

See Gratwicke and Foltynie (doi:10.1093/brain/awx333) for a scientific commentary on this article.
Cognitive impairments in Parkinson’s disease show variable onset, severity and progression. Ray et al. demonstrate that the future cognitive status of newly diagnosed patients can be predicted from the volume of the nucleus basalis of Meynert, with implications for the development of interventions for cognitive decline in Parkinson’s disease dementia.
Keywords: mild cognitive impairment, Parkinson’s disease, dementia, structural MRI
Abstract
See Gratwicke and Foltynie (doi:10.1093/brain/awx333) for a scientific commentary on this article.
Cognitive impairments are a prevalent and disabling non-motor complication of Parkinson’s disease, but with variable expression and progression. The onset of serious cognitive decline occurs alongside substantial cholinergic denervation, but imprecision of previously available techniques for in vivo measurement of cholinergic degeneration limit their use as predictive cognitive biomarkers. However, recent developments in stereotactic mapping of the cholinergic basal forebrain have been found useful for predicting cognitive decline in prodromal stages of Alzheimer’s disease. These methods have not yet been applied to longitudinal Parkinson’s disease data. In a large sample of people with de novo Parkinson’s disease (n = 168), retrieved from the Parkinson’s Progressive Markers Initiative database, we measured cholinergic basal forebrain volumes, using morphometric analysis of T1-weighted images in combination with a detailed stereotactic atlas of the cholinergic basal forebrain nuclei. Using a binary classification procedure, we defined patients with reduced basal forebrain volumes (relative to age) at baseline, based on volumes measured in a normative sample (n = 76). Additionally, relationships between the basal forebrain volumes at baseline, risk of later cognitive decline, and scores on up to 5 years of annual cognitive assessments were assessed with regression, survival analysis and linear mixed modelling. In patients, smaller volumes in a region corresponding to the nucleus basalis of Meynert were associated with greater change in global cognitive, but not motor scores after 2 years. Using the binary classification procedure, patients classified as having smaller than expected volumes of the nucleus basalis of Meynert had ∼3.5-fold greater risk of being categorized as mildly cognitively impaired over a period of up to 5 years of follow-up (hazard ratio = 3.51). Finally, linear mixed modelling analysis of domain-specific cognitive scores revealed that patients classified as having smaller than expected nucleus basalis volumes showed more severe and rapid decline over up to 5 years on tests of memory and semantic fluency, but not on tests of executive function. Thus, we provide the first evidence that volumetric measurement of the nucleus basalis of Meynert can predict early cognitive decline. Our methods therefore provide the opportunity for multiple-modality biomarker models to include a cholinergic biomarker, which is currently lacking for the prediction of cognitive deterioration in Parkinson’s disease. Additionally, finding dissociated relationships between nucleus basalis status and domain-specific cognitive decline has implications for understanding the neural basis of heterogeneity of Parkinson’s disease-related cognitive decline.
Introduction
Cognitive impairment is a major non-motor complication of Parkinson’s disease and is associated with significant disability and reduced quality of life (Lawson et al., 2016). The estimated one-third of patients with Parkinson’s disease who have cognitive impairment even very early in the disease (Foltynie et al., 2004) have an increased risk of developing Parkinson’s disease dementia within a short timeframe (Foltynie et al., 2004; Janvin et al., 2006). However, early cognitive impairments in Parkinson’s disease are complex and multifarious; individual patients experience impairments in different cognitive domains, with variable onset, severity and progression rates (Kehagia et al., 2013). Yet, the development of interventions that could delay the onset of Parkinson’s disease dementia depends on accurate prognostication of individual patients at early disease stages, making the development of predictive biomarkers a critical area of Parkinson’s disease research.
Fortunately, longitudinal clinical data—in part collected through large-scale, multicentre initiatives—are beginning to reveal clinical, genetic, molecular, neuroimaging and electrophysiological biomarkers that can predict the rate and severity of cognitive decline in Parkinson’s disease (Dujardin et al., 2004; Williams-Gray et al., 2009a, b, 2013; Siderowf et al., 2010; Bohnen et al., 2011; Chen-Plotkin et al., 2011; Klassen et al., 2011; Weintraub et al., 2011; Mak et al., 2015; Schrag et al., 2017). Most recently, Schrag et al. (2017), using data collected as part of the Parkinson’s Progressive Marker’s Initiative (PPMI), an ongoing multicentre longitudinal study in Parkinson’s disease, reported that a simple algorithm combining age, sensory, behavioural, biological, and neuroimaging markers at the time of Parkinson’s disease diagnosis can predict global cognitive decline after 2 years. Progress in this field has huge implications for understanding the pathophysiology of cognitive decline and Parkinson’s disease dementia, and for stratification of at-risk participants in clinical trials. However, given the known involvement of the cholinergic system in Parkinson’s disease-related cognitive impairments (Candy et al., 1983; Liu et al., 2015), the absence of cholinergic biomarkers in these studies is a recognized omission (Caspell-Garcia et al., 2017).
Previous work has aimed to understand the pattern of cognitive impairments across different cognitive domains in Parkinson’s disease, and whether these relate differentially to the development of Parkinson’s disease dementia (Williams-Gray et al., 2009a). A synthesis of research in this field led to a ‘dual-syndrome’ hypothesis (Kehagia et al., 2013). This posits that a distinction can be made between fronto-striatal executive impairments that are dopaminergically mediated, and which may not herald the progression of serious cognitive decline, and a dementia syndrome typified by cholinergically-mediated memory and visuospatial deficits associated with posterior cortical and temporal lobe dysfunction. Evidence for this idea is accumulating from large-scale longitudinal studies, which have additionally implicated distinct genetic contributions to the two syndromes (Williams-Gray et al., 2009a, b, 2013). Importantly, these findings imply that effective biomarkers will predict different long-term cognitive outcomes for dissociated cognitive syndromes.
Structural and functional neuroimaging can be usefully incorporated with genetic and neuropsychological markers to better understand cognitive symptom heterogeneity in Parkinson’s disease (for reviews see Shi et al., 2010; Baggio et al., 2015; Strafella et al., 2017). In particular, imaging of grey matter volume reductions in Parkinson’s disease offers a relatively low cost, widely available and non-invasive procedure that can reveal the cortical and subcortical changes associated with cognitive symptom progression (Weintraub et al., 2011; Hanganu et al., 2014; Lee et al., 2014a). Recent longitudinal work has revealed that mild cognitive impairment (MCI) in Parkinson’s disease is associated with the development of more widespread cortical atrophy than in those with normal cognition (Mak et al., 2015). In that study, the MCI group had atrophy extending from frontal regions to posterior temporal cortices over 18 months of follow-up, potentially reflecting additional pathophysiological processes including multisystem neurotransmitter decline.
In early work on cognitive decline in Parkinson’s disease, autopsy studies identified degeneration of the cholinergic basal forebrain (cBF), in particular the nucleus basalis of Meynert (NBM), and ensuing cortical cholinergic depletion as major determinants of cognitive complications. This represents a neuropathological feature Parkinson’s disease shares with Alzheimer’s disease dementia (Candy et al., 1983; Liu et al., 2015). Previous efforts to study the structural status of the cBF in vivo on MRI scans have relied on manual delineation of a relatively gross anatomical region identified by the grey matter ventral to the crossing of the anterior commissure as an external landmark, typically termed ‘substantia innominata’. In Parkinson’s disease dementia, cBF degeneration has been hitherto detected in vivo as volumetric reduction of the substantia innominata (Choi et al., 2012). Structural changes of this region have also been identified in non-demented Parkinson’s disease patients, and shown to be predictive of imminent conversion from MCI to Parkinson’s disease dementia (Lee et al., 2014b). However, there is little exploration of how volume reductions in the cBF and its functionally and anatomically distinct subdivisions are related to current or subsequent cognitive decline in newly diagnosed Parkinson’s disease patients.
