

Abstract
Hepatotoxicity is of major concern for humans exposed to industrial chemicals and drugs. Disruption of farnesoid X receptor (FXR), a master regulator of bile acid (BA) metabolism, enhanced the sensitivity to liver injury in mice after toxicant exposure, but the precise mechanism remains unclear. In this study, the interconnection between BA metabolism, FXR, and chemically induced hepatotoxicity was investigated using metabolomics, Fxr-null mice (Fxr−/−) and hepatocytes, and recombinant adenoviruses. A single low-dose intraperitoneal injection of carbon tetrachloride (CCl4), an inducer of acute hepatitis in mice, resulted in more severe hepatocyte damage and higher induction of pro-inflammatory mediators, such as chemokine (C–C motif) ligand 2 (Ccl2), in Fxr−/−. Serum metabolomics analysis revealed marked increases in circulating taurocholate (TCA) and tauro-β-muricholate (T-β-MCA) in these mice, and forced expression of bile salt export protein (BSEP) by recombinant adenovirus in Fxr−/− ameliorated CCl4-induced liver damage. Treatment of Fxr-null hepatocytes with TCA, but not T-β-MCA, significantly increased c-Jun-N-terminal kinase (JNK) activation and Ccl2 mRNA levels, and up-regulation of Ccl2 mRNA was attenuated by co-treatment with a JNK inhibitor SP600125, indicating that TCA directly amplifies hepatocyte inflammatory signaling mainly mediated by JNK under FXR-deficiency. Additionally, pretreatment with SP600125 or restoration of FXR expression in liver by use of recombinant adenovirus, attenuated CCl4-induced liver injury. Collectively, these results suggest that the TCA–JNK axis is likely associated with increased susceptibility to CCl4-induced acute liver injury in Fxr−/−, and provide clues to the mechanism by which FXR and its downstream gene targets, such as BSEP, protects against chemically induced hepatotoxicity.
Keywords: farnesoid X receptor, c-Jun-N-terminal kinase, taurocholate, CCl4, bile acids
Drug-induced liver injury (DILI) is the leading cause of drug withdraws from the market and is also among the main reasons for drug candidate attrition. DILI is a major challenge for drug development and clinical use (Tujios and Fontana, 2011). Because of the significant impact of DILI, there is clearly a need to determine the mechanisms of DILI in order to aid in both the drug developmental process and clinical treatment. Untargeted metabolomic analyses on rats treated with thirteen known hepatotoxins causing various types of DILI revealed that the most consistent change induced by the hepatotoxins was significant elevations of bile acids (BAs) in the plasma, suggesting that the perturbation of BA homeostasis is an early event of DILI (Yamazaki et al., 2013). It was also reported that pro-inflammatory cytokines alter the expression of gene related with BA metabolism (Tanaka et al., 2012). However, the actual and direct contribution of BAs to DILI remains undetermined.
The nuclear receptor farnesoid X receptor (FXR) is the primary BA receptor, acting as a master regulator of BA homeostasis, that modulates BA synthesis, transport, and enterohepatic circulation (Matsubara et al., 2013). Farnesoid X receptor is a central regulator of lipid/glucose metabolism. Fxr-null mice (Fxr−/−) develop severe fatty liver and have elevated circulating free fatty acids, which was associated with increased serum glucose and impaired glucose and insulin tolerance (Ma et al., 2006). FXR regulates normal liver repair by promoting regeneration and preventing cell death (Huang et al., 2006; Meng et al., 2010), indicating that it protects against chronic liver disease. However, the role of FXR and BAs in the pathogenesis of acute liver injury remains unclear.
Carbon tetrachloride (CCl4) is a common solvent and hepatotoxin widely used to study the mechanisms of drug-induced acute liver injury in experimental models. To investigate whether disruption of BA homeostasis renders susceptibility to hepatotoxins, Fxr−/− mice were treated with low-dose of CCl4 and the severity of liver injury and various histological and biochemical parameters was compared with those in wild-type (WT) mice.
MATERIALS AND METHODS
Animal maintenance and treatments
All animal studies and procedures were carried out in accordance with Institute of Laboratory Animal Resources guidelines and approved by the National Cancer Institute Animal Care and Use Committee. Fxr−/− mice were generated as previously described (Sinal et al., 2000) and WT mice with the same genetic background were used as controls. Mice were housed in a pathogen-free animal facility under a standard 12-h light/dark cycle and given pelleted NIH-31 chow diet and water ad libitum. Male mice between 8 and 12 weeks old (n = 5–8) were used in each group of experiments. After overnight fasting, the mice were intraperitoneally injected with 0.25 ml/kg body weight of CCl4 (Sigma-Aldrich, St. Louis, MO) or vehicle (corn oil, Sigma-Aldrich). Although the conventional dose of CCl4 to induce acute hepatitis is 1.0 ml/kg (Avasarala et al., 2006), the lower dose regimen was adopted in this study to delineate a clear difference in susceptibility to this hepatotoxicant between Fxr−/− and WT mice. The c-Jun-N-terminal kinase (c-Jun) N-terminal kinase (JNK) inhibitor SP600125 was prepared as 3 mg/ml with 10% Solutol-HS15 (Sigma-Aldrich) in phosphate-buffered saline (PBS) and administered (30 mg/kg, single intraperitoneal injection) 30 min prior to CCl4 injection. At the prescribed time points after CCl4 injection, the mice were anesthetized and killed by CO2 asphyxiation 3 h after food withdrawal and blood collected using Serum Separator Tubes (Becton Dickinson and Company, Franklin Lakes, NJ) and centrifuged for 10 min at 8000 × g at 4 °C to isolate serum. Tissues were divided into two parts: One part was immediately frozen in liquid nitrogen and the other part soaked in 10% neutral formalin for histological evaluation. Serum and tissues were kept at −80 °C until use.
Genotyping protocol for Fxr
Genomic DNA was extracted from tail and deletion of Fxr gene was examined by polymerase chain reaction (PCR) using the following primers: FXR-F1: 5’-TCTCTTAAGTGATGACGGAATCT-3’, FXR-F2: 5’-GCTCTAAGGAGAGTCACTTGTGCA-3’ and FXR-R1: 5’-GCATGCTCTGTTCATAAACGCCAT-3’. The amplicon with 249 and 291 bp is detected in WT and Fxr−/− mice, respectively.
Quantitative real-time PCR analysis (qPCR)
Total RNA of liver was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA) and 1 µg of total RNA was reverse-transcribed with qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD) to generate cDNA. qPCRs were carried out using SYBR green PCR master mix (Thermo Fisher Scientific) in an ABI Prism 7900HT Sequence Detection System. The primer pairs were designed using qPrimerDepot. Values were quantified with the comparative CT method, normalized to 18S ribosomal RNA, and subsequently normalized to those of control mice or hepatocytes.
