

Abstract
Toxicology has made steady advances over the last 60+ years in understanding the mechanisms of toxicity at an increasingly finer level of cellular organization. Traditionally, toxicological studies have used animal models. However, the general adoption of the principles of 3R (Replace, Reduce, Refine) provided the impetus for the development of in vitro models in toxicity testing. The present commentary is an attempt to briefly discuss the transformation in toxicology that began around 1980. Many genes important in cellular protection and metabolism of toxicants were cloned and characterized in the 80s, and gene expression studies became feasible, too. The development of transgenic and knockout mice provided valuable animal models to investigate the role of specific genes in producing toxic effects of chemicals or protecting the organism from the toxic effects of chemicals. Further developments in toxicology came from the incorporation of the tools of “omics” (genomics, proteomics, metabolomics, interactomics), epigenetics, systems biology, computational biology, and in vitro biology. Collectively, the advances in toxicology made during the last 30–40 years are expected to provide more innovative and efficient approaches to risk assessment. A goal of experimental toxicology going forward is to reduce animal use and yet be able to conduct appropriate risk assessments and make sound regulatory decisions using alternative methods of toxicity testing. In that respect, Tox21 has provided a big picture framework for the future. Currently, regulatory decisions involving drugs, biologics, food additives, and similar compounds still utilize data from animal testing and human clinical trials. In contrast, the prioritization of environmental chemicals for further study can be made using in vitro screening and computational tools.
Keywords: toxicology, Tox21, in vitro, nuclear receptors, transcription factors, adverse outcome pathway
Toxicology is an experimental laboratory science that has traditionally utilized various animal models to study the effects of chemicals. Historically, toxicology studies relied heavily on various histopathological and biochemical endpoints in whole animals to draw conclusions on the effects of a chemical on living systems. The theoretical and experimental foundations of toxicology are derived from a number of basic and clinical sciences, such as chemistry, anatomy, cell biology, physiology, biochemistry, pathology, and biostatistics.
The development of molecular biology tools in the late 1970s, such as gene cloning, DNA sequencing and gene expression techniques transformed all branches of biological sciences, including toxicology. The transformation in toxicology began in the 1980s. Briefly, the availability of molecular biology tools and techniques enabled toxicologists to clone genes regulating toxic response, investigate the effects of a toxicant on DNA structure, function, and (target) gene expression, and also manipulate DNA for developing genetically modified animal models for use in toxicological studies. Toxicology has been steadily integrating knowledge gained from the advances in “omics” (genomics, proteomics, metabolomics, interactomics), epigenetics, systems biology, computational science and in vitro biology. Consequently, toxicology has been rapidly transforming itself into an even more integrative scientific discipline.
The initial transformation of toxicology from a science concerned with describing tissue injury in the target organ of toxicity, to investigating the mechanisms of toxicity at the level of nucleic acids, and specific biochemical and molecular pathways was a major advancement. Toxicology entered into another transformative phase with the release of the National Research Council (NRC) report Toxicity Testing in the 21st Century: A Vision and a Strategy (NRC-NAS, 2007). The proposition of NRC’s vision and strategy is approaching its 10th year. Therefore, it is an appropriate time to look back and trace the evolution of toxicology from its tradition of toxicity testing in animals to its promised future of toxicity prediction based on computational and in vitro methods and less dependence on animal testing.
It is difficult to write an “ideal” review on this topic. The selection of important events and facts is somewhat subjective and could be shaped by the authors’ personal views. With that realization, effort has been made in keeping this review a neutral assessment on the evolution of toxicology during the last 30−40 years and also going forward. This commentary does not attempt to document all the discoveries or the name of each discoverer in the context of a relevant technique or concept.
THE PERIOD BETWEEN 1930 AND 1980 WITNESSED CONSIDERABLE DEVELOPMENTS AND AWARENESS RELATED TO TOXICOLOGY; AND THE FOCUS OF TOXICOLOGICAL STUDIES EVOLVED FROM IDENTIFYING TARGET ORGANS OF CHEMICAL TOXICITY TO IDENTIFYING TARGETS OF TOXICITY AT AN INCREASINGLY FINER LEVEL OF CELLULAR ORGANIZATION
The period between 1930 and 1980 witnessed considerable developments and awareness related to toxicology, including environmental toxicology. For example, in 1937 the marketing of a therapeutic potion containing sulfanilamide resulted in the death of more than 100 individuals. This unfortunate event accelerated the passage of the 1938 Food, Drug, and Cosmetic Act (FD&C Act). FDA established an organized approach to evaluating the safety of chemical constituents of foods, drugs, and cosmetics (Swan, 1998). After World War II, there was a significant growth in the chemical industry that led to the development of chemicals, such as pesticides. Concerns also grew about chemical contamination of the environment and its effects on human health and wildlife. These concerns were captured in the now classic book Silent Spring (Carson, 1962), which inspired a grassroots environmental movement across the country. Pioneering studies on the metabolism of environmental carcinogens were undertaken in the 1950s in the laboratory of J.A. and E.C. Miller at the University of Wisconsin. These studies paved the way for many important discoveries in toxicology in the following years. The U.S. Society of Toxicology was founded in 1961. The U.S. National Institute of Environmental Health Sciences was founded in the mid1960s, and the U.S. Environmental Protection Agency (EPA) was created in early 1970s.
Thus, one would conclude that toxicology made impressive advances within a span of about 30 years beginning around 1950, thanks to the ability of toxicologists to integrate the knowledge and the tools from multiple disciplines. In the middle of the 20th Century, toxicologists were mainly concerned with understanding the target organ toxicity of chemicals. Therefore, histopathology was a very important tool. Some biochemical markers of toxicity were also useful, such as the quantification of liver enzymes in the serum for hepatotoxicity, urinary creatinine clearance for nephrotoxicity, etc. The focus of studies quickly moved towards understanding the mechanisms of toxicity, particularly hepatotoxicity. This is because the liver metabolizes chemicals into more water soluble forms that are readily eliminated from the body, but some chemicals or their metabolites produce hepatotoxicity.
Early attempts to investigate the cellular/biochemical mechanisms of toxicity focused on different organelles of the cell that are important in cellular survival. Predictably, several studies published from 1940s onwards focused on cell membrane integrity and mitochondrial function as potential targets of chemical toxicity. The uncoupling of oxidative phosphorylation was identified as an important biochemical mechanism of toxicity by many chemicals (eg, 2,4-dinitrophenol). Beginning from the late 1960s, studies also focused on cytoskeleton as another possible target of chemicals including antineoplastic drugs (eg, vinca alkaloids) and neurotoxicants (n-hexane). Metabolism studies involving chemical carcinogens had been initiated in 1940s, but further understanding of the role of metabolism in mediating toxicity was gained in 1960s with the advances in cytochromes P450 research. It was realized that P450-mediated metabolism detoxified some chemicals but made others more reactive and toxic. The use of high-speed centrifugation opened many doors for biochemical studies related to cellular functions, drug metabolism and toxicity. The development of 3 new instruments—the spectrophotometer, mass spectrometer, and oxygen electrode played a key role in P450 research (Estabrook, 2003).
In 1971, Brodie and coworkers reported that halogenated aromatic hydrocarbons (eg, bromobenzene) could potentiate hepatic necrosis in rats. Using in vitro experiments and autoradiography, the authors inferred that the hepatotoxic effects were mediated by chemically active metabolites (glutathione-conjugates) formed in the hepatocytes at the site of necrosis. This study highlighted the importance of metabolism-mediated bioactivation (ie, formation of active metabolites), covalent binding, and glutathione depletion as some of the key mechanisms of toxicity. Within a few years, similar findings on acetaminophen-mediated hepatotoxicity further elucidated the role of glutathione depletion and oxidative stress in toxicity. It was reported that hepatic necrosis after acetaminophen administration was preceded by the depletion of glutathione in the liver and the covalent binding of a reactive metabolite of acetaminophen to macromolecules (Potter et al., 1974). By then, it had also been reported that carcinogens formed DNA adducts (Maher et al., 1971; Tada and Tada, 1971). These findings opened up the possibility that depletion of glutathione resulted in oxidative stress that may lead to macromolecular damage, including damage to the genetic material. Thus, DNA damage, DNA and protein adduct formation, and DNA repair became the focus of numerous studies trying to understand the mechanisms of toxicity at the level of biological macromolecules, including the structural integrity and functioning of the genetic material.
The discovery of aryl hydrocarbon receptor (AhR) in the 1970s was yet another breakthrough in toxicology and cancer research. The history of AhR is long and complex. It indirectly began in the 1950s in the laboratory of J.A. and E.C. Miller at the University of Wisconsin, with studies on the induction of benzo[a]pyrene hydroxylase (later renamed aryl hydrocarbon hydroxylase [AHH]). Beginning in the late 1960s, it was shown that AHH could be induced by several polycyclic aromatic hydrocarbons in some inbred mouse strains (eg, C57BL/6) but not in others (eg, DBA/2) (Nebert and Gelboin, 1969; Nebert and Bausserman, 1970). The differential AHH inducibility was shown to be inherited primarily as an autosomal dominant trait, suggesting the existence of a genetic locus (the Ah locus) (Gielen et al., 1972; Nebert et al., 1972). Shortly thereafter, Poland and Glover (1974) demonstrated that 2,3,7,8-tetrachlorodibenzodioxin (TCDD) was 30 000 times more potent than 3-methylcholanthrene as an inducer of AHH activity, and TCDD could induce high AHH activity in nonresponsive mice as well (Poland et al., 1974). From further studies on the induction of AHH activity in nonresponsive mice, Poland and Glover (1975) suggested that the nonresponsive mice had a mutation in the induction receptor. In a subsequent landmark paper, Poland and coworkers demonstrated, using radiolabeled TCDD, that the product of the Ah locus was a receptor that could bind TCDD in the cytosol with very high affinity. This receptor was responsible for the induction of AHH activity, and it was mutated in nonresponsive mice, thereby altering the ligand binding affinity (Poland et al., 1976). Interestingly, the AhR cDNA was cloned in the early 1990s (discussed later). Thus, among nuclear receptors of toxicological significance, the discovery of AhR predates the age of reverse genetics (clone first, define function later).
In the early 1970s, Bruce Ames and colleagues published a series of papers describing the development of bacterial reverse mutation assays to detect chemical mutagens. The use of liver homogenates to metabolically activate procarcinogens into carcinogens that are potent mutagens proved to be a useful tool. This was hailed as a major technical development benefiting toxicology, carcinogenesis, and mutation research.
Experimental toxicology utilizes analytical techniques to understand the concentrations, distribution, metabolism, and excretion of a toxicant in the body following exposure. Various analytical chemistry methods (eg, gas chromatography, high-performance-liquid chromatography, mass spectrometry, atomic-absorption spectroscopy, nuclear magnetic resonance spectroscopy) have been successfully used to detect toxicants in body fluids, tissues, and excreta. These toxicokinetic studies enabled the transformation of the administered dose to the concentration of the toxicant at the target tissue that produced toxicity. The analytical chemistry methods also helped identify various metabolites, and characterize DNA and protein adducts. Currently, these methods have become indispensable in metabolomic studies. Two other techniques—confocal microscopy, and flow cytometry are being widely used in toxicology and cancer research. Confocal microscopy is becoming a crucial technique in nanotoxicology.
In the mid1950s, efforts were made to reduce the number of animals and introduce humane techniques in experiments that used animals. These efforts led to the development of the principles of 3R (Replace, Reduce, Refine) published in 1959. Gradually, these principles became part of the essential considerations when animals were used in research. In the United Kingdom, these principles were formally adopted into animal procedures legislation in 1986 (Flecknell, 2002). The widespread adoption of the principles of 3R provided the impetus for the development of in vitro models in toxicity testing.
However, the use of in vitro systems to reproduce the toxicity seen in whole animals had a less than desirable beginning. Early cell culture systems could not maintain the structural and functional integrity, as well as the highly complex and dynamic 3D in vivo environments. Consequently, the expression of many xenobiotic metabolizing enzymes and transport activities were not retained well. Some additional common confounding factors in traditional in vitro studies were (1) the lack of tissue-specific cell type diversity and cell-cell communication, that is, most traditional cell culture systems contain only 1 cell type although the intact tissue that is the source of the cell contains many cell types; (2) high level of chemical exposure of cells, that is, the concentrations of the chemical to be tested are often much higher in the in vitro system than in intact animals; and (3) altered biochemical and genetic characteristics of immortal cells in culture; for example, cancer cells frequently used in cell culture continuously evolve due to ongoing selection through numerous cycles of growth, division, and passaging that result in major biochemical and genetic changes compared with the original cell that was isolated from the cancerous tissue. This was demonstrated by Gillet et al. (2011) who studied the known multidrug resistance (MDR) transcriptome of 6 cancer types in established cancer cell lines (grown in monolayer, 3D scaffold, or in xenograft) and clinical samples. Upregulation of genes that would facilitate survival across all cultured cancer cell lines was observed as expected but no correlation was found between clinical samples and established cancer cell lines. The authors also found that all of the cell lines, grown either in vitro or in vivo, bear more resemblance to each other, regardless of the tissue of origin, than to the clinical samples they are supposed to model.