Recent developments in stereotactic mapping of the cBF nuclei in the human brain have rendered this region accessible to detailed MRI-based morphometric assessments, providing an in vivo surrogate marker of cBF structural integrity (Kilimann et al., 2014). These novel methods have been informative in Alzheimer’s disease research, revealing that subregional cBF degeneration (in particular posterior parts of the NBM) can already be detected at predementia stages of the disease, where it is associated with emerging cognitive deficits (Grothe et al., 2010, 2012, 2013, 2016; Schmitz and Spreng, 2016). Such findings offer hope that a more detailed analysis of cBF structure than has been available so far may provide an early marker for cognitive impairments and future dementia even in de novo Parkinson’s disease.
In non-demented Parkinson’s disease, as expected given considerable heterogeneity of cognitive symptoms, cholinergic denervation revealed by in vivo PET imaging is variable (Bohnen et al., 2012, 2015). Thus, in analogy to the approach in Bohnen et al. (2015) we use a binary classification method to define participants with newly diagnosed Parkinson’s disease with reduced cBF volumes (based on a normative sample), measured using detailed MRI-based morphometry based on stereotactic mappings of cBF nuclei. Additionally, we track the cognitive status of participants classified as having reduced or within normal range cBF volumes at Parkinson’s disease diagnosis over a period of up to 5 years to determine the predictive utility of this distinction for the development of cognitive decline.
Patients and methods
Study design
The PPMI is a longitudinal, multicentre study to assess progression of clinical features, imaging and biological markers in patients with Parkinson’s disease (de novo Parkinson’s disease, prodromal and subjects with specific genetic mutations) and healthy controls. It is a public-private partnership funded by the Michael J. Fox Foundation for Parkinson’s research and funding partners, a full list of whom can be found at www.ppmi-info.org/fundingpartners.
To enrol in the study, participants with Parkinson’s disease must have had a diagnosis of Parkinson’s disease for 2 years or less at the screening visit, and be at Hoehn and Yahr stage I or II at the baseline visit. Participants are also not expected to require Parkinson’s disease medication within at least 6 months from the baseline visit. Control participants were excluded if they had a first-degree relative with idiopathic Parkinson’s disease or a Montreal Cognitive Assessment (MoCA) score <26. The inclusion criteria for the PPMI database are described in detail in the PPMI study documents available at http://www.ppmi-info.org/data. For up-to-date information on the study, visit www.ppmi-info.org.
Participants
Participants selected from the PPMI for the current study were all those with T1 MRI scans available at 3 T, acquired on Tim Trio 3 T scanners (Siemens), and with acquisition parameters as described below. These were 172 participants with Parkinson’s disease and 76 control participants, who had been recruited at 11 centres worldwide.
Following manual investigation of brain images (see ‘MRI preprocessing’ section), four Parkinson’s disease brain images were removed. Thus, of the 168 Parkinson’s disease patients with baseline neuroimaging and at least partial cognitive data (i.e. scores available on at least the MoCA at baseline), 143, 149, 144, 122 and 53 had at least partial cognitive data at 1-, 2-, 3-, 4- and 5-year follow-up visits, respectively.
Motor, cognitive and clinical assessments
Alongside the Unified Parkinson’s Disease Rating Scale (UPDRS) for motor symptom severity, and the MoCA, the cognitive status of participants in the PPMI was categorized by study clinicians as normal cognition, MCI, or Parkinson’s disease dementia based on Level 1 criteria developed by the Movement Disorder Society (Litvan et al., 2012). In brief, MCI was determined by the following factors: cognitive complaint by either the patient or informant (spouse, family member or friend), at least two test scores (of the six reported below) from at least one domain with >1.0 standard deviation (SD) below the standardized mean, and no functional impairment as a result of cognitive impairment. Parkinson’s disease dementia was determined as cognitive impairment defined as at least one test score from two domains >1.5 SD below the standardized mean along with functional impairment due to cognitive impairment. A full description of these methods is available at http://www.ppmi-info.org/study-design/research-documents-and-sops/.
This element of the PPMI was implemented at a late stage, meaning that many patients only have cognitive categorization from 2-year follow-up visit onwards. Thus, to compare cBF volumes between patients with and without evidence of cognitive impairment at baseline, we applied the thresholding criteria for MCI to patients’ baseline cognitive task scores. Since we do not have information on patient/informant complaint of cognitive impairment at this time point, we applied a more stringent threshold of >1.5 SD below the control sample’s mean. Patients falling below these criteria were defined as ‘suspected MCI’, to reflect the lower level of confidence in this classification.
In secondary analyses we assessed the cognitive domain specificity of reduced cBF volume by examining scores on each of the separate cognitive tests employed in the PPMI. These were the Hopkins Verbal Learning Test (HVLT) (immediate and delayed recall memory), semantic fluency (language), the Benton test of line orientation (visuospatial ability), the Symbol Digit Substitution test (attention/working memory) and the Letter Number Sequencing task (attention/working memory). Scores on these tests were collected at baseline and all available annual follow-up visits up to 5 years from enrolment.
MRI acquisition
MRI data acquisition was performed, across multiple centres, on Tim Trio 3 T scanners (Siemens). High-resolution structural images were collected by using a 3D magnetization-prepared rapid acquisition gradient echo (MP-RAGE) T1-weighted sequence with the following parameters: sagittal section thickness, 1.0 mm; no gap; repetition time, 2300 ms; echo time, 2.98 ms; flip angle, 9°; field of view, 240 × 256 mm; matrix size, 240 × 256; inversion time, 900 ms; voxel size, 1 × 1 × 1 mm3. The image plane aligned in the sagittal plane along the hemispheric fissure and axially along the anterior/posterior commissure plane. The 176 sections covered the entire brain from ear to ear and the bottom of the cerebellum to the vertex.
Stereotactic map of the cholinergic basal forebrain
The cBF map was created using stereotactic mapping of cBF nuclei as described in Kilimann et al. (2014). In brief, the map was derived from combined post-mortem MRI and subsequent histological preparation of a brain specimen of a 56-year-old male who had died without any evidence of cognitive decline or psychiatric illnesses. Cerebral MRI scans were performed in situ 15 h after death as well as after the brain was removed from the skull and dehydrated for histological preparation. Cholinergic nuclei were identified and delineated on digital pictures of the stained brain slices following Mesulam’s nomenclature (Mesulam et al., 1983), and the cBF regions identified on the histological slices were manually transferred from the digital pictures into the corresponding magnetic resonance slices of the dehydrated brain. Transformation of the delineations from the space of the dehydrated brain into the space of the in situ brain scan was based on an initial 12-parameter affine transformation followed by a high-dimensional non-linear registration between the two brain scans (Ashburner and Friston, 1999). The improved grey–white matter contrast of the in situ brain scan enabled the use of the highly accurate high-dimensional DARTEL (Diffeomorphic Anatomic Registration using Exponentiated Lie algebra) registration method (Ashburner, 2007; Klein et al., 2009) for the final transformation from in situ space into Montreal Neurological Institute (MNI) standard space. The stereotactic cBF map distinguishes different cholinergic subdivisions within the basal forebrain, including cell clusters corresponding to the medial septum, vertical and horizontal limb of the diagonal band, and NBM (Ch1–4 according to Mesulam’s nomenclature; Mesulam et al., 1983) (Fig. 1).
Figure 1.

