

Abstract
Aims
Sudden cardiac death (SCD) is a major public health burden. Mitochondrial dysfunction has been implicated in a wide range of cardiovascular diseases including cardiomyopathy, heart failure, and arrhythmias, but it is unknown if it also contributes to SCD risk. We sought to examine the prospective association between mtDNA copy number (mtDNA-CN), a surrogate marker of mitochondrial function, and SCD risk.
Methods and results
We measured baseline mtDNA-CN in 11 093 participants from the Atherosclerosis Risk in Communities (ARIC) study. mtDNA copy number was calculated from probe intensities of mitochondrial single nucleotide polymorphisms (SNP) on the Affymetrix Genome-Wide Human SNP Array 6.0. Sudden cardiac death was defined as a sudden pulseless condition presumed due to a ventricular tachyarrhythmia in a previously stable individual without evidence of a non-cardiac cause of cardiac arrest. Sudden cardiac death cases were reviewed and adjudicated by an expert committee. During a median follow-up of 20.4 years, we observed 361 SCD cases. After adjusting for age, race, sex, and centre, the hazard ratio for SCD comparing the 1st to the 5th quintiles of mtDNA-CN was 2.24 (95% confidence interval 1.58–3.19; P-trend <0.001). When further adjusting for traditional cardiovascular disease risk factors, prevalent coronary heart disease, heart rate, QT interval, and QRS duration, the association remained statistically significant. Spline regression models showed that the association was approximately linear over the range of mtDNA-CN values. No apparent interaction by race or by sex was detected.
Conclusion
In this community-based prospective study, mtDNA-CN in peripheral blood was inversely associated with the risk of SCD.
Keywords: Mitochondrial DNA, Mitochondrial copy number, Sudden cardiac death, Risk factor, Cardiovascular disease
Introduction
Sudden cardiac death (SCD) is a major clinical and public health burden, with 200 000 to 450 000 events in the USA annually.1 Despite major advances in understanding the mechanisms underlying SCD, prediction of SCD remains a challenge, as known clinical factors and biomarkers have only demonstrated limited prognostic power of SCD risk.2 Consequently, it is of critical importance to identify novel markers of SCD, particularly among general population samples in which the majority of SCD cases occur.3
Mitochondria are cellular organelles specialized in energy production and responsible for generating nearly all cellular adenosine triphosphate (ATP) through oxidative phosphorylation.4 Mitochondrial dysfunction may contribute to a wide range of conditions including cardiovascular diseases (CVD) such as cardiomyopathy, heart failure, and arrhythmias.5–7 Each cell has multiple mitochondria, and each mitochondrion has 2–10 copies of mtDNA.8 Mitochondrial DNA copy number (mtDNA-CN), which measures the amount of mtDNA relative to the amount of nuclear DNA, has been established as an indirect marker of mitochondrial function.9 Furthermore, mtDNA-CN is measured using a low-cost scalable assay and allows for rapid determination of mitochondrial function in a large number of samples. Indeed, recent studies have shown that low levels of mtDNA-CN were associated with diabetes, chronic kidney disease, CVD, and mortality in general population samples.10–14 However, it is unknown if mtDNA-CN is also a risk marker of SCD.
In the present study, we sought to examine the prospective association between baseline mtDNA-CN and the risk of SCD among participants from the Atherosclerosis Risk in Communities (ARIC) study. We hypothesized that participants with lower levels of mtDNA-CN would be at increased risk of SCD.
Methods
Study design and population
The ARIC study is a population based, prospective cohort study of 15 792 individuals 45–64 years of age from four US communities (Forsyth County, NC; Jackson, MS; suburban Minneapolis, MN; and Washington County, MD).15 The first visit was carried out during 1987–1989, with four subsequent in-person visits and annual telephone interviews (a 5th-follow-up visit is currently underway). The present analysis was restricted to 11 455 Caucasian or African American participants who had DNA available for mtDNA-CN measurements. We further excluded 362 participants with missing CVD risk factor information or other covariates. The final sample size was 11 093 (4971 men and 6122 women). All centres obtained approval from their respective institutional review boards and all participants provided written informed consent.
