

Abstract
Lewisite (LEW), a potent arsenical vesicating chemical warfare agent, poses a continuous risk of accidental exposure in addition to its feared use as a terrorist weapon. Ocular tissue is exquisitely sensitive to LEW and exposure can cause devastating corneal lesions. However, detailed pathogenesis of corneal injury and related mechanisms from LEW exposure that could help identify targeted therapies are not available. Using an established consistent and efficient exposure system, we evaluated the pathophysiology of the corneal injury in New Zealand white rabbits following LEW vapor exposure (at 0.2 mg/L dose) for 2.5 and 7.5 min, for up to 28 day post-exposure. LEW led to an increase in total corneal thickness starting at day 1 post-exposure and epithelial degradation starting at day 3 post-exposure, with maximal effect at day 7 postexposure followed by recovery at later time points. LEW also led to an increase in the number of blood vessels and inflammatory cells but a decrease in keratocytes with optimal effects at day 7 postexposure. A significant increase in epithelial-stromal separation was observed at days 7 and 14 post 7.5 min LEW exposure. LEW also caused an increase in the expression levels of cyclooxygenase-2, IL-8, vascular endothelial growth factor, and matrix metalloproteinase-9 at all the study time points indicating their involvement in LEW-induced inflammation, vesication, and neovascularization. The outcomes here provide valuable LEW-induced corneal injury endpoints at both lower and higher exposure durations in a relevant model system, which will be helpful to identify and screen therapies against LEW-induced corneal injury.
Keywords: lewisite, arsenical vesicant, corneal injury, inflammation, vesication, neovascularization
Lewisite (2-chlorovinyldichloroarsine; C2H2AsCl3; L, LEW) is an extremely toxic organic arsenical vesicant agent that induces rapid inception of severe pain and blistering upon exposure (King et al., 1992; Kehe et al., 2001; Li et al., 2016a; Lindsay et al., 2004; Sahu et al., 2013). First synthesized in 1918, LEW is not reported to be deployed in warfare; however, it may have been used by the Japanese army against China (Beebe, 1960) and mixed with the vesicating agent sulfur mustard (2,2′-dichloroethylsulfide; SM) to achieve greater effectiveness in conflicts (Rice and Brown, 1999). In addition to its possible use in warfare, large stockpiles of LEW with several nations since World War I poses accidental exposure risk apart from its possible use in terrorist attacks (Li et al., 2016a; Mouret et al., 2013). since LEW is a threat to both civilians (in an accidental and terrorist attack scenarios) and armed-forces, there is a need for the identification of safe and effective targeted therapy against LEW exposure (Hughes, 1947; Mann et al., 1947; Nguon et al., 2014; Vilensky and Redman, 2003). Notably, the use of a specific antidote, British anti-LEW (BAL, dimercaprol) has shown efficacy in minimizing the tissue damage but it is associated with a number of drawbacks.
Due to its rapid absorption as compared with SM, exposure to even small quantities of LEW can result in pain, rapid onset of symptoms including vesication, and significant systemic toxicity (Hotta, 1997; Mouret et al., 2013; Nguon et al., 2014). The eye is the most sensitive organ to LEW exposure and causes instant irritation, pain, swelling, and tearing, which can be severe with inflammation, edema of eyelids, massive corneal necrosis, and blindness (Olajos et al., 1998). These effects could be due to LEW’s vesicating properties, or additional effects due to its breakdown into arsenic oxide, reaction with biological sulfhydryls resulting in additional injuries related to arsenic compounds, and liberation of hydrochloric acid which lowers the pH of the eye and causes superficial opacity (Hughes, 1946; Lindsay et al., 2004; Sahu et al., 2013). There are a few early reports from 1940s on clinical and pathologic characteristics of ocular injuries following LEW exposure (Hughes, 1946, 1947; Mann et al., 1946); however, ocular injuries are not well-characterized and quantitative evaluations to establish valuable biomarkers for efficacy studies are limited. Although the pathophysiology of ocular injury from mustard vesicating agent SM, which was used extensively in warfare for over 100 years has been extensively studied, the efforts to study LEW-induced ocular injury progression to identify therapeutic targets are limited or undocumented.
