paterni et al., 2014; Warner and Gustafsson, 2015). While various ERβ-ligands were protective in EAE, it remained unclear whether in vivo effects could occur directly through binding to ERβ expressed by immune cells. Studies included the classic ERβ-ligand DPN [with an β/α affinity of 70:1 (Meyers et al., 2001; Du et al., 2011)], indazole-Cl [β/α, 107:1 (De Angelis et al., 2005; Moore et al., 2014)], LY3201 [β/α, 19:1 (Richardson et al., 2007; Wu et al., 2013)], and AC-186 [β/α, 830:1 (McFarland et al., 2013; Itoh et al., 2017)]. Also, 5-androstene-3β,17β-diol (β/α, 4:1) (Kuiper et al., 1997) reduced iNOS, IL-1β, IL-6 and IL-23 expression in cultured activated microglia and astrocytes through recruitment of inhibitory transcription factors to inflammatory promotors, and was protective in EAE, with this protection lost in global ERβ knockouts (Saijo et al., 2011). However, CKO studies of ERβ in astrocytes previously (Spence et al., 2013) and in CD11c+ microglia here did not show direct effects on these cells in vivo during EAE, underscoring important differences between in vivo versus in vitro studies of these cells. Here, we show direct effects in vivo of ERβ-ligand treatment on myeloid dendritic cells and macrophages during EAE.
Investigations of neuroprotection induced by oestrogen treatment can inform the development of other neuroprotective treatments. The optimal receptor for ligation during treatment, ERβ, has not only been identified, it has now been selectively deleted in neurons, astrocytes, oligodendrocytes, microglia and peripheral myeloid cells to determine functional effects in vivo during disease. This comprehensive approach has not been applied to any other neuroprotective treatment for any neurological disease. Other putative neuroprotective treatments have used cell-specific conditional knockouts in one, and rarely in two, CNS cell types. The comprehensive approach used with ERβ-ligand treatment permits critical insights herein regarding in vivo mechanisms of neuroprotection by contrasting effects in some cells (myeloid dendritic cells and macrophages and OLCs) with lack of effects in others (neurons, astrocytes, resident microglia). Specifically, the mechanism of neuroprotection during ERβ-ligand treatment in EAE involves a combination of effects on two developmentally different populations: myeloid dendritic cells and macrophages and OLCs. ERβ in myeloid dendritic cells and macrophages is immunomodulatory in the CNS microenvironment, while ERβ in OLCs enhances OPC differentiation and maturation to increase remyelination (Khalaj et al., 2013; Kumar et al., 2013; Lariosa-Willingham et al., 2016). The maturation of oligodendrocytes by ERβ-ligand treatment during EAE cannot occur without immunomodulation of myeloid-derived dendritic cells and macrophages in the CNS. Indeed, when each cell type (CD11c+ or Olig1+) is targeted for ERβ specific deletion independently, neuroprotective treatment effects are lost. Thus, each is necessary, but not sufficient. This emphasizes the importance of developing treatment strategies targeting not only remyelination, but also inhibition of pro-inflammatory bone marrow-derived dendritic cells and macrophages. Targeting ERβ signalling pathways in cells of these two distinct lineages may be ideally suited to achieve neuroprotection in multiple sclerosis and perhaps other neurodegenerative diseases.
Supplementary Material
Acknowledgements
We thank the members of the R.V. laboratory, particularly M. Peng, R. Alejandro-Ramirez, and K. Herrera for technical assistance. We thank Dr Michael Sofroniew in the Department of Neurobiology at UCLA for consultation with conditional knockout mouse generation and validation. We thank UCLA Brain Research Institute Electron Microscopy Core Facility, especially the Core Supervisor Marianne Cilluffo for her consultation and assistance on tissue preparation and electron microscopy. We thank UCLA Center for AIDS Research Humanized Mouse Core, supported by National Institutes of Health (NIH) grant P30AI28697, especially Dr Scott G. Kitchen for his support and technical advices on creating the bone marrow chimeras. We thank the entire staff at the UCLA Flow Cytometry Core Laboratory, supported by National Institutes of Health (NIH) grants P30CA016042 and P30AI028697.