Clinical data collection
ARIC participants underwent a comprehensive medical history and cardiovascular examination by centrally trained clinical teams during each standardized clinical visit.15 Age, sex, race/ethnicity, and smoking status were self-reported. Height and weight were measured and body mass index was calculated. Hypertension was defined as systolic blood pressure ≥140 mmHg, diastolic blood pressure ≥90 mmHg, or current use of anti-hypertension medication.
Fasting blood samples were drawn and sent to a central laboratory for measurement of glucose, lipids, and creatinine. Diabetes was defined as fasting glucose ≥126 mg/dL, non-fasting glucose ≥200 mg/dL, use of hypoglycaemic medication, or self-reported physician diagnosis of diabetes. Estimated glomerular filtration rate (eGFR) was calculated using the chronic kidney disease epidemiology collaboration (CKD-EPI) equation.16 Prevalent coronary heart disease (CHD) was defined as a history of definite or probable myocardial infarction or cardiac procedure (heart or arterial surgery, coronary bypass, or angioplasty). Heart rate, QT interval, and QRS duration were measured by standard 12-lead electrocardiogram (ECG).
Sudden cardiac death events
Sudden cardiac death was defined as a sudden pulseless condition presumed due to a ventricular tachyarrhythmia in a previously stable individual without evidence of a non-cardiac cause of cardiac arrest. To identify SCD, cases of fatal cardiovascular death that occurred by 31 December 2012 were reviewed and adjudicated by a committee of electrophysiologists, general cardiologists, and internists in two phases. In the first phase, CVD deaths occurring on or before 31 December 2001 were adjudicated by five physicians. In the second phase, CVD deaths occurring between 1 January 2002 and 31 December 2012 were adjudicated by a committee of 11 physicians. All cases of fatal CVD that occurred out of the hospital or in the emergency room were reviewed. In the first phase, all in-hospital CVD deaths were also reviewed. In the second phase, in-hospital deaths were reviewed only if cardiac arrest with cardiopulmonary resuscitation occurred prior to hospital admission.
The committee reviewed all available data from death certificates, informant interviews, physician questionnaires, coroner reports, prior medical history, and hospital discharge summaries, in addition to circumstances surrounding the event, to help classify whether the subject had experienced SCD. For witnessed cases, SCD was operationally defined as a sudden pulseless condition without evidence of a non-cardiac cause of cardiac arrest. For unwitnessed cases, the patient had to have been known to be in a stable condition in the 24 h prior to cardiac arrest without evidence of a non-cardiac cause of the cardiac arrest. Study participants were not classified as sudden death if there was evidence of an acute non-cardiac morbidity that could account for the arrest, such as drug overdose, stroke, aortic aneurysm rupture, other acute bleeding, pulmonary embolism, acute respiratory failure, or trauma. To minimize misclassification of phenotype, we excluded cases with chronic terminal illnesses, including terminal cancer and end-stage liver disease. Severe dementia or severely debilitated individuals who required long-term care were excluded. Similarly, nursing home deaths were excluded. Inter-reviewer agreement for SCD adjudication was 83.2%, and there was 92.5% agreement between the two phases of SCD adjudication.
Measurement of mtDNA copy number
The method for measuring mtDNA-CN has been described previously.13 Briefly, DNA samples derived from buffy coat were used for array-based genotyping. mtDNA-CN was calculated from probe intensities of mitochondrial single nucleotide polymorphisms (SNP) on the Affymetrix Genome-Wide Human SNP Array 6.0 using the Genvisis software package (www.genvisis.org). This method uses median mitochondrial probe intensity of 25 high-quality mitochondrial probes as an initial raw measure of mtDNA-CN. To correct for batch effects, DNA quality, and starting DNA quantity, surrogate variable analysis was applied to probe intensities of 43 316 autosomal SNPs to adjust for different amounts of total DNA hybridized to the arrays and additional technical artifacts.17 We used linear regression model to adjust the effect of age, sex, enrolment centre, and the surrogate variables on the initial raw estimate of mtDNA-CN. In the present analysis, the standardized residuals from this linear model were used as the measurement for mtDNA-CN (i.e. with a mean of 0 and standard deviation of 1).
