

Abstract
Background
Selective cyclooxygenase-2 inhibitors and conventional non-selective non-steroidal anti-inflammatory drugs (nsNSAIDs) have been associated with adverse cardiovascular (CV) effects. We compared the CV safety of switching to celecoxib vs. continuing nsNSAID therapy in a European setting.
Method
Patients aged 60 years and over with osteoarthritis or rheumatoid arthritis, free from established CV disease and taking chronic prescribed nsNSAIDs, were randomized to switch to celecoxib or to continue their previous nsNSAID. The primary endpoint was hospitalization for non-fatal myocardial infarction or other biomarker positive acute coronary syndrome, non-fatal stroke or CV death analysed using a Cox model with a pre-specified non-inferiority limit of 1.4 for the hazard ratio (HR).
Results
In total, 7297 participants were randomized. During a median 3-year follow-up, fewer subjects than expected developed an on-treatment (OT) primary CV event and the rate was similar for celecoxib, 0.95 per 100 patient-years, and nsNSAIDs, 0.86 per 100 patient-years (HR = 1.12, 95% confidence interval, 0.81–1.55; P = 0.50). Comparable intention-to-treat (ITT) rates were 1.14 per 100 patient-years with celecoxib and 1.10 per 100 patient-years with nsNSAIDs (HR = 1.04; 95% confidence interval, 0.81–1.33; P = 0.75). Pre-specified non-inferiority was achieved in the ITT analysis. The upper bound of the 95% confidence limit for the absolute increase in OT risk associated with celecoxib treatment was two primary events per 1000 patient-years exposure. There were only 15 adjudicated secondary upper gastrointestinal complication endpoints (0.078/100 patient-years on celecoxib vs. 0.053 on nsNSAIDs OT, 0.078 vs. 0.053 ITT). More gastrointestinal serious adverse reactions and haematological adverse reactions were reported on nsNSAIDs than celecoxib, but more patients withdrew from celecoxib than nsNSAIDs (50.9% patients vs. 30.2%; P < 0.0001).
Interpretation
In subjects 60 years and over, free from CV disease and taking prescribed chronic nsNSAIDs, CV events were infrequent and similar on celecoxib and nsNSAIDs. There was no advantage of a strategy of switching prescribed nsNSAIDs to prescribed celecoxib. This study excluded an increased risk of the primary endpoint of more than two events per 1000 patient-years associated with switching to prescribed celecoxib.
Clinical Trial Registration
https://clinicaltrials.gov/show/NCT00447759; Unique identifier: NCT00447759.
Keywords: NSAIDs, Celecoxib, Cardiovascular, Arthritis
Introduction
The safety of non-steroidal anti-inflammatory drugs (NSAIDs) has been the topic of much debate. Hospitalization for NSAID-associated upper gastrointestinal (GI) tract ulceration was previously a common serious adverse event and selective cyclo-oxygenase 2 (COX-2) inhibitors gained popularity because they caused a lower incidence of these complications compared with non-selective NSAIDs (nsNSAIDs).1 However, COX-2 inhibitors were associated with higher cardiovascular (CV) risks than nsNSAIDs.2–4 This led the US Food and Drug Administration to strengthen warnings on all NSAIDs5 and the European Medicines Agency (EMA) to contraindicate COX-2 selective drugs in subjects with established CV disease.6,7
We conducted the Standard care vs. Celecoxib Outcome Trial (SCOT), which randomized patients free from established CV disease to switch to celecoxib or to remain on their prescribed nsNSAID, to clarify the risk–benefit balance of switching to celecoxib.
Methods and analysis
Trial design
The SCOT trial used a Prospective, Randomized, Open label, Blinded Endpoint evaluation (PROBE) design8 to compare the CV and GI safety of continuing prescribed nsNSAID therapy vs. switching to prescribed celecoxib in individuals with osteoarthritis or rheumatoid arthritis. The study was unusual in incorporating many pragmatic features: there were no study visits after randomization; study treatments were prescribed by primary care physicians and dispensed in community pharmacies; blood samples to assess suitability for inclusion were analysed in health service laboratories; follow-up used electronic medical records of hospitalizations and deaths; and there were no special study-specific procedures to ensure adherence to study medication. The aim was to mimic normal clinical practice and normal patient behaviour. The protocol has been published.9
The trial was mandated by the EMA with a requirement that it be conducted in at least two member states. It was conducted in the UK, Denmark, and the Netherlands and included patients aged 60 years or over, free from significant CV disease (a history of coronary or cerebrovascular disease or New York Heart Association class III or IV heart failure). The trial inclusion and exclusion criteria are listed in the Supplementary material online, Tables S1 and S2, respectively.
Potential participants were identified in primary care. Those who responded to an invitation letter to participate, gave consent, and satisfied inclusion and exclusion criteria were randomly assigned to switch to prescribed celecoxib (the celecoxib group) or to continue their usual prescribed nsNSAID (the nsNSAID group) using a central randomization system. All medication was either free to patients or fully reimbursed (see Supplementary material online, Supplementary methods). Subjects could optionally provide a sample of serum and blood for later analyses of lipids and uric acid levels.
Prescribed nsNSAIDs with an estimated frequency of usage of >12% (ibuprofen and diclofenac) were assigned to unique strata and other NSAIDs were pooled into a single stratum for the purpose of randomization. Randomization was also stratified by osteoarthritis or rheumatoid arthritis status. Within strata, treatment groups were allocated randomly in equal numbers in permuted blocks of 4.