Newer methods, such as the combination of microfluidics technology with 3D cell culture offer better in vitro models, although it is still far from being a perfect replacement of the in vivo environment. In a recent article, Antoni et al. (2015) concluded that the current 3D cell culture technology has limitations of “performance, sensitivity and compatibility with high-throughput screening instruments”. Technological advances, such as the 3D primary human hepatocyte spheroid system, the so-called “liver spheroids”, represents an in vitro system that allows the co-culture of hepatocytes with nonparenchymal cells, such as biliary cells, stellate cells and Kupffer cells. Therefore, these liver spheroids represent yet another improved in vitro system to study chemical toxicity (Bell et al., 2016).
Progress in understanding of the mechanisms of toxicity from cell membrane and cytoskeletal integrity to organelle function down to the level of DNA integrity, transcription and translation was dependent on advances in technology as well as an increased understanding of the science of molecular biology.
The collection of analytical, biochemical, histopathological, toxicokinetics, and in vitro methods used in toxicology studies remain indispensable. However, the emergence of molecular biology methods provided toxicologists with yet another set of tools to study the mechanisms of toxicity. Applications of molecular biology concepts and tools have helped us understand cellular responses to toxic insults in 2 major ways; (1) the effects of low-dose exposure to toxicants, and (2) the detection of early cellular responses before any pathophysiologic effects are observed. Because early cellular responses may also be adaptive in nature, additional studies may be designed to further characterize the effect of low-dose exposure to toxicants. Thus, an understanding of the molecular targets of toxicants and the molecular mechanism of toxicity represented a major paradigm shift in toxicology research.
CLONING AND CHARACTERIZATION OF GENES ASSOCIATED WITH CELLULAR PROTECTION AND BIOTRANSFORMATION OF XENOBIOTICS, ALONG WITH THE AVAILABILITY OF TECHNIQUES TO STUDY GENE EXPRESSION, BROUGHT ABOUT A TRANSFORMATION IN EXPERIMENTAL TOXICOLOGY BEGINNING IN THE 1980s
Technological inventions in molecular biology had to precede the transformation of toxicology and other disciplines that began incorporating gene expression data in publications. The year 1977 was very important in the annals of molecular biology. The publication of 3 major technological developments in 1977 enabled experimental molecular biology to move in a new direction. These were: (1) the development of suitable cloning vectors (eg, pBR322), (2) the development of 2 direct DNA sequencing techniques (Sanger’s dideoxy technique and Gilbert’s chemical cleavage technique), and (3) the development of blotting techniques (eg, northern blot) to study gene expression (see Choudhuri, 2006). These technological developments simplified cloning and expression studies that became increasingly common, including in the field of toxicology.
Throughout the 1980s and 90s, several genes involved in cellular protection, as well as phases I and II xenobiotic metabolism were cloned from various species including mice, rats and humans and their expression studied. A few examples are discussed below.
Metallothioneins (MTs) are a group of small, cysteine-rich proteins. MT binds cadmium (Cd) and other metals in cells and limits their distribution to the mitochondria; thus MT plays an important role in protecting against metal toxicity, particularly Cd toxicity. MT is induced by many chemicals, such as various heavy metals, glucocorticoids, bacterial exotoxins and endotoxin Lipopolysaccharide, inflammatory cytokines, as well as oxidative stress-inducing chemicals (see review by Klaassen et al., 1999). MT is a protein of interest to toxicologists due to its protective role against a wide variety of toxic insults. Because MT expression is upregulated by a number of inducers, the MT gene was of great interest to molecular biologists studying gene regulation. Such converging interest on MT proved beneficial to toxicologists in understanding the organization of the MT gene and its regulation in response to xenobiotics. The first MT cloned was the mouse MT-I cDNA and the gene (Glanville et al., 1981). The cloning of various MT isoforms from other species soon followed.
The induction of hepatic microsomal enzymes by xenobiotics and the role of these enzymes in xenobiotic metabolism began to be reported in the mid1950s by J.A. and E.C. Miller and their research team at the University of Wisconsin (see Miller and Miller, 1981). The molecular basis of how xenobiotics induce these enzymes was not fully appreciated until the genes encoding these enzymes were cloned and their expression studied. These enzymes were cytochromes P450, a term coined by Omura and Sato in 1962. Major efforts in the cloning of various cytochrome P450 (CYP) genes involved in xenobiotic metabolism began in the 1980s. Several P450 genes in rodents and humans were cloned and their regulation studied (see Nebert and Gonzalez, 1987). A comprehensive account of the progress for the last half-a-century in the field of toxicology and metabolism with emphasis on CYP enzymes has been provided by Guengerich (2014). The cloning of a number of phase II xenobiotic metabolizing enzymes was also reported beginning from the mid1980s. The cloning of Uridine 5’-diphospho-glucuronosyltransferase, cDNA from rat liver was first reported by 2 groups a few months apart (Mackenzie et al., 1984; Jackson et al., 1985). The cloning of other phase II enzymes and from different species soon followed. The cloning and characterization of various phases I and II enzymes helped elucidate the multiplicity of drug and xenobiotic metabolizing enzymes and their substrates.
Many investigators have studied oxidative stress, its adverse health effects and the cellular antioxidant defense systems, since the 1970s. The cloning and characterization of some of the major players involved in the cellular antioxidant defense system were reported in the early 1980s, such as heme oxygenase (HO), Cu/Zn superoxide dismutase (Cu/Zn-SOD), Mn-SOD, and catalase, of which the cloning and expression of the cDNA for rat HO was reported in 1985 (Shibahara et al., 1985). This was followed by the cloning and characterization of other enzymes. The expression of these genes is generally induced by exposure to oxidative stress-inducing agents, hypoxia, and many other cellular stressors. The discovery of SOD led to the superoxide theory, which proposes that the superoxide radical is a major source of oxygen toxicity.
ONLY A FEW TOXICOLOGISTS WERE INVOLVED IN CLONING EXPERIMENTS BUT AN INCREASING NUMBER OF TOXICOLOGISTS UNDERTOOK GENE EXPRESSION STUDIES
Gene expression studies in toxicology became possible only after the relevant genes/cDNAs were cloned and their sequences determined. Although not many toxicologists were involved in cloning experiments, an increasing number of toxicological investigations undertook gene expression studies. The development of early techniques to study gene expression, such as northern blot (Alwine et al., 1977) and dot blot (Kafatos et al., 1979) resulted in their widespread use in toxicology. Beginning from the 1980s, gene expression studies became economically and technically feasible for many laboratories, and many investigators started publishing gene expression data as part of their toxicological studies. By the early 1990s, gene expression studies became common in experimental toxicology, using techniques like filter hybridization (northern blot, dot blot), solution hybridization for quantifying mRNA expression, and in situ hybridization for tissue localization of mRNA expression. A method that revolutionized molecular biology and was quickly adopted by toxicologists, was the polymerase chain reaction (PCR) developed in the mid1980s by Kary Mullis (Saiki et al., 1985), who was awarded the Nobel Prize for this invention. Various applications of PCR, including rapid amplification of cDNA ends for cDNA cloning, were harnessed by an increasing number of toxicologists beginning in the 1990s.
WITH THE DEVELOPMENT OF MORE ADVANCED GENE EXPRESSION TECHNIQUES, MAINLY IN THE 1990s, THE EFFECT OF A TOXICANT ON THE TRANSCRIPTIONAL REGULATION OF MULTIPLE TARGET GENES COULD BE STUDIED MORE COMPREHENSIVELY
The earlier gene expression techniques, such as the blotting techniques, largely determined the expression of 1 gene in multiple samples from 1 experiment, such as dose response and time response in different tissues. With the development of more advanced techniques, such as subtractive hybridization (Sargent and Dawid 1983), differential display (Liang and Pardee, 1992), branched DNA signal amplification assay (Pachl et al., 1995), and serial analysis of gene expression (SAGE) (Velculescu et al., 1995), the scale and scope of gene expression studies greatly expanded. Simultaneous quantification of the expression of many genes in response to treatment by 1 toxicant, as well as high-throughput expression analysis of the constitutive expression of many genes soon became possible. Many toxicology studies reported impressive results utilizing these techniques. The use of SAGE in typical rodent toxicology studies is relatively limited in the published literature. Interestingly, both SAGE and microarray were reported at the same time (October 20 issue of Science, 1995); subsequently microarray was rapidly adopted by the scientific community for transcriptomics studies. An example of a more recent rodent study using SAGE is that of Kurachi et al. (2002).
CLONING AND CHARACTERIZATION OF VARIOUS DRUG AND XENOBIOTIC TRANSPORTERS ESTABLISHED THE MOLECULAR UNDERPINNINGS OF THE ABSORPTION (A), DISTRIBUTION (D), AND EXCRETION (E) ELEMENTS OF THE CLASSICAL ABSORPTION, DISTRIBUTION, METABOLISM, EXCRETION CONCEPT
During the 1990s another series of very important discoveries, relevant for toxicology, were made. These include the cloning and characterization of xenobiotic transporters, and a number of “orphan” nuclear receptors (discussed later) that regulate the expression of various xenobiotic transporters and xenobiotic metabolizing enzymes. Many uptake and efflux transporter cDNAs and genes in mice, rats and humans were cloned and characterized, their expression and regulation were studied, and their substrate spectrum and transport kinetics were investigated. These studies were largely published in the 1990s (see review by Klaassen and Aleksunes, 2010). Interestingly, the most widely studied of these transporters is the MDR gene (encoding P-glycoprotein or P-gp), which was first cloned and characterized in the mid1980s (Riordan et al., 1985).
An important group of efflux drug transporters identified, in addition to P-gp, was the MDR-associated protein family (MRPs). Natural functional inactivation of the MRP2 transporter is responsible for the Dubin-Johnson Syndrome (DJS) in humans. DJS involves chronic conjugated hyperbilirubinemia (hence lifelong mild jaundice) because of the inability of the liver to excrete conjugated bilirubin into bile. The animal model for DJS is the TR‒ (transport deficient) rat. Paulusma et al. cloned the MRP2 cDNA from TR‒ rat (1996) and from a DJS patient (1997) and found that the coding sequence of the gene is disrupted by a 1-base pair deletion in rats, and a C→T transition mutation at codon 1066 in humans. In both cases, the mutation creates a premature stop codon that produces a truncated and inactivated MRP2 protein.
Many xenobiotic uptake transporters (solute carrier or SLCO family) were also cloned, their expression studied, and their transport properties characterized in the 1990s. Some examples include organic anion transporting polypeptides (OATPs) (see review by Hagenbuch and Stieger, 2013), organic anion transporters and organic cation transporters (see review by Koepsell, 2013), and bile acid transporters NTCP and ASBT (see review by Da Silva et al., 2013). Studies on some of these transporters produced interesting findings. For example, Oatp1b2 null mice were completely resistant to the hepatotoxicity induced by phalloidin and the blue-green algal toxin microcystin-LR, but were sensitive to α-amanitin-induced hepatotoxicity, compared with the wild-type mice (Lu et al., 2008). This transporter is also important for the uptake of unconjugated bile acids (Csanaky et al., 2011).
DISCOVERY OF A GROUP OF LIGAND-DEPENDENT TRANSCRIPTION FACTORS REVEALED THE EXISTENCE OF A COMMON MOLECULAR MECHANISM REGULATING AND COORDINATING XENOBIOTIC TRANSPORT AND METABOLISM
A number of transcription activating proteins (transcription factors) that provide a clear molecular underpinning of the cellular response to xenobiotics and endogenous compounds were discovered and characterized largely in the 1980s and 1990s. Several important studies on these proteins were also conducted in the first decade of the 21st Century. An important group of these transcription factors belongs to the nuclear receptor superfamily, whereas some belong to the Per-ARNT-Sim (PAS) domain superfamily. There are other transcription factors that do not belong to either of these 2 superfamilies, but are important mediators of cellular response to toxic insults. The following discussion focuses on the nuclear receptor and PAS domain superfamilies.