Statistical analyses
DNA for mtDNA-CN analysis was collected in Visit 1 (1987–1989) for 470 participants (4.2%), in Visit 2 (1990–1992) for 8810 participants (79.4%), in Visit 3 (1993–1995) for 1752 participants (15.8%), and in Visit 4 for (1996–1998) for 61 participants (0.6%). The visit of DNA collection in each participant was considered the baseline visit and all covariates were obtained from that visit. Follow-up for events started from the baseline visit and continued until death or through 31 December 2012, whichever came first.
Baseline characteristics of the study population were shown by quintiles of mtDNA-CN. We used one-way analysis-of-variance to compare means for continuous variables, and χ2 tests to compare proportions for categorical variables. We used a Cox proportional hazards model to estimate hazard ratios (HR) and 95% confidence intervals (CI) for the association between mtDNA-CN and SCD. In the primary analysis, we categorized mtDNA-CN into quintiles based on the sample distribution. Tests for linear trend across quintiles were conducted by including a variable with the median mtDNA-CN level of each quintile in the models. In secondary analysis, we modelled mtDNA as a continuous variable and estimated the HR for SCD comparing the 10th to the 90th percentile of mtDNA-CN. Additionally, we modelled mtDNA-CN as restricted quadratic splines with knots at the 5th, 50th, and 95th percentiles of its distribution to provide a smooth yet flexible description of the dose–response relationship between mtDNA-CN and SCD.
To adjust for potential confounders, we used four multivariate models with progressive degrees of adjustment. The first model was adjusted for age, race, sex, and enrolment centre. The second model was further adjusted for body mass index, smoking, total and HDL cholesterol, triglycerides, hypertension, and diabetes. The third model was further adjusted for heart rate, QT interval, and QRS duration. The fourth model was further adjusted for prevalent CHD at baseline. The proportional hazards assumption was checked by plotting the log(−log(survival)) vs. log(survival time) and by using Schoenfeld residuals.
Additionally, we performed pre-specified subgroup analyses by race and sex, and tested for potential interactions. We also performed several sensitivity analyses. First, we excluded 802 participants with prevalent CHD at baseline. Second, since mtDNA-CN from peripheral blood is associated with white blood cell count (WBC), we further adjusted for log-transformed WBC count among 9435 participants in whom WBC measurements were available. Third, we adjusted for eGFR among 9341 participants in whom creatinine measurements were available. Finally, we performed competing risk analyses treating deaths other than SCD as competing events, with similar results (data not shown). All statistical analyses were performed using STATA version 12 (StataCorp LP, College Station, TX, USA).
Results
The average age (standard deviation) of study participants at baseline was 57.9 (6.0) years (Table 1); 44.8% of participants were men and 79.4% were white. Participants with lower mtDNA-CN were more likely to be current smokers, hypertensive, diabetic, and to have lower HDL cholesterol, higher triglycerides, faster heart rate, slightly shorter QT interval, and prevalent CHD at baseline.
Table 1.
| Characteristic | Total (n = 11 093) | Q1 (n = 2219) | Q2 (n = 2219) | Q3 (n = 2218) | Q4 (n = 2219) | Q5 (n = 2218) | P value |
|---|---|---|---|---|---|---|---|