Funding
This work was funded by NIH grants R01NS096748 (to R.V.); National Research Service Award predoctoral Fellowship F31NS096906 and UCLA Laboratory of Neuroendocrinology training fellowship T32HD07228 (to R.Y.K); the Conrad N. Hilton Foundation grant #20150232 (to R.V.); the California Community Foundation #BAPP-15-118094 (to R.V.) and the Tom Sherak MS Hope Foundation.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- CKO
conditional knock out
- βAPP
beta amyloid precursor protein
- DPN
diarylpropionitrile
- EAE
experimental autoimmune encephalomyelitis
- ERα/β
oestrogen receptors α/β
- GSTπ
glutathione-S transferase π
- iNOS
inducible nitrogen oxide synthase
- MHCII
major histocompatibility class II
- NF200
neurofilament 200
- NG2
neural/glial antigen 2
- OLC
oligodendrocyte lineage cells
- OPC
oligodendrocyte precursor cells
- SMI32
neurofilament H non-phosphorylated
References
- Antal MC, Krust A, Chambon P, Mark M. Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERbeta-null mutant. Proc Natl Acad Sci USA 2008; 105: 2433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid dendritic cells presenting endogenous myelin peptides ‘preferentially' polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol 2007; 8: 172–80. [DOI] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA 2016; 113: E1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, et al. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol 2008; 508: 687–710. [DOI] [PubMed] [Google Scholar]
- Clarkson BD, Walker A, Harris MG, Rayasam A, Sandor M, Fabry Z. CCR2-dependent dendritic cell accumulation in the central nervous system during early effector experimental autoimmune encephalomyelitis is essential for effector T cell restimulation in situ and disease progression. J Immunol 2015; 194: 531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis M, Stossi F, Carlson KA, Katzenellenbogen BS, Katzenellenbogen JA. Indazole estrogens: highly selective ligands for the oestrogen receptor beta. J Med Chem 2005; 48: 1132–44. [DOI] [PubMed] [Google Scholar]
- Dogan RN, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF- and iNOS-expressing macrophages and myeloid dendritic cells. J Immunol 2008; 180: 7376–84. [DOI] [PubMed] [Google Scholar]
- Du S, Sandoval F, Trinh P, Umeda E, Voskuhl R. Oestrogen receptor-beta ligand treatment modulates dendritic cells in the target organ during autoimmune demyelinating disease. Eur J Immunol 2011; 41: 140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasor J, Barnett DH, Danes JM, Hess R, Parlow AF, Katzenellenbogen BS. Response-specific and ligand dose-dependent modulation of oestrogen receptor (ER) alpha activity by ERbeta in the uterus. Endocrinology 2003; 144: 3159–66. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330: 841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006; 129(Pt 8): 1953–71. [DOI] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 2005; 11: 328–34. [DOI] [PubMed] [Google Scholar]
- Harris HA, Katzenellenbogen JA, Katzenellenbogen BS. Characterization of the biological roles of the oestrogen receptors, ERalpha and ERbeta, in oestrogen target tissues in vivo through the use of an ERalpha-selective ligand. Endocrinology 2002; 143: 4172–7. [DOI] [PubMed] [Google Scholar]
- Itoh N, Kim R, Peng M, DiFilippo E, Johnsonbaugh H, MacKenzie-Graham A, et al. Bedside to bench to bedside research: oestrogen receptor beta ligand as a candidate neuroprotective treatment for multiple sclerosis. J Neuroimmunol 2017; 304: 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Voskuhl RR. Cell specificity dictates similarities in gene expression in multiple sclerosis, Parkinson's disease, and Alzheimer's disease. PLoS One 2017; 12: e0181349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karman J, Ling C, Sandor M, Fabry Z. Initiation of immune responses in brain is promoted by local dendritic cells. J Immunol 2004; 173: 2353–61. [DOI] [PubMed] [Google Scholar]
- Khalaj AJ, Yoon J, Nakai J, Winchester Z, Moore SM, Yoo T, et al. Estrogen receptor (ER) beta expression in oligodendrocytes is required for attenuation of clinical disease by an ERbeta ligand. Proc Natl Acad Sci USA 2013; 110: 19125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RY, Hoffman AS, Itoh N, Ao Y, Spence R, Sofroniew MV, et al. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J Neuroimmunol 2014; 274: 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Liva SM, Dalal MA, Verity MA, Voskuhl RR. Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology 1999; 52: 1230–8. [DOI] [PubMed] [Google Scholar]
- King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009; 113: 3190–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997; 138: 863–70. [DOI] [PubMed] [Google Scholar]