Due to these limitations, our earlier reported study evaluated LEW vapor-induced clinical progression of corneal injury in New Zealand white rabbits employing an innovative ocular exposure system for identical and controlled LEW vapor (right eye) exposure and diluent vapor (left eye; control) for consistent injury (Tewari-Singh et al., 2016). There are only few studies reported for LEW exposure of the eye. Based on a study by Hughes (1946), where LEW was directly applied or eyes were exposed to saturated vapor of LEW for 30 s, we used a range of doses and selected a dose of 0.2 mg/L LEW for 2.5 and 7.5 min exposure to give us a mild and severe ocular injury paralleling effect of LEW in the human eye.
Corneal wounding, ulceration, inflammation, neovascularization, and their progression were observed clinically for the first time and quantified by us in our previous study following LEW exposure. In this study, we examined in detail the histopathological changes in rabbit cornea and related mechanisms following LEW vapor exposure for shorter (2.5 min) and longer (7.5 min) durations. Our ongoing efforts in understanding the structural changes and mechanism of ocular injury progression following LEW exposure will help in outlining pathways that could be targeted to develop effective therapies against LEW-induced ocular injuries.
MATERIALS AND METHODS
Exposure of rabbit eyes to LEW
New Zealand white rabbits (n = 5 per group) were purchased from Charles River Laboratories (2.5 to 4.0 kg and not less than 3 months of age at time of arrival) anesthetized and the eyes were exposed to LEW (0.2 mg/l) for 2.5 or 7.5 min using vapor exposure system for exposing up to 6 rabbits simultaneously as reported earlier (Tewari-Singh et al., 2016). LEW synthesis and characterization, and pain management was carried out as detailed earlier (Tewari-Singh et al., 2016). Right eye of each animal was exposed to LEW using modified goggles with a flow through design while the left eye was exposed to dilution air and served as control. Flow rates of LEW vapor were controlled through the goggles with critical orifice control meters to assure that all flow rates were equilibrated. All exposures were conducted within the chemical agent hood line, in compliance with MRIGlobal standard operating procedures. Standard safety procedures and Institutional Animal Care and Use Committee protocols were strictly followed. Animals were sacrificed at days 1, 3, 7, 14, and 28 post-exposure and the cornea was dissected and was either snap frozen or fixed in formalin for further studies.
Histopathological evaluation of corneal sections and measurement of corneal thickness, epithelial thickness, epithelial degradation, and epithelial-stromal separation
The formalin fixed corneas were processed, sectioned, and 5 μm sections were stained with hematoxylin and eosin (H&E) as reported earlier (Tewari-Singh et al., 2012). The H&E stained sections were evaluated microscopically for corneal thickness, epithelial thickness, epithelial-degradation, and epithelial-stromal separation as reported earlier (Goswami et al., 2016; Tewari-Singh et al., 2012). Corneal thickness from 10 to 12 randomly selected corneal areas was measured (approximately 1.0 mm away from both the sides of the cornea and the limbus region). The epithelial thickness was measured in at least 5 randomly selected fields throughout the length of the cornea and 5 measurements were carried out from each field. Epithelial degradation and epithelial-stromal separation was measured in approximately 7 mm length of the cornea.
Measurement of inflammatory cells, blood vessels, and keratocytes in the stroma
The H&E stained rabbit cornea sections were evaluated microscopically and quantification of number of keratocytes, blood vessels, and inflammatory cells in the stroma was carried out. Keratocyte quantification was carried out in approximately 7 mm2 of the stroma and average in 0.5 mm2 was calculated. The number of blood vessels and inflammatory cells were quantified from whole stromal region of the cornea. The density of inflammatory cells was scored as 1, < 50; 2, 50–100; 3, 100–500; and 4, > 500 inflammatory cells.
Immunohistochemistry for VEGF, COX-2, and MMP-9
The formalin fixed corneas were processed, sectioned, and 5 μm sections were subjected to antigen retrieval and blocking of endogenous peroxidase activity. Thereafter, the sections were incubated with anti-vascular endothelial growth factor (VEGF) (Abcam, Cambridge, Massachusetts), anti-cyclooxygenase-2 (COX-2) (Cayman Chemicals, Ann Arbor, Michigan), or anti-matrix metalloproteinase-9 (MMP-9) (Abcam, Cambridge, Massachusetts) antibodies, followed by incubation with biotinylated secondary antibody and streptavidin-HRP conjugated antibody as detailed earlier (Goswami et al., 2016). Rabbit IgG antibody (N-Universal, DAKO) was used as negative control. The sections were then incubated in 3, 3′-diaminobenzidine (DAB) and counterstained with diluted hematoxylin. The brown colored cytoplasmic staining was scored as positivity score in 10 randomly selected fields. The intensity of brown color was scored as 0 (no staining), + 1 (weak staining), + 2 (moderate staining), + 3 (strong staining), and + 4 (very strong staining).