| Age (year) | 57.9 ± 6.0 | 57.9 ± 6.0 | 58.0 ± 6.0 | 57.9 ± 6.0 | 57.9 ± 5.9 | 57.9 ± 5.8 | NAc |
| Sex | NAc | ||||||
| Male | 4971 (44.8) | 980 (44.2) | 998 (45.0) | 992 (44.7) | 1002 (45.2) | 999 (45.0) | |
| Female | 6122 (55.2) | 1239 (55.8) | 1221 (55.0) | 1226 (55.3) | 1217 (54.8) | 1219 (55.0) | |
| Race | 0.43 | ||||||
| White | 8810 (79.4) | 1772 (79.9) | 1786 (80.5) | 1755 (79.1) | 1762 (79.4) | 1735 (78.2) | |
| Black | 2283 (20.6) | 447 (20.1) | 433 (19.5) | 463 (20.9) | 457 (20.6) | 483 (21.8) | |
| Enrolment centre | NAc | ||||||
| Forsyth County, NC | 2933 (26.4) | 608 (27.4) | 597 (26.9) | 562 (25.3) | 582 (26.2) | 584 (26.3) | |
| Jackson, MS | 1998 (18.0) | 394 (17.8) | 373 (16.8) | 409 (18.4) | 392 (17.7) | 430 (19.4) | |
| Minneapolis, MN | 3269 (29.5) | 636 (28.7) | 668 (30.1) | 698 (31.5) | 669 (30.1) | 598 (27.0) | |
| Washington County, MD | 2893 (26.1) | 581 (26.2) | 581 (26.2) | 549 (24.8) | 576 (26.0) | 606 (27.3) | |
| Smoking | <0.001 | ||||||
| Never | 4280 (38.6) | 760 (34.2) | 843 (38.0) | 871 (39.3) | 882 (39.7) | 924 (41.7) | |
| Former | 4260 (38.4) | 780 (35.2) | 812 (36.6) | 840 (37.9) | 924 (41.6) | 904 (40.8) | |
| Current | 2553 (23.0) | 679 (30.6) | 564 (25.4) | 507 (22.9) | 413 (18.6) | 390 (17.6) | |
| Body mass index (kg/m2) | 28.0 ± 5.4 | 28.0 ± 5.6 | 28.0 ± 5.5 | 28.2 ± 5.4 | 27.7 ± 5.2 | 27.9 ± 5.4 | 0.09 |
| Total cholesterol (mg/dL) | 209.7 ± 39.6 | 209.6 ± 41.8 | 208.7 ± 39.6 | 209.7 ± 38.1 | 209.6 ± 38.9 | 211.1 ± 39.4 | 0.38 |
| HDL cholesterol (mg/dL) | 50.2 ± 17.1 | 48.7 ± 17.0 | 49.7 ± 16.8 | 50.0 ± 16.9 | 51.2 ± 17.6 | 51.2 ± 17.2 | <0.001 |
| Triglycerides (mg/dL) | 137.3 ± 91.3 | 147.4 ± 113.7 | 138.7 ± 86.6 | 135.7 ± 79.6 | 133.8 ± 84.9 | 131.1 ± 86.9 | <0.001 |
| Heart rate (beat/min) | 65.5 ± 10.2 | 66.6 ± 10.7 | 65.5 ± 10.1 | 65.3 ± 10.2 | 65.0 ± 10.0 | 65.1 ± 9.8 | <0.001 |
| QT interval (ms) | 403.7 ± 31.2 | 401.9 ± 32.1 | 403.6 ± 30.8 | 404.5 ± 31.7 | 404.8 ± 31.0 | 404.0 ± 30.3 | 0.02 |
| QRS duration (ms) | 92.9 ± 13.2 | 93.0 ± 13.7 | 93.1 ± 13.5 | 92.7 ± 12.9 | 92.9 ± 12.8 | 92.6 ± 13.0 | 0.65 |
| Hypertension | 4014 (36.2) | 880 (39.7) | 845 (38.1) | 772 (34.8) | 746 (33.6) | 771 (34.8) | <0.001 |
| Diabetes | 1635 (14.7) | 433 (19.5) | 354 (16.0) | 293 (13.2) | 280 (12.6) | 275 (12.4) | <0.001 |
| Prevalent CHD | 802 (7.2) | 227 (10.2) | 167 (7.5) | 142 (6.4) | 139 (6.3) | 127 (5.7) | <0.001 |
SD, standard deviation; mtDNA-CN, mtDNA copy number.
Values are mean ± SD or number (%).
The cut-points for mtDNA-CN quintiles are −0.71, −0.15, 0.29, and 0.78.
P-value is not reported as mtDNA-CN is adjusted for age, sex, and enrolment centre.
During a median follow-up of 20.4 years, 361 participants had a SCD event (Table 2, Figure 1). In a model adjusted for age, race, sex, and enrolment centre, the HR for SCD comparing the 1st quintile of mtDNA-CN to the 5th quintile was 2.24 (95% CI 1.58–3.19; P-trend <0.001). When further adjusted for additional CVD risk factors, heart rate, QT interval, QRS duration, and prevalent CHD, the HR was attenuated but remained statistically significant (HR 1.76, 95% CI 1.22–2.53; P-trend = 0.002). Further adjustment for WBC (HR = 1.66, 95% CI 1.11–2.48; P-trend = 0.01) or eGFR (HR = 1.66, 95% CI 1.12–2.48; P-trend = 0.01) did not appreciably change the magnitude of the association. Spline regression analysis also confirmed that mtDNA-CN was inversely associated with the risk of SCD, with an approximately linear dose–response relationship (P-value for non-linear spline terms >0.85; Figure 2).