Nuclear Receptor Superfamily of Transcription Factors
The discovery of nuclear receptors has its historical roots in endocrinology. The glucocorticoid receptor was the first nuclear receptor cloned in 1985 (Hollenberg et al., 1985), followed by the cloning of estrogen receptor (Green et al., 1986) and thyroid hormone receptor (Sap et al., 1986; Weinberger et al., 1986). The androgen, estrogen and thyroid hormone receptors are important in toxicology because they are frequent targets of endocrine disruptors. Consequently, receptor binding assays are used for the identification of chemicals that are potential endocrine disruptors. Many other structurally related transcription factors were eventually discovered. Because the ligands of these proteins were unknown at the time, they were termed “orphan” receptors and were adopted into the nuclear receptor superfamily. These receptors were conserved throughout metazoan evolution (except in plants). Figures 1A and 1B show the generalized structure and function of nuclear receptors, respectively.
Figure 1.
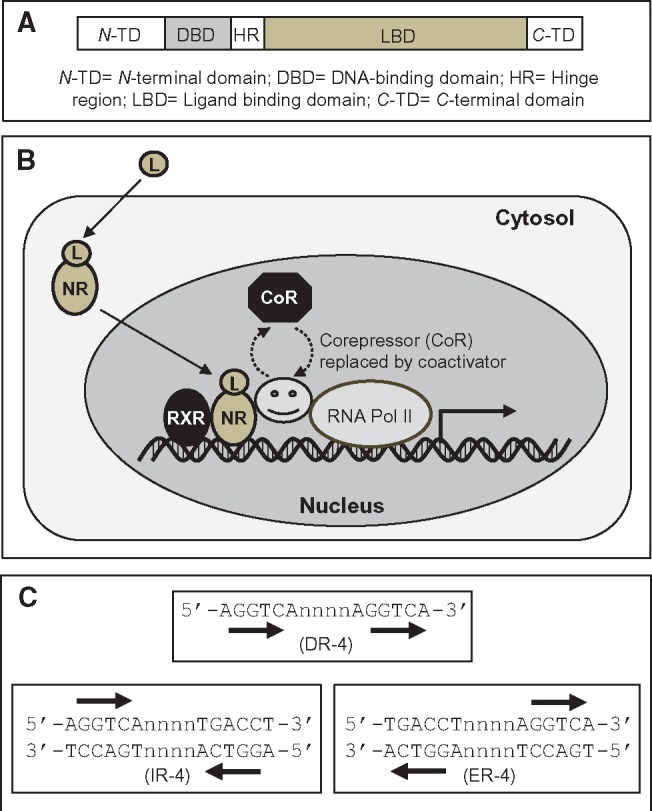
Structure, function and regulation of nuclear receptor superfamily of transcription factors. A, The structure of a typical nuclear receptor transcription factor. Structural divergence of the LBD determines the species-specific differences in the response to different ligands. B, nuclear receptor function. On entering the cell, the ligand (L) binds to the nuclear receptor, forming the NR-L complex. The NR-L complex translocates to the nucleus and heterodimerizes with the RXR. Formation of the heterodimer results in the dissociation of the transcriptional corepressor complex and recruitment of the coactivator complex, which enhances transcription. C, The organization of the direct repeat, inverted repeat, and everted repeat binding motifs. In this example the number of intervening bases shown is 4 (DR-4, IR-4, and ER-4); it usually varies between 1 and 6. The DR usually involves one strand whereas the IR and ER involve both strands.
The discovery of orphan nuclear receptors began with the cloning of the retinoid-X-receptor α (RXRα) (Mengelsdorf et al., 1990). RXR was also the first orphan nuclear receptor to have its endogenous ligand identified, 9-cis retinoic acid, a metabolite of vitamin A. The cloning of the peroxisome proliferator-activated receptor α (PPARα) was also reported in 1990 (Issemann and Green, 1990). Other orphan members of the nuclear receptor superfamily cloned and characterized in the 1990s included the liver-X-receptor (LXR) (Apfel et al., 1994; Shinar et al., 1994; Song et al., 1994); constitutive androstane receptor (CAR) (Baes et al., 1994; Choi et al., 1997); farnesoid-X-receptor (FXR) (Forman et al., 1995); and pregnane-X-receptor (PXR) (Kliewer et al., 1998). Many of these nuclear receptors (eg, RXR, PPAR, LXR) have multiple isoforms (eg, α,β,γ). In each case, the discovery of the first isoform was soon followed by the discovery of the other isoforms.
Functional characterization revealed that RXR is a promiscuous heterodimerization partner of the adopted orphan nuclear receptors, thereby defining a novel feature of multiple intertwined signaling pathways (Evans and Mengelsdorf, 2014). The ligands of many (but not all) of the orphan nuclear receptors were eventually identified. The physiological roles that various nuclear receptors play include fatty acid metabolism (PPARα), sterol homeostasis (LXR), bile acid homeostasis (FXR), and endobiotic/xenobiotic metabolism (PXR and CAR). Retinoic acid plays an important role during normal embryogenesis, but high doses of retinoic acid are teratogenic. The teratogenic effects of retinoic acids are mediated by retinoic acid receptors (RARs) and the RXRs. The ligand-bound RAR-RXR heterodimer regulates the proper spatio-temporal expression of developmental genes. Just as high doses of retinoic acid are teratogenic, overexpression of RAR also causes developmental abnormalities, underscoring the importance of RAR in developmental toxicology and teratology.
The nuclear receptors (transcription factors) bind to the conserved half-site sequence 5′-AGGTCA-3′ or some variants of it (viz, 5′-AGTTCA-3′) with different affinities. The specific DNA-binding site for each nuclear receptor involves the half site sequence oriented as a direct repeat, inverted repeat or everted repeats, and separated by 1, 2, 3, 4 or 6 spacer nucleotides (Figure 1C). For example, the designation DR-4 in this context means that the binding site consists of half sites that are in direct repeat orientation (hence DR) and separated by 4 spacer nucleotides (hence DR-4).
PAS Domain Superfamily of Transcription Factors
The proteins belonging to the PAS domain superfamily act as sensors of environmental and developmental signals. The AhR belongs to the PAS domain superfamily. The AhR is an intracellular ligand-dependent transcription factor that controls the expression of a diverse set of genes, including several genes involved in biotransformation of xenobiotics. The AhR lacks the Zn-finger domain typical of nuclear receptors; instead it contains the basic helix-loop-helix domain. Unlike the nuclear receptors that heterodimerize with RXR, AhR’s dimerization partner is the AhR-nuclear-translocator (ARNT) protein. Both ARNT and AhR cDNAs were cloned in the early 1990s (Hoffman et al., 1991; Burbach et al., 1992).
Studies on AhR have identified various AhR ligands that include man-made chemicals in the environment, naturally occurring phytochemicals as well as physiological compounds (Table 1). Toxicological consequences of AhR activation in response to dioxins (eg, TCDD) have been widely investigated, and these include various impairments affecting development; reproductive, nervous, and immune systems; and carcinogenesis. Studies using AhR knockout mice indicate that the AhR plays an important role in the development of the immune system (Tian et al., 2015).
Table 1.
Some Toxicologically Relevant Ligand-Dependent Transcription Factors, Their DNA-Binding Sites (Response Elements or REs), and Ligands
| Some toxicologically relevant transcription factors | Some ligands and activators of these transcription factors |
|---|---|
|
Glucocorticoids (eg, Dexamethasone, Prednisolone, Cortisone) |
|
Retinoic acid (Vitamin A) |
|
9-cis-retinoic acid |
|
|
|
Oxysterols |
|
Bile acids |
|
|
|
Androstanol (inhibitor in mouse), Meclizine (inhibitor in human, but activator in mouse), Bilirubin, TCPOBOP (mouse), CITCO (human), PB (does not bind to CAR but activates by an indirect mechanism), Clotrimazole (activator in human) |
|
|
|
Cadmium, Zinc |
|
|
|
Activator: Phorbol ester |
|
Activator: Sulforaphane, Phorbol ester, Endotoxin |
Other Transcription Factors
Several other transcription factors regulating gene expression in response to toxic insults include Nrf1, Nrf2, p53, NF-kB, STAT, HIF, MTF, and HSF (Jennings et al., 2013). The p53 protein was independently discovered by several groups in 1979 studying SV40-derived tumor antigens; NF-κB (Nuclear factor-kappa B) was discovered in mid1980s (Sen and Baltimore, 1986). One of the widely studied responders to chemical exposure is Nrf2, which was first reported in 1994 (Moi et al., 1994). Nrf2 is the primary mediator of gene expression through the antioxidant response element (ARE). It is involved in the upregulation of phase II xenobiotic metabolizing genes and also genes that protect against oxidative stress (Wu et al., 2012).
Nuclear Receptor Transcription Factors and the Regulation of Xenobiotic Transport and Metabolism
The classical inducers of xenobiotic metabolizing enzymes activate the nuclear receptor transcription factors. For example, in rodents TCDD and 3-methylcholanthrene activate AhR and induce CYP1A1/2 expression; phenobarbital (PB) and TCPOBOP activate CAR and induce CYP2B1/2 expression (PB activates CAR in a ligand-independent, ie, indirect mechanism); dexamethasone (DEX) and pregnenolone-16α-carbonitrile (PCN) activate PXR and induce CYP3A1/2 expression; fibrate (eg, clofibrate), WY-14,643, perfluoroalkyl acids (PFOA, PFDA) activate PPARα and induce CYP4A1/2. The nuclear receptors also play pivotal roles in regulating transporter expression in the intestine and liver (Staudinger et al., 2013). Table 1 shows some of these toxicologically relevant transcription factors (not a complete list). The DNA binding sites of the nuclear receptors are located in the promoter or the enhancer region of the target genes. Recent genome-wide analysis indicates that most nuclear receptors binding sites are found in the enhancer elements located far away from the transcriptional start site (Sever and Glass, 2013).
The identification of a plethora of transcription factors involved in the regulation of xenobiotic transport and metabolism, as well as in the regulation of various cytoprotective proteins has provided a clear molecular basis of understanding how xenobiotics affect an organism by directly impinging on the genes and modulating their expression. These transcription factors often function as molecular links to fine-tune the transport-metabolism coordination.
DEVELOPMENT OF TRANSGENIC AND KNOCKOUT MICE MODELS AND PARTICULARLY HUMANIZED TRANSGENIC MICE MODELS BENEFITED TOXICOLOGISTS WITH POWERFUL TOOLS TO STUDY THE FUNCTIONS OF SPECIFIC GENES IN MEDIATING CELLULAR TOXICITY/PROTECTION AS WELL AS XENOBIOTIC METABOLISM
The wider availability to experimental toxicologists of various transgenic and gene knockout (or null) mice starting in the 1990s further aided in studies on the mechanisms of toxicity. For example, several studies demonstrated that MT-TG mice were more resistant to Cd-induced hepatotoxicity (Palmiter et al., 1993). In contrast and expectedly, MT-null mice were highly susceptible to Cd-induced hepatotoxicity and chronic nephrotoxicity (Klaassen and Liu, 1998). Studies using AhR-null mice, generated independently by 2 laboratories (Fernandez-Salguero et al., 1995; Schmidt et al., 1996) demonstrated that the induction of Cyp1a1, Cyp1a2, and Cyp1b1 by TCDD is abolished in these mice. The constitutive expression of Cyp1a2 is also decreased. Although AhR is best known to toxicologists for its role in TCDD-mediated toxicity and teratogenicity, as well as benzo[a]pyrene-mediated carcinogenicity, studies using AhR-null mice demonstrated that AhR plays an important role in normal cell physiology (Tian et al., 2015).
Several TG and null mice models (eg, TG.AC, TgrasH2, p53+/−, XPA−/−) are often used to screen for carcinogenic potential of test compounds. The TG.AC mouse, developed by Leder et al. (1990), contains the transgene v-Ha-ras oncogene, which has 2 activating mutations in codons 12 and 59. Because tumor initiation is highly correlated with these mutations, these activating mutations act as initiators in place of mutagens. Therefore, the TG.AC mouse model is useful in identifying potential tumor promoters. The TgrasH2 mouse was developed by Saitoh et al. (1990) in which the introduced transgene is a c-Ha-ras protooncogene. The p53+/− mouse was developed by Donehower et al. (1992). FDA’s adoption of ICH S1B in 1997 (ICH, 1997) “Testing for the Carcinogenicity of Pharmaceuticals,” opened the door for the use of such transgenic models in regulatory toxicology (Jacobson-Kram et al., 2004).