The trial treatments were prescribed at approved doses and adjusted as clinically indicated. If pain-relief was inadequate, non-NSAID analgesics could be added and antacid or ulcer healing therapy was prescribed as required. All prescriptions in the UK were provided free of charge and in Denmark and The Netherlands subjects were reimbursed any prescription costs.
The primary endpoint was the composite of hospitalization for non-fatal myocardial infarction or other biomarker positive acute coronary syndrome, non-fatal stroke or CV death.
Secondary outcomes were hospitalization or death for upper GI ulcer complications (bleeding, perforation, or obstruction); hospitalized upper GI ulcer complications or primary outcome; hospitalization for heart failure; hospitalization for heart failure or primary outcome; death from any cause; new or worsening renal failure; hospitalization for critical limb ischemia; hospitalization for pulmonary embolism.
The principal method of follow-up in the SCOT was by record-linkage10 (see Supplementary material online, Supplementary methods). Treatment-related adverse events and all serious adverse events reported by study sites were recorded and reconciled with the record-linkage data. For potential endpoints, support documentation was retrieved from hospital records, de-identified and reviewed by CV or GI endpoint committees.
Statistical methods
Baseline characteristics were compared using two-sample t-tests (or Mann–Whitney tests) and χ2 (or Fisher’s exact) tests, as appropriate. Annual incidence rate per 100 patient-years at risk is reported and Cox proportional hazards models were used to analyse time-to-first-event data, including the baseline NSAID and the osteoarthritis/rheumatoid arthritis strata and the randomized treatment group as covariates. Where the number of events was <30, the Cox model was replaced by an exact Poisson regression model, adjusting for the same variables with the logarithm of time as an offset. Statistical significance was based on the Wald statistic, and two-sided 95% confidence intervals (CIs) for the estimated hazard ratio (HR) (or rate ratio for the Poisson model). Absolute treatment effects were estimated from additive Poisson regression models adjusted for over-dispersion.
The main non-inferiority analysis for the primary and secondary outcomes used on-treatment comparisons. The final study design (see Supplementary material online, Supplementary methods) had a non-inferiority limit HR of 1.4 for the primary CV endpoint requiring 277 first primary endpoints for 80% power. On-treatment analyses censored subjects after the first of, discontinuation from the randomized treatment group (for the nsNSAID group this involved withdrawal from any nsNSAID; see Supplementary material online, Supplementary methods), death, withdrawal of consent, or end-of-study date. These analyses were supported by a modified intention-to-treat (ITT) analysis censoring on the first of death, withdrawal of consent for follow-up, or end-of-study date.
Treatment-by-subgroup interactions were tested in Cox models (or by exact Poisson regression analysis) incorporating subgroup-by-treatment interactions. Subgroups where the SAS programs of tests for interaction or for estimation of risk ratios failed are not reported. Subgroups with inadequate events are not reported. Time-to-event curves were estimated by the Kaplan–Meier method.
Role of the funding source
SCOT was an academic study, funded through an Investigator Initiated Research Grant, by Pfizer. The University of Dundee was the study sponsor and monitored and quality assured the study. The protocol was developed and owned by the Trial Steering Committee. The sponsor and the Trial Steering Committee were wholly responsible for data collection, trial management, analysis and interpretation of data, writing of the report and submission of the report for publication and Pfizer played no role in any of these activities. Study data were collected, managed, and analysed at the Robertson Centre for Biostatistics, Glasgow Clinical Trials Unit, University of Glasgow.
Participants provided written informed consent. The trial was approved by the UK Multi-Centre Research Ethics Committee (reference number: 2006-005735-24) and by relevant authorities in Denmark and the Netherlands. Record linkage was approved by information governance committees in the participating countries. It was registered on ClinicalTrials.gov (reference number: NCT00447759).
Results
We screened and gained consent from 8872 subjects between 29 January 2008 and 27 March 2013 and randomized 7297 across 9 trial centres and 706 primary care practices (see Supplementary material online, Figure S1). Thirty-three per cent of subjects judged to be eligible by their usual family physician and invited to participate actually agreed to be screened and 14% randomized into the study. The median ITT follow-up for the primary outcome was 3.0 years (maximum 6.3 years, total 22 600 person-years). Table 1 shows the baseline characteristics. A total of 39% of participants were taking diclofenac, 32% ibuprofen, and 94% had osteoarthritis. The mean age at entry was 68 years, 59% were female, 15.5% current smokers, 8% had a history of diabetes, and 44% a history of hypertension. Mean systolic and diastolic blood pressures were 141 and 78 mmHg, respectively, and the mean body mass index was 29.7 kg/m2.
Table 1.