Cholinergic basal forebrain regions of interest. Full details of how the subregional cBF map was generated can be found in Kilimann et al. (2014). The regions of interest have been superimposed on the T1-weighted high-resolution ICBM 2009a Nonlinear Asymmetric MNI152 standard space template available at http://www.bic.mni.mcgill.ca/ServicesAtlases/ICBM152NLin2009. Slices from left to right are coronal slices −8, 3, 6, 9. Red represents the Ch1-2 region of interest corresponding to the medial septum and vertical limb of the diagonal band, green is the nucleus subputaminalis/anterior-lateral NBM and the horizontal limb of the diagonal band (not selected as an region of interest in the current paper), Light blue + dark blue represents the Ch4 region of interest corresponding to the NBM, and dark blue represents the Ch4p region of interest corresponding to the posterior NBM.
MRI preprocessing
Scans acquired at baseline were downloaded from the PPMI in May 2016. Following our previously established method for MRI-based cBF volumetry (Grothe et al., 2012, 2014, 2016), the T1 scans were automatically segmented into grey matter, white matter, and CSF partitions of 1.5-mm isotropic voxel size using the segmentation routine of the VBM8 toolbox (http://www.neuro.uni-jena.de/vbm/download/) running under SPM8 (http://www.fil.ion.ucl.ac.uk/spm/software/spm8/). The resulting grey and white matter partitions of each subject in native space were then high-dimensionally registered to MNI space using DARTEL (Ashburner, 2007). The grey matter segments were then warped using the individual flow fields resulting from the DARTEL registration, and voxel values were modulated for volumetric changes introduced by the high-dimensional normalization. Thus, the total amount of grey matter volume present before warping was preserved. All preprocessed grey matter maps were required to pass a visual inspection for overall segmentation and registration accuracy. Four participants (in the Parkinson’s disease group) did not pass this inspection, and therefore their data were removed from further analyses.
Subregional cBF grey matter volumes were calculated by summing up the modulated grey matter voxel values within the respective region of interest masks in template space (Ashburner, 2009). These volumes and global grey matter were further scaled (by ANCOVA) to total intracranial volume, calculated as the sum of total grey matter, white matter and CSF volumes, as a proxy for head size. We considered two distinct subdivisions of the cBF that can be clearly separated anatomically, and are characterized by differential cortical projection preferences and associations with specific cognitive and behavioural functions (Mesulam et al., 1983; Muir, 1997; Zaborszky et al., 2015). These correspond to an anterior-medial cBF region of interest that combines the medial septum (Ch1) and the vertical limb of the diagonal band (Ch2), and a more posterior region of interest covering the NBM (Ch4). Additionally, hypothesis-driven analyses were applied to a posterior NBM subdivision (Ch4p) given that previous in vivo analyses in MCI and Alzheimer’s disease have found a particular involvement of this region (Grothe et al., 2012, 2013; Kilimann et al., 2014), which is consistent with neuropathological evidence for a subregion-specific vulnerability to Alzheimer’s disease-related neurodegeneration (Vogels et al., 1990).
Identification of participants with cholinergic basal forebrain volumes below the range in controls
To define participants with cBF volumes outside the range of controls, linear regression models were constructed to predict regional cBF volumes from total intracranial volume and age in the control sample (sex was not a significant contributor in any of these models and was therefore not included in the final models). These models were able to significantly predict volumes in all cBF regions, with R2 values between 0.69 and 0.74. Beta weights from these regressions were used to calculate expected cBF volumes in the Parkinson’s disease sample, and participants with cBF volumes below/above the lower 68% confidence limit (corresponding to −1 SD) of the control sample were classified as having reduced/normal-range cBF volumes, subsequently referred to as the cBF− and cBF+ Parkinson’s disease groups, respectively.
Statistical analysis
General linear model-based statistical analyses were conducted in SPSS statistics 22 and MatLab r2016a. Linear mixed model analyses were carried out in STATA/SE 14.2.
Baseline cholinergic basal forebrain volumes, demographics and clinical scores
T-tests and univariate analyses (controlling for age, sex and disease duration) were used to compare demographics, clinical and cognitive measures, and total intracranial volume-normalized cBF region of interest volumes between control and Parkinson’s disease participants, and between control subjects and patients with Parkinson’s disease with and without suspected MCI. Pearson’s partial correlations controlling for age, sex and disease duration were computed for the relationship between volume of each cBF region of interest and motor assessment.
Baseline cholinergic basal forebrain volumes, MoCA scores and cognitive categorization with longitudinal follow-up
Hierarchical multiple regression models were constructed to determine the predictive utility of baseline cBF volumes on change in MoCA scores (from baseline) at the 2-year follow-up visit, with age, sex, disease duration and baseline MoCA scores entered simultaneously in the first step, and cBF volumes entered in the second.
In addition, Cox proportional hazards regression analysis was performed to model the risk of MCI or Parkinson’s disease dementia over the available follow-up period as a function of being classified as either cBF− or cBF+ at baseline (with each region of interest included in separate Cox regressions). Time to event (categorization of MCI or Parkinson’s disease dementia) was calculated as the number of months since Parkinson’s disease diagnosis until an MCI or Parkinson’s disease dementia classification was made. MCI classifications based on Level 1 MDS criteria can fluctuate in early disease stages (Lawson et al., 2017), with patients frequently being later classified as normal cognition. To mitigate the potential for misdiagnosis, patients with MCI classifications on a single visit, who were subsequently and consistently classified as normal cognition (provided at least two subsequent visits were available to confirm normal cognition classification) were deemed to be normal cognition at the end of their available visits. Nine patients without full cognitive testing on visits following baseline could not be included. Additional covariates entered into the model were age, gender, baseline MoCA scores, disease duration and total intracranial volume-normalized global grey matter volumes. Due to many patients’ first cognitive categorizations being made at the 2-year visit, there were 30 patients with MCI or Parkinson’s disease dementia at their first categorization, meaning we could not be sure that patients did not already meet the criteria for MCI at baseline. Thus, the Cox regression was repeated in a more restricted sample to ensure classification as cBF+/cBF− was predictive of future cognitive decline, and did not depend on patients who may have deteriorated cognitively very early in their disease. In the repeated analysis, the criteria for suspected MCI (defined above) were applied at the baseline visit, and only participants without suspected MCI were included. Subject numbers contributing to each of the Cox regressions are reported in the ‘Results’ section to avoid confusion.
Baseline cholinergic basal forebrain volumes and domain-specific cognitive assessments with longitudinal follow-up
For cBF regions of interest showing a significant effect for predicting changes in MoCA or cognitive categorization (MCI/Parkinson’s disease dementia), linear mixed modelling was used to investigate the mean decline in scores on the full PPMI cognitive battery over time (up to 5 years) in the cBF− and cBF+ groups, controlling for global grey matter, age, sex and disease duration. Mixed effects models with maximum likelihood estimators were used. Such an approach is preferred as it can effectively handle the hierarchical nature of longitudinal, repeated-measures data, and does not require list-wise deletion in cases with missing values, as is the case with standard general linear models. Separate models for each cognitive test used test scores as outcome variables, and a first order polynomial (linear time), group (cBF− or cBF+), and the interaction between the two as fixed effects predictors. The random part of all models included participant ID and a first order polynomial. All models allowed for the intercept (cognition at baseline) and slope (the rate of change) to vary across individuals. Values reported are unstandardized beta values with 95% confidence intervals (CI).
Correction for multiple comparisons
All analyses that included the Ch1-2 and Ch4 regions of interest were Benjamini-Hochberg corrected for multiple comparisons across the two regions. For Ch4p, which has previously been implicated in Alzheimer’s disease-related degeneration (Grothe et al., 2012, 2013; Kilimann et al., 2014), results were considered significant at P < 0.05 (two-tailed). We did not have region-specific predictions in control subjects, therefore Benjamini-Hochberg corrections were applied for multiple comparisons across the three cBF regions of interest. For between-group (cBF+/cBF−) comparisons of progressive decline on the domain-specific cognitive tasks, Benjamini-Hochberg corrections were applied for multiple comparisons across the six tasks assessed. To avoid confusion, corrected P-values are noted in the ‘Results’ section.