Humanized transgenic mice models provide a unique opportunity to study the functions of a human gene in vivo without having to conduct human studies, and not relying upon animal-to-human extrapolation. A humanized mouse is created by first inactivating (knocking out) the endogenous mouse gene, followed by the introduction (knocking in) of the human gene, whose expression remains under the control of a promoter that is functional in mouse cells.
The humanized transgenic mice models for toxicological and metabolism studies became available from the early 2000s. Xie et al. (2000) engineered a humanized mouse model of PXR by replacing the endogenous mouse PXR gene with the human PXR gene. These PXR-humanized transgenic mice produce human CYP3A in response to rifampicin that is a human-specific PXR activator, but not dexamethasone and PCN that are mouse-specific PXR activators. In contrast, dexamethasone and PCN activate mouse PXR but not human PXR, and induce mouse Cyp3a but not human CYP3A. Thus, this humanized mouse model can be used to directly predict CYP3A-mediated metabolism of drugs and xenobiotics in humans, rather than trying to extrapolate mouse data to humans. Within 2–3 years, other humanized mice models were developed, such as the CAR-humanized mouse (Zhang et al., 2002, 2013), AhR-humanized mouse (Moriguchi et al., 2003), and PPARα-humanized mouse (Cheung et al., 2004).
ADVENT OF MICROARRAY AND GLOBAL GENE EXPRESSION PROFILING USHERED IN THE ERA OF TOXICOGENOMICS AND TRANSFORMED THE SCALE AND SCOPE OF “DATA”, THEREBY CHANGING THE RESEARCH PARADIGM IN TOXICOLOGY
Following the development of microarray for global gene expression profiling (Schena et al., 1995), toxicology underwent another major technological transformation in its ability to generate global gene expression profiling data on a massive scale. Such a global view of the changes in genome expression (instead of a few genes) ushered in the experimental paradigm of data-driven hypothesis formulation for further research.
The term transcriptome had already been used by Velculescu et al. (1997) to indicate the entire collection of transcripts. By quantifying the transcriptome, microarray ushered in the era of transcriptomics. Transcriptomics became interchangeably used by toxicologists in the context of toxicogenomics, a term coined by Nuwaysir et al. (1999) and was described as a subdiscipline concerned with the identification of potential human and environmental toxicants, and their putative mode of action (MOA), through the use of genomics resources (Nuwaysir et al., 1999). The term quickly became mainstream in toxicology. The idea that early expression profiling of specific genes might be used as a biomarker for potential adverse effects long before any pathological changes are observed, gained a lot of traction among toxicologists. The idea of public health benefits from the use of genomics data was important enough that regulatory agencies became involved in toxicogenomic initiatives by partnering with scientists from academia and industry. The concerted effort was expected to help facilitate the integration of genomics data in order to improve, for instance, drug development as well as drug safety and efficacy assessment (Lesko and Woodcock, 2004). Global gene expression profiling has been further refined by the development of RNA sequencing, which utilizes massively parallel sequencing for transcriptome analyses at a much higher resolution than microarray-based methods (eg, Cui et al., 2012; van Delft et al., 2012).
In keeping with the high-throughput genomic techniques, high-throughput proteomic and metabolomic techniques contributed to the mapping of the global protein expression and cellular small molecule identification, respectively. Although toxicogenomics intuitively means global analysis of mRNA, its definition has broadened over time. Currently, it is defined as a conceptually broad term to include global analysis of mRNAs (transcriptomics), proteins (proteomics) and metabolites (metabolomics) to study the effects of chemicals on organisms (Hamadeh et al., 2002; NRC-NAS, 2007).
Metabolomics has also been gaining increasing attention because of its ability to characterize the exposome. Metabolomics deals with the characterization of small molecule metabolites in biological systems. Therefore, metabolomics can help identify chemical fingerprints that cellular processes produce following exposure to a chemical. It is being increasingly realized that metabolomics can be a great tool to characterize the exposome, that is, the totality of environmental exposure of an individual in a lifetime and how those exposures relate to health.
Toxicogenomics appeared on the scientific stage with enormous promise. The capability of generating huge volumes of genomic data in a short period of time triggered a lot of enthusiasm among scientists. In 2010, ILSI/HESI conducted an online survey among scientists and scientific decision/policy makers actively engaged in the field of toxicogenomics from the industrial, academic, and regulatory sectors of the United States, Europe, and Japan. The goal of the survey was to evaluate the (current and future) applications and broader scientific impact of toxicogenomics. The survey revealed that scientists were optimistic on the utility of toxicogenomics for basic and mechanistic research. In contrast, not as many scientists were enthusiastic on the wider impact of toxicogenomics in influencing safety assessment and policy decision making (Pettit et al., 2010).
Advances in toxicogenomics also led to the development of the concept of systems toxicology. The term was introduced by Waters and Fostel (2004) after the concept of systems biology (Ideker et al., 2001). Systems toxicology aims to describe all of the toxicological interactions within a living system and operates on the premise that global genomic data can be integrated and modeled computationally (Waters and Fostel, 2004). Thus, systems toxicology, like systems biology, involves the analysis of interactions of a large network of biological molecules and macromolecules and their perturbations following exposure to a chemical.
THE ROLE OF EPIGENETIC CHANGES IN MEDIATING TOXICITY CREATED A NEW PARADIGM OF EPIGENETIC REGULATION OF TOXICITY SUPERIMPOSED ON THE GENOME-CENTRIC VIEW OF TOXIC RESPONSE
The selective expression of certain genes and the silencing of others in specific tissues constitute the genetic basis of tissue-specific functional differences. Advances in understanding of the epigenetic regulation of gene expression superimposed an extra layer of complexity on the DNA sequence-centric view of genome function and regulation.
Epigenetics is defined as the study of changes in gene function without alterations in the DNA sequence. Epigenetic marks are dynamic horizontally (in the life span of an organism) as well as vertically (through inheritance). It is the heritability of epigenetic marks that makes the study of epigenetics important from a developmental perspective. Epigenetic regulation is achieved through processes, such as (1) DNA methylation, (2) histone modifications and (3) noncoding RNA (ncRNA)-mediated regulation. Epigenomics can be stated as “epigenetics of the genome”, and it refers to the study of genome-wide epigenetic changes. Toxicoepigenomics studies the genome-wide epigenetic changes as a result of exposure to drugs or environmental toxicants.
Holliday (1979) proposed that altered DNA methylation status could be associated with carcinogenesis. This was confirmed in 1983 by 2 publications co-authored by Feinberg and Vogelstein (1983a,b). Since then, many studies have been conducted to document the epigenetic changes associated with various cancers. Thus, knowledge of the epigenetic landscape of many cancers became an integral part of understanding the molecular mechanism of carcinogenesis beyond genetic perturbations (epigenetic mechanisms). The research on epigenetics and carcinogenesis eventually led to studies, conducted in the 1990s, investigating the role of epigenetic changes in mediating toxicity (eg, Lee et al., 1995; Wagner and Blevins, 1993; Zhao et al., 1997). These toxicoepigenetic studies rapidly expanded in number and scope, thereby making a bigger impact within the first decade of the 21st Century. Most of the initial toxicoepigenetic studies focused on DNA methylation. Wagner and Blevins (1993) first demonstrated using butyric acid, tetradecanoylphorbol-13-acetate (TPA) and propane sultone that histone modification could also be an epigenetic mechanism of carcinogenesis, in addition to DNA methylation. Even though Wagner and Blevins focused on carcinogenesis, their study demonstrated xenobiotic-induced histone modifications as an epigenetic mechanism of toxic response. Up until then, DNA methylation was the main focus of epigenetics.
It is becoming increasingly recognized, based on both in vitro and in vivo determinations of epigenetic changes, that there is a significant potential for epigenetic modifications to be the underlying cause of toxicity. An ever-increasing number of studies are documenting the effects of epigenetic modifications on the expression of genes associated with xenobiotic transport, metabolism, and toxicity (see reviews by Cheng et al., 2012; Choudhuri et al, 2010; Klaassen et al., 2011).
THE “DATA EXPLOSION” ALSO HELPED REFINE THE ALREADY EXISTING NONTEST PREDICTIVE APPROACHES IN TOXICOLOGY, SUCH AS COMPUTATIONAL TOXICOLOGY
The nontest approaches involve the use of predictive models that are developed using available data (eg, computational toxicology, read-across) and not direct laboratory experiments. Computational (in silico) prediction of toxicity of an unknown compound based on its structural similarity with that of a known toxicant (structure-activity relationship or SAR) was among the first applications of computers in biology. More than fifty years have passed since the field of SAR was founded in the 1960s as a logical extension of physical organic chemistry (see Cherkasov et al., 2014). The initial SAR-based predictions had limited success because the prediction algorithms were developed based on a limited number of parameters. Over time, an exponential increase in toxicology data, advances in computational power and predictive model building, and further improvements in refining the chemical structure into descriptors (eg, substructure, physico-chemical properties, 3D properties) have collectively improved the accuracy of prediction and led to the introduction of the quantitative aspect into SAR (often referred to as QSAR). Therefore, in silico predictive toxicology has become a useful tool that can complement toxicology data and information in decision making (see Winkler, 2002). Computational toxicology focuses on applying computational tools across many scales, from chemical characterization to macromolecular interactions to systems behavior and beyond.
A good predictive model of computational toxicology is heavily dependent on a good training dataset that should ideally include the entire spectrum of features of all different classes of chemicals associated with a toxicological endpoint, such as receptor binding, mutagenicity, or genotoxicity assays, to name a few. Unfortunately, developing such a good training set is often confounded by data gaps. Data bias in the training dataset may result in prediction bias or inaccurate prediction. In a recent publication Kirchmair et al. (2015) discussed the scope and limitations of computational methods for various types of prediction purposes associated with drug metabolism and toxicity. However, the prediction capability for bioactivity and toxicological effects appears inadequate due to data gaps in the training dataset. Greene and Pennie (2015) expressed similar thoughts while discussing the pros and cons of the current state of computational toxicology.
Kirchmair et al. (2015) also pointed out that the quantitative structure-toxicity relationship models serve primarily as hazard identification tools to support general risk assessment. Only rarely are they derived for exposure-response relationships that allow the prediction of absolute toxicity in isolation. Developing high-quality predictive computational models, particularly complex models like dose-response models, require large volumes of high-quality data, preferably in vivo data. Because of the dynamic nature of the computational tools and techniques, computational approaches in toxicology are expected to undergo further refinements as more in vivo and in vitro data become available.
The binary prediction scheme is useful for hazard identification. An example is the screening for receptor-ligand interaction and consequent transcriptional activation of the toxicant-target gene(s). Such in vitro tests for hazard identification are useful in prioritizing chemicals in the environment (environmental chemicals) for further testing, including in vivo toxicity testing, to build the detailed toxicological profile.
An important ongoing endeavor in computational toxicology is the creation of virtual tissues. A “virtual liver” is currently being developed by U.S. EPA. The overall goal is to develop a multiscale computational model of the liver that incorporates anatomical and biochemical information relevant to toxicological mechanisms and responses. This virtual liver is expected to serve as the biologically based predictive model of chronic toxicity of various environmental chemicals (Kavlock et al., 2008).
A REPORT BY THE U.S. NATIONAL ACADEMY OF SCIENCES IN 2007 PROPOSED FUNDAMENTALLY NEW DIRECTIONS FOR TOXICITY TESTING THAT RECOMMENDED MOVING AWAY FROM GENERATING TRADITIONAL APICAL TOXICITY ENDPOINTS THROUGH ANIMAL TESTING, BUT INSTEAD USING IN VITRO METHODS TO IDENTIFY TOXICITY PATHWAYS
In 2005, the U.S. EPA along with the U.S. National Toxicology Program (NTP) funded a project at the NRC of the National Academies of Science (NAS) to develop a long-range vision for toxicity testing. The goal was to develop a toxicity testing paradigm for the future, using advances in toxicogenomics, in vitro biology, computational sciences, and information technology, to rely increasingly on human data as opposed to animal data, and to offer increased efficiency in design and costs (Collins et al., 2008).