Characteristics of the patients at baseline
| Characteristic | Celecoxib | nsNSAID |
|---|---|---|
| (N = 3647) | (N = 3650) | |
| Male sex, no. (%) | 1527 (41.9) | 1432 (39.2) |
| Current smoker, no. (%) | 547 (15.0) | 583 (16.0) |
| Ethnic group (White), no. (%) | 3638 (99.8) | 3636 (99.6) |
| Age, years | 68.6 ± 6.2 | 68.2 ± 6.1 |
| Systolic blood pressurea, mmHg | 140.5 ± 17.8 | 140.8 ± 17.9 |
| Diastolic blood pressurea, mmHg | 77.6 ± 10.7 | 77.7 ± 10.7 |
| Heart ratea beats/min | 70.8 ± 11.3 | 70.4 ± 11.1 |
| Body-mass indexb,c | 29.6 ± 5.7 | 29.8 ± 5.6 |
| Waist/Hip ratiod,e | 0.91 ± 0.1 | 0.91 ± 0.1 |
| Histories of, no. (%) | ||
| Heart failure (NYHA I/II) | 4 (0.1) | 10 (0.3) |
| Diabetes | 301 (8.3) | 285 (7.8) |
| High blood pressure | 1634 (44.8) | 1607 (44.0) |
| Raised cholesterol | 1268 (34.8) | 1211 (33.2) |
| Renal disease | 133 (3.6) | 125 (3.4) |
| Peptic ulcer | 254 (7.0) | 239 (6.5) |
| Upper GI hospitalization | 168 (4.6) | 180 (4.9) |
| Upper GI surgery | 88 (2.4) | 96 (2.6) |
| Gout | 305 (8.4) | 300 (8.2) |
| Helicobacter pylori eradication | 170 (4.7) | 154 (4.2) |
| Asthma | 365 (10.0) | 362 (9.9) |
| Chronic obstructive pulmonary disease | 158 (4.3) | 159 (4.4) |
| Randomization strataf, no. (%) | ||
| Arthritis indication—Osteoarthritis | 3421 (93.8) | 3422 (93.8) |
| NSAID strata (Ibuprofen) | 1149 (31.5) | 1153 (31.6) |
| NSAID strata (Diclofenac) | 1413 (38.7) | 1412 (38.7) |
| Baseline medication, no. (%) | ||
| Statins | 774 (21.1) | 748 (20.5) |
| Aspirin | 421 (11.5) | 433 (11.9) |
| Ulcer healing drug | 1401 (38.4) | 1357 (37.2) |
| Total cholesterolg, mg/dL | 202.3 ± 42.5 | 203.4 ± 42.5 |
| HDL cholesterolg, mg/dL | 55.6 ± 15.4 | 55.6 ± 15.4 |
| Triglyceridesh (IQR), mg/dL | 143.4 (104.4–204.4) | 145.1 (105.3–206.2) |
| Uric acidi, mg/dL | 5.5 ± 1.5 | 5.5 ± 1.5 |
Values are mean ± SD. There were no statistically significant differences between the two groups except for age (P = 0.006) and sex (P = 0.02), with values not adjusted for multiple testing. Percentages may not total 100 because of rounding.
IQR, denotes interquartile range.
aData on systolic blood pressure, diastolic blood pressure, and heart rate were not available for one participant in the celecoxib group and three participants in the nsNSAID group.
bData on body-mass index were not available for two participants in the celecoxib group and one participant in the nsNSAID group.
cThe body-mass index is the weight in kilograms divided by the square of the height in meters.
dData on waist/hip ratio were not available for four participants in the celecoxib group and one participant in the nsNSAID group.
eThe waist/hip ratio is the waist circumference in centimetres divided by the hip circumference in centimetres.
fNo P-values were calculated for the arthritis indication or NSAID strata as randomization was stratified according to these factors.
gData on total cholesterol and HDL cholesterol were not available for 386 participants in the celecoxib group and 385 participants in the nsNSAID group.
hData on triglycerides were not available for 385 participants in the celecoxib group and 385 participants in the nsNSAID group.
iData on uric acid were not available for 323 participants in the celecoxib group and 323 participants in the nsNSAID group.
In the prescribed celecoxib group, 50.9% withdrew from the randomized therapy compared with 30.2% not continuing with any prescribed nsNSAID therapy (P < 0.0001, Figure 1). A total of 6.7% withdrew informed consent in the celecoxib group compared with 3.9% in the nsNSAID group (P < 0.0001). The reasons recorded for withdrawal from the drug a participant was initially randomized to are listed in Table 2.
Figure 1.
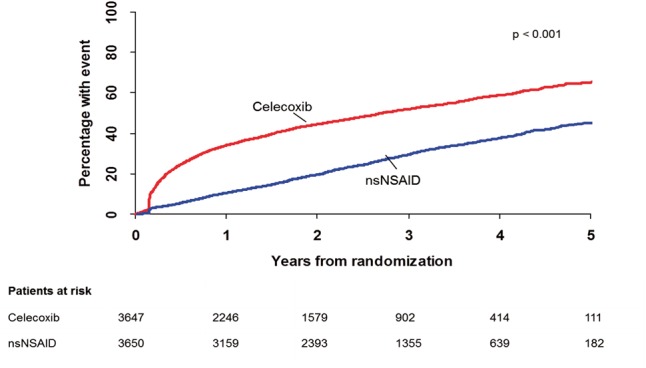
Withdrawal from randomized treatment group. For the non-selective non-steroidal anti-inflammatory drugs group this means complete withdrawal from any non-selective non-steroidal anti-inflammatory drug. P-values reported are for superiority.
Table 2.
Recorded reasons for withdrawal from the specific drug to which the participant was initially randomized
| Parameter | Celecoxib | nsNSAID |
|---|---|---|
| (N = 3647) | (N = 3650) | |
| Withdrawal from randomized medication, no. (%) | 1759 (48.2) | 1150 (31.5) |
| Reason for first withdrawal from randomized medication as noted at follow-up visits, no. (%) | ||
| Adverse event | 304 (17.3) | 162 (14.1) |
| Serious adverse event | 95 (5.4) | 71 (6.2) |
| Onset of symptoms that limit tolerability | 144 (8.2) | 42 (3.7) |
| Lack of efficacy | 409 (23.3) | 111 (9.7) |
| Switch to non-allocated therapy or discontinuation | 242 (13.8) | 296 (25.7) |
| Doctor’s recommendation (non-AE) | 172 (9.8) | 218 (19.0) |
| Protocol violation | 7 (0.4) | 4 (0.3) |
| Patient’s request | 220 (12.5) | 85 (7.4) |
| Other | 166 (9.4) | 161 (14.0) |
The mean doses of NSAIDs taken per day in Scotland where full data were available were 169.8 (SD 80.6) mg for celecoxib, 79.4 (38.3) mg for diclofenac, 675.9 (345.9) mg for ibuprofen, and 581.0 (263.4) mg for naproxen (see Supplementary material online, Table S7 for these and other NSAIDs).