Results
Control subjects versus patients with Parkinson’s disease at baseline
There were no significant differences in age or gender between participants with Parkinson’s disease and healthy control subjects (Table 1). As expected, in univariate comparisons controlling for age and sex, participants with Parkinson’s disease had significantly greater total scores on the UPDRS [F(1,238) = 22.25, P < 0.001], and had a greater degree of cognitive impairment as measured on the MoCA [F(1,240) = 7.58, P = 0.006].
Table 1.
Participant characteristics
| Healthy controls | Parkinson’s disease | |||
|---|---|---|---|---|
| n = 76, female = 26 | n = 168, female = 63 | |||
| Mean | SD | Mean | SD | |
| Age, years | 59.89 | 10.18 | 61.52 | 9.42 |
| Disease duration, months | 7.23 | 7.52 | ||
| UPDRS-MDS* | 3.47 | 3.67 | 30.79 | 13.06 |
| MoCA | ||||
| At baseline* | 28.25 | 1.16 | 27.49 | 2.13 |
| At 2-year follow-up* | 27.46 | 2.18 | 26.66 | 2.74 |
| HVLT (immediate recall) | 25.66 | 4.42 | 25.21 | 5.25 |
| HVLT (delayed recall) | 9.08 | 2.29 | 8.67 | 2.49 |
| Letter number sequencing | 11.39 | 2.46 | 10.65 | 2.79 |
| Symbol digit substitution* | 46.59 | 9.96 | 41.78 | 9.7 |
| Semantic fluency | 51.72 | 10.53 | 49.52 | 11.3 |
| Benton JLO | 13.41 | 1,57 | 12.85 | 2.01 |
HVLT = Hopkins Verbal Learning Test; JLO = judgement of line orientation; UPDRS = Unified Parkinson’s Disease Rating Scale-Movement Disorder Society.
*Significantly different between participants with Parkinson’s disease and healthy controls at P < 0.05 (age corrected).
On domain-specific assessments at baseline, Parkinson’s disease patients had worse scores on the Symbol Digit Modalities task: [F(1,237) = 11.43, P = 0.001]; and a tendency for poorer scores on the Letter-Number Sequencing [F(1,237) = 3.03, P = 0.08] and Benton task [F(1,237) = 3.58, P = 0.06]. Performance was not significantly different to controls on all other tests: memory [immediate recall, F(1,237) = 0.26, P = 0.61; delayed recall, F(1,237) = 0.12, P = 0.28], semantic fluency [F(1,237) = 2.04, P = 0.16].
In both groups, age was significantly correlated with all total intracranial volume-normalized cBF regional volumes (Controls: r > −0.46, P < 0.001 in all regions; Parkinson’s disease: r > −0.49; P < 0.001 in all regions). After controlling for age and sex, univariate tests revealed there were no significant differences in Ch1-2, Ch4 or Ch4p volume between controls and participants with Parkinson’s disease as a whole [Ch1-2: F(1,240) = 0.41, P = 0.52 corrected; Ch4: F(1,240) = 0.09, P = 0.76 corrected; Ch4p: F(1,240) = 1.81, P = 0.19]. However, in univariate tests comparing cBF region of interest volumes between controls and Parkinson’s disease patients with, and without suspected MCI at baseline, we found smaller Ch4p [F(2,236) = 3.09 P = 0.047], but not Ch4 or Ch1-2 volumes [Ch1-2: F(2,236) = 0.72, P = 0.48; Ch4: F(2,236) = 2.25, P = 0.11]. Post hoc tests revealed group differences in the Ch4p region were restricted to comparisons between controls and Parkinson’s disease patients with suspected MCI [F(1,98) = 6.95, P = 0.01; Fig. 2].
Figure 2.
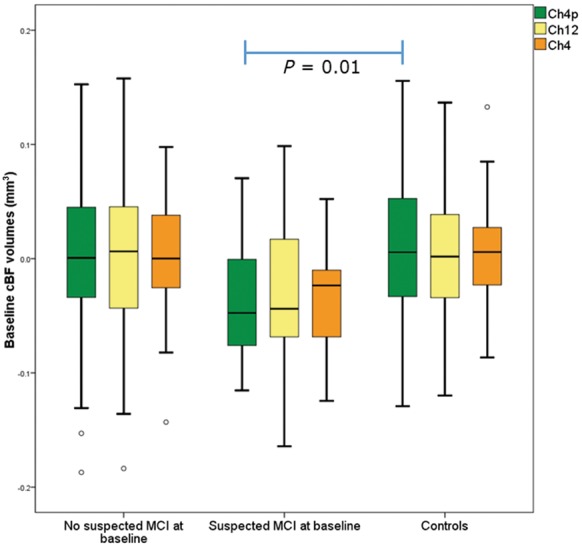
Box plot of cBF region of interest volumes at baseline in controls and Parkinson’s disease patients with and without suspected MCI. CBF region of interest volumes in controls, patients with Parkinson’s disease with and without suspected MCI, with the Ch4p region (corresponding to the posterior NBM) being significantly different between controls and Parkinson’s disease patients with suspected MCI.
There were no significant partial correlations (controlling for age, sex and disease duration) between cBF volumes and Parkinson’s disease patients’ scores on the UPDRS part III (r < −0.12, P > 0.22 in all regions).
Baseline cholinergic basal forebrain volumes and longitudinal MoCA scores
In controls, a hierarchical linear regression analysis with age, sex and baseline MoCA entered as a first step revealed there was no significant relationship between change in MoCA scores after 2 years and volumes of any cBF region of interest (β < 0.22, P > 0.12 in all regions).
In contrast, in Parkinson’s disease, Ch4 and Ch4p volumes, but not Ch1-2 volumes, were a significant predictor of 2-year change in MoCA score after controlling for age, sex, disease duration and baseline MoCA score (Ch4: β = −0.23, ΔR2 = 0.26, P = 0.04 corrected; Ch4p: β = −0.20, P = 0.04; Ch1-2: β = −0.15, P = 0.07 corrected).
Binary cBF−/cBF+ classifications at baseline and longitudinal cognitive categorizations
For each of the examined cBF regions of interest, Ch1-2, Ch4 and Ch4p, there were 32, 33, and 32 participants with Parkinson’s disease, respectively, who had regional volumes that were smaller than expected given the control range. As well as smaller regional cBF volumes, global grey matter volumes were lower in the cBF− groups for each region (Ch1-2: t = 6.12; P < 0.001 corrected; Ch4: t = 4.97; P < 0.001 corrected; Ch4p: t = 3.07; P = 0.003). As such, global grey matter is included as a covariate in all subsequent analyses. Different, but partially overlapping, participants were identified as cBF− and cBF+ when classifications were based on the different regions of interest: participants with smaller volumes in all three regions: n = 14; in two regions: n = 14; in one region only: n = 27.
Cox proportional hazards models revealed that age and global grey matter were independently associated with increased risk of being categorized as MCI or Parkinson’s disease dementia (age: Wald = 4.93, P = 0.03; global grey matter: Wald = 14.02, P < 0.001). Classification as cBF−/cBF+ in the Ch1-2 (n = 29/129) and Ch4p regions did not significantly alter risk (Ch1-2: Wald = 0.25, P = 0.87 corrected; Ch4p (n = 29/129): Wald = 0.56, P = 0.45). However, addition of cBF+/cBF− classification to the model revealed that having smaller than expected Ch4 volume (n = 30/128) did add independently to risk of being diagnosed with MCI or Parkinson’s disease dementia (Wald = 10.36, P = 0.002 corrected, hazard ratio = 3.51) (Fig. 3). Restricting this analysis to only patients without suspected MCI at baseline revealed that classification as Ch4 ± (n = 23/112) continued to incur risk for future MCI (Wald = 5.60, P = 0.02, hazard ratio = 3.00).