Measurement of biochemical parameters
Serum activities of aspartate and alanine aminotransferase (AST and ALT, respectively) were measured with the assay kits (CATACHEM INC. Oxford, CT).
Histological analysis
Formalin-fixed tissues were dehydrated by graded ethanol and xylene and embedded in paraffin. Sections (4 µm thick) were stained with the hematoxylin and eosin (H&E) (Tanaka et al., 2012).
Ultra-performance liquid chromatography-electrospray ionization quadrupole time-of-flight mass spectrometry (UPLC-ESI-QTOFMS) analysis
Six microliters of serum were diluted with 114 µl of 66% acetonitrile containing chlorpropamide as an internal standard and centrifuged twice at 18 000 × g for 25 min at 4 °C for removal of precipitated proteins and other particulates. The UPLC-ESI-QTOFMS method was described (Matsubara et al., 2011). Briefly, the eluted sample (5 µl/injection) was introduced by electrospray ionization into the mass spectrometer Q-TOF Premier (Waters Corporation, Milford, MA) operating in either negative or positive electrospray ionization modes. All samples were analyzed in a randomized fashion to avoid complications caused by artifacts related to injection order and changes in instrument efficiency. MassLynx software (Waters Corporation) was used to acquire the chromatogram and mass spectrometric data. Centroided and integrated chromatographic mass data were processed by MarkerLynx software (Waters Corporation) to generate a multivariate data matrix. Pareto-scaled MarkerLynx matrices including information on sample identity were analyzed by principal components analysis (PCA) and supervised orthogonal projection to latent structures (OPLS) analysis using SIMCA-version 14 (Umetrics, Kinnelon, NJ). The OPLS loadings scatter S-plot was used to determine those ions that contributed significantly to the separation between CCl4-treated Fxr−/− and CCl4-treated WT mice. The identity of ions with a correlation of 0.8 or higher to the model was further investigated. The metabolite structures were determined using the METLIN metabolite and LIPID MAPS database, and the identity was confirmed by tandem mass spectrometry MS/MS fragmentation patterns.
Quantification of BAs
Fifty microliters of mouse serum or 50 mg of mouse liver was mixed with 100 μl or 500 µl of 100% acetonitrile containing 1 μM d5-taurocholate (TCA) (Sigma-Aldrich) as an internal standard, respectively, and centrifuged twice at 15 000 × g for 25 min at 4 °C for removal of precipitated proteins and other particulates. The supernatant was diluted by an equal volume of HPLC grade water (Thermo Fisher Scientific) containing 0.1% formic acid. Quantification of BA metabolites was conducted as described previously (Jiang et al., 2015; Li et al., 2013; Takahashi et al., 2016). Unconjugated α-muricholate (α-MCA), β-MCA, cholic acid (CA), chenodeoxycholic acid, deoxycholic acid (DCA), hyodeoxycholic acid (HDCA), ursodeoxycholic acid (UDCA), and their taurine-conjugated derivatives were measured and quantified because taurine-conjugated BAs are the predominant conjugated BAs in mice whereas humans produce glycine-conjugated BAs. LC–MS was performed on a Waters Acquity H-Class UPLC system using a Waters Acquity BEH C18 column (2.1 × 100 mm) coupled to a Waters Xevo G2 QTOFMS. BA standards were purchased from Steraloids Inc. (Newport, RI) or Sigma-Aldrich, and the identity of BAs was confirmed based on MS/MS fragmentation pattern.
Western blotting
Approximately 50 mg of liver or primary mouse hepatocyte protein was homogenized in RIPA buffer containing a Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). The homogenates were centrifuged at 10 000 × g for 10 min at 4 °C and the supernatants (20 μg of protein) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% bovine serum albumin or skim milk and incubated overnight with primary antibodies against cytochrome P450 (CYP) 2E1 (Abnova, MAB3817, 1:1000 dilution), stress-activated protein kinase (SAPK)/JNK (Cell Signaling, #9252, 1:1000 dilution), phosphorylated SAPK/JNK (Cell Signaling, Danvers, MA, #9251, 1:1000 dilution), extracellular signal-regulated kinase 1/2 (ERK, Cell Signaling, #9102, 1:1000 dilution), and phosphorylated ERK (Cell Signaling, #9101, 1:1000 dilution). Immunoblotting for nuclear factor-kappa B (NF-κB) p65 component (Cell Signaling, #8242, 1:1000 dilution) and phosphorylated p65 (Cell Signaling, #3033, 1:1000 dilution) was performed as described previously (Tanaka et al., 2014). As a loading control, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) staining was carried out by re-probing the membranes with antibody against GAPDH (Merck Millipore, Billerica, MA, #MAB374, 1:10 000 dilution).
For detecting the expression of bile salt export pump (BSEP), mouse liver membrane fractions were prepared according to published protocol with a minor modification (Hoekstra et al., 2006). Briefly, approximately 100 mg of liver tissue was homogenized in 900 μl of sucrose–Tris buffer composed of 0.25 M sucrose and 25 mM Tris-HCl with pH 7.4 and containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). The homogenates were centrifuged at 39 000 × g for 60 min at 4 °C, the supernatant discarded, and the remaining pellet suspended with 150 μl of sucrose-Tris buffer which was used as the crude membrane fraction. Fifty micrograms of membrane protein were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membrane, blocked with 5% Blotto Non-Fat Dry Milk (Santa Cruz Biotechnology, Dallas, TX, sc-2325) and incubated overnight with primary antibodies against BSEP (Santa Cruz Biotechnology, sc-74500, 1:200 dilution) with Can Get Signal Immunoreaction Enhancer Solution (Toyobo, Osaka, Japan). An antibody against calnexin (Santa Cruz Biotechnology, sc-6465, 1:10 000 dilution) was used as a loading control.
To measure the FXR protein levels, mouse liver nuclear fractions were prepared according to a published protocol with a minor modification (Shinzawa et al., 2015). One hundred milligrams of mouse liver were homogenized and lysed in hypotonic cytosol extraction buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1.5 mM dithiothreitol [DTT], 0.05% Nonidet P-40, and containing Halt Protease and Phosphatase Inhibitor Cocktail [Thermo Fisher Scientific]. The suspension was incubated on ice for 20 min and then disrupted by repetitive pipetting. Nuclei were pelleted by centrifugation at 15 000 rpm for 1 min at 4 °C and re-suspended with nuclear extraction buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 420 mM NaCl, 25% glycerol, 0.2 mM EDTA, 0.5 mM DTT, and a Halt Protease and Phosphatase Inhibitor Cocktail [Thermo Fisher Scientific] and 25% glycerol). The suspension was kept on ice for 20 min, and centrifuged at 15 000 rpm for 5 min at 4 °C, and the supernatant collected as the nuclear protein extract. Fifty micrograms of nuclear protein were separated and transferred to the membrane in the same manner. The membrane was blocked with 5% skim milk and incubated with primary antibody against FXR (Santa Cruz Biotechnology, sc-13063, 1:200 dilution) for 3 h at room temperature. The membrane was re-probed with an antibody against proliferating cell nuclear antigen (Santa Cruz Biotechnology, sc-56, 1:10 000 dilution) used as a loading control.