Amongst patients randomized to celecoxib those who were taking diclofenac prior to study entry were more likely to withdraw from celecoxib than patients taking ibuprofen at baseline [HR (95% CI) of withdrawing from celecoxib by ibuprofen stratum vs. diclofenac stratum = 0.84 (0.75, 0.95)].
A total of 278 primary endpoints occurred in 249 (1.12 per 100 patient-years) participants in the ITT analysis, 146 (0.90 per 100-patient years) of these during the on-treatment period (Table 3 and Figure 2). In the ITT analysis, 125 participants (1.14 per 100 patient-years) in the prescribed celecoxib group had a primary outcome compared with 124 (1.10 per 100 patient-years) in the prescribed nsNSAID group (HR 1.04; 95% CI, 0.81–1.33; P = 0.75). Statistically significant non-inferiority was demonstrated in the ITT analysis. In the on-treatment analysis, 65 participants (0.95 per 100 patient-years) in the celecoxib group had a primary outcome compared with 81 (0.86 per 100 patient-years) in the nsNSAID group (HR 1.12; 95% CI, 0.81–1.55; P = 0.50) (see Supplementary material online, Figure S3). Results for the primary outcome for the initial nsNSAID strata are shown in Figure 3 and corresponding results for other pre-specified subgroups in the Supplementary material online, Figure S2. There were no statistically significant subgroup interactions. Absolute differences in the rates (celecoxib − nsNSAID group) of the primary endpoint were 0.8, 95% CI (−0.5, 2.0) events per 1000 patient-years for the on-treatment analysis and 0.4, 95% CI (−1.1, 1.8) events per 1000 patient-years for the ITT analysis.
Table 3.
Treatment comparisons (celecoxib vs. non-selective non-steroidal anti-inflammatory drugs) for the primary outcome (and its subcomponents) and all secondary endpoints
|
On-treatment analysis
|
Intention-to-treat analysis
|
|||||
|---|---|---|---|---|---|---|
| Celecoxib | nsNSAID | Celecoxib | nsNSAID | |||
| Numbers of subjects | 3647 | 3650 | 3647 | 3650 | ||
| Follow-up, years (primary outcome) | 6842 | 9460 | 10 993 | 11 318 | ||
| n (n/100PY) | n (n/100PY) | HR (95% CI); P | n (n/100PY) | n (n/100PY) | HR (95% CI); P | |
| Primary endpoint | 65 (0.95) | 81 (0.86) | 1.12 (0.81, 1.55); 0.50 | 125 (1.14) | 124 (1.10) | 1.04 (0.81, 1.33); 0.75 |
| Hospitalization for non-fatal MI | 38 (0.56) | 40 (0.42) | 1.34 (0.86, 2.09); 0.20 | 70 (0.63) | 56 (0.49) | 1.29 (0.91, 1.84); 0.15 |
| Non-fatal stroke | 16 (0.23) | 25 (0.26) | 0.89 (0.47, 1.67); 0.71 | 31 (0.28) | 36 (0.32) | 0.89 (0.55, 1.44); 0.63 |
| CV death | 15 (0.22) | 17 (0.18) | 1.22 (0.61, 2.46); 0.57 | 32 (0.29) | 39 (0.34) | 0.85 (0.53, 1.35); 0.49 |
| Biomarker positive ACS | 0 (0) | 1 (0.01) | — | 0 (0) | 1 (0.01) | — |
| Secondary endpoints | ||||||
| (a) Hospitalization or death for upper GI ulcer complications | 7 (0.10) | 5 (0.05) | 1.96 (0.54, 7.84); 0.38 | 10 (0.09) | 5 (0.04) | 2.08 (0.65, 7.74); 0.27 |
| (b) Secondary endpoint (a) or Primary endpoint | 72 (1.05) | 86 (0.91) | 1.16 (0.85, 1.59); 0.34 | 132 (1.20) | 129 (1.14) | 1.06 (0.83, 1.35); 0.65 |
| (c) Hospitalization for heart failure | 7 (0.10) | 10 (0.11) | 0.96 (0.31, 2.78); 1.00 | 11 (0.10) | 15 (0.13) | 0.76 (0.31, 1.76); 0.61 |
| (d) Secondary endpoint (c) or Primary endpoint | 70 (1.02) | 86 (0.91) | 1.14 (0.83, 1.56); 0.43 | 130 (1.18) | 132 (1.17) | 1.02 (0.80, 1.29); 0.90 |
| (e) All-cause mortality | 35 (0.51) | 41 (0.43) | 1.20 (0.76, 1.88); 0.43 | 99 (0.89) | 111 (0.97) | 0.92 (0.70, 1.21); 0.56 |
| (f) Hospitalization for new or worsening renal failure | 4 (0.06) | 3 (0.03) | 1.83 (0.31, 12.49); 0.67 | 7 (0.06) | 8 (0.07) | 0.90 (0.28, 2.83); 1.00 |
Figure 2.

Primary composite endpoint: (A) on-treatment and (B) intention-to-treat analyses. All-cause mortality: (C) on-treatment and (D) intention-to-treat analyses. P-values reported are for superiority.
Figure 3.

Forest plot for primary endpoint by subgroups of baseline non-steroidal anti-inflammatory drug medication use for the on-treatment and the intention-to-treat analyses.
Results by arthritis sub-type are given in the Supplementary mate-rial online, Table S6.