Figure 3.
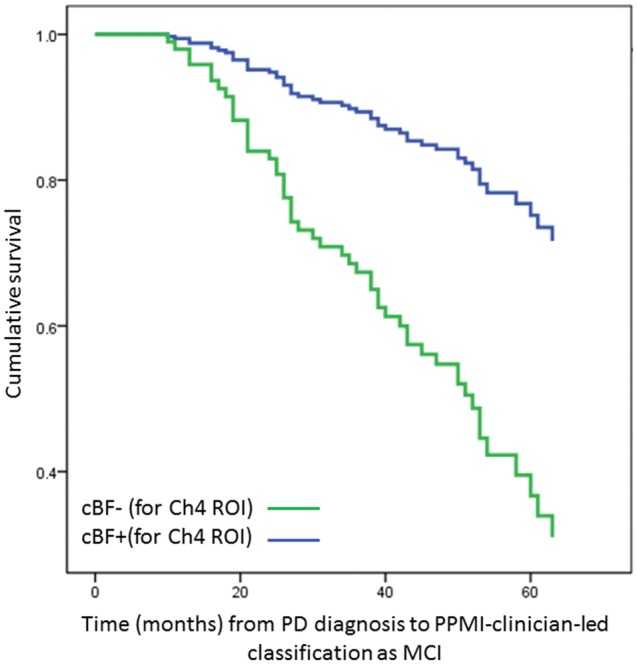
Survival function for categorization as MCI or Parkinson’s disease dementia in cBF− and cBF+ patients. Cox proportional hazards survival functions for months between diagnosis of Parkinson’s disease and categorization as MCI or Parkinson’s disease dementia, separated by cBF− (green) versus cBF+ (blue) status for the Ch4 region (i.e. patients with Parkinson’s disease with reduced volumes in the NBM versus those with volumes within range of controls, respectively). PD = Parkinson’s disease; ROI = region of interest.
Given these results, in all further analyses, classifications as cBF− or cBF+ are based only on volumes in the Ch4 region.
Domain-specific cognitive tests in cBF− and cBF+ patients
The cBF− and cBF+ groups were of similar age (t = 0.56, P = 0.58). Linear growth curve models evaluated dissociations in progressive decline over 5 years in cognitive scores between the cBF− and cBF+ groups. When controlling for global grey matter, age, sex and disease duration, and following false discovery rate correction for the six tests measured, these analyses revealed that scores on all tasks were not significantly different between the groups at baseline [MoCA: b = 0.08 (−0.81, 0.97), P = 0.868; delayed recall: b = 0.30 (−0.77, 1.37), P = 0.577; immediate recall: b = 0.42 (−1.72, 2.57), P = 0.700; semantic fluency: b = −1.34 (−5.81, 3.13), P = 0.556; Benton: b = −0.75 (−1.58, −0.08), P = 0.075; Symbol-digit Substitution: b = −4.16 (−7.77, −0.55), P = 0.168; letter number sequencing b = 0.29 (−0.67, 1.24), P = 0.558]. However, scores on the MoCA, delayed and immediate recall, and semantic fluency declined significantly faster in the cBF− group [MoCA: b = −0.45 (−0.74, −0.15), P = 0.003; immediate recall: b = −0.37 (−0.59, −0.14), P = 0.002; delayed recall: β = −0.74 (−1.17, −0.31), P = 0.001; semantic fluency: β = −0.99 (−1.78, −0.19), P = 0.015] (Fig. 4A–D).
Figure 4.

Linear growth curves in cBF− and cBF+ patients. Linear growth curves modelled in the cBF− versus cBF+ groups (patients with Parkinson’s disease with reduced volumes in the NBM versus those with volumes within range of controls, respectively). (A–D) A faster decline over 5 years on the MoCA, delayed and immediate recall on the HVLT, and semantic fluency.
On the Symbol-Digit Substitution task, the patients got progressively worse on the task over time [β = −0.79 (−1.14, −0.43), P < 0.001], at a similar rate between groups [β = −0.11 (−0.92, 0.70), P = 0.788]. Finally, on the Letter-Number Sequencing task and the Benton task, the patients did not show any decline over time [Letter-Number Sequencing: β = −0.09 (−0.18, 0.01), P = .070], Benton test [β = −0.05 (−0.23, 0.14), P = 0.627].
Discussion
The present study is the first to assess regional cBF atrophy in very early stage de novo Parkinson’s disease participants alongside longitudinal evaluation of cognitive status and multi-domain cognitive assessment. Our findings broadly confirm previous autopsy and in vivo PET studies that implicate the cBF in progressive cognitive decline leading to dementia in Parkinson’s disease (Bohnen et al., 2003; Hilker et al., 2005; Shimada et al., 2009). Moreover, we provide first evidence that degeneration in a region corresponding to the NBM can be detected even at very early disease stages in patients with suspected MCI, and can be used to predict future development of MCI even in patients without current cognitive impairment. These findings, in the context of a lack of group-level differences in NBM volumes between Parkinson’s disease patients and controls at baseline, confirm substantial heterogeneity of cholinergic involvement in de novo Parkinson’s disease. Thus, while such heterogeneity may obscure differences between control and Parkinson’s disease patients as a whole, our findings suggest it can be useful for dissociating Parkinson’s disease patients at risk for future cognitive impairment and Parkinson’s disease dementia. Thus, volumetric measurement of the NBM should be evaluated as an additional parameter alongside combined clinical, genetic, and other established neuroimaging biomarkers to improve prognostication in Parkinson’s disease-related cognitive decline. Future work will therefore aim to extend the current analysis to determine the additional contribution of NBM volumetry to the recently reported predictive models (Caspell-Garcia et al., 2017; Schrag et al., 2017) using the entire PPMI sample.
Given the known involvement of the cBF in Parkinson’s disease and Parkinson’s disease-related cognitive decline (Bohnen and Albin, 2009) there is a clear interest in developing methods to measure degeneration in this brain region in vivo. Our methods were based on a recently developed automated morphometry approach that allows comprehensive assessment of cBF volume based on reference to a stereotactic map of the cBF nuclei (Grothe et al., 2012, 2016; Kilimann et al., 2014). In previous studies, volumetry of the cBF has relied on measurement of the substantia innominata as a proxy measure. These studies have revealed reduced substantia innominata volumes from Hoehn and Yahr stage 2 (Ziegler et al., 2013), in patients with Parkinson’s disease dementia compared to those with Parkinson’s disease and MCI (Choi et al., 2012), and in Parkinson’s disease patients with MCI who subsequently developed Parkinson’s disease dementia (Lee et al., 2014b). However, substantia innominata volume does not appear to reflect atrophy occurring in patients with MCI compared to those without cognitive impairment (Choi et al., 2012), limiting its use as a predictive marker for cognitive decline at early disease stages. Thus, our finding that atrophy in the NBM can be detected in newly diagnosed Parkinson’s disease patients with suspected MCI, can predict changes in MoCA scores after 2 years across a heterogeneously progressing Parkinson’s disease group, and can be used to prospectively classify patients at risk for progressive cognitive symptoms, will be of considerable interest to Parkinson’s disease clinicians and researchers.