Table 2.
Hazard ratio for sudden cardiac death by quintiles of mtDNA copy number
| Events/total N | HR (95% CI) |
||||
|---|---|---|---|---|---|
| Model 1a | Model 2b | Model 3c | Model 4d | ||
| mtDNA-CN quintiles | |||||
| Q1 | 93/2219 | 2.24 (1.58, 3.19) | 1.94 (1.36, 2.77) | 1.86 (1.30, 2.66) | 1.76 (1.22, 2.53) |
| Q2 | 78/2219 | 1.83 (1.27, 2.63) | 1.66 (1.15, 2.40) | 1.59 (1.10, 2.31) | 1.62 (1.11, 2.36) |
| Q3 | 78/2218 | 1.78 (1.24, 2.56) | 1.77 (1.22, 2.56) | 1.73 (1.19, 2.50) | 1.70 (1.17, 2.48) |
| Q4 | 65/2219 | 1.43 (0.98, 2.08) | 1.49 (1.03, 2.17) | 1.43 (0.98, 2.08) | 1.39 (0.94, 2.05) |
| Q5 | 47/2218 | Reference | Reference | Reference | Reference |
| P-trend | <0.001 | <0.001 | 0.001 | 0.002 | |
| 10th vs. 90th percentile | 361/11093 | 1.82 (1.50, 2.21) | 1.55 (1.27, 1.88) | 1.53 (1.25, 1.86) | 1.47 (1.20, 1.81) |
HR, hazard ratio; CI, confidence interval; CHD, coronary heart disease.
Model 1: Adjusted for age, race, sex, and enrolment centre.
Model 2: Further adjusted for body mass index, smoking, total and HDL cholesterol, triglycerides, hypertension, and diabetes.
Model 3: Further adjusted for heart rate, QT interval, and QRS duration.
Model 4: Further adjusted for prevalent CHD at baseline.
Figure 1.
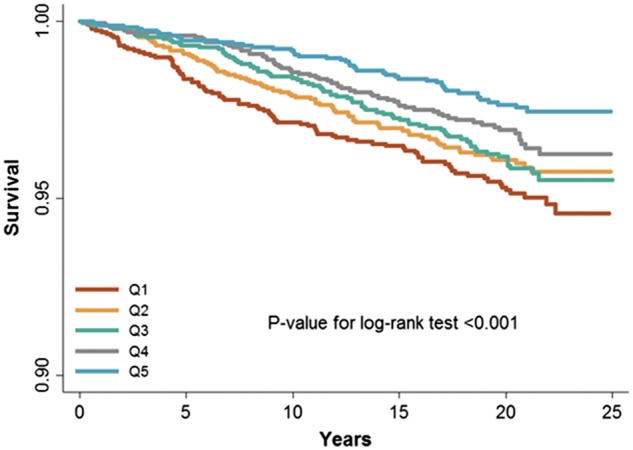
Kaplan–Meier curve of sudden cardiac death by quintiles of mtDNA copy number.
Figure 2.
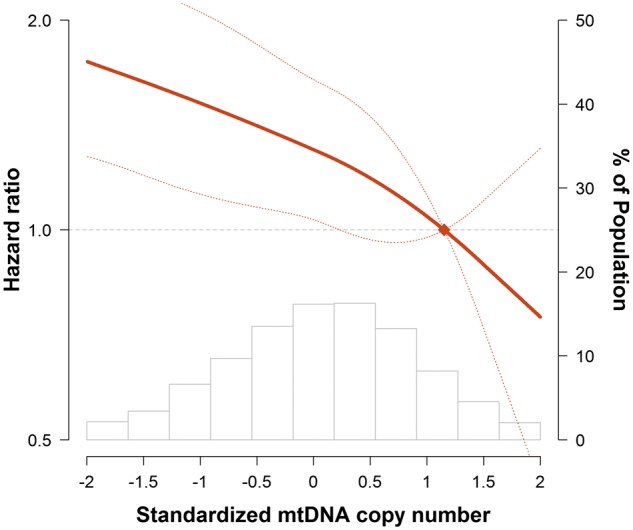
Hazard ratios for sudden cardiac death by levels of mtDNA copy number. The curves represent adjusted hazard ratios (solid line) and their 95% confidence intervals (dashed lines) based on restricted quadratic splines of mtDNA copy number with knots at the 5th, 50th, and 95th percentiles of its distribution. The reference value (diamond dot) was set at the 90th percentile of the distribution. Results were obtained from a Cox model adjusted for age, race, sex, enrolment centre, body mass index, smoking, total and HDL cholesterol, triglycerides, hypertension, diabetes, heart rate, QT interval, QRS duration, and prevalent coronary heart disease at baseline. Histograms represent the frequency distribution of mtDNA copy number at baseline.