Results for secondary outcomes are reported in Table 3. There were no statistically significant differences between the randomized treatment groups for any of the secondary outcomes. Ulcer-related upper GI complications were uncommon and for the on-treatment analysis there were only seven patients with events in the prescribed celecoxib group and five in the prescribed nsNSAID group (P = 0.38), and for the ITT analysis, 10 and 5, respectively (P = 0.27).
There were 35 deaths in the celecoxib group and 41 in subjects prescribed nsNSAIDs in the on-treatment analysis (HR 1.20; 95% CI, 0.76–1.88; P = 0.43), and 99 deaths in the celecoxib group compared with 111 in the nsNSAIDs group by ITT analysis (HR 0.92; 95% CI, 0.70–1.21; P = 0.56). There were no statistically significant differences in cause-specific deaths [CV causes (33.3% celecoxib, 35.3% nsNSAIDs), neoplasia (39.2% and 29.3%), and non-malignant respiratory causes (14.7% vs. 12.9%)] with only two attributed to upper GI bleeding (both on celecoxib).
Serious and non-serious adverse events are tabulated by the Medical Dictionary for Regulatory Activities11 system organ class in the Supplementary material online, Tables S3–S5. There were more non-serious adverse reactions (ARs) attributed to trial treatment in the prescribed celecoxib group: 804 (22%) participants with events vs. 586 (16.1%) in the prescribed nsNSAID group (P < 0.001); however in an open study, attribution to study drug may be prone to bias. Overall, more GI ARs were reported on celecoxib than nsNSAIDs (10.56% vs. 9.12%). There was no statistically significant difference in serious ARs: 190 (5.2%) participants with events in the celecoxib group vs. 213 (5.8%) in the nsNSAID group. However, there were more GI serious ARs on nsNSAIDs than celecoxib (1.81% vs. 1.04%, P = 0.007) with 10 vs. 2 reports of rectal haemorrhage and 13 vs. 3 of gastritis, respectively. Haematological ARs were reported in more nsNSAID than celecoxib patients (1.34% vs. 0.66%) largely attributable to more patients with anaemia or iron deficiency anaemia (1.34% vs. 0.58%). Some (non-serious) ARs appeared to be more common on celecoxib than nsNSAIDs such as dermatological, central nervous system (CNS), musculoskeletal, and GI.
Discussion
In this study, we collected data to compare the extent to which switching prescribed nsNSAIDs to prescribed celecoxib affected the risk of CV, GI, and other adverse outcomes. We found low CV and upper GI adverse event rates with no statistically significant difference between prescribed nsNSAIDs and those switched to prescribed celecoxib for these outcomes or for any other pre-specified secondary outcome. Only 34% of deaths were CV and only two were due to GI haemorrhage and there was no evidence of a treatment group difference in death rates. Results for the primary outcome were consistent across subgroups. The rates of serious adverse events were similar in both groups. The pre-specified non-inferiority limit of 1.4 for the HR for the primary outcome was achieved only using the intention-to treat-analysis. Despite this, because of the low event rate we were able to exclude an increased risk of more than two primary outcomes per 1000 patient-years in on-treatment analysis and 1.8 events per 1000 patient-years by ITT analysis.
The original power calculation was for 13 682 patients to be followed up for 2 years or 27 364 patient-years of exposure to generate 611 primary endpoints with an event rate of about 2% per year. Fewer subjects than expected signed up to take part in the study so to compensate for this we followed up those randomized for much longer. In fact, 7297 subjects were followed for an average of 3.2 years giving 23 670 patient-years of exposure (87% of planned). The adjudicated primary CV event rate was substantially lower than expected at (observed 0.9% expected > 2%).
Had the study recruited all 13 682 subjects as envisaged, the original non-inferiority limit of 1.3 would have required an average of 5 years follow-up rather than the 2 years envisaged. With 7297 subjects, 611 endpoint would have taken 9.4 years of average follow-up. The steering committee was of the opinion that abandoning the trial in the face of these findings would have been nihilistic and unethical and that adjusting the non-inferiority limit to 1.4 was the more responsible option.
The low event rates for both CV and GI endpoints are in themselves of some importance. The trial participants were older subjects with typical CV risk factors taking chronic NSAID therapy but the low observed CV event rates make it unlikely that there could be substantial attributable NSAID toxicity either from nsNSAIDs or celecoxib.
Overall withdrawal rates in the trial were relatively high, reflecting the length of our study but these were actually similar to those seen at comparable time points in the shorter Celecoxib vs. Omeprazole and Diclofenac in patients with Osteoarthritis and Rheumatoid arthritis (CONDOR)12 and Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET) studies.13
We found a higher withdrawal rate from those switched to prescribed celecoxib compared with prescribed nsNSAIDs. It is important to note that whilst the trial was open, the steering committee remained blinded to this differential drop-out rate until after database lock. The dominant reason recorded for the higher withdrawal in those allocated to switch to celecoxib was lack of efficacy. We found that those patients who were prescribed diclofenac prior to randomization were more likely to discontinue celecoxib after randomization than those prescribed ibuprofen prior to randomization. Despite studies suggesting that celecoxib is of equivalent effectiveness as diclofenac14 and that nsNSAIDs for knee arthritis are similarly effective,15 this study provides some indirect evidence that diclofenac may be more effective than ibuprofen. A recent network meta-analysis supports this finding.16 Other reasons for celecoxib discontinuation were poor tolerability and adverse effects suggesting that celecoxib was not as well tolerated as nsNSAIDs.