Immunostaining of BSEP
Frozen mouse liver sections were cut at 12 µm at −20°C with a Leica Cryotome CM 3050S (Leica, Bensheim, Germany) and placed on Colorfrost plus slides (Thermo Fisher Scientific). The sections were air-dried (45 min) and thereafter stored at −45°C until staining. Immunostaining of BSEP was conducted as described elsewhere (Aleksunes et al., 2013). Briefly, frozen sections were fixated with 4% paraformaldehyde (room temperature, 10 min), soaked with 0.1% Triton-X/PBS in a Coverplate Chamber System (Thermo Fisher Scientific), rinsed twice in 0.1% Triton-X/PBS, and blocked for 1 h with 5% goat serum in 0.1% Triton-X/PBS. Sections were incubated for 2 h with anti-BSEP (K44) primary antibody (1:200 in 5% goat serum/0.1% Triton-X/PBS). The same concentrations of goat serum in 0.1% Triton-X/PBS was used for negative control staining. After rinsing 3 times with 0.1% Triton-X/PBS, the sections were incubated with a secondary antibody (Molecular Probes goat anti-rabbit Alexa Fluor 594, 1:750 in 5% goat serum in 0.1% Triton-X/PBS) for 1 h. After a series of rinses, the sections were cover-slipped with ProLong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific).
Preparation and treatment of mouse primary hepatocytes
Primary hepatocytes were isolated from C57BL/6J mice as described previously (Takahashi et al., 2016). The number of hepatocytes were counted and then seeded in collagen-coated 12-well plates (Becton Dickinson and Company) at a density of 4 × 105 cells/well. Primary hepatocytes were cultured in William’s E medium (Thermo Fisher Scientific) with 10% fetal bovine serum, and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin). Four to six hours after seeding, cells were treated with 100 µM DCA (Sigma-Aldrich), 100 µM TCA (Sigma-Aldrich), 100 µM T-β-MCA (Sigma-Aldrich) and 20 µM SP600125 (Sigma-Aldrich). At the prescribed time points, cells were harvested and subjected to qPCR and immunoblot analysis.
Production of recombinant adenoviruses and injection to mice
cDNAs of FXR and ATP-binding cassette subfamily B member 11 (Abcb11), which encodes BSEP, were obtained by PCR using cDNA derived from C57BL6 mouse liver using the forward and reverse primers for Fxr, 5’-CACCATGAATCTGATTGGGCA-3’ and 5’-TCACTGCACATCCCAGATCTC-3’; those for Abcb11, 5’-CACCATGTCTGACTCAGTGATTCTTCG-3’ and 5’-TCAACTGATGGGGGCTCCAGT-3’. The fragment was inserted into pENTR-TOPO to generate pENTR-Fxr and pENTR-Abcb11, respectively. The producing process was carried out in accordance with the protocol of ViraPower Adenoviral Expression System, pENTR Directional TOPO Cloning Kits and GATEWAY LR clonase II MS Enzyme MIX. The titer of recombinant adenoviruses was determined using Adeno-X Rapid Titer Kit. The recombinant adenovirus overexpressing FXR (Ad-Fxr) or BSEP (Ad-Abcb11) was purified using CsCl gradient centrifugation. These viruses were dissolved in 200 µl of physiological saline and were injected from tail vein using 27G needle (2.0 × 109 pfu/mouse). Recombinant adenovirus overexpressing green fluorescent protein gene (Ad-Ctrl) was injected in the same manner as a control.
Statistical analysis
Statistical analysis was performed with Prism version 7 (GraphPad Software). Appropriate statistical analysis was applied, assuming a normal sample distribution. When comparing two groups, statistical significance was determined using two-tailed Student's t-test. When more than two groups and two factors were investigated, two-way ANOVA followed by Tukey’s post hoc correction was applied for comparisons. A P value of less than .05 was considered as significant difference. Results are expressed as the means ± standard deviation (SD).
RESULTS
Fxr−/− Mice Are More Susceptible to CCl4-Induced Liver Injury Than WT Mice
To examine whether disruption of BA homeostasis renders susceptibility to the hepatotoxins, CCl4 at lower dose than conventionally used one was administered to WT and Fxr−/− mice. Serum AST and ALT were significantly increased in Fxr−/− mice compared with WT mice 24 h after low-dose CCl4 injection with only modest increases noted in WT mice (Figure 1A). In H&E staining, hepatocyte degeneration was present in both mouse lines but more severe in Fxr−/− mice (Figure 1B), although CYP2E1 protein, the main enzyme responsible for CCl4 detoxification, was found at similar levels to WT mice in CCl4 treatment (Figure 1C). The mRNAs encoding typical acute-phase proteins induced by inflammation, chemokine (C–C motif) ligand 2 (Ccl2, encoding monocyte chemotactic protein [MCP] -1), serine (or cysteine) peptidase inhibitor, clade E, member 1 (Pai1, encoding serpin E1) and tumor necrosis factor (Tnf encoding tumor necrosis factor alpha [TNFα]), were significantly elevated in CCl4-treated Fxr−/− mice, the mRNA levels of these genes in vehicle-treated Fxr−/− mice were comparable to those in similarly treated WT mice (Figure 1D). The expressions of Abcb11 and small heterodimer partner (Shp) mRNAs, encoded by FXR target genes, were significantly suppressed in Fxr−/− mice (Figure 1D).
Figure 1.