When stratified by race and sex, the associations between mtDNA-CN and SCD were similar among men and women, and slightly stronger in whites than in blacks, but the interactions by race or by sex were not statistically significant (all P-interactions >0.1; Supplementary material online, Tables S1 and S2). In sensitivity analysis, the association between mtDNA-CN and SCD persisted after excluding participants with prevalent CHD at baseline (see Supplementary material online, Table S3).
Discussion
In this large community-based prospective cohort of white and black Americans, we found that mtDNA-CN was inversely associated with the risk of SCD. This novel association was approximately linear and similar among men and women, as well as among whites and blacks. Additionally, the association between mtDNA-CN and SCD remained after adjusting for traditional CVD risk factors and prevalent CHD, suggesting that mtDNA-CN may be a risk marker for SCD independent of CHD, traditional CHD risk factors, heart rate, QT interval, and QRS duration.
mtDNA dysfunction has been associated with several CVD outcomes including heart failure, cardiomyopathy, long QT syndrome, and arrhythmias.5,6,18–20 However, most of the evidence was obtained in small studies of patients with genetically confirmed mtDNA abnormalities (i.e. mtDNA deletions and/or mutations), and the clinical implication of variations in mtDNA function in the general population is largely unknown. mtDNA-CN, which measures the average level of mtDNA per cell, correlates with mitochondrial enzyme activities and ATP production,21 and is a surrogate marker of mtDNA function.9,11,13 The cost for measuring mtDNA-CN in a typical research setting is <$2 per sample using qPCR. mtDNA copy number could represent a low-cost approach to improving risk prediction of SCD in the general population. Indeed, recent studies in general population samples have shown associations of mtDNA-CN with aging, frailty, chronic kidney disease, and all-cause mortality.8,11,13
The precise mechanism underlying the association between mtDNA-CN and SCD is unclear. Cardiac cells rely heavily on mitochondrial oxidative energy to maintain myocardial contractility and electrical activity.7 It is estimated that 30% of the cardiac ATP generated by mitochondria is used for sarcolemmal and sarcoplasmic reticulum ion channels and transporters, which are required for the electrical activity of the myocytes.7,22 It is possible that decreased mtDNA-CN measured in blood reflects mitochondrial dysfunction in cardiac cells, which could compromise ATP production and energy supply to ion channels and transporters, leading to altered ion homeostasis, membrane excitability, and cardiac arrhythmias.7
In addition to ATP, mitochondria generate reactive oxygen species (ROS) as a by-product of oxidative phosphorylation.7 Excessive ROS production can alter action potential propagation and impair cardiac excitability through a number of ion channels and transporters.7,23,24 Furthermore, an increase in ROS above a threshold level triggers the opening of mitochondrial channels such as the mitochondrial permeability transition pore or the inner membrane anion channel, leading to the collapse of mitochondrial membrane potential and a further increase in ROS generation, a process known as mitochondrial ROS-induced ROS release.25–27 mtDNA is highly susceptible to oxidative damage due to its close proximity to mitochondrial ROS, lack of protective histone proteins, and limited DNA repair capabilities. As a consequence, oxidative stress-related mitochondrial damage may further contribute to a decreased mtDNA-CN and an increased risk of SCD.8,28 Additionally, regional mitochondrial depolarization triggered by oxidative stress activates sarcolemmal ATP-sensitive K+ currents to form a metabolic sink, contributing to the development of re-entry arrhythmias.29 Mitochondrial oxidative stress also plays an important role in angiotensin II-induced gap junction remodelling and arrhythmogenesis.30