Adverse reactions attributed to celecoxib were more common in some categories, such as dermatological, CNS, musculoskeletal, and GI. This likely reflects, at least in part, exposure to a new drug compared with continuing one that subjects had found to suit them. During the trial there was significant public debate about COX-2 inhibitor safety which may have dissuaded primary care physicians from up-titrating celecoxib and influenced overall withdrawal rates. In addition, EMA warnings about the CV risks of diclofenac resulted in primary care physicians being unhappy to continue to prescribe diclofenac,17 resulting in a protocol amendment that allowed patients in the nsNSAID group to switch to other prescribed nsNSAIDs.
There have been a number of meta-analyses describing the CV risk related to NSAIDs.3,18 Studies of celecoxib have been observational or have examined CV toxicity that occurred in randomized efficacy trials of short duration.19,20 Network meta-analyses have considered selective COX-2 inhibitors as a group and included trials with heterogeneous risk groups and included trials of COX-2 agents with doses much higher than approved for arthritis.3 In contrast, the biggest observational study of NSAID toxicity found celecoxib to have one of the lowest CV event rates when used in routine clinical care.21 In Europe, selective COX-2 inhibitors may only be prescribed to subjects with no significant CV disease so a study in this low-risk population with celecoxib as prescribed in normal care was required.
We are confident that the lower than expected CV and GI event rates in our study are robust. The use of record-linkage to registries of hospitalizations and deaths maximized event detection and we had a low threshold for sending events for adjudication.
Falling event rates have been reported for coronary disease across Europe22 and the USA23 and the low event rates we observed were similar to those seen in primary prevention studies.24
At baseline, only about 38% of subjects took concurrent ulcer healing drugs but during the study the UK National Institute for Health and Care Excellence published two guidelines that advocated the co-prescription of proton pump inhibitors with chronic NSAID therapy for osteoarthritis.25,26 This may account for the very low rate of ulcer-related complications.
A strength of SCOT is that it was a randomized study and had a number of pragmatic features. Follow-up was primarily by record-linkage and NSAID therapy was evaluated in a normal primary care setting. SCOT is thus likely to have good external validity as it reflects outcomes that occur in usual care. The low CV event rate resulted in reduced power to establish non-inferiority in the on-treatment analysis than we had anticipated at the design stage, but also supports the view that any enhancement of CV risk with celecoxib or nsNSAID is modest. Increasing the size of the trial once the low event rate had become apparent was not feasible. The differential discontinuation rate that was not known until investigators were un-blinded further affected the on-treatment analyses. The reasons for this differential withdrawal are complex and associated with both patient and prescriber preference, permitted within the ethical constraints of the study, on a background of adverse publicity about NSAIDs in general at the time the study was being conducted. We also found lower rates of NSAID-associated GI complications than previous studies1 possibly reflecting the use of relatively low doses in this setting and the increasing co-prescription of anti-ulcer drugs. Our results reduce the rationale for switching patients established on nsNSAIDs to celecoxib, particularly in view of the high withdrawal rate of patients undergoing this strategy.
SCOT studied the clinical use of NSAIDs in standard (or usual) care in a primary care setting. The results of SCOT are thus externally valid and highly likely to reflect the real world toxicity and use of NSAIDs. In order to achieve such externally valid results, SCOT inevitably had to trade internal validity as might be obtained from a double-blind study where subjects were given high doses of NSAIDs. Despite being an open study, the SCOT investigators were blinded to the results of the study until after database lock. The fact that there was a differential drop out between celecoxib and standard NSAIDs is an externally valid finding of this study. This indicates that subjects were less likely to stay on celecoxib after the switch. This is important information and whilst it did result in the non-inferiority margin being non-significant, it did reflect what is likely to happen in clinical care. The ITT analysis, which looked at events based only on randomized intention, was non-inferior. In a pragmatic trial of safety such a result is not unimportant. The strategy of switching was non-inferior to staying on therapy in the trial as a whole. Although the purist might regard a per-protocol analysis the gold standard for safety, a pragmatic study does not force a treatment protocol so per-protocol has less meaning. This is why we called our analysis ‘on-treatment’. From a philosophical standpoint, an ITT analysis might be argued to be the only sensible analysis for a pragmatic trial so we should not denigrate the importance of this finding.
We believe that the SCOT study provides important information on the safety of celecoxib vs. nsNSAIDs in clinical practice in subjects free from established CV. The low event rates seen in this trial suggest that the attributable risk of nsNSAID or celecoxib therapy is likely to be acceptably small.
Summary
In individuals with arthritis and without known CV disease, CV event rates were low and serious ulcer-related complication rates were very low, and neither outcome differed significantly between nsNSAIDs and celecoxib. This study could exclude an increase in the primary CV event rate of more than two events per 1000 patient-years associated with switching to prescribed celecoxib. However, a strategy of switching prescribed nsNSAIDs to prescribed celecoxib in the setting of primary care had no advantage.
Authors’ contributions
The original study protocol was written by T.M., I.F., C.H., and E.F. with contributions from L.W.; all authors contributed to the final study design and were co-investigators and all authors participated in the interpretation of the study results, critical review of the manuscript, and approval of the final version; the following authors also contributed to patient recruitment: T.M.M., C.J.H., J.H., E.F., D.E.G., F.D.R.H., S.H.R., D.M.R., M.T.W., J.W., I.S.M. The manuscript text, tables and figures were prepared by T.M., I.F., J.M., C.H., I.M., and N.G., who had unrestricted access to the data, and were reviewed and edited by all authors. All authors agreed to submit the manuscript for publication and assume responsibility for the accuracy and completeness of the analyses.
SCOT Study Group collaborators:
Executive Committee: Thomas M. MacDonald (Chief Investigator), Ian Ford, and Chris Hawkey.