Fxr−/− mice are more susceptible to CCl4-induced liver injury. Fxr−/− and WT mice were injected with CCl4 or vehicle (corn oil) (0.25 ml/kg). Twenty-four hours after single CCl4 injection, these samples were collected and measured. A, Serum AST and ALT levels. B, H&E staining of liver tissue. C, CYP2E1 protein level in liver. D, Expression of FXR and FXR-regulated genes and pro-inflammatory modulator mRNAs. n=5–8 mice per group. Data are presented as the means±SD and one-way ANOVA with Tukey’s correction was adopted for statistical analysis. **P <.01; ***P<.001
Identification of Serum Metabolites Significantly Altered in Fxr−/− Mice 24 h After CCl4 Administration
To find metabolites associated with the phenotypic changes in CCl4-treated Fxr−/− mice, serum metabolomic analysis was performed. Principal components analysis showed clear separation into three groups, especially between the CCl4-treated WT and Fxr−/− mouse groups (Figure 2A). Based on OPLS, S-plot, and loadings scatter analysis, the top 3 serum metabolites significantly altered between CCl4-treated WT and Fxr−/− mice are listed in Supplementary Table 1. Two taurine-conjugated BAs, TCA and T-β-MCA, were selected as principal metabolites significantly increased in CCl4-treated Fxr−/− mice with the highest confidence and greatest contribution to the separation between the CCl4-injected Fxr−/− and WT mice (Figure 2B and Supplementary Table 1). Quantitation of BA metabolites confirmed the marked increases in TCA and T-β-MCA both in serum (Figure 2C) and liver tissue (Supplementary Figure 1). Additionally, palmitoyl-lysophosphatidylcholine (LPC, 16:0-LPC), linoleoyl-LPC (18:2-LPC), and oleoyl-LPC (18:1-LPC) were identified as metabolites that were significantly lower in CCl4-treated Fxr−/− mice compared with similarly treated WT mice (Supplementary Table 1), and these reductions were verified by quantification (Supplementary Figure 2).
Figure 2.

Identification of serum metabolites significantly altered in Fxr−/− mice 24 h after CCl4 injection. A, Principal component analysis (PCA) of serum metabolites using SIMCA software for metabolomics analysis. Metabolite profiles were different between WT and Fxr−/− mice after CCl4 injection. B, The identities of taurine-conjugated BAs having the highest confidence and greatest contribution to separation in S-plot between CCl4-treated WT and Fxr−/− mice. C, Bile acids levels in the serum. Data are presented as the means±SD and two-way ANOVA with Tukey’s correction was adopted for statistical analysis. ***P<.001 versus the other groups. α-MCA, α-muricholate; β-MCA, β-muricholate; CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; HDCA, hyodeoxycholic acid; UDCA, ursodeoxycholic acid; T-, taurine-conjugated; N, not detected.
Recombinant Adenovirus Overexpressing BSEP Attenuates CCl4-Induced Hepatotoxicity in Fxr−/− Mice
Although increased serum TCA and T-β-MCA levels were found specifically in CCl4-treated Fxr−/− mice, it was unclear whether the elevation of these BAs are the cause or result of more severe liver injury. BSEP is a main transporter for the movement of conjugated BAs from liver to the bile and intestine and its expression is tightly regulated by FXR. The basal expression of Abcb11 mRNA encoding BSEP is significantly lower in Fxr−/− mice and the levels were further suppressed by CCl4 administration, leading to disrupted BA homeostasis. Hypothesizing that BSEP recovery might attenuate CCl4-induced increases in TCA and T-β-MCA and hepatotoxicity in Fxr−/− mice, Ad-Abcb11 was injected to the tail vein of Fxr−/− mice and CCl4 was given 5 days after adenovirus delivery. Immunofluorescence and immunoblot analyses confirmed that BSEP expression was elevated and in the natural cellular membrane compartment in Fxr−/− mouse liver after Ad-Abcb11 injection (Supplementary Figure 3A and B). Serum levels of AST, ALT, TCA, and T-β-MCA were significantly reduced in the Ad-Abcb11-injected group compared with the Ad-Ctrl group (Figure 3A and B). Moreover, Ccl2, Pai1, and Tnf mRNAs were significantly decreased in the Ad-Abcb11-treated group compared with the Ad-Ctrl-treated group (Figure 3C). Forced Abcb11 expression in hepatocytes attenuated circulating BA concentrations and liver injury after CCl4 injection without altering FXR expression (Figure 3B and D).
Figure 3.

Hepatocyte BSEP recovery by adenovirus decreases CCl4-induced hepatotoxicity in Fxr−/− mice. Adenovirus carrying the mouse Abcb11 cDNA (Ad-Abcb11) expressing BSEP or empty vector (Ad-Ctrl) was injected to Fxr−/− mice (2.0×109 pfu/mouse) 5 days before CCl4 injection (0.25 ml/kg). A, Serum AST and ALT levels. B, Serum TCA and T-β-MCA concentrations. C, Hepatic mRNA levels of Abcb11, Fxr, and pro-inflammatory modulators. D, H&E staining of liver tissue. Data are presented as the means±SD and two-tailed Student’s t-test was used for statistical analysis. **P<.01; ***P<.001; ND, not detected.
Taurocholate Increases mRNA encoding Pro-Inflammatory Modulators in Primary Hepatocytes Isolated From Fxr−/− Mice
To examine the possibility that certain BAs might amplify the CCl4-induced hepatotoxicity in Fxr−/− mice, primary hepatocytes isolated from Fxr−/− and WT mice were treated for 12 h with 100 μM DCA, TCA, and T-β-MCA. Taurocholate treatment, but not DCA and T-β-MCA, increased Ccl2 and Pai1 mRNA levels in Fxr-null hepatocytes (Figure 4A). Increased mRNA levels of these pro-inflammatory modulators in Fxr-null hepatocytes by TCA treatment were concentration-dependent and the increased mRNAs were not detected in WT hepatocytes (Figure 4B). These results suggest that TCA directly induces the expression of Ccl2 and Pai1 mRNAs specifically in Fxr-null hepatocytes, which may be associated with hypersensitivity to CCl4-induced liver injury in Fxr−/− mice.
Figure 4.

TCA increases mRNA levels of pro-inflammatory modulators in primary hepatocytes isolated from Fxr−/− mice. A, Primary hepatocytes isolated from Fxr−/− and WT mice were treated with DCA (100 μM), TCA (100 μM), T-β-MCA (100 μM), or the same volume of vehicle (dimethyl sulfoxide) for 6 h. Levels of Ccl2, Pai1, and Tnf mRNAs were measured by qPCR. The mRNA levels were expressed as the relative values to those of vehicle-treated WT hepatocytes. B, Primary hepatocytes isolated from Fxr−/− and WT mice were treated with different concentrations of TCA (10, 30, and 100 µM) or vehicle for 6 h. The mRNA levels were expressed as the relative values to those of vehicle-treated WT hepatocytes. *P<.05; **P<.01; ***P<.001.
Taurocholate Activates JNK in Primary Hepatocytes Isolated From Fxr−/− Mice
To assess the mechanism of TCA-induced enhancement of pro-inflammatory mediators in Fxr-null hepatocytes, activation of typical transcription factors regulating inflammation/cell death signaling, such as JNK, ERK, and NF-κB p65, was examined. Compared with WT hepatocytes, Fxr-null hepatocytes showed greater phosphorylation of JNK1/2 (Figure 5A and B). On the other hand, the levels of phosphorylated p65 and ERK were not altered in TCA-treated Fxr-null hepatocytes (Figure 5A). Total JNK1/2, ERK, and p 5 in TCA-treated Fxr-null hepatocytes were comparable to those in similarly treated WT hepatocytes (Figure 5A). Moreover, co-treatment with the JNK inhibitor SP600125 significantly suppressed TCA-induced increases in Ccl2 mRNA in Fxr-null hepatocytes (Figure 5C). These results indicate that the TCA-induced up-regulation of Ccl2 in Fxr-null hepatocytes is mediated by JNK activation.