A few limitations of this study need to be considered. Due to the observational nature of our study, we could identify an association but not establish a causal link between mtDNA-CN and SCD. Our study measured mtDNA-CN in DNA derived from peripheral blood, which may not necessarily be the relevant tissue with respect to SCD. However, mtDNA-CN in peripheral blood has been shown to correlate with that of cardiomyocytes (Pearson correlation = 0.72),10 suggesting that mtDNA-CN in peripheral blood may serve as a marker for mitochondrial function in the heart. Additionally, we measured mtDNA-CN at a single time point and did not account for the dynamic nature of mtDNA-CN throughout the life course. Serial measures of mtDNA-CN may provide additional insight into the relationship between mitochondrial function and SCD. Although an extensive amount of information was used in the adjudication of SCD cases, autopsy data were often not available and therefore the exact cause of SCD could not be firmly established in many patients. The ARIC study enrolled patients 45–64 at study entry, and the average age of the study participants in our analysis was 58 years. Therefore, we were not able to examine if the association between mtDNA-CN and SCD was similar for SCD cases occurring among young adults where SCD may be more likely due to underlying congenital cardiac abnormalities or electrical disorders than due to CHD.
The major strengths of this study include a large sample size of both white and black participants, prospective cohort study design and long follow-up period for SCD events, stringent SCD adjudication, as well as the use of state-of-the-art tools for mtDNA-CN measurement.
In conclusion, in this community-based prospective study, mtDNA-CN in peripheral blood was inversely associated with SCD risk independent of CHD, CHD risk factors, heart rate, QT interval, and QRS duration. These findings may provide a novel risk marker for SCD among general population samples and uncover new therapeutic targets for reducing SCD. Future studies are needed to confirm our findings in other study populations and to elucidate underlying mechanisms.
Supplementary material
Supplementary material is available at European Heart Journal online.
Supplementary Material
Acknowledgements
We would like to acknowledge the sudden cardiac death mortality classification committee members: Nona Sotoodehnia (lead), Selcuk Adabag, Sunil Agarwal, Lin Chen, Raj Deo, Leonard Ilkhanoff, Liviu Klein, Saman Nazarian, Ashleigh Owen, Kris Patton, and Larisa Tereschchenko. The authors thank the staff and participants of the ARIC study for their important contributions.
Funding
US National Institutes of Health grants (R01HL131573 to Y.Z., E.G., F.N.A., R.J.L., C.A.C., D.E.A.; R01HL111267 to F.N.A., R.J.L., D.E.A.; and R01HL116747 to N.S.). The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), R01HL087641, R01HL59367, and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. Adjudication of sudden cardiac death cases was funded by R01 HL111089. Infrastructure was partly supported by grant number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
Conflict of interest: none declared.
References
- 1. Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al Samara M, Mariani R, Gunson K, Jui J.. Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis 2008;51:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fishman GI, Chugh SS, Dimarco JP, Albert CM, Anderson ME, Bonow RO, Buxton AE, Chen PS, Estes M, Jouven X, Kwong R, Lathrop DA, Mascette AM, Nerbonne JM, O'rourke B, Page RL, Roden DM, Rosenbaum DS, Sotoodehnia N, Trayanova NA, Zheng ZJ.. Sudden cardiac death prediction and prevention: report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation 2010;122:2335–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Deo R, Albert CM.. Epidemiology and genetics of sudden cardiac death. Circulation 2012;125:620–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor RW, Turnbull DM.. Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005;6:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marin-Garcia J, Goldenthal MJ.. Understanding the impact of mitochondrial defects in cardiovascular disease: a review. J Card Fail 2002;8:347–361. [DOI] [PubMed] [Google Scholar]
- 6. Wahbi K, Bougouin W, Behin A, Stojkovic T, Becane HM, Jardel C, Berber N, Mochel F, Lombes A, Eymard B, Duboc D, Laforet P.. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Eur Heart J 2015;36:2886–2893. [DOI] [PubMed] [Google Scholar]
- 7. Yang KC, Bonini MG, Dudley SC Jr.. Mitochondria and arrhythmias. Free Radic Biol Med 2014;71:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mengel-From J, Thinggaard M, Dalgard C, Kyvik KO, Christensen K, Christiansen L.. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum Genet 2014;133:1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malik AN, Czajka A.. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013;13:481–492. [DOI] [PubMed] [Google Scholar]