In 2007, the NRC released its expert panel report titled, Toxicity Testing in the 21st Century: A Vision and a Strategy (NRC-NAS, 2007), which laid out a vision for the future of toxicity testing to support human health risk assessments. The NRC report called for transforming toxicology utilizing advances in toxicogenomics, bioinformatics, systems biology and computational toxicology. The report also called for moving away from whole-animal testing to one founded primarily on in vitro methods that evaluate changes in biological processes using cells, cell lines, or cellular components, preferably of human origin. The NRC report envisioned a new toxicity testing system that relies on our understanding of toxicity pathways. The report also emphasized that animal studies often involve higher doses than would be expected for typical human exposure, thereby requiring assumptions about low-dose exposure. Implicit in this vision is the importance of toxicity testing using low-dose exposure that typically occurs in the general population.
In order to meet the challenge of toxicology in the 21st Century, a number of national and international collaborations were initiated to develop the ability to utilize and model the large-scale datasets on the perturbations of genes, proteins, signaling, and network functions. The goal is to advance the molecular understanding and predictability of toxic responses that in turn could aid in risk assessment. In the United States, NTP, EPA, and the National Institutes of Health Chemical Genomics Center (NCGC) established a collaborative research program in 2008 to meet this challenge. The 3 partners in this collaboration had unique expertise, such as experimental toxicology at the NTP, computational toxicology at the EPA, and high-throughput technologies at the NCGC (Collins et al., 2008). The NCGC is now part of the National Center for Advancing Translational Sciences (NCATS) of NIH (Tice et al., 2013). The collaboration came to be known as Tox21. In 2010, the U.S. Food and Drug Administration (FDA) joined the Tox21 collaboration through a 5-year memorandum of understanding (MOU). In June, 2015, the Tox21 collaborators renewed their commitment to the program through another 5-year MOU between NTP, NCATS, EPA, and FDA (https://ntp.niehs.nih.gov/results/tox21/history-index.html; last accessed September 11, 2017).
The Tox21 initiative was launched with 4 major goals: (1) to better understand the biological processes and pathways operating in normal life functions; (2) to develop rapid, cost-effective, high-throughput assays (using in vitro systems or phylogenetically lower animals, such as fish, worms, etc.) that will help determine how these pathways and processes are adversely affected by chemicals; (3) to develop predictive modeling to prognosticate the in vivo hazard of a chemical; and (4) to prioritize chemicals for in-depth toxicological evaluations. In keeping with the 4 major goals of Tox21, 4 working groups—Compound Selection, Assays and Pathways, Informatics, and Targeted Testing—were established. One of the initial goals was to characterize hits from the primary screening of 10,000 compounds (Tox21 10 K library) in cellular models.
The collaborative research in Tox21 has developed, validated and optimized more than 100 in vitro cell-based assays using quantitative high-throughput screening. The collaboration has produced several publications thus far (see https://ntp.niehs.nih.gov/results/tox21/pubs-factsheets-index.html; last accessed September 11, 2017). In a recent publication Huang et al. (2016) reported the testing of approximately 10 000 chemicals in triplicates at 15 concentrations against a panel of NR and stress-response pathway assays, producing more than 50 million data points. The authors were able to cluster compounds by structural similarity and activity profile and concluded that the structure–activity relationships could be useful for the generation of mechanistic hypotheses. Using the cluster-based approach the authors built predictive models for 72 in vivo toxicity end points. The authors concluded that in vitro activity profiles can be applied as signatures associated with a compound’s toxicity profile and used in prioritization for more in-depth toxicological testing. The authors, however, emphasized the continued need for quality in vivo toxicity data to further improve the predictability.
The NAS’s report has been welcomed by many in the scientific community with great enthusiasm and interest. The Committee’s proposal of moving away from animal testing to in vitro methods represents a radical paradigm shift in toxicity testing that has been practiced for well over half-a-century. Consequently, there are dissenting voices as well (see http://www.belleonline.com/newsletters/volume15/vo15-3.pdf; last accessed September 11, 2017; Gillet et al., 2011; Nebert and Ingelman-Sundberg, 2016). It is evident from Figure 2 (as first described by Gregus and Klaassen, 1996) that the development and validation of the in vitro assays predictive of the steps/events involved in the mechanism of toxicity in vivo is no small task.
Figure 2.

Potential stages in the development of toxicity following chemical exposure. Toxicity involves the delivery of the toxicant to the target tissue where an interaction with its target molecule(s) may result in cellular dysfunction, which is primary mediated by aberrant cell signaling and inadequate cell maintenance. The process may involve the toxicant itself or its metabolism product, which could be more reactive than the parent compound. Damaged cells could die off or attempt to repair, recover, and restore homeostasis. Cells that attempt to repair and recover tend to undergo apoptosis if repair and recovery fails. The surviving damaged cells could trigger the development of an adverse outcome, including carcinogenesis.
FORMULATION OF THE ADVERSE OUTCOME PATHWAY FRAMEWORK PRODUCED A FURTHER REFINEMENT OF THE CONCEPT OF TOXICITY PATHWAY FOR UNDERSTANDING AND PREDICTING TOXICITY AS A LINEAR SEQUENCE OF EVENTS FROM EXPOSURE TO THE MANIFESTATION OF TOXICITY AT A BIOLOGICAL LEVEL OF ORGANIZATION THAT COULD BE USEFUL FOR RISK ASSESSMENT
The underlying message of the NRC report is that a thorough understanding of the toxicity pathways (TPs) would enable toxicologists to profile the potential hazards and conduct risk assessment. The TP was defined as a cellular response pathway that would result in an adverse health effect when sufficiently perturbed.
Ankley et al. (2010) argued that NRC’s definition of TP focuses almost exclusively on initiating events and proximal cellular responses (overt toxic effects) that can be quantified in vitro. The authors suggested that the TP framework should utilize the data and knowledge collected at many levels of biological organization so that it is useful for risk assessors. They introduced the concept of adverse outcome pathway (AOP). Although otherwise normal and biologically essential pathways, the AOP terminology describes a sequence of quantifiable key events (KEs) triggered by a molecular initiating event (MIE). In the AOP pathway, the MIE is the most upstream event caused by the stressor interacting with the biological target. This interaction (MIE) then triggers a cascade of KEs that are the quantifiable molecular, cellular, structural and functional changes in biological systems. Triggering of these downstream KEs ultimately results in a quantifiable adverse outcome at a biological level of organization relevant to risk assessment (Ankley et al., 2010). The causal links between the KEs are called the key event relationships (KERs) (Figure 3A). Therefore, the AOP framework is a conceptual construct. The AOP as depicted in Figure 3A certainly relates to the ecotoxicological effects of environmental chemicals.
Figure 3.

A, The difference between TP and AOP is depicted. A TP focuses on initiating events and proximal cellular responses that can be measured in vitro. In contrast, an AOP is defined as a conceptual construct that portrays existing knowledge concerning the linkage between a direct MIE and an adverse outcome at a biological level of organization relevant to risk assessment. The steps (events) that lie between the MIE and the adverse outcome constitute the KEs, and their causal links are called the KERs. B, The exposure-to-outcome-continuum model discussed in the recent NAS report. The interaction with biological molecules step is similar to the MIE step in Ankley’s AOP pathway model. Elaboration of the exposure events as external exposure, internal exposure, and target exposure reflects the recent developments in the exposure science.
In a recent publication, the NAS summarized the scientific advances made since the original proposal of Tox21 (NAS, 2017). These advances, it is hoped, will support the development of further research to ensure that the full potential of 21st century science is realized to help solve the complex environmental and public-health problems. In this report, NAS further elaborated on the concept of TP as the exposure-to-outcome-continuum. The KEs in this model have many similarities to those discussed in the AOP pathway. Elements in 2 of the KEs interaction with biological macromolecule and cell response are discussed as well (Figure 3B). In the exposure-to-outcome-continuum model, the interaction with biological molecules step is similar to the MIE step in the AOP pathway model. Elaboration of the exposure events as external exposure, internal exposure, and target exposure further reflects the recent developments in exposure science.
The AOP concept (and now the exposure-to-outcome-continuum model) overlaps considerably with the traditional concept of MOA. However, there are subtle differences—the MOAs are chemical-specific, whereas AOP represents a chemical-independent conceptual framework. Therefore, an MOA can be constructed from the AOP by using chemical-specific toxicological information (Edwards et al., 2016). In essence, all these terms focus on understanding the underlying biological mechanisms, pathways or interaction network associated with the development of adverse effects (Burden et al., 2015).
The term AOP or TP could be misleading because they may lead one to think that natural selection has fixed the pathways to evolve as effector pathways for mediating adverse outcome/toxicity. In that sense the term AOP or TP is a misnomer. The pathways are not intrinsically adverse; it is only when they are perturbed, adverse outcome/toxicity ensues.
DEVELOPMENT OF GENOME EDITING TOOLS OFFERS GREAT POSSIBILITIES OF CREATING NEW IN VITRO AND IN VIVO MODELS OF TOXICITY TESTING THAT WAS NOT POSSIBLE PREVIOUSLY
A recent technological development expected to further advance toxicology research is the collection of genome editing tools, such as Zn-Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR associated (Cas9) system (CRISPR-Cas9). Whereas ZFN and TALEN are protein-guided genome editing tools, CRISPR-Cas9 is an RNA-guided genome editing tool. The use of ZFN for genome editing was first reported in 1996, that of TALEN in 2010, while that of CRISPR-Cas9 in 2013.
Originally, gene function was inactivated by mutating a gene through deletion of a large part of the coding sequence, introduced by means of homologous recombination. This was the so-called gene knockout technique. The knockout animal models were all mice because the knockout technique required the use of mouse embryonic stem (ES) cells, which can be cultured, maintained and manipulated more easily. In contrast, the manipulation of rat ES cells was not possible, making the generation of knockout rats technically challenging.
In 1998, the discovery of RNA interference (RNAi) mediated by double-stranded RNA opened the possibility of in vivo and in vitro knockdown (instead of knockout) of gene expression. Both mouse and rat models of RNAi have been generated (Hasuwa et al., 2002; Akai et al. 2007). Nevertheless, functional knockdown most often does not completely abolish the expression of the target gene; rather it significantly reduces it. Additionally, the extent of reduction is not identical or fully predictable for every single insert and genomic integration scenario.
The development of genome editing tools and techniques paved the way to obtain null animal models that could not be obtained using the standard knockout or knockdown technologies, such as rat, which is the animal model of choice for many toxicological studies commonly submitted to the regulatory agencies. Genome editing using ZFN has made the generation of rat knockout models possible (Geurts et al., 2009). Likewise, there have been several reports on the generation of knockout rat models using CRISPR-Cas9 genome editing technology (eg, Ma et al., 2014a,b; Shao et al., 2014).
Several knockout rat models have been generated by individual investigators in their laboratories. Also, many knockout rat models are currently commercially available. A web search using the search term “knockout rats” retrieves the information from a number of vendors. The genome editing tools can also be used to generate appropriate in vitro models to study the molecular mechanisms of toxicity following exposure to specific toxicants.
CONCLUSIONS: GOING FORWARD
There Is No Perfect Experimental Model to Predict Systemic Toxicity in Humans
Traditionally, whole-animal toxicity testing has been the source of most of the available toxicity data that have helped develop our current knowledge base in toxicology. However, the long history of experimental animal toxicology has also demonstrated that despite their utility, no animal model is perfect in predicting the fate and effect of a chemical in humans. Thus, animal-to-human extrapolation of experimental results could be questionable or challenging. Even humans are not perfect models for all humans. For example, the toxicity and metabolic fate of a compound in adults is often not useful in predicting the same in infants. Likewise, due to inter-individual variability, the toxicity and metabolic fate of a compound in 1 human subpopulation may not be reflective of the same for the entire human population. For example, it is well known that drugs that pass Phase III clinical trials may show toxicity in certain human subpopulations that was not predicted from the fate of the drug in human volunteers who participated in clinical trials.
Therefore, the More Experimental and Predictive Models Toxicologists Have, the Better the Risk Assessment Is Likely to Be
Currently, in vitro toxicity assays can be used to investigate the toxicity potential of a compound, screen and rank chemicals for their toxicity potential, and improve subsequent study design. In vitro toxicity assays are often conducted to evaluate endpoints like cytotoxicity, protein binding, enzyme induction/inhibition, membrane permeability etc. Nevertheless, as discussed before, the available in vitro models are not necessarily optimal for in vitro-to-in vivo extrapolation.