Classification of at-risk patients based on NBM volumes at baseline was based on a technique using regression in a healthy control group to describe expected relationships between total intracranial volume, age and NBM volumes alongside a binary classification procedure. This allowed us to define a group of Parkinson’s disease patients with smaller than expected (given their age and head size) NBM volumes. Using an arbitrary threshold of 1 SD below the control range, we found that this group had ∼3-fold elevated risk of developing MCI or Parkinson’s disease dementia. Additionally, a linear mixed modelling analysis of longitudinal year-wise cognitive assessments could corroborate a faster decline on the MoCA over 5-years of follow-up in those classified as having reduced NBM volume. Further work is now necessary in independent samples, both to corroborate our findings and to determine the optimal thresholds for defining reduced NBM volumes.
It is interesting to note that the region corresponding to the NBM as whole was most closely associated with future cognitive impairments, while in Alzheimer’s disease initial degeneration at the MCI stage (when compared to controls) seems to be in a region corresponding to the posterior NBM (Vogels et al., 1990; Grothe et al., 2010, 2012, 2013; Kilimann et al., 2014). Additionally, in the current paper, Parkinson’s disease patients with suspected MCI, when compared with controls, had significantly different cBF volumes only in the posterior NBM. This implies that more severe degeneration in the posterior NBM region is characteristic of MCI in both Alzheimer’s disease and Parkinson’s disease, but that future cognitive decline, at least in Parkinson’s disease, is best predicted by variability in the degree of degeneration throughout a more widespread cBF region. Indeed, in Alzheimer’s disease degeneration spreads to more anterior-medial regions of the cBF at the time at which patients develop clinically manifest dementia (Grothe et al., 2012, 2013). On a related note, the PPMI cognitive battery does not allow subtyping of MCI types, and the current sample likely includes patients with forms of MCI that may not herald the onset of Parkinson’s disease dementia. In future work, it will be interesting to determine if degeneration in the NBM is significantly greater at early disease stages in Parkinson’s disease patients who develop different forms of MCI, as well as those who go on to develop Parkinson’s disease dementia. In this regard, our methods may also be informative for using the degree of cBF degeneration to predict future cognitive decline in patients with prodromal Alzheimer’s disease.
Whether the involvement of NBM in Parkinson’s disease reflects a shared pathophysiological process with Alzheimer’s disease warrants further investigation. Schmitz and Spreng (2016) have recently provided compelling evidence that NBM atrophy precedes degeneration of the medial temporal lobe in Alzheimer’s disease (a hallmark of the disease), challenging the prevailing view that Alzheimer’s disease has a cortical origin, and raising the possibility that degeneration of the cBF contributes causatively to hippocampal volume losses. Given the known involvement of the hippocampus in Parkinson’s disease-related cognitive decline (Weintraub et al., 2011), it would be useful (both for biomarker development and for improving our understanding of the disease) to gain similar insight into the temporal relationships between NBM and hippocampal degeneration in Parkinson’s disease. Additionally, in Alzheimer’s disease, there are spatial and temporal relationships between NBM atrophy and pathological changes in the wider brain (Grothe et al., 2014, 2016; Teipel et al., 2014; Kerbler et al., 2015; Kilimann et al., 2017) that are also evident in Parkinson’s disease patients with cognitive decline (Jokinen et al., 2009; Apostolova et al., 2012; Gomperts et al., 2013; González-Redondo et al., 2014; Pereira et al., 2014). Thus, investigation of regional cBF degeneration in Parkinson’s disease and its relationship with cortical brain changes and Parkinson’s disease-related cognitive impairments is of high importance.
In previous work semantic fluency, memory and visuospatial function in early disease stages have been shown to be associated with later progression to Parkinson’s disease dementia (Williams-Gray et al., 2009a; Domellöf et al., 2015). Using a linear mixed model approach, we also revealed the domain-specificity of NBM-related changes in cognitive function over 5 years. While we did not observe any changes in the group as a whole on the visuospatial task, individuals classified as having reduced NBM volumes did decline more quickly and to a greater degree on the other preferentially temporal- and posterior cortex-dependant tasks (recall memory and semantic fluency). In contrast, these patients did not show any differences (to those with NBM volumes within the expected range) over 5 years on frontal tasks. Thus, our findings imply that while not identical, there are strong similarities between the type of cognitive function affected over time in patients with reduced NBM volumes at baseline and in patients who eventually progress to Parkinson’s disease dementia (Williams-Gray et al., 2009a; Domellöf et al., 2015). It is important therefore that a more fine-grained analysis of the relationship between NBM degeneration and cognitive function than is permitted by the PPMI is carried out.
The extent to which our findings can be reconciled with previous research on genotype–phenotype associations that may underlie the cognitive heterogeneity in early Parkinson’s disease warrants further investigation. Such research has revealed a strong influence of risk alleles in the genes encoding for the apolipoprotein E (APOE) and the microtubule-associated protein tau (MAPT) on cognitive decline in Parkinson’s disease and the development of dementia (Williams-Gray et al., 2009a, b; Morley et al., 2012; Mata et al., 2014; Huertas et al., 2017). Additionally, functional MRI studies in patients with early Parkinson’s disease show an association between these genotypes and reduced brain activation in medial temporal and parietal regions during memory and visuospatial tasks, respectively, but no association with fronto-striatal function (Nombela et al., 2014; Winder-Rhodes et al., 2015). Notably, both genotypes have also been linked to faster cognitive decline and development of dementia in Alzheimer’s disease (Samaranch et al., 2010; Desikan et al., 2015; Lacour et al., 2017). However, whether this genetic overlap between Alzheimer’s disease and Parkinson’s disease links to a common neuropathological pathway that affects cognitive decline in both diseases is currently largely unknown. The finding that both cBF degeneration and APOE and MAPT genotypes influence cognitive decline and progression to dementia in a transdiagnostic manner warrants further investigation into their possible neuropathological interrelation. Additionally, given the widespread connectivity of the cBF, it will be of great interest to understand the link between genetic risks for Parkinson’s disease dementia, cBF status, and functional brain networks, analogous to ongoing work aimed at understanding how brain networks are altered in those with a genetic risk for Alzheimer’s disease (Teipel et al., 2016).
Our approach for classifying patients with NBM volumes below the normal range has the potential to translate into clinical applications. Recent work on multiple modality biomarkers for Parkinson’s disease-related cognitive decline has not included a cholinergic biomarker (Caspell-Garcia et al., 2017; Schrag et al., 2017); yet the current study implies volumetry of the NBM could be usefully incorporated. The ability to define a group of patients at Parkinson’s disease diagnosis with increased risk of early development of progressive cognitive impairment leading to Parkinson’s disease dementia has important implications for stratification of patients in clinical trials of cholinomimetics and other treatments, and for understanding the disease-modifying potential of targeted interventions (Freund et al., 2009; Rolinski et al., 2012; Gratwicke et al., 2013; Wang et al., 2015). Further, similar cognitive performance in patients with reduced cBF volumes at baseline implies that structural degeneration precedes progressive cognitive deficits, and the ability to detect this degeneration as described here offers the opportunity to study Parkinson’s disease dementia at very early stages of the disease process.
Funding
M.J.G. received funding from the Alzheimer Forschung Initiative e. V. PPMI - a public-private partnership - is funded by the Michael J. Fox Foundation for Parkinsons Research and funding partners, including Abbvie, Avid, Biogen, Bristol-Myers Squibb, Covance, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Lundbeek, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Servier, Teva, UCB, and Golub Capital.