- 10. Huang J, Tan L, Shen R, Zhang L, Zuo H, Wang DW.. Decreased peripheral mitochondrial DNA copy number is associated with the risk of heart failure and long-term outcomes. Medicine (Baltimore) 2016;95:e3323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ashar FN, Moes A, Moore AZ, Grove ML, Chaves PH, Coresh J, Newman AB, Matteini AM, Bandeen-Roche K, Boerwinkle E, Walston JD, Arking DE.. Association of mitochondrial DNA levels with frailty and all-cause mortality. J Mol Med (Berl) 2015;93:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R.. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res 2010;106:1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tin A, Grams ME, Ashar FN, Lane JA, Rosenberg AZ, Grove ML, Boerwinkle E, Selvin E, Coresh J, Pankratz N, Arking DE.. Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the atherosclerosis risk in communities study. J Am Soc Nephrol 2016;27:2467–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee HK, Song JH, Shin CS, Park DJ, Park KS, Lee KU, Koh CS.. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract 1998;42:161–167. [DOI] [PubMed] [Google Scholar]
- 15. The ARIC investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol 1989;129:687–702. [PubMed] [Google Scholar]
- 16. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, Ckd EPI.. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD.. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012;28:882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ng YS, Grady JP, Lax NZ, Bourke JP, Alston CL, Hardy SA, Falkous G, Schaefer AG, Radunovic A, Mohiddin SA, Ralph M, Alhakim A, Taylor RW, McFarland R, Turnbull DM, Gorman GS.. Sudden adult death syndrome in m.3243A>G-related mitochondrial disease: an unrecognized clinical entity in young, asymptomatic adults. Eur Heart J 2016;37:2552–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Opdal SH, Vege A, Egeland T, Musse MA, Rognum TO.. Possible role of mtDNA mutations in sudden infant death. Pediatr Neurol 2002;27:23–29. [DOI] [PubMed] [Google Scholar]
- 20. Ruppert V, Nolte D, Aschenbrenner T, Pankuweit S, Funck R, Maisch B.. Novel point mutations in the mitochondrial DNA detected in patients with dilated cardiomyopathy by screening the whole mitochondrial genome. Biochem Biophys Res Commun 2004;318:535–543. [DOI] [PubMed] [Google Scholar]
- 21. Jeng JY, Yeh TS, Lee JW, Lin SH, Fong TH, Hsieh RH.. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J Cell Biochem 2008;103:347–357. [DOI] [PubMed] [Google Scholar]
- 22. Schramm M, Klieber HG, Daut J.. The energy expenditure of actomyosin-ATPase, Ca(2+)-ATPase and Na+,K(+)-ATPase in guinea-pig cardiac ventricular muscle. J Physiol 1994;481(Pt 3):647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aggarwal NT, Makielski JC.. Redox control of cardiac excitability. Antioxid Redox Signal 2013;18:432–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu M, Liu H, Dudley SC Jr.. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res 2010;107:967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zorov DB, Juhaszova M, Sollott SJ.. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 2014;94:909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zorov DB, Juhaszova M, Sollott SJ.. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta 2006;1757:509–517. [DOI] [PubMed] [Google Scholar]
- 27. Brown DA, O'rourke B.. Cardiac mitochondria and arrhythmias. Cardiovasc Res 2010;88:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsutsui H, Kinugawa S, Matsushima S.. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 2011;301:H2181–H2190. [DOI] [PubMed] [Google Scholar]
- 29. Zhou L, Solhjoo S, Millare B, Plank G, Abraham MR, Cortassa S, Trayanova N, O'rourke B.. Effects of regional mitochondrial depolarization on electrical propagation: implications for arrhythmogenesis. Circ Arrhythm Electrophysiol 2014;7:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sovari AA, Rutledge CA, Jeong EM, Dolmatova E, Arasu D, Liu H, Vahdani N, Gu L, Zandieh S, Xiao L, Bonini MG, Duffy HS, Dudley SC Jr.. Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circ Arrhythm Electrophysiol 2013;6:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.