Figure 5.

JNK activation is important for up-regulation of pro-inflammatory modulators by TCA in Fxr-null hepatocytes. A, Primary hepatocytes isolated from Fxr−/− and WT mice were treated with 100 μM TCA or vehicle (Veh) for 6 h. Whole cell lysates (20 μg of proteins) were subjected to immunoblot analysis for determining hepatic levels of total (T-) and phosphorylated (P-) JNK, ERK, and p65. The band of GAPDH was used as a loading control. B, Quantification of P-JNK. The band intensities of P-JNK and GAPDH were quantified using ImageJ software, and the ratio of P-JNK to GAPDH was calculated. Data are presented as the means±SD and two-way ANOVA with Tukey’s correction was adopted for statistical analysis. *P<.05; **P<.01. C, Primary hepatocytes isolated from Fxr−/− and WT mice were treated with 100 µM TCA or vehicle (Veh) with or without 10 µM JNK inhibitor (SP600125) for 6 h. The mRNA levels of pro-inflammatory modulators were measured. n=3 samples per group. Data are presented as the means±SD and two-tailed Student's t-test was used for statistical analysis; ***P<.001.
Suppression of JNK Activation Attenuates CCl4-Induced Acute Liver Injury in Fxr−/− Mice
In order to further determine the role of JNK signaling for the more severe CCl4-induced hepatotoxicity in Fxr−/− mice, SP600125 or vehicle was administered to Fxr−/− mice 30 min before CCl4 injection and the liver phenotypes were examined. Serum AST and ALT levels were significantly decreased in SP600125-cotreated Fxr−/− than in vehicle-treated mice (Figure 6A), which was in accordance with histological findings (Figure 6C). A lack of changes in serum TCA levels by co-treatment with SP600125 suggested only a minor impact of JNK signaling on TCA metabolism (Figure 6A). The expression of Ccl2 and Pai1 mRNAs were significantly decreased in SP600125-treated Fxr−/− mice (Figure 6B). Collectively, JNK activation plays a key role in increased susceptibility during CCl4-induced acute liver injury in Fxr−/− mice.
Figure 6.

Pretreatment with a JNK inhibitor attenuates CCl4-induced hepatotoxicity in Fxr−/− mice. Fxr−/− mice were injected with SP600125 (30 mg/kg) or vehicle 30 min before CCl4 injection (0.25 ml/kg). A, Serum AST, ALT, and TCA levels. B, Hepatic mRNA levels of pro-inflammatory modulators. C, H&E staining of liver tissue. Data are presented as the means±SD and two-tailed Student’s t-test was used for statistical analysis. n=6 mice per group. **P<.01; ***P<.001.
Recovery of Hepatocyte FXR by Adenovirus Ameliorates CCl4-Induced Hepatotoxicity in Fxr−/− Mice
Studies on cultured hepatocytes suggested a close relationship between hepatocyte FXR disruption, TCA-induced JNK activation and hypersensitivity to CCl4 in Fxr−/− mouse livers. To confirm these mechanistic links in vivo, Ad-Fxr was injected to Fxr−/− mice 5 days prior to CCl4 administration. Immunoblot analysis verified that nuclear FXR expressed from Ad-Fxr was at levels in Fxr−/− that are comparable to that found in WT mouse liver nuclei (Supplementary Figure 4). Forced expression of FXR in CCl4-treated Fxr−/− mice significantly decreased serum concentrations of AST, ALT, and TCA (Figure 7A), reversed the increased expression of Fxr, Abcb11, and Ccl2 mRNAs (Figure 7B), and moderated JNK phosphorylation (Figure 7C and D). These results demonstrate that hepatocyte FXR protects against CCl4-induced hepatotoxicity, likely due to maintaining BA metabolism, limiting TCA increases, and inhibiting TCA-induced JNK activation after CCl4 challenge (Figure 8).
Figure 7.

Forced expression of FXR ameliorates CCl4-induced hepatotoxicity in Fxr−/− mice. Adenovirus carrying mouse Fxr cDNA (Ad-Fxr) or empty vector (Ad-Ctrl) was injected to Fxr−/− mice and wild-type (WT) mice (2.0×109 pfu/mouse) 5 days before CCl4 injection (0.25 ml/kg). A, Serum AST, ALT, and TCA levels. B, Hepatic mRNA levels of genes encoding FXR-related proteins and genes encoding proinflammatory modulators. C, Western blot analysis of JNK. Whole liver lysates (20 μg of proteins) were electrophoresed and transferred to PVDF membrane for determining hepatic levels of total (T-) and phosphorylated (P-) JNK. The band of GAPDH was used as a loading control. D, Quantification of P-JNK. The band intensities of P-JNK and GAPDH were quantified using ImageJ software, and the ratio of P-JNK to GAPDH was calculated. Data are presented as the means ± SD and one-way ANOVA followed by Tukey’s post hoc correction was applied for comparisons. *P < .05; ***P < .001 versus all other groups.
Figure 8.
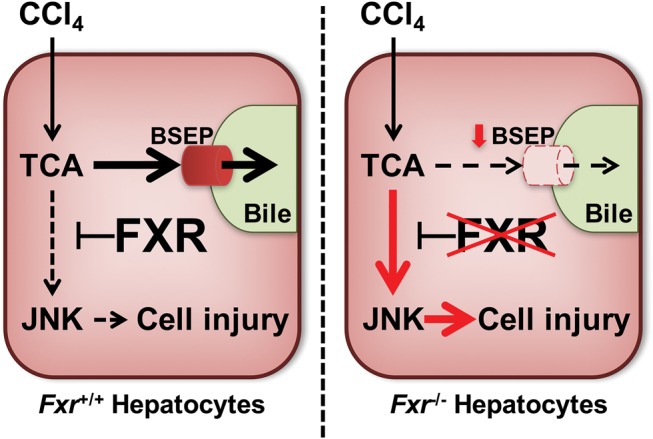
Proposed mechanism of higher susceptibility to CCl4-induced liver toxicity in Fxr−/− mice. In mice, hepatocyte FXR protects livers from low-dose CCl4 toxicity due to maintaining BSEP function, limiting the increase in TCA, and inhibiting JNK activation. However, disruption of FXR in hepatocytes impairs BSEP function and increases TCA levels, even at low-dose CCl4 exposure. Increased TCA activates JNK in FXR-disrupted hepatocytes, leading to induction of cell injury.