However, in recent years there have been some positive developments in this field (Bale et al., 2014). The “liver spheroids” in vitro model appears to hold promise. Recently Bell et al. (2016) developed and extensively characterized a 3D primary human hepatocyte spheroid system in chemically defined, serum-free conditions. The culture conditions allowed co-culture of the hepatocyte spheroids with nonparenchymal cells, such as biliary cells, stellate cells and Kupffer cells and supported their long-term viability. Using whole proteome analyses, the authors found that the hepatocyte spheroids were similar to the liver in vivo; phenotypically stable; retained morphology, viability, hepatocyte-specific functions; and even retained their inter-individual variability. The authors also reported that under chronic exposure, the sensitivity of the hepatocytes drastically increased and the toxicity of a set of hepatotoxicants was detected at clinically relevant concentrations. The use of appropriate in vitro models can also overcome certain limitations of animal experiments (eg, number of animals for statistical power, repetitions of the experiment, etc.) and can expand the scope of data collection efforts. An increase in high-quality toxicology and metabolism data obtained by in vitro experiments is expected to eventually fill the data gap and significantly improve computational prediction of toxicity and metabolism of new molecular entities. The acceptability of in vitro models as surrogates for animal toxicity testing will require, in addition to extensive validation, consensus by experts in the field. Nevertheless, it is undeniable that the collection of in vitro models currently available to experimental toxicologists is superior to what existed even a decade ago.
The Existing in Vitro Models Can Be Used for Hazard Identification for Environmental Chemicals and Also for the Safety Evaluation of Certain Dermal Products
Advances in in vitro methodologies could facilitate rapid hazard identification using high-throughput screening, such as the identification of environmental chemicals (chemicals in the environment) that may interact with hormone receptors and cause endocrine disruption. The high-throughput screening of environmental chemicals can help prioritize them for further toxicity testing including animal testing, to build detailed toxicological profiles. Thus, the initial use of various in vitro toxicity testing methods could significantly reduce the use of animals in the long run. Therefore, regulatory decision-making process in environmental toxicology will likely benefit from the advances in in vitro toxicity testing. Already in the field of toxicology of cosmetic ingredients, regulatory agencies accept in vitro tests for dermal absorption, irritation and corrosivity, as well as ocular irritation and corrosivity. In addition, in vitro genotoxicity and mutagenicity tests are also accepted.
In Contrast to Chemicals in the Environment, the Risk Assessment of Chemicals That Are Directly Introduced Into the Body Through Oral, Intravenous and Other Routes Routinely Utilizes Animal Toxicity Testing
Humans may be inadvertently exposed to chemicals in the environment, usually at a low-level. In contrast, food ingredients, drugs, and biologics are directly introduced into the body through oral, intravenous and other routes, and the exposure is usually not low-level. The risk assessment of food ingredients, drugs, biologics, etc. requires the use of animal testing. Certain in vitro tests, such as in vitro genotoxicity and mutagenicity tests are accepted as part of the toxicology data package for regulatory evaluation. Therefore, the utility of in vivo toxicity data still remains undeniable. This is reflected in a statement by Huang et al. (2016), mentioned earlier, in which the authors emphasized “the continued need for quality in vivo toxicity data” to further improve the predictability of the alternative toxicology methods. Preclinical animal toxicology studies are also used to determine the dose for human clinical trials. Therefore, toxicity testing in animals and clinical trials in humans are expected to remain useful in the foreseeable future for the practicing toxicologists in conducting risk assessments. This seems to be more relevant for food ingredients, drugs, biologics, devices, and similar compounds than for chemicals in the environment.
An Important Challenge for Toxicologists Is to Address Many Observations From in Vivo Studies That Might Be Difficult to Explain Using Alternative Methods of Toxicity Prediction
Addressing the observations from in vivo experimental toxicology that seem challenging to explain using alternative methods of toxicity testing will help build the expert consensus with regard to the utility and acceptability of alternative methods of toxicity testing and prediction. For example, Nebert and Ingelman-Sundberg (2016) expressed concern regarding the value of using cell lines, instead of laboratory animals, for the assessment of clinical toxicity. They stated, “It is evident from thousands of publications that in vitro systems—although being developed in the format of organ-on-a chip and 3D models—cannot predict the fate of chemicals in an in vivo setting. Initiatives for new complex 3D in vivo tissue systems, such as tissue buds and integrated tissue combinations in different formats, are reasonable to pursue because they have the advantage of being focused on the human, which should lead to using fewer animals for toxicity testing. However, these approaches still remain far from being able to predict human toxicity in vivo. Due to the need for such challenges as taking pharmacokinetic factors into account, and requiring true in vivo conditions for mimicking toxicity, animal testing is thus still necessary”. The authors provided the examples of fibrate drug-treated PPARα-humanized mice, and benzo[a]pyrene-treated CYP1A1 and CYP1B1 null mice and underscored that the findings from these animal studies could never have been made using in vitro studies alone. (Nebert and Ingelman-Sundberg (2016) have been quoted here as is from their publication in order to maintain the emphasis of their statement.)
A recent publication (Cantor et al., 2017) demonstrated how the conditions widely used for in vitro studies may lead to erroneous conclusions on in vivo metabolism. The authors developed a culture medium with polar metabolite concentrations comparable to those of adult human plasma (human plasma-like medium [HPLM]). They found that culture in HPLM, relative to that in traditional media, had widespread effects on cellular metabolism, including on the metabolome, redox state, and glucose utilization. The authors concluded that media that better mimic the composition of human plasma reveal unforeseen metabolic wiring and regulation. Unfortunately, the traditional synthetic cell culture media do not mimic human plasma in their composition.
In a recent article, Nei and Malloy (2017) voiced similar concerns and highlighted certain inherent problems of the current state of alternative testing strategies (ATS). They stated that cells do not exhibit the biological complexity of the intact organism, unwanted false-positives and false-negatives may adversely impact the outcome of the experiments, ATS addresses acute rather than the chronic conditions, and the role of metabolism is not addressed by ATS.
The publications by Cantor et al. (2017), and Nei and Malloy (2017) corroborate an early report by Knutson and Poland (1980) who demonstrated that TCDD, one of the most potent small molecule toxicants known, failed to produce toxicity in 23 cultured cell types that included primary culture, as well as cells from established and transformed cell lines. The markers of cellular toxicity studied were altered morphology, decreased viability, altered growth rate and pattern, and AHH activity in TCDD-treated cells compared with control cells. Collectively, these findings demonstrate that a sole dependence on in vitro toxicity studies may lead to erroneous conclusion (including underestimation) on the toxicity potential of chemicals in vivo.
In a review of the clinical relevance of the cancer cell lines, Gillet et al. (2013) stated that human cancer-derived cell lines are the most widely used models to study the biology of cancer and to test hypotheses to improve cancer treatment. However, the clinical relevance of these models has been continuously questioned. The use of “-omics” and high-throughput analytical methods showed that at the genomic level driver mutations are retained, but a drift occurs at the transcriptome level, leading to the conclusion that cancer cell lines bear more resemblance to each other, regardless of the tissue of origin, than to the clinical samples that they are supposed to model. In other words, the evolution of different types of cancer cells under the same selection pressure to rapidly grow and divide may lead to the development of similar characters irrespective of their origin (convergent evolution in vitro). Without knowledge of such heterogeneity of the in vitro experimental model, it is easy to be misled and draw erroneous conclusions from the experimental outcome.
There are other examples, too. Reactive metabolite-mediated toxicity is frequently limited to the organ where the electrophilic metabolites are generated. Some reactive metabolites however, translocate to a different tissue from their site of formation. Irving and Elfarra (2012) discussed such examples where the reactive metabolites are generated in the liver but they translocate to the kidney and cause nephrotoxicity.
Oxidative stress is known to play a significant role in carcinogenicity. Interestingly, paraquat, which is one of the strongest oxidative-stress inducing agents known, is noncarcinogenic. There are other questions as well. For example, can the toxicity of a chemical that exerts an adverse effect only in a small window of time during development (eg, as seen in the case of thalidomide) be predicted by the existing alternative methods of toxicity testing? These examples (and many more) underscore the fact that despite the recent developments in alternative toxicity testing, there is a long road ahead before these developments are directly translated into regulatory decision-making, particularly for food additives, drugs, biologics, devices and similar compounds. In contrast, the screening of environmental chemicals in order to prioritize them for further study, may already benefit from the developments in alternative toxicity testing, including in vitro testing, computational methods and read across.
The ultimate goal of toxicology is to protect human, animal and environmental health. Therefore, advances in our understanding of the molecular underpinnings of health and disease and how environmental stressors affect them lies at the core of innovative, informed, and more efficient approaches to risk assessment.
FUNDING
This work was supported in part by NIH grant ES025708
REFERENCES
- Akai S., Hosomi H., Minami K., Tsuneyama K., Katoh M., Nakajima M., Yokoi T. (2007). Knock down of gamma-glutamylcysteine synthetase in rat causes acetaminophen-induced hepatotoxicity. J. Biol. Chem 282, 23996–24003. [DOI] [PubMed] [Google Scholar]
- Alwine J. C., Kemp D. J., Stark G. R. (1977). Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc. Natl. Acad. Sci. U.S.A. 74, 5350–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankley G. T., Bennett R. S., Erickson R. J., Hoff D. J., Hornung M. W., Johnson R. D., Mount D. R., Nichols J. W., Russom C. L., Schmieder P. K., et al. (2010). Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environment. Toxicol. Chem. 29, 730–741. [DOI] [PubMed] [Google Scholar]
- Antoni D., Burckel H., Josset E., Noel G. (2015). Three-dimensional cell culture: A breakthrough in vivo. Int. J. Mol. Sci. 16, 5517–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfel R., Benbrook D., Lernhardt E., Ortiz M. A., Salbert G., Pfahl M. (1994). A novel orphan receptor specific for a subset of thyroid hormone-responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol. Cell. Biol. 14, 7025–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baes M., Gulick T., Choi H. S., Martinoli M. G., Simha D., Moore D. D. (1994). A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol. Cell. Biol. 14, 1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale A. S., Kenyon E., Flynn T. J., Lipscomb J. C., Mendrick D. L., Hartung, T., and Patton, G.W. (2014). Correlating in vitro data to in vivo findings for risk assessment. Altex 31, 79–90. [DOI] [PubMed] [Google Scholar]