Steering Committee: Thomas M. MacDonald (Chair), Ian Ford, Chris Hawkey, Jesper Hallas, John Webster, David Reid, Isla Mackenzie, Stuart Ralston, Matthew Walters, F. D. Richard Hobbs, Sir Lewis Ritchie, Mark Davis, Susana Perez-Gutthann, Frank Ruschitzka, James Scheiman, Evelyn Findlay, John McMurray, and Diederick Grobbee.
Independent Data and Statistical Centre (Robertson Centre for Biostatistics, University of Glasgow)
Sharon Kean, Jane Aziz, Robbie Wilson and Bernard Kelly (IT and data management), Nicola Greenlaw and Michele Robertson (Biostatistics).
Consultant Epidemiologist: Li Wei.
Independent Data Monitoring Committee
Kim Fox (Chair), Gordon Murray, Frank Murray.
Endpoint Adjudication Committees
Cardiovascular: John McMurray (Chair), Pardeep Jhund, Mark Petrie, Michael MacDonald.
Gastrointestinal: James Scheiman (Chair), John Dillon, Jane Moeller, Angel Lanas.
Supplementary material
Supplementary material is available at European Heart Journal online.
Supplementary Material
Acknowledgements
We would like to express our sincere gratitude to all the patients who participated in the study and all the doctors and nurses from each of the centres who contributed to the SCOT study. Recruiting Centres: Dundee: Wendy Saywood, Claudine Jennings, Alison McGinnis, Irene Donald, Emma Gellatly, Caroline Hall, Dawn Ross, Fiona Gowans, Kate Cowan, Wendy Urquhart, Patricia Robertson, Lesley Riley, Pamela Goodman, Moira Dryburgh, Johan McGill, Avril Donaldson. Aberdeen: Jacqueline Furnace, Joan Henderson, Frances Rentoul, Mandy Thompson, Emma Wilson, Heather Lawrence, Helen Keith, Julie Shotton. Edinburgh: Janet Thomson, Susan Begg, Julia Boyd, Theresa Harper, Guen Innes, Debra Kerr, Helen Reynolds, Lorraine Petrie, Janet Connelly, Morag McLean. Glasgow: Iain McInnes, Roger Sturrock, Linda Wilson, Geraldine Campbell, Rhona McKay, Kirsty Simpson, Joanne Flynn, Anne Benson, June Innes. Birmingham: Rachel Iles, Clare Taylor. Oxford: Ben Thompson, Sabrina Petersen, Pippa Whitbread, Marie-Lucie Gibbons, Mina Davoudianfar, Faye Alexander. Nottingham: Jen Dumbleton, Diane Stevenson, Vic Shepherd, David Goddard, Angela Andrew, Alice Cotton. Denmark: Michael Dall, Kasper Soltoft Larsen; Morten Rix Hansen, Viv Toft Lie, Ellen Kathrine Arve, Anita Hagelskaer, Susanne Leed Henriksen, Charlotte Enok Poulsen, Trine Ammentorp Gregersen, Birgith Kjaergaard, Dorthe Karup Holm, Brit Jorgensen, Anja Holmgaard, Vibeke Karlsen, Birgitte Vajsbaek. Amsterdam: Nils Visser, Helen van den Heuvel, Astrid Suiker, Nathalie Groetelaers, Aline Veurink-Westrik.
Funding
An unrestricted Investigator Initiated Research Grant from Pfizer USA to the University of Dundee. The University of Dundee was the legal sponsor of the study (responsible for all aspects of the trial). Some of the funding received from Pfizer USA went towards salaries for Dundee staff (TMM, IM AW, EF) and staff at other Universities. The University of Dundee disbursed study funding to all other investigator institutions so, for example, IF received research funding for the SCOT study indirectly from Pfizer (via the University of Dundee) as did other investigators. F.D.R.H. is partially supported by NIHR School for Primary Care Research, NIHR CLAHRC Oxford, NIHR Oxford BRC and Harris Manchester College, Oxford.
Conflict of interest: The University of Dundee received reimbursement from Pfizer for ISM and TMM in connection with the SCOT. In the last 5 years TMM has received honoraria for lectures and consulting from NiCox, Novartis, and Astra Zeneca in relation to NSAID therapies and from other companies regarding other therapeutic areas. The Medicines Monitoring Unit holds research grants from Pfizer, Menarini, Novartis, Shire, RTI, GSK, and Amgen. I.F. received research funding for the SCOT indirectly from Pfizer (via the University of Dundee).
References
- 1. Schnitzer TJ, Burmester GR, Mysler E, Hochberg MC, Doherty M, Ehrsam E, Gitton X, Krammer G, Mellein B, Matchaba P, Gimona A, Hawkey CJ; TARGET Study Group. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet 2004;364: 665–74. [DOI] [PubMed] [Google Scholar]
- 2. Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C.. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ 2006;332:1302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, Farkouh ME, FitzGerald GA, Goss P, Halls H, Hawk E, Hawkey C, Hennekens C, Hochberg M, Holland LE, Kearney PM, Laine L, Lanas A, Lance P, Laupacis A, Oates J, Patrono C, Schnitzer TJ, Solomon S, Tugwell P, Wilson K, Wittes J, Baigent C.. Coxib and traditional NSAID Trialists' (CNT) Collaboration, Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 2013;382:769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mukherjee D, Nissen SE, Topol EJ.. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 2001;286:954–9. [DOI] [PubMed] [Google Scholar]
- 5. http://www.fda.gov/Drugs/DrugSafety/ucm451800.htm (10 July 2015).
- 6. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM383180.pdf (10 July 2015).