DISCUSSION
The present study demonstrated that disruption of hepatic FXR led to more severe acute liver injury after low-dose CCl4 administration. Serum metabolomic analysis showed that the conjugated BAs, TCA and T-β-MCA, were dramatically increased in CCl4-treated Fxr−/− mice. Studies with isolated hepatocytes revealed that TCA, but not T-β-MCA, up-regulated Ccl2 mRNA through JNK activation specifically in Fxr-null hepatocytes, and suppressed the JNK pathway attenuated CCl4-induced liver injury in Fxr−/− mice. Finally, restoration of hepatocyte FXR in Fxr−/− mice ameliorated the increases in circulating TCA concentrations and hepatic Ccl2 mRNA levels, JNK activation, and liver injury after low-dose CCl4 administration. Based on these results, the FXR–TCA–JNK axis may be a novel mechanism to protect liver against toxicant-induced acute hepatitis (Figure 8).
It was documented that Fxr−/− mice exhibited more severe inflammation under several diseased conditions, and the mechanism was explained as counteracting the effect of FXR against NF-κB signaling in Kupffer cells (Wang et al., 2008). However, phosphorylation of p65 and significant induction of Tnf mRNA, a typical target gene of NF-κB, were not detected in TCA-treated Fxr-null hepatocytes. Although ERK is sometimes activated during toxicant-induced acute liver damage (Wang et al., 2010), significant phosphorylation of ERK was not seen in TCA-treated Fxr-null hepatocytes. JNK phosphorylates c-Jun, leading to enhanced inflammatory signaling (Reimold et al., 2001; Woo et al., 2008; Ziraldo et al., 2013). JNK activation was found in several mouse models of acute liver injury, such as acetaminophen (APAP)-induced hepatotoxicity where inhibition of JNK by SP600125 protected hepatocytes against APAP-induced necrosis (Gunawan et al., 2006). The current study revealed a novel contributor other than NF-κB to the higher susceptibility to CCl4-induced acute liver injury in Fxr−/− mice.
Several mechanisms on how JNK activation causes liver damage have been proposed (Seki et al., 2012). In the current study, TCA directly activated JNK and increased Ccl2 mRNA levels in primary hepatocytes of Fxr−/− mice, and this effect was suppressed by co-treatment with a JNK inhibitor. MCP-1, a potent chemokine encoded by Ccl2, recruits and activates macrophages/monocytes resulting in potentiation of inflammatory cell injury (Baeck et al., 2012). Indeed, disruption of MCP-1 affords protection from liver damage and development of oxidative stress in an acute toxic model using CCl4 (Zamara et al., 2007), indicating a critical role for MCP-1 in this context. Additionally, it was documented that Ccl2 mRNA levels were induced by JNK activation in liver ischemia/reperfusion injury (Uehara et al., 2005). Furthermore, RIP3-dependent JNK activation promotes the release of MCP-1, thereby attracting macrophages to the injured liver and cell death, and liver fibrosis (Gautheron et al., 2014). These findings indicate that JNK-induced liver injury is partially mediated by MCP-1, corroborating results of the present study.
The mechanism by which TCA-induced JNK activation occurs preferentially in Fxr-null hepatocytes remains undetermined. One possible explanation is that JNK activation is mediated by sphingosine-1-phosphate receptor 2 (S1PR2). Hydrophobic BAs, such as glycochenodeoxycholate, can activate JNK and promote hepatocyte cell death, partially via S1PR2 (Webster and Anwer, 2016). Additionally, TCA induces pro-inflammatory cyclooxygenase-2 expression through S1PR2 in a human cholangiocellular carcinoma cell line (Liu et al., 2015). The contribution of S1PR2 to TCA–JNK–Ccl2 axis and chemically induced hepatotoxicity deserves further investigation.
PAI1 is a key acute-phase reactant promoting thrombosis and its induction in the liver disrupts microcirculation in sinusoids, ie, congestion and ischemia, which may accelerate hepatocyte apoptosis/necrosis. Indeed, Pai1 mRNA was initially induced after a single injection of concanavalin A, a potent inducer of immune-mediated acute hepatitis, and was correlated with the severity of liver injury (Kato et al., 2013). Considering the changes in Pai1 mRNA expression in the present study, this molecule may partially contribute to the pathogenesis of acute hepatitis in Fxr−/− mice after CCl4 challenge.
Untargeted serum metabolomic analysis revealed a significant decreases in some LPCs in CCl4-treated Fxr−/− mice. Because hepatic inflammation is known to reduce serum LPC levels (Tanaka et al., 2012), these decreases may reflect more severe pathologies in CCl4-injected Fxr−/− mice.
Another intriguing finding in the present study was that forced expression of BSEP in hepatocytes of Fxr−/− mice suppressed CCl4-induced hepatotoxicity. Moreover, expression of recombinant FXR in Fxr-null hepatocytes could reverse hepatic Abcb11 expression and CCl4-induced hepatotoxicity. It was reported that Fxr−/− mice were highly susceptible to α-naphthyl isothiocyanate-induced liver injury because of impaired induction of the hepatobiliary efflux transporter BSEP (Cui et al., 2009). However, there are marked species differences in BSEP function between humans, rats, and mice. Abcb11-disrupted C57BL/6J mice recapitulated the condition of human BSEP deficiency (Zhang et al., 2012), ie, severe progressive cholestatic liver disease from early infancy, but the Abcb11-null mice on a mixed genetic background showed no evidence of progressive liver injury (Wang et al., 2001). In BSEP-disrupted rats, biliary excretion of endogenous BAs was impaired but a relatively normal liver function was maintained through physiological compensation by other transporters (Cheng et al., 2016). Children and adults possessing genetic variants of ABCB11 suffer from intrahepatic cholestasis due to BSEP deficiency (Byrne et al., 2009), indicating a crucial function of BSEP in comprehensive BA metabolism (Fuchs et al., 2017). Patients with the ABCB11 c.1331T > C polymorphism are at increased risk of developing hepatocellular type of DILI (Ulzurrun et al., 2013). Indeed, several drugs were shown to inhibit BSEP in humans, and the pharmaceutical industry has proposed minimization of BSEP inhibition during drug discovery to help reduce DILI risk (Kenna, 2014). Based on the results in this study, therapeutic interventions targeting BSEP restoration in the liver may be a promising strategy against human liver disease.