- Bell C. C., Hendriks D. F. G., Moro S. M. L., Ellis E., Walsh J., Renblom, A., Fredriksson Puigvert, L., Dankers, A.C., Jacobs, F., Snoeys, J., et al. (2016). Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 6, 25187.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie B. B., Reid W. D., Cho A. K., Sipes G., Krishna G., and Gillette, J.R. (1971). Possible mechanism of liver necrosis caused by aromatic organic compounds. Proc. Natl. Acad. Sci. U.S.A. 68, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach K., Poland A., Bradfield C. A. (1992). Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. U.S.A. 89, 8185–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burden N., Sewell F., Andersen M. E., Boobis A., Chipman J. K., Cronin, M.T., Hutchinson, T.H., Kimber, I., and Whelan, M. (2015). Adverse Outcome Pathways can drive non-animal approaches for safety assessment. J. Appl. Toxicol. 35, 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor J. R., Abu-Remaileh M., Kanarek N., Freinkman E., Gao X., Louissaint A. Jr., Lewis C. A., Sabatini D. M. (2017). Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169, 258–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson R. 1962. Silent Spring. Houghton Mifflin, Boston. [Google Scholar]
- Cheng T. F., Choudhuri S., Muldoon-Jacobs K. (2012). Epigenetic targets of some toxicologically relevant metals: A review of the literature. J. Appl. Toxicol. 32, 643–653. [DOI] [PubMed] [Google Scholar]
- Cherkasov A., Muratov E. N., Fourches D., Varnek A., Baskin I. I., Cronin, M., Dearden, J., Gramatica, P., Martin, Y.C., Todeschini, R., et al. (2014). QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 57, 4977–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C., Akiyama T. E., Ward J. M., Nicol C. J., Feigenbaum L., Vinson, C., and Gonzalez, F.J. (2004). Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res. 64, 3849–3854. [DOI] [PubMed] [Google Scholar]
- Choi H. S., Chung M., Tzameli I., Simha D., Lee Y. K., Seol, W., and Moore, D.D. (1997). Differential transactivation by two isoforms of the orphan nuclear hormone receptor CAR. J. Biol. Chem. 272, 23565–23571. [DOI] [PubMed] [Google Scholar]
- Choudhuri S. (2006). Some major landmarks in the path from nuclein to human genome. Toxicol. Mech. Methods 16, 137–159. [DOI] [PubMed] [Google Scholar]
- Choudhuri S., Cui Y., Klaassen C. D. (2010). Molecular targets of epigenetic regulation and effectors of environmental influences. Toxicol. Appl. Pharmacol. 245, 378–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F. S., Gray G. M., Bucher J. R. (2008). Toxicology. Transforming environmental health protection. Science 319, 906–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanaky I. L., Lu H., Zhang Y., Ogura K., Choudhuri S., Klaassen C. D. (2011). Organic anion-transporting polypeptide 1b2 (Oatp1b2) is important for the hepatic uptake of unconjugated bile acids: Studies in Oatp1b2-null mice. Hepatology 53, 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. Y., Gunewardena S. S., Yoo B., Liu J., Renaud H. J., Lu, H., Zhong, X.B., and Klaassen, C.D. (2012). RNA-Seq reveals different mRNA abundance of transporters and their alternative transcript isoforms during liver development. Toxicol. Sci. 127, 592–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva T. C., Polli J. E., Swaan P. W. (2013). The solute carrier family 10 (SLC10): Beyond bile acid transport. Mol. Asp. Med. 34, 252–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower L. A., Harvey M., Slagle B. L., McArthur M. J., Montgomery C. A. Jr, Butel, J.S., and Bradley, A. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221. [DOI] [PubMed] [Google Scholar]
- Edwards S. W., Tan Y.-M., Villeneuve D. L., Meeks M. E., McQueen C. A. (2016). Adverse outcome pathways− organizing toxicological information to improve decision making. J. Pharmacol. Exp. Therap. 356, 170–181. [DOI] [PubMed] [Google Scholar]
- Estabrook R. (2003). A passion for P450s (remembrances of the early history of research on cytochrome P450). Drug Metab. Disp. 31, 1461–1473. [DOI] [PubMed] [Google Scholar]
- Evans R. M., Mengelsdorf D. J. (2014). Nuclear Receptors, RXR, and the Big Bang. Cell 157, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A. P., Vogelstein B. (1983a). Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301, 89–92. [DOI] [PubMed] [Google Scholar]
- Feinberg A. P., Vogelstein B. (1983b). Hypomethylation of ras oncogenes in primary human cancers. Biochem. Biophys. Res. Commun. 111, 47–54. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P., Pineau T., Hilbert D. M., McPhail T., Lee S. S., Kimura, S., Nebert, D.W., Rudikoff, S., Ward, J.M., and Gonzalez, F.J. (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726. [DOI] [PubMed] [Google Scholar]
- Flecknell P. (2002). Replacement, reduction and refinement. Altex 19, 73–78. [PubMed] [Google Scholar]
- Forman B. M., Goode E., Chen J., Oro A. E., Bradley D. J., Perlmann, T., Noonan, D.J., Burka, L.T., McMorris, T., Lamph, W.W., et al. (1995). Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 81, 687–693. [DOI] [PubMed] [Google Scholar]
- Gielen J. E., Goujon F. M., Nebert D. W. (1972). Genetic regulation of aryl hydrocarbon hydroxylase induction. II. Simple Mendelian expression in mouse tissues in vivo. J. Biol. Chem 247, 1125–1137. [PubMed] [Google Scholar]
- Gillet J. P., Calcagno A. M., Varma S., Marino M., Green, L.J., Vora, M.I., Patel, C., Orina, J.N., Eliseeva, T.A., Singal, V., et al. (2011). Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 18708–18713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet J. P., Varma S., Gottesman M. M. (2013). The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 105, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S., Walter P., Kumar V., Krust A., Bornert J. M., Argos, P., and Chambon, P. (1986). Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 320, 134–139. [DOI] [PubMed] [Google Scholar]
- Greene N., Pennie W. (2015). Computational toxicology: Friend or foe? Toxicol. Res. 4, 1159–1162. [Google Scholar]
- Gregus Z., Klaassen C. D. (1996). Mechanisms of toxicity (Chapter 3). In Casarett and Doull’s Toxicology: The Basic Science of Poisons, 5th ed (Klaassen C. D., Amdur M. O., Doull J., Eds.). McGraw-Hill, New York, pp. 35–75. [Google Scholar]
- Geurts A. M., Cost G. J., Freyvert Y., Zeitler B., Miller J. C., Choi, V.M., Jenkins, S.S., Wood, A., Cui, X., Meng, X., et al. (2009). Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325, 433.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanville N., Durnam D. M., Palmiter R. D. (1981). Structure of mouse metallothionein-I gene and its mRNA. Nature 292, 267–269. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. (2014). Fifty years of progress in drug metabolism and toxicology: What do we still need to know about cytochrome P450 enzymes? In Fifty Years of Cytochrome P450 Research (Yamazaki H. Ed.). DOI 10.1007/978-4-431-54992-5_2,© Springer Japan 2014, pp. 17–41. [Google Scholar]
- Hagenbuch B., Stieger B. (2013). The SLCO (former SLC21) superfamily of transporters. Mol. Asp. Med. 34, 396–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamadeh H. K., Amin R. P., Paules R. S., Afshari C. A. (2002). An overview of toxicogenomics. Curr. Issues Mol. Biol. 4, 45–56. [PubMed] [Google Scholar]
- Hasuwa H., Kaseda K., Einarsdottir T., Okabe M. (2002). Small interfering RNA and gene silencing in transgenic mice and rats. FEBS Lett. 532, 227–230. [DOI] [PubMed] [Google Scholar]
- Hoffman E. C., Reyes H., Chu F. F., Sander F., Conley L. H., Brooks, B.A., and Hankinson, O. (1991). Cloning of a factor required for activity of the Ah (dioxin) receptor. Science 252, 954–958. [DOI] [PubMed] [Google Scholar]
- Hollenberg S. M., Weinberger C., Ong E. S., Cerelli G., Oro A., Lebo, R., Thompson, E.B., Rosenfeld, M.G., and Evans, R.M. (1985). Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. (1979). A new theory of carcinogenesis. Br. J. Cancer. 40, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R., Xia M., Sakamuru S., Zhao J., Shahane S. A., Attene-Ramos, M., Zhao, T., Austin, C.P., and Simeonov, A. (2016). Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat. Commun. 7, 10425.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICH 1997. Testing for Carcinogenicity of Pharmaceuticals (S1B). ICH Harmonised Tripartite Guideline. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S1B/Step4/S1B_Guideline.pdf, last accessed September 11, 2017. [Google Scholar]
- Ideker T., Galitski T., Hood L. (2001). A new approach to decoding life: Systems biology. Annu. Rev. Genomics Hum. Genet. 2, 343–372. [DOI] [PubMed] [Google Scholar]
- Irving R. M., Elfarra A. A. (2012). Role of reactive metabolites in the circulation in extrahepatic toxicity. Expert Opin. Drug Metab. Toxicol. 8, 1157–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issemann I., Green S. (1990). Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347, 645–650. [DOI] [PubMed] [Google Scholar]
- Jacobson-Kram D., Sistare F. D., Jacobs A. (2004). Use of transgenic mice in carcinogenicity hazard assessment. Toxicologic Pathol. 32(Suppl. 1), 49–52. [DOI] [PubMed] [Google Scholar]
- Jackson M. R., McCarthy L. R., Corser R. B., Barr G. C., Burchell B. (1985). Cloning of cDNAs coding for rat hepatic microsomal UDP-glucuronyltransferases. Gene 34, 147–153. [DOI] [PubMed] [Google Scholar]
- Jennings P., Limonciel A., Felice L., Leonard M. O. (2013). An overview of transcriptional regulation in response to toxicological insult. Arch. Toxicol. 87, 49–72. [DOI] [PubMed] [Google Scholar]
- Kafatos F. C., Jones C. W., Efstratiadis A. (1979). Determination of nucleic acid sequence homologies and relative concentrations by a dot hybridization procedure. Nucl. Acids Res. 7, 1541–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavlock R. J., Ankley G., Blancato J., Breen M., Conolly R., Dix, D., Houck, K., Hubal, E., Judson, R., Rabinowitz, J., et al. (2008). Computational toxicology−a state of the science mini review. Toxicol. Sci. 103, 14–27. [DOI] [PubMed] [Google Scholar]
- Kirchmair J., Göller A. H., Lang D., Kunze J., Testa B., Wilson, I.D., Glen, R.C., and Schneider, G. (2015). Predicting drug metabolism: Experiment and/or computation. Nat. Rev. Drug Discov. 14, 387–404. [DOI] [PubMed] [Google Scholar]
- Klaassen C. D., Liu J. (1998). Metallothionein transgenic and knock-out mouse models in the study of cadmium toxicity. J. Toxicol. Sci. 23(Suppl. II), 97–108. [DOI] [PubMed] [Google Scholar]
- Klaassen C. D., Liu J., Choudhuri S. (1999). Metallothionein: An intracellular protein to protect against cadmium toxicity. Annu. Rev. Pharmacol. Toxicol. 39, 267–294. [DOI] [PubMed] [Google Scholar]
- Klaassen C. D., Aleksunes L. M. (2010). Xenobiotic, bile acid, and cholesterol transporters: Function and regulation. Pharmacol. Rev. 62, 1–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen C. D., Lu H., Cui J. Y. (2011). Epigenetic regulation of drug processing genes. Toxicol. Mech. Methods 21, 312–324. [DOI] [PubMed] [Google Scholar]
- Kliewer S. A., Moore J. T., Wade L., Staudinger J. L., Watson M. A., Jones, S.A., McKee, D.D., Oliver, B.B., Willson, T.M., Zetterström, R.H., et al. (1998). An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92, 73–82. [DOI] [PubMed] [Google Scholar]
- Knutson J. C., Poland A. (1980). 2,3,7,8-Tetrachlorodibenzo-p-dioxin: Failure to demonstrate toxicity in twenty-three cultured cell types. Toxicol. Appl. Pharmacol. 54, 377–383. [DOI] [PubMed] [Google Scholar]
- Koepsell H. (2013). The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Asp. Med. 34, 413–435. [DOI] [PubMed] [Google Scholar]
- Kurachi M., Hashimoto S., Obata A., Nagai S., Nagahata T., Inadera, H., Sone, H., Tohyama, C., Kaneko, S., Kobayashi, K., et al. (2002). Identification of 2,3,7,8-tetrachlorodibenzo-p-dioxin-responsive genes in mouse liver by serial analysis of gene expression. Biochem. Biophys. Res. Commun. 292, 368–377. [DOI] [PubMed] [Google Scholar]
- Leder A., Kuo A., Cardiff R. D., Sinn E., Leder P. (1990). v-Ha-ras transgene abrogates the initiation step in mouse skin tumorigenesis: Effects of phorbol esters and retinoic acid. Proc. Natl. Acad. Sci. U.S.A. 87, 9178–9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. W., Klein C. B., Kargacin B., Salnikow K., Kitahara J., Dowjat, K., Zhitkovich, A., Christie, N.T., and Costa, M. (1995). Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: A new model for epigenetic carcinogens. Mol. Cell. Biol. 15, 2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesko L. J., Woodcock J. (2004). Translation of pharmacogenomics and pharmacogenetics: A regulatory perspective. Nat. Rev. Drug Discov. 3, 763–769. [DOI] [PubMed] [Google Scholar]
- Liang P., Pardee A. B. (1992). Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257, 967–971. [DOI] [PubMed] [Google Scholar]