Glossary
Abbreviations
- cBF
cholinergic basal forebrain
- MCI
mild cognitive impairment
- MoCA
Montreal Cognitive Assessment
- NBM
nucleus basalis of Meynert
- PPMI
Parkinson’s Progressive Marker’s Initiative
References
- Apostolova L, Alves G, Hwang KS, Babakchanian S, Bronnick KS, Larsen JP, et al. Hippocampal and ventricular changes in Parkinson’s disease mild cognitive impairment. Neurobiol Aging 2012; 33: 2113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage 2007; 38: 95–113. [DOI] [PubMed] [Google Scholar]
- Ashburner J. Computational anatomy with the SPM software. Magn Reson Imaging 2009; 27: 1163–74. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. Nonlinear spatial normalization using basis functions. Hum Brain Mapp 1999; 7: 254–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio HC, Segura B, Junque C. Resting-state functional brain networks in Parkinson’s disease. CNS Neurosci Ther 2015; 21: 793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Albin RL. Cholinergic denervation occurs early in Parkinson disease. Neurology 2009; 73: 256–7. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Albin RL, Müller ML, Petrou M, Kotagal V, Koeppe RA, et al. Frequency of cholinergic and caudate nucleus dopaminergic deficits across the predemented cognitive spectrum of Parkinson disease and evidence of interaction effects. JAMA Neurol 2015; 72: 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, Koeppe RA, Davis JG, et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol 2003; 60: 1745–8. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Koeppe RA, Minoshima S, Giordani B, Albin RL, Frey KA, et al. Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J Nucl Med 2011; 52: 848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Müller ML, Kotagal V, Koeppe RA, Kilbourn MR, Gilman S, et al. Heterogeneity of cholinergic denervation in Parkinson’s disease without dementia. J Cereb Blood Flow Metab 2012; 32: 1609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candy JM, Perry RH, Perry EK, Irving D, Blessed G, Fairbairn AF, et al. Pathological changes in the nucleus of Meynert in Alzheimer’s and Parkinson’s diseases. J Neurol Sci 1983; 59: 277–89. [DOI] [PubMed] [Google Scholar]
- Caspell-Garcia C, Simuni T, Tosun-Turgut D, Wu IW, Zhang Y, Nalls M, et al. Multiple modality biomarker prediction of cognitive impairment in prospectively followed de novo Parkinson disease. PLoS One 2017; 12: e0175674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Hu WT, Siderowf A, Weintraub D, Goldmann Gross R, Hurtig HI, et al. Plasma epidermal growth factor levels predict cognitive decline in Parkinson disease. Ann Neurol 2011; 69: 655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Jung TM, Lee JE, Lee SK, Sohn YH, Lee PH. Volumetric analysis of the substantia innominata in patients with Parkinson’s disease according to cognitive status. Neurobiol Aging 2012; 33: 1265–72. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Schork AJ, Wang Y, Witoelar A, Sharma M, McEvoy LK, et al. Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus. Mol Psychiatry 2015; 20: 1588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domellöf ME, Ekman U, Forsgren L, Elgh E. Cognitive function in the early phase of Parkinson’s disease, a five-year follow-up. Acta Neurol Scand 2015; 132: 79–88. [DOI] [PubMed] [Google Scholar]
- Dujardin K, Defebvre L, Duhamel A, Lecouffe P, Rogelet P, Steinling M, et al. Cognitive and SPECT characteristics predict progression of Parkinson’s disease in newly diagnosed patients. J Neurol 2004; 251: 1383–92. [DOI] [PubMed] [Google Scholar]
- Foltynie T, Brayne CE, Robbins TW, Barker RA. The cognitive ability of an incident cohort of Parkinson’s patients in the UK. The CamPaIGN study. Brain J Neurol 2004; 127: 550–60. [DOI] [PubMed] [Google Scholar]
- Freund HJ, Kuhn J, Lenartz D, Mai JK, Schnell T, Klosterkoetter J, et al. Cognitive functions in a patient with Parkinson-dementia syndrome undergoing deep brain stimulation. Arch Neurol 2009; 66: 781–5. [DOI] [PubMed] [Google Scholar]
- Gomperts SN, Locascio JJ, Rentz D, Santarlasci A, Marquie M, Johnson KA, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology 2013; 80: 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Redondo R, García-García D, Clavero P, Gasca-Salas C, García-Eulate R, Zubieta JL, et al. Grey matter hypometabolism and atrophy in Parkinson’s disease with cognitive impairment: a two-step process. Brain J Neurol 2014; 137: 2356–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratwicke J, Kahan J, Zrinzo L, Hariz M, Limousin P, Foltynie T, et al. The nucleus basalis of Meynert: a new target for deep brain stimulation in dementia? Neurosci Biobehav Rev 2013; 37: 2676–88. [DOI] [PubMed] [Google Scholar]
- Grothe M, Heinsen H, Teipel S. Longitudinal measures of cholinergic forebrain atrophy in the transition from healthy aging to Alzheimer’s disease. Neurobiol Aging 2013; 34: 1210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe M, Heinsen H, Teipel SJ. Atrophy of the cholinergic basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol Psychiatry 2012; 71: 805–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe M, Zaborszky L, Atienza M, Gil-Neciga E, Rodriguez-Romero R, Teipel SJ, et al. Reduction of basal forebrain cholinergic system parallels cognitive impairment in patients at high risk of developing Alzheimer’s disease. Cereb Cortex 2010; 20: 1685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe MJ, Ewers M, Krause B, Heinsen H, Teipel SJ; Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain atrophy and cortical amyloid deposition in nondemented elderly subjects. Alzheimers Dement 2014; 10: S344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe MJ, Heinsen H, Amaro E, Grinberg LT, Teipel SJ. Cognitive correlates of basal forebrain atrophy and associated cortical hypometabolism in mild cognitive impairment. Cereb Cortex 2016; 26: 2411–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanganu A, Bedetti C, Degroot C, Mejia-Constain B, Lafontaine AL, Soland V, et al. Mild cognitive impairment is linked with faster rate of cortical thinning in patients with Parkinson’s disease longitudinally. Brain J Neurol 2014; 137: 1120–9. [DOI] [PubMed] [Google Scholar]
- Hilker R, Thomas AV, Klein JC, Weisenbach S, Kalbe E, Burghaus L, et al. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology 2005; 65: 1716–22. [DOI] [PubMed] [Google Scholar]
- Huertas I, Jesús S, García-Gómez FJ, Lojo JA, Bernal-Bernal I, Bonilla-Toribio M, et al. Genetic factors influencing frontostriatal dysfunction and the development of dementia in Parkinson’s disease. PLoS One 2017; 12: e0175560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janvin CC, Larsen JP, Aarsland D, Hugdahl K. Subtypes of mild cognitive impairment in Parkinson’s disease: progression to dementia. Mov Disord 2006; 21: 1343–9. [DOI] [PubMed] [Google Scholar]
- Jokinen P, Brück A, Aalto S, Forsback S, Parkkola R, Rinne JO. Impaired cognitive performance in Parkinson’s disease is related to caudate dopaminergic hypofunction and hippocampal atrophy. Parkinsonism Relat Disord 2009; 15: 88–93. [DOI] [PubMed] [Google Scholar]
- Kehagia AA, Barker RA, Robbins TW. Cognitive impairment in Parkinson’s disease: the dual syndrome hypothesis. Neurodegener Dis 2013; 11: 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbler GM, Fripp J, Rowe CC, Villemagne VL, Salvado O, Rose S, et al. Basal forebrain atrophy correlates with amyloid β burden in Alzheimer’s disease. Neuroimage Clin 2015; 7: 105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilimann I, Grothe M, Heinsen H, Alho EJ, Grinberg L, Amaro E, et al. Subregional basal forebrain atrophy in Alzheimer’s disease: a multicenter study. J Alzheimers Dis 2014; 40: 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilimann I, Hausner L, Fellgiebel A, Filippi M, Würdemann TJ, Heinsen H, et al. Parallel atrophy of cortex and basal forebrain cholinergic system in mild cognitive impairment. Cereb Cortex 2017; 27: 1841–8. [DOI] [PubMed] [Google Scholar]