- Kumar S, Patel R, Moore S, Crawford DK, Suwanna N, Mangiardi M, et al. Estrogen receptor beta ligand therapy activates PI3K/Akt/mTOR signaling in oligodendrocytes and promotes remyelination in a mouse model of multiple sclerosis. Neurobiol Dis 2013; 56: 131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lariosa-Willingham KD, Rosler ES, Tung JS, Dugas JC, Collins TL, Leonoudakis D. A high throughput drug screening assay to identify compounds that promote oligodendrocyte differentiation using acutely dissociated and purified oligodendrocyte precursor cells. BMC Res Notes 2016; 9: 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen C. Results of a 5 years prospective study of estriol succinate treatment in patients with climacteric complaints. Horm Metab Res 1987; 19: 579–84. [DOI] [PubMed] [Google Scholar]
- Lelu K, Laffont S, Delpy L, Paulet PE, Perinat T, Tschanz SA, et al. Estrogen receptor alpha signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J Immunol 2011; 187: 2386–93. [DOI] [PubMed] [Google Scholar]
- Lindberg BS, Johansson ED, Nilsson BA. Plasma levels of nonconjugated oestrone, oestradiol-17beta and oestriol during uncomplicated pregnancy. Acta Obstet Gynecol Scand Suppl 1974; 32: 21–36. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, et al. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 2002; 109: 75–86. [DOI] [PubMed] [Google Scholar]
- Mackenzie-Graham AJ, Rinek GA, Avedisian A, Morales LB, Umeda E, Boulat B, et al. Estrogen treatment prevents gray matter atrophy in experimental autoimmune encephalomyelitis. J Neurosci Res 2012; 90: 1310–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Price DL, Davis CN, Ma JN, Bonhaus DW, Burstein ES, et al. AC-186, a selective nonsteroidal estrogen receptor beta agonist, shows gender specific neuroprotection in a Parkinson's disease rat model. ACS Chem Neurosci 2013; 4: 1249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F, Fancy SP, Shen YA, Niu J, Zhao C, Presley B, et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat Med 2014; 20: 954–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem 2001; 44: 4230–51. [DOI] [PubMed] [Google Scholar]
- Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci 2007; 1103: 179–91. [DOI] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 2013; 16: 1211–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra MK, Yong VW. Myeloid cells - targets of medication in multiple sclerosis. Nat Rev Neurol 2016; 12: 539–51. [DOI] [PubMed] [Google Scholar]
- Moore SM, Khalaj AJ, Kumar S, Winchester Z, Yoon J, Yoo T, et al. Multiple functional therapeutic effects of the estrogen receptor beta agonist indazole-Cl in a mouse model of multiple sclerosis. Proc Natl Acad Sci USA 2014; 111: 18061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales LB, Loo KK, Liu HB, Peterson C, Tiwari-Woodruff S, Voskuhl RR. Treatment with an estrogen receptor alpha ligand is neuroprotective in experimental autoimmune encephalomyelitis. J Neurosci 2006; 26: 6823–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson S, Koehler KF, Gustafsson JA. Development of subtype-selective oestrogen receptor-based therapeutics. Nat Rev Drug Discov 2011; 10: 778–92. [DOI] [PubMed] [Google Scholar]
- Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol 2004; 172: 1426–36. [DOI] [PubMed] [Google Scholar]
- Palaszynski KM, Liu H, Loo KK, Voskuhl RR. Estriol treatment ameliorates disease in males with experimental autoimmune encephalomyelitis: implications for multiple sclerosis. J Neuroimmunol 2004; 149: 84–9. [DOI] [PubMed] [Google Scholar]
- Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERalpha) and beta (ERbeta): subtype-selective ligands and clinical potential. Steroids 2014; 90: 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodinger C, Bunse J, Kruger M, Schiefenhovel F, Brandt C, Laman JD, et al. CD11c-expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol 2011; 121: 445–58. [DOI] [PubMed] [Google Scholar]
- Prokai L, Nguyen V, Szarka S, Garg P, Sabnis G, Bimonte-Nelson HA, et al. The prodrug DHED selectively delivers 17beta-estradiol to the brain for treating estrogen-responsive disorders. Sci Transl Med 2015; 7: 297ra113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawji KS, Yong VW. The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin Dev Immunol 2013; 2013: 948976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson TI, Dodge JA, Durst GL, Pfeifer LA, Shah J, Wang Y, et al. Benzopyrans as selective estrogen receptor beta agonists (SERBAs). Part 3: synthesis of cyclopentanone and cyclohexanone intermediates for C-ring modification. Bioorg Med Chem Lett 2007; 17: 4824–8. [DOI] [PubMed] [Google Scholar]
- Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell 2011; 145: 584–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, Matzelle DD, Ray SK, Banik NL. Physiological low dose of estrogen-protected neurons in experimental spinal cord injury. Ann N Y Acad Sci 2010; 1199: 86–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen FW, Saga Y, Litman G, Freeman G, Tung JS, Cantor H, et al. Cloning of Ly-5 cDNA. Proc Natl Acad Sci USA 1985; 82: 7360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicotte NL, Liva SM, Klutch R, Pfeiffer P, Bouvier S, Odesa S, et al. Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann Neurol 2002; 52: 421–8. [DOI] [PubMed] [Google Scholar]
- Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ, et al. Neuroprotection mediated through estrogen receptor-{alpha} in astrocytes. Proc Natl Acad Sci USA 2011; 108: 8867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence RD, Voskuhl RR. Neuroprotective effects of estrogens and androgens in CNS inflammation and neurodegeneration. Front Neuroendocrinol 2012; 33: 105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence RD, Wisdom AJ, Cao Y, Hill HM, Mongerson CR, Stapornkul B, et al. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERalpha signaling on astrocytes but not through ERbeta signaling on astrocytes or neurons. J Neurosci 2013; 33: 10924–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Friedman BA, Larson JL, Lauffer BE, Goldstein LD, Appling LL, et al. Untangling the brain's neuroinflammatory and neurodegenerative transcriptional responses. Nat Commun 2016; 7: 11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 1996; 85: 299–302. [DOI] [PubMed] [Google Scholar]
- Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity 2007; 26: 629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Brown CM, Wise PM. Neuroprotective effects of estrogens following ischemic stroke. Front Neuroendocrinol 2009; 30: 201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Manabe A, Okada M, Kurioka H, Kanasaki H, Miyazaki K. Efficacy and safety of oral estriol for managing postmenopausal symptoms. Maturitas 2000; 34: 169–77. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff S, Morales LB, Lee R, Voskuhl RR. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)alpha and ERbeta ligand treatment. Proc Natl Acad Sci USA 2007; 104: 14813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulchinsky D, Hobel CJ, Yeager E, Marshall JR. Plasma estrone, estradiol, estriol, progesterone, and 17-hydroxyprogesterone in human pregnancy. I. Normal pregnancy. Am J Obstet Gynecol 1972; 112: 1095–100. [DOI] [PubMed] [Google Scholar]
- Uchoa MF, Moser VA, Pike CJ. Interactions between inflammation, sex steroids, and Alzheimer's disease risk factors. Front Neuroendocrinol 2016; 43: 60–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein ID, Vinet J, Brouwer N, Brendecke S, Biagini G, Biber K, et al. In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia 2014; 62: 1724–35. [DOI] [PubMed] [Google Scholar]
- Voskuhl RR, Wang H, Wu TC, Sicotte NL, Nakamura K, Kurth F, et al. Estriol combined with glatiramer acetate for women with relapsing-remitting multiple sclerosis: a randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2016; 15: 35–46. [DOI] [PubMed] [Google Scholar]
- Warner M, Gustafsson JA. Estrogen receptor beta and Liver X receptor beta: biology and therapeutic potential in CNS diseases. Mol Psychiatry 2015; 20: 18–22. [DOI] [PubMed] [Google Scholar]
- Wisdom AJ, Cao Y, Itoh N, Spence RD, Voskuhl RR. Estrogen receptor-beta ligand treatment after disease onset is neuroprotective in the multiple sclerosis model. J Neurosci Res 2013; 91: 901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk A, Cedile O, Jensen KN, Jasson A, Mony JT, Khorooshi R, et al. Pathologic and protective roles for microglial subsets and bone marrow- and blood-derived myeloid cells in central nervous system inflammation. Front Immunol 2015; 6: 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk A, Lobner M, Cedile O, Owens T. Comparison of microglia and infiltrating CD11c(+) cells as antigen presenting cells for T cell proliferation and cytokine response. J Neuroinflammation 2014; 11: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WF, Tan XJ, Dai YB, Krishnan V, Warner M, Gustafsson JA. Targeting estrogen receptor beta in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2013; 110: 3543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehn MO, Avedisian AA, Dervin SM, O'Dell TJ, Voskuhl RR. Estriol preserves synaptic transmission in the hippocampus during autoimmune demyelinating disease. Lab Invest 2012; 92: 1234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.