- 7. http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2010/01/news_detail_000969.jsp&mid=WC0b01ac058004d5c1 (31 July 2015).
- 8. Hansson L, Hedner T, Dahlof B. Prospective randomised open blinded end-point (PROBE) study. A novel design for intervention trials. Prospective Randomised Open Blinded End-Point. Blood Press 1992;1:113–9. [DOI] [PubMed] [Google Scholar]
- 9. MacDonald TM, Mackenzie IS, Wei L, Hawkey CJ, Ford, SCOT study group collaborators. Methodology of a large prospective, randomised, open, blinded endpoint, streamlined safety study of celecoxib versus conventional non-steroidal anti-inflammatory drugs in patients with osteoarthritis or rheumatoid arthritis: protocol of the standard care versus celecoxib outcome trial (SCOT). BMJ Open 2013;3:e002295. doi:10.1136/bmjopen-2012-002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ford I, Murray H, Shepherd J, Packard CJ, Macfarlane PW, Cobbe SM.. Long-term follow-up of the West of Scotland Coronary Prevention Study. N Eng J Med 2007;357:1477–1486. [DOI] [PubMed] [Google Scholar]
- 11. http://www.meddra.org/ (11 August 2015).
- 12. Chan FK, Lanas A, Scheiman J, Berger MF, Nguyen H, Goldstein JL. Celecoxib versus omeprazole and diclofenac in patients with osteoarthritis and rheumatoid arthritis (CONDOR): a randomised trial. Lancet 2010;376:173–9. [DOI] [PubMed] [Google Scholar]
- 13. Farkouh ME, Kirshner H, Harrington RA, Ruland S, Verheugt FW, Schnitzer TJ, Burmester GR, Mysler E, Hochberg MC, Doherty M, Ehrsam E, Gitton X, Krammer G, Mellein B, Gimona A, Matchaba P, Hawkey CJ, Chesebro JH; TARGET Study Group. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: randomised controlled trial. Lancet 2004;364:675–84. [DOI] [PubMed] [Google Scholar]
- 14. Singh G1, Fort JG, Goldstein JL, Levy RA, Hanrahan PS, Bello AE, Andrade-Ortega L, Wallemark C, Agrawal NM, Eisen GM, Stenson WF, Triadafilopoulos G. ; SUCCESS-I Investigators. Celecoxib versus naproxen and diclofenac in osteoarthritis patients: SUCCESS-I Study. Am J Med 2006;119:255–66. [DOI] [PubMed] [Google Scholar]
- 15. Bannuru RR, Schmid CH, Kent DM, Vaysbrot EE, Wong JB, McAlindon TE.. Comparative effectiveness of pharmacologic interventions for knee osteoarthritis: a systematic review and network meta-analysis. Ann Intern Med 2015;162:46–54. [DOI] [PubMed] [Google Scholar]
- 16. da Costa BR, Reichenbach S, Keller N, Nartey L, Wandel S, Jüni P, Trelle S.. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: a network meta-analysis. Lancet 2016;387:2093–105. [DOI] [PubMed] [Google Scholar]
- 17. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/referrals/Diclofenac-containing_medicines/human_referral_prac_000009.jsp&mid=WC0b01ac05805c516f (14 August 2015).
- 18. Varas-Lorenzo C, Riera-Guardia N, Calingaert B, Castellsague J, Salvo F, Nicotra F, Sturkenboom M, Perez-Gutthann S.. Myocardial infarction and individual nonsteroidal anti-inflammatory drugs meta-analysis of observational studies. Pharmacoepidemiol Drug Saf. 2013;22:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. White WB, West CR, Borer JS, Gorelick PB, Lavange L, Pan SX, Weiner E, Verburg KM.. Risk of cardiovascular events in patients receiving celecoxib: a meta-analysis of randomized clinical trials. Am J Cardiol 2007;99:91–8. [DOI] [PubMed] [Google Scholar]
- 20. Moore RA, Derry S, Makinson GT, McQuay HJ.. Tolerability and adverse events in clinical trials of celecoxib in osteoarthritis and rheumatoid arthritis: systematic review and meta-analysis of information from company clinical trial reports. Arthritis Res Ther 2005;7:R644–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. http://cordis.europa.eu/publication/rcn/14115_en.html (15 August 2015).
- 22. Dégano IR, Salomaa V, Veronesi G, Ferriéres J, Kirchberger I, Laks T, Havulinna AS, Ruidavets JB, Ferrario MM, Meisinger C, Elosua R, Marrugat J; Acute Myocardial Infarction Trends in Europe (AMITIE) Study Investigators. 25-year trends in myocardial infarction attack and mortality rates, and case-fatality, in 6 European populations. Heart 2015;101:1413–20. [DOI] [PubMed] [Google Scholar]
- 23. Gerber Y, Weston SA, Jiang R, Roger VL.. The changing epidemiology of myocardial infarction in Olmsted County, Minnesota, 1995–2012. Am J Med 2015;128:144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Estruch R, Ros E, Salas-Salvadó J, Covas MI, Corella D, Arós F, Gómez-Gracia E, Ruiz-Gutiérrez V, Fiol M, Lapetra J, Lamuela-Raventos RM, Serra-Majem L, Pintó X, Basora J, Muñoz MA, Sorlí JV, Martínez JA, Martínez-González MA; PREDIMED Study Investigators. Primary prevention of cardiovascular disease with a mediterranean diet. N Engl J Med 2013;368:1279–90. [DOI] [PubMed] [Google Scholar]
- 25. http://www.nice.org.uk/guidance/cg59 (14 August 2015).
- 26. http://www.nice.org.uk/guidance/cg177 (14 August 2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.