- Lu H., Choudhuri S., Ogura K., Csanaky I. L., Lei X., Cheng, X., Song, P.Z., and Klaassen, C.D. (2008). Characterization of organic anion transporting polypeptide 1b2-null mice: Essential role in hepatic uptake/toxicity of phalloidin and microcystin-LR. Toxicol. Sci. 103, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Zhang X., Shen B., Lu Y., Chen W., Ma, J., Bai, L., Huang, X., and Zhang, L. (2014a). Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 24, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Shen B., Zhang X., Lu Y., Chen W., Ma, J., Huang, X., and Zhang, L. (2014b). Heritable multiplex genetic engineering in rats using CRISPR/Cas9. PLoS One 9, e89413.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie P. I., Gonzalez F. J., Owens I. S. (1984). Cloning and Characterization of DNA complementary to rat liver UDP-glucuronosyltransferase mRNA. J. Biol. Chem. 259, 12153–12160. [PubMed] [Google Scholar]
- Maher V. M., Lesko S. A. Jr, Straat P. A., Ts'o P. O. (1971). Mutagenic action, loss of transforming activity, and inhibition of deoxyribonucleic acid template activity in vitro caused by chemical linkage of carcinogenic polycyclic hydrocarbons to deoxyribonucleic acid. J. Bacteriol. 108, 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengelsdorf D. J., Ong E. S., Dyck J. A., Evans R. M. (1990). Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 345, 224–229. [DOI] [PubMed] [Google Scholar]
- Miller E. C., Miller J. A. (1981). Searches for ultimate chemical carcinogens and their reactions with cellular macromolecules. Cancer 47, 2327–2345. [DOI] [PubMed] [Google Scholar]
- Moi P., Chan K., Asunis I., Cao A., Kan Y. W. (1994). Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. U.S.A. 91, 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi T., Motohashi H., Hosoya T., Nakajima O., Takahashi S., Ohsako, S., Aoki, Y., Nishimura, N., Tohyama, C., Fujii-Kuriyama, Y., et al. (2003). Distinct response to dioxin in an arylhydrocarbon receptor (AHR)-humanized mouse. Proc. Natl. Acad. Sci. U.S.A. 100, 5652–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Academy of Sciences. 2017. Using 21st Century Science to Improve Risk-Related Evaluations. The National Academies Press, Washington, DC, pp. 216. [PubMed] [Google Scholar]
- National Research Council of the National Academies (NRC-NAS). (2007). Applications of Toxicogenomic Technologies to Predictive Toxicology and Risk Assessment. The National Academies Press, Washington DC, pp. 300. [PubMed] [Google Scholar]
- Nebert D. W., Gelboin H. V. (1969). The in vivo and in vitro induction of aryl hydrocarbon hydroxylase in mammalian cells of different species, tissues, strains, and developmental and hormonal status. Arch. Biochem. Biophys. 134, 76–89. [DOI] [PubMed] [Google Scholar]
- Nerbert D. W., Bausserman L. L. (1970). Genetic differences in the extent of aryl hydrocarbon hydroxylase induction in mouse fetal cell culture. J. Biol. Chem. 245, 6373–6382. [PubMed] [Google Scholar]
- Nebert D. W., Gonzalez F. J. (1987). P450 genes: Structure, evolution and regulation. Annu. Rev. Biochem. 56, 945–993. [DOI] [PubMed] [Google Scholar]
- Nebert D. W., Goujon F. M., Gielen J. E. (1972). Aryl hydrocarbon hydroxylase induction by polycyclic hydrocarbons: Simple autosomal dominant trait in the mouse. Nat. New Biol. 236, 107–110. [DOI] [PubMed] [Google Scholar]
- Nebert D. W., Ingelman-Sundberg M. (2016). What do animal experiments tell us that in vitro systems cannot? The human toxome project. Regul. Toxicol. Pharmacol. 75, 1–4. [DOI] [PubMed] [Google Scholar]
- Nei A. E., Malloy T. F. (2017). Policy reforms to update chemical safety testing; TSCA reform empowers EPA to use modernized safety testing in the United States. Science 355, 1016–1018. [DOI] [PubMed] [Google Scholar]
- Nuwaysir E. F., Bittner M., Trent J., Barrett J. C., Afshari C. A. (1999). Microarrays and toxicology: The advent of toxicogenomics. Mol. Carcinogen. 24, 153–159. [DOI] [PubMed] [Google Scholar]
- Pachl C., Todd J. A., Kern D. G., Sheridan P. J., Fong S. J., Stempien, M., Hoo, B., Besemer, D., Yeghiazarian, T., Irvine, B., et al. (1995). Rapid and precise quantification of HIV-1 RNA in plasma using a branched DNA signal amplification assay. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 8, 446–454. [DOI] [PubMed] [Google Scholar]
- Palmiter R. D., Sandgren E. P., Koeller D. M., Brinster R. L. (1993). Distal regulatory elements from the mouse metallothionein locus stimulate gene expression in transgenic mice. Mol. Cell Biol. 13, 5266–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulusma C. C., Bosma P. J., Zaman G. J., Bakker C. T., Otter M., Scheffer, G.L., Scheper, R.J., Borst, P., and Oude Elferink, R.P. (1996). Congenital jaundice in rats with a mutation in a multidrug resistance-associated protein gene. Science 271, 1126–1128. [DOI] [PubMed] [Google Scholar]
- Paulusma C. C., Kool M., Bosma P. J., Scheffer G. L., ter Borg F., Scheper, R.J., Tytgat, G.N., Borst, P., Baas, F., and Oude Elferink, R.P. (1997). A mutation in the human canalicular multispecific organic anion transporter gene causes the Dubin-Johnson syndrome. Hepatology 25, 1539–1542. [DOI] [PubMed] [Google Scholar]
- Pettit S., des Etages S. A., Mylecraine L., Snyder R., Fostel J., Dunn, R.T. 2nd, Haymes, K., Duval, M., Stevens, J., Afshari, C., et al. (2010). Current and future applications of toxicogenomics: Results summary of a survey from the HESI Genomics State of Science Subcommittee. Environ. Health Perspect. 118, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland A. P., Glover E. (1974). Comparison of 2,3,7,8-tetrachlorodibenzo-p-dioxin, a potent inducer of aryl hydrocarbon hydroxylase, with 3-methylcholanthrene. Mol. Pharmacol. 10, 349–359. [PubMed] [Google Scholar]
- Poland A., Glover E., Robinson J. R., Nebert D. W. (1974). Genetic expression of aryl hydrocarbon hydroxylase activity: Induction of monooxygenase activities and cytochrome P1-450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons. J. Biol. Chem. 249, 5599–5606. [PubMed] [Google Scholar]
- Poland A., Glover E. (1975). Genetic expression of aryl hydrocarbon hydroxylase by 2,3,7,8-tetrachlorodibenzo-p-dioxin: Evidence for a receptor mutation in genetically non-responsive mice. Mol. Pharmacol. 11, 389–398. [Google Scholar]
- Poland A., Glover E., Kende A. S. (1976). Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-pdioxin by hepatic cytosol. J. Biol. Chem. 251, 4936–4946. [PubMed] [Google Scholar]
- Potter W. Z., Thorgeirsson S. S., Jollow D. J., Mitchell J. R. (1974). Acetaminophen-Induced hepatic necrosis V. correlation of hepatic necrosis, covalent binding and glutathione depletion in hamsters. Pharmacology 12, 129–143. [DOI] [PubMed] [Google Scholar]
- Riordan J. R., Deuchars K., Kartner N., Alon N., Trent J., Ling V. (1985). Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature 316, 817–819. [DOI] [PubMed] [Google Scholar]
- Saiki R. K., Scharf S., Faloona F., Mullis K. B., Horn G. T., Erlich, H.A., and Arnheim, N. (1985). Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230, 1350–1354. [DOI] [PubMed] [Google Scholar]
- Sargent T. D., Dawid I. B. (1983). Differential gene expression in the gastrula of Xenopus laevis. Science 222, 135–139. [DOI] [PubMed] [Google Scholar]
- Saitoh A., Kimura M., Takahashi R., Yokoyama M., Nomura T., Izawa, M., Sekiya, T., Nishimura, S., and Katsuki, M. (1990). Most tumors in transgenic mice with human c-Ha-ras gene contained somatically activated transgenes. Oncogene 5, 1195–1200. [PubMed] [Google Scholar]
- Sap J., Munoz A., Damm K., Goldberg Y., Ghysdael J., Leutz, A., Beug, H., and Vennström, B. (1986). The c-erb-A protein is a high affinity receptor for thyroid hormone. Nature 324, 635–640. [DOI] [PubMed] [Google Scholar]
- Schena M., Shalon D., Davis R. W., Brown P. O. (1995). Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470. [DOI] [PubMed] [Google Scholar]
- Schmidt J. V., Su G. H., Reddy J. K., Simon M. C., Bradfield C. A. (1996). Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. U.S.A. 93, 6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R., Baltimore D. (1986). Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46, 705–716. [DOI] [PubMed] [Google Scholar]
- Sever R., Glass C. K. (2013). Signaling by nuclear receptors. CSH Perspect. Biol. 5, a016709.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y., Guan Y., Wang L., Qiu Z., Liu M., Chen, Y., Wu, L., Li, Y., Ma, X., Liu, M., et al. (2014). CRISPR/Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nat. Protocol 9, 2493–2512. [DOI] [PubMed] [Google Scholar]
- Shibahara S., Müller R., Taguchi H., Yoshida T. (1985). Cloning and expression of cDNA for rat heme oxygenase. Proc. Natl. Acad. Sci. U.S.A. 82, 7865–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinar D. M., Endo N., Rutledge S. J., Vogel R., Rodan G. A., Schmidt A. (1994). NER, a new member of the gene family encoding the human steroid hormone nuclear receptor. Gene 147, 273–276. [DOI] [PubMed] [Google Scholar]
- Song C., Kokontis J. M., Hiipakka R. A., Liao S. (1994). Ubiquitous receptor: A receptor that modulates gene activation by retinoic acid and thyroid hormone receptors. Proc. Natl. Acad. Sci. U.S.A. 91, 10809–10813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger J. L., Woody S., Sun M., Cui W. (2013). Nuclear receptor-mediated regulation of drug- and bile acid-transporter proteins in gut and liver. Drug Metab. Rev. 45, 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan J. P. 1998. FDA’s Origin In Adapted from A Historical Guide to the U.S. Government (George K., Ed.) Oxford University Press, NY: Available at: http://www.fda.gov/AboutFDA/WhatWeDo/History/Origin/ucm124403.htm, last accessed September 11, 2017. [Google Scholar]
- Tada M., Tada M. (1971). Interaction of a carcinogen, 4-nitroquinoline-1-oxide, with nucleic acids: Chemical degradation of the adducts. Chem. Biol. Interact. 3, 225–229. [DOI] [PubMed] [Google Scholar]
- Tian J., Feng Y., Fu H., Xie H. Q., Jiang J. X., Zhao B. (2015). The aryl hydrocarbon receptor: A key bridging molecule of external and internal chemical signals. Environ. Sci. Technol. 49, 9518–9531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice R. R., Austin C. P., Kavlock R. J., Bucher J. R. (2013). Improving the human hazard characterization of chemicals: A Tox21 update. Environ. Health Perspect. 121, 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft J., Gaj S., Lienhard M., Albrecht M. W., Kirpiy A., Brauers, K., Claessen, S., Lizarraga, D., Lehrach, H., Herwig, R., et al. (2012). RNA-Seq provides new insights in the transcriptome responses induced by the carcinogen benzo[a]pyrene. Toxicol. Sci. 130, 427–439. [DOI] [PubMed] [Google Scholar]
- Velculescu V. E., Zhang L., Vogelstein B., Kinzler K. W. (1995). Serial analysis of gene expression. Science 270, 484–487. [DOI] [PubMed] [Google Scholar]
- Velculescu V. E., Zhang L., Zhou W., Vogelstein J., Basrai M. A., Bassett, D.E. Jr, Hieter, P., Vogelstein, B., and Kinzler, K.W. (1997). Characterization of the yeast transcriptome. Cell 88, 243–251. [DOI] [PubMed] [Google Scholar]
- Wagner V. O., Blevins R. D. (1993). Chemically-induced histone modification as a predictor of carcinogenicity. Arch. Environ. Contam. Toxicol. 25, 260–266. [DOI] [PubMed] [Google Scholar]
- Waters M. D., Fostel J. M. (2004). Toxicogenomics and systems toxicology: Aims and prospects. Nat. Rev. Genet. 5, 936–948. [DOI] [PubMed] [Google Scholar]
- Weinberger C., Thompson C. C., Ong E. S., Lebo R., Gruol D. J., Evans R. M. (1986). The c-erb-A gene encodes a thyroid hormone receptor. Nature 324, 641–646. [DOI] [PubMed] [Google Scholar]
- Winkler D. A. (2002). The role of quantitative structure−activity relationships (QSAR) in biomolecular discovery. Brief. Bioinform. 3, 73–86. [DOI] [PubMed] [Google Scholar]
- Wu K. C., Liu J. J., Klaassen C. D. (2012). Nrf2 activation prevents cadmium-induced acute liver injury. Toxicol. Appl. Pharmacol. 263, 14–20. [DOI] [PubMed] [Google Scholar]
- Xie W., Barwick J. L., Downes M., Blumberg B., Simon C. M., Nelson, M.C., Neuschwander-Tetri, B.A., Brunt, E.M., Guzelian, P.S., and Evans, R.M. (2000). Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature 406, 435–439. [DOI] [PubMed] [Google Scholar]
- Zhang J., Huang W., Chua S. S., Wei P., Moore D. D. (2002). Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science 298, 422–424. [DOI] [PubMed] [Google Scholar]
- Zhang Y. K., Lu H., Klaassen C. D. (2013). Expression of human CAR splicing variants in BAC-transgenic mice. Toxicol. Sci. 132, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C. Q., Young M. R., Diwan B. A., Coogan T. P., Waalkes M. P. (1997). Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. U.S.A. 94, 10907–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]