Lastly, studies with isolated hepatocytes and recombinant adenoviruses revealed an important role for hepatocyte FXR in CCl4-induced hepatotoxicity. Several possible molecular mechanisms of DILI have been reported, including direct toxicity of reactive metabolites, metabolic idiosyncrasy and aberrant immune response. It was documented that nonsteroidal anti-inflammatory drugs, well-known causal agents of DILI, have FXR-antagonizing properties as revealed using a systems pharmacology approach (Lu et al., 2015), and several detrimental stimuli, such as hepatotoxicants and pro-inflammatory signals/cytokines, with resultant lower FXR signaling in the liver (Fang et al., 2004). Additionally, constitutive FXR expression was reported to be reduced in aged mice and overfed mice (Kim et al., 2015; Xiong et al., 2014). Under such physiological conditions, intake of some pharmacological agents may further down-regulate FXR and cause DILI, resembling the setting of the present study. Previous murine experiments have demonstrated the beneficial effects of FXR stimulation in various diseases, but unfavorable effects of FXR agonists, such as pruritus, jaundice, worsened hepatic insulin resistance, and increased concentrations of total cholesterol and low-density-lipoprotein cholesterol, were reported in humans (Ali et al., 2015; Neuschwander-Tetri et al., 2015). Indeed, the fact that hepatic FXR agonism or intestinal FXR antagonism can attenuate hepatosteatosis suggests tissue/cell-specific FXR action on whole-body homeostasis. The present study demonstrated that restoration of hepatocyte FXR levels in Fxr−/− mice was sufficient to attenuate CCl4-induced hepatotoxicity. These results suggest that strategies to reverse or correct hepatocyte FXR levels in damaged livers may be beneficial for the treatment/prevention of acute DILI.
In conclusion, this study demonstrated that TCA–JNK axis is associated with increased susceptibility for CCl4-induced acute liver injury in Fxr−/− mice. These results provide novel insight for considering the pathogenesis of acute hepatitis/DILI and the possible therapeutic targets.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank Michael Goedken, Rutgers University, for his valuable advice. We thank Linda Byrd and John Buckley for help with the animal management and protocols.
FUNDING
This work was supported by the National Cancer Institute Intramural Research Program, Center for Cancer Research and U54 ES16015 (F.J.G). ST was supported by a Japanese Society for the Promotion of Science Research Fellowship for Japanese Biomedical and Behavioral Researcher at NIH (KAITOKU-NIH). T.F. and Y.M. were supported by the program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (No. S2601) from the Japanese Society for the Promotion of Science.
REFERENCES
- Aleksunes L. M., Xu J., Lin E., Wen X., Goedken M. J., Slitt A. L. (2013). Pregnancy represses induction of efflux transporters in livers of type I diabetic mice. Pharm. Res. 30, 2209–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A. H., Carey E. J., Lindor K. D. (2015). Recent advances in the development of farnesoid X receptor agonists. Ann. Transl. Med. 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avasarala S., Yang L., Sun Y., Leung A. W., Chan W. Y., Cheung W. T., Lee S. S. (2006). A temporal study on the histopathological, biochemical and molecular responses of CCl(4)-induced hepatotoxicity in Cyp2e1-null mice. Toxicology 228, 310–322. [DOI] [PubMed] [Google Scholar]
- Baeck C., Wehr A., Karlmark K. R., Heymann F., Vucur M., Gassler N., Huss S., Klussmann S., Eulberg D., Luedde T., et al. (2012). Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 61, 416–426. [DOI] [PubMed] [Google Scholar]
- Byrne J. A., Strautnieks S. S., Ihrke G., Pagani F., Knisely A. S., Linton K. J., Mieli-Vergani G., Thompson R. J. (2009). Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology 49, 553–567. [DOI] [PubMed] [Google Scholar]
- Cheng Y., Freeden C., Zhang Y., Abraham P., Shen H., Wescott D., Humphreys W. G., Gan J., Lai Y. (2016). Biliary excretion of pravastatin and taurocholate in rats with bile salt export pump (Bsep) impairment. Biopharm. Drug Dispos. 37, 276–286. [DOI] [PubMed] [Google Scholar]
- Cui Y. J., Aleksunes L. M., Tanaka Y., Goedken M. J., Klaassen C. D. (2009). Compensatory induction of liver efflux transporters in response to ANIT-induced liver injury is impaired in FXR-null mice. Toxicol. Sci. 110, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C., Yoon S., Tindberg N., Jarvelainen H. A., Lindros K. O., Ingelman-Sundberg M. (2004). Hepatic expression of multiple acute phase proteins and down-regulation of nuclear receptors after acute endotoxin exposure. Biochem. Pharmacol. 67, 1389–1397. [DOI] [PubMed] [Google Scholar]
- Fuchs C. D., Paumgartner G., Wahlstrom A., Schwabl P., Reiberger T., Leditznig N., Stojakovic T., Rohr-Udilova N., Chiba P., Marschall H. U., et al. (2017). Metabolic preconditioning protects BSEP/ABCB11-/- mice against cholestatic liver injury. J. Hepatol. 66, 95–101. [DOI] [PubMed] [Google Scholar]
- Gautheron J., Vucur M., Reisinger F., Cardenas D. V., Roderburg C., Koppe C., Kreggenwinkel K., Schneider A. T., Bartneck M., Neumann U. P., et al. (2014). A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 6, 1062–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawan B. K., Liu Z. X., Han D., Hanawa N., Gaarde W. A., Kaplowitz N. (2006). c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 131, 165–178. [DOI] [PubMed] [Google Scholar]
- Hoekstra H., Porte R. J., Tian Y., Jochum W., Stieger B., Moritz W., Slooff M. J., Graf R., Clavien P. A. (2006). Bile salt toxicity aggravates cold ischemic injury of bile ducts after liver transplantation in Mdr2+/- mice. Hepatology 43, 1022–1031. [DOI] [PubMed] [Google Scholar]
- Huang W., Ma K., Zhang J., Qatanani M., Cuvillier J., Liu J., Dong B., Huang X., Moore D. D. (2006). Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 312, 233–236. [DOI] [PubMed] [Google Scholar]
- Jiang C., Xie C., Li F., Zhang L., Nichols R. G., Krausz K. W., Cai J., Qi Y., Fang Z. Z., Takahashi S., et al. (2015). Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Invest. 125, 386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J., Okamoto T., Motoyama H., Uchiyama R., Kirchhofer D., Van Rooijen N., Enomoto H., Nishiguchi S., Kawada N., Fujimoto J., et al. (2013). Interferon-gamma-mediated tissue factor expression contributes to T-cell-mediated hepatitis through induction of hypercoagulation in mice. Hepatology 57, 362–372. [DOI] [PubMed] [Google Scholar]
- Kenna J. G. (2014). Current concepts in drug-induced bile salt export pump (BSEP) interference. Curr. Protoc. Toxicol. 61, 23.7.1–23.7.15. [DOI] [PubMed] [Google Scholar]