- Klassen BT, Hentz JG, Shill HA, Driver-Dunckley E, Evidente VG, Sabbagh MN, et al. Quantitative EEG as a predictive biomarker for Parkinson disease dementia. Neurology 2011; 77: 118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, Andersson J, Ardekani BA, Ashburner J, Avants B, Chiang MC, et al. Evaluation of 14 nonlinear deformation algorithms applied to human brain MRI registration. Neuroimage 2009; 46: 786–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacour A, Espinosa A, Louwersheimer E, Heilmann S, Hernández I, Wolfsgruber S, et al. Genome-wide significant risk factors for Alzheimer’s disease: role in progression to dementia due to Alzheimer’s disease among subjects with mild cognitive impairment. Mol Psychiatry 2017; 22: 153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson RA, Yarnall AJ, Duncan GW, Breen DP, Khoo TK, Williams-Gray CH, et al. Cognitive decline and quality of life in incident Parkinson’s disease: the role of attention. Parkinsonism Relat Disord 2016; 27: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson RA, Yarnall AJ, Duncan GW, Breen DP, Khoo TK, Williams-Gray CH, et al. Stability of mild cognitive impairment in newly diagnosed Parkinson’s disease. J Neurol Neurosurg Psychiatry 2017; 88: 648–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HM, Kwon KY, Kim MJ, Jang JW, Suh SI, Koh SB, et al. Subcortical grey matter changes in untreated, early stage Parkinson’s disease without dementia. Parkinsonism Relat Disord 2014a; 20: 622–6. [DOI] [PubMed] [Google Scholar]
- Lee JE, Cho KH, Song SK, Kim HJ, Lee HS, Sohn YH, et al. Exploratory analysis of neuropsychological and neuroanatomical correlates of progressive mild cognitive impairment in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2014b; 85: 7–16. [DOI] [PubMed] [Google Scholar]
- Litvan I, Goldman JG, Tröster AI, Schmand BA, Weintraub D, Petersen RC, et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov Disord 2012; 27: 349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AK, Chang RC, Pearce RK, Gentleman SM. Nucleus basalis of Meynert revisited: anatomy, history and differential involvement in Alzheimer’s and Parkinson’s disease. Acta Neuropathol 2015; 129: 527–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak E, Su L, Williams GB, Firbank MJ, Lawson RA, Yarnall AJ, et al. Baseline and longitudinal grey matter changes in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain J Neurol 2015; 138: 2974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Hurtig HI, Van Deerlin VM, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol 2014; 71: 1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6). Neuroscience 1983; 10: 1185–201. [DOI] [PubMed] [Google Scholar]
- Morley JF, Xie SX, Hurtig HI, Stern MB, Colcher A, Horn S, et al. Genetic influences on cognitive decline in Parkinson’s disease. Mov Disord 2012; 27: 512–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir JL. Acetylcholine, aging, and Alzheimer’s disease. Pharmacol Biochem Behav 1997; 56: 687–96. [DOI] [PubMed] [Google Scholar]
- Nombela C, Rowe JB, Winder-Rhodes SE, Hampshire A, Owen AM, Breen DP, et al. Genetic impact on cognition and brain function in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain J Neurol 2014; 137: 2743–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JB, Svenningsson P, Weintraub D, Brønnick K, Lebedev A, Westman E, et al. Initial cognitive decline is associated with cortical thinning in early Parkinson disease. Neurology 2014; 82: 2017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2012; CD006504. doi: 10.1002/14651858.CD006504.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Cervantes S, Barabash A, Alonso A, Cabranes JA, Lamet I, et al. The effect of MAPT H1 and APOE ɛ4 on transition from mild cognitive impairment to dementia. J Alzheimers Dis 2010; 22: 1065–71. [DOI] [PubMed] [Google Scholar]
- Schmitz TW, Spreng RN. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat Commun 2016; 7: 13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag A, Weintraub D, Schott JM. Cognitive decline before diagnosis of Parkinson’s disease - Authors’ reply. Lancet Neurol 2017; 16: 262. [DOI] [PubMed] [Google Scholar]
- Shi M, Huber BR, Zhang J. Biomarkers for cognitive impairment in Parkinson disease. Brain Pathol Zurich Switz 2010; 20: 660–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada H, Hirano S, Shinotoh H, Aotsuka A, Sato K, Tanaka N, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 2009; 73: 273–8. [DOI] [PubMed] [Google Scholar]
- Siderowf A, Xie SX, Hurtig H, Weintraub D, Duda J, Chen-Plotkin A, et al. CSF amyloid {beta} 1-42 predicts cognitive decline in Parkinson disease. Neurology 2010; 75: 1055–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strafella AP, Bohnen NI, Perlmutter JS, Eidelberg D, Pavese N, Van Eimeren T, et al. Molecular imaging to track Parkinson’s disease and atypical parkinsonisms: new imaging frontiers. Mov Disord 2017; 32: 181–92. [DOI] [PubMed] [Google Scholar]
- Teipel S, Grothe MJ, Zhou J, Sepulcre J, Dyrba M, Sorg C, et al. Measuring cortical connectivity in Alzheimer’s disease as a brain neural network pathology: toward clinical applications. J Int Neuropsychol Soc 2016; 22: 138–63. [DOI] [PubMed] [Google Scholar]
- Teipel S, Heinsen H, Amaro E, Grinberg LT, Krause B, Grothe M, et al. Cholinergic basal forebrain atrophy predicts amyloid burden in Alzheimer’s disease. Neurobiol Aging 2014; 35: 482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels OJ, Broere CA, ter Laak HJ, ten Donkelaar HJ, Nieuwenhuys R, Schulte BP. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol Aging 1990; 11: 3–13. [DOI] [PubMed] [Google Scholar]
- Wang HF, Yu JT, Tang SW, Jiang T, Tan CC, Meng XF, et al. Efficacy and safety of cholinesterase inhibitors and memantine in cognitive impairment in Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies: systematic review with meta-analysis and trial sequential analysis. J Neurol Neurosurg Psychiatry 2015; 86: 135–43. [DOI] [PubMed] [Google Scholar]
- Weintraub D, Doshi J, Koka D, Davatzikos C, Siderowf AD, Duda JE, et al. Neurodegeneration across stages of cognitive decline in Parkinson disease. Arch Neurol 2011; 68: 1562–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Gray CH, Evans JR, Goris A, Foltynie T, Ban M, Robbins TW, et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain J Neurol 2009a; 132: 2958–69. [DOI] [PubMed] [Google Scholar]
- Williams-Gray CH, Goris A, Saiki M, Foltynie T, Compston DA, Sawcer SJ, et al. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s disease. J Neurol 2009b; 256: 493–8. [DOI] [PubMed] [Google Scholar]
- Williams-Gray CH, Mason SL, Evans JR, Foltynie T, Brayne C, Robbins TW, et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry 2013; 84: 1258–64. [DOI] [PubMed] [Google Scholar]
- Winder-Rhodes SE, Hampshire A, Rowe JB, Peelle JE, Robbins TW, Owen AM, et al. Association between MAPT haplotype and memory function in patients with Parkinson’s disease and healthy aging individuals. Neurobiol Aging 2015; 36: 1519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Csordas A, Mosca K, Kim J, Gielow MR, Vadasz C, et al. Neurons in the basal forebrain project to the cortex in a complex topographic organization that reflects corticocortical connectivity patterns: an experimental study based on retrograde tracing and 3D reconstruction. Cereb Cortex 2015; 25: 118–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler DA, Wonderlick JS, Ashourian P, Hansen LA, Young JC, Murphy AJ, et al. Substantia nigra volume loss before basal forebrain degeneration in early Parkinson disease. JAMA Neurol 2013; 70: 241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]