- Kim D. H., Xiao Z., Kwon S., Sun X., Ryerson D., Tkac D., Ma P., Wu S. Y., Chiang C. M., Zhou E., et al. (2015). A dysregulated acetyl/SUMO switch of FXR promotes hepatic inflammation in obesity. EMBO J. 34, 184–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Jiang C., Krausz K. W., Li Y., Albert I., Hao H., Fabre K. M., Mitchell J. B., Patterson A. D., Gonzalez F. J. (2013). Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 4, 2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R., Li X., Qiang X., Luo L., Hylemon P. B., Jiang Z., Zhang L., Zhou H. (2015). Taurocholate induces cyclooxygenase-2 expression via the sphingosine 1-phosphate receptor 2 in a human cholangiocarcinoma cell line. J. Biol. Chem. 290, 30988–31002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Cheng F., Jiang J., Zhang C., Deng X., Xu Z., Zou S., Shen X., Tang Y., Huang J. (2015). FXR antagonism of NSAIDs contributes to drug-induced liver injury identified by systems pharmacology approach. Sci. Rep. 5, 8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K., Saha P. K., Chan L., Moore D. D. (2006). Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Invest. 116, 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara T., Li F., Gonzalez F. J. (2013). FXR signaling in the enterohepatic system. Mol. Cell Endocrinol. 368, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara T., Tanaka N., Patterson A. D., Cho J. Y., Krausz K. W., Gonzalez F. J. (2011). Lithocholic acid disrupts phospholipid and sphingolipid homeostasis leading to cholestasis in mice. Hepatology 53, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z., Wang Y., Wang L., Jin W., Liu N., Pan H., Liu L., Wagman L., Forman B. M., Huang W. (2010). FXR regulates liver repair after CCl4-induced toxic injury. Mol. Endocrinol. 24, 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuschwander-Tetri B. A., Loomba R., Sanyal A. J., Lavine J. E., Van Natta M. L., Abdelmalek M. F., Chalasani N., Dasarathy S., Diehl A. M., Hameed B., et al. (2015). Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold A. M., Kim J., Finberg R., Glimcher L. H. (2001). Decreased immediate inflammatory gene induction in activating transcription factor-2 mutant mice. Int. Immunol. 13, 241–248. [DOI] [PubMed] [Google Scholar]
- Seki E., Brenner D. A., Karin M. (2012). A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 143, 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa M., Konno H., Qin J., Akiyama N., Miyauchi M., Ohashi H., Miyamoto-Sato E., Yanagawa H., Akiyama T., Inoue J. (2015). Catalytic subunits of the phosphatase calcineurin interact with NF-kappaB-inducing kinase (NIK) and attenuate NIK-dependent gene expression. Sci. Rep. 5, 10758.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinal C. J., Tohkin M., Miyata M., Ward J. M., Lambert G., Gonzalez F. J. (2000). Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744. [DOI] [PubMed] [Google Scholar]
- Takahashi S., Fukami T., Masuo Y., Brocker C. N., Xie C., Krausz K. W., Wolf C. R., Henderson C. J., Gonzalez F. J. (2016). Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 57, 2130–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N., Matsubara T., Krausz K. W., Patterson A. D., Gonzalez F. J. (2012). Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 56, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N., Takahashi S., Fang Z. Z., Matsubara T., Krausz K. W., Qu A., Gonzalez F. J. (2014). Role of white adipose lipolysis in the development of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta 1841, 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tujios S., Fontana R. J. (2011). Mechanisms of drug-induced liver injury: From bedside to bench. Nat. Rev. Gastroenterol. Hepatol. 8, 202–211. [DOI] [PubMed] [Google Scholar]
- Uehara T., Bennett B., Sakata S. T., Satoh Y., Bilter G. K., Westwick J. K., Brenner D. A. (2005). JNK mediates hepatic ischemia reperfusion injury. J. Hepatol. 42, 850–859. [DOI] [PubMed] [Google Scholar]
- Ulzurrun E., Stephens C., Crespo E., Ruiz-Cabello F., Ruiz-Nunez J., Saenz-Lopez P., Moreno-Herrera I., Robles-Diaz M., Hallal H., Moreno-Planas J. M., et al. (2013). Role of chemical structures and the 1331T>C bile salt export pump polymorphism in idiosyncratic drug-induced liver injury. Liver Int. 33, 1378–1385. [DOI] [PubMed] [Google Scholar]
- Wang A. Y., Lian L. H., Jiang Y. Z., Wu Y. L., Nan J. X. (2010). Gentiana manshurica Kitagawa prevents acetaminophen-induced acute hepatic injury in mice via inhibiting JNK/ERK MAPK pathway. World J. Gastroenterol. 16, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Salem M., Yousef I. M., Tuchweber B., Lam P., Childs S. J., Helgason C. D., Ackerley C., Phillips M. J., Ling V. (2001). Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc. Natl. Acad. Sci. U.S.A. 98, 2011–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. D., Chen W. D., Wang M., Yu D., Forman B. M., Huang W. (2008). Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster C. R., Anwer M. S. (2016). Hydrophobic bile acid apoptosis is regulated by sphingosine-1-phosphate receptor 2 in rat hepatocytes and human hepatocellular carcinoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G865–G873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. W., Siow Y. L., Karmin O. (2008). Homocysteine induces monocyte chemoattractant protein-1 expression in hepatocytes mediated via activator protein-1 activation. J. Biol. Chem. 283, 1282–1292. [DOI] [PubMed] [Google Scholar]
- Xiong X., Wang X., Lu Y., Wang E., Zhang Z., Yang J., Zhang H., Li X. (2014). Hepatic steatosis exacerbated by endoplasmic reticulum stress-mediated downregulation of FXR in aging mice. J. Hepatol. 60, 847–854. [DOI] [PubMed] [Google Scholar]
- Yamazaki M., Miyake M., Sato H., Masutomi N., Tsutsui N., Adam K. P., Alexander D. C., Lawton K. A., Milburn M. V., Ryals J. A., et al. (2013). Perturbation of bile acid homeostasis is an early pathogenesis event of drug induced liver injury in rats. Toxicol. Appl. Pharmacol. 268, 79–89. [DOI] [PubMed] [Google Scholar]
- Zamara E., Galastri S., Aleffi S., Petrai I., Aragno M., Mastrocola R., Novo E., Bertolani C., Milani S., Vizzutti F., et al. (2007). Prevention of severe toxic liver injury and oxidative stress in MCP-1-deficient mice. J. Hepatol. 46, 230–238. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Li F., Patterson A. D., Wang Y., Krausz K. W., Neale G., Thomas S., Nachagari D., Vogel P., Vore M., et al. (2012). Abcb11 deficiency induces cholestasis coupled to impaired beta-fatty acid oxidation in mice. J. Biol. Chem. 287, 24784–24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziraldo C., Vodovotz Y., Namas R. A., Almahmoud K., Tapias V., Mi Q., Barclay D., Jefferson B. S., Chen G., Billiar T. R., et al. (2013). Central role for MCP-1/CCL2 in injury-induced inflammation revealed by in vitro, in silico, and clinical studies. PLoS One 8, e79804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.