Cytokine array
Frozen corneal tissue sample lysates (n = 3) from 7.5 min LEW exposure time at 7 day post-exposure were subjected to G-series Rabbit Cytokine Array (Ray Biotech, Norcross, Georgia). The array was carried out using the manufacturers’ instructions. Briefly, the array surface was blocked on the provided glass slides with the sample diluent and incubated with the samples. Thereafter, biotinylated detection antibody was added followed by incubation with Cy3 equivalent dye-conjugated streptavidin; the slides were then scanned and data were extracted using Raybiotech scanning and data extraction services. The data were analyzed using Raybiotech array specific data analysis software and plotted as relative fluorescence units.
Statistical analysis
Data were analyzed using 1-way analysis of variance (1-way ANOVA) to get the statistically significant difference in control versus LEW exposed groups, with Tukey or Bonferroni t test for multiple comparisons (Sigma Stat 2.03). Differences were considered significant if the P value was < 0.05. Data are presented as the mean ± SEM.
RESULTS
LEW Exposure Caused an Increase in Thickness of the Rabbit Cornea
Our recently reported study showed that apart from corneal opacity, ulceration, and neovascularization, an increase in corneal thickness was recorded by pachymetry following ocular LEW exposure (Tewari-Singh et al., 2016). Hence, we further examined the corneal thickness in the H&E stained corneal sections following LEW exposure. LEW exposure resulted in an increase in corneal thickness, which was more pronounced at 3–7 days post-exposure (Figure 1A). Although a significant (P < 0.05) increase in the corneal thickness following LEW exposure at both the exposure durations was observed at day 1 postexposure, there was a dose-dependent effect of LEW at all the study time points out to day 28 post-exposure (Figure 1B). The longer (7.5 min) LEW exposure induced increase in corneal thickness was maximal (2.0-fold increase) at 7 day post-exposure and remained prominent up to day 28 post-exposure; however, the corneal thickness decreased to baseline measurements at this time point in 2.5 min LEW exposed corneas (Figs. 1A and 1B).
Figure 1.
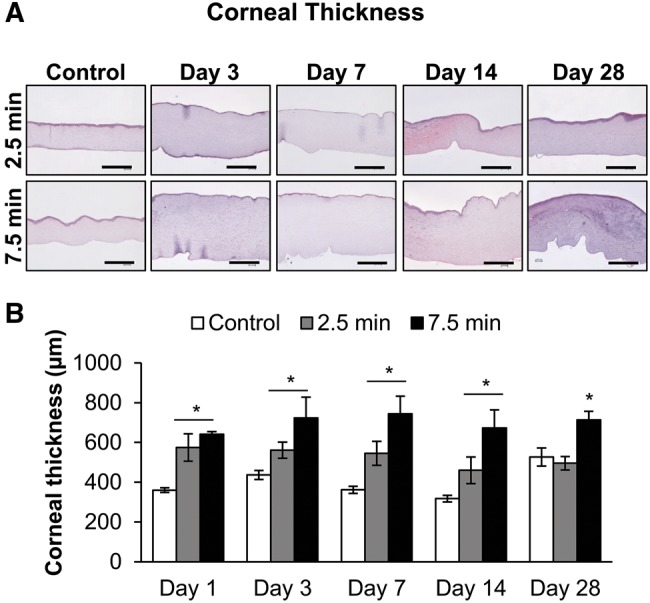
Ocular lewisite (LEW) exposure causes an increase in rabbit corneal thickness. New Zealand white rabbits were exposed to either dilution air (control; left eye) or LEW (0.2 mg/L; right eye) vapor for either 2.5 or 7.5 min. The rabbits were euthanized; the corneal tissue was dissected from eye at days 1, 3, 7, 14, and 28 post-LEW exposure and fixed for histopathologic evaluation. The total corneal thickness in the hematoxylin and eosin (H&E) stained corneal sections was measured from the control and LEW exposed sections as detailed under the “Materials and Methods” section. Representative images showing corneal thickness of control and LEW exposed corneal sections (A) and bar diagram showing quantitative data from the measurement of the corneal thickness (B). Data presented are mean ± SEM (n = 3–5). *P < 0.05 compared with the control group; size bar in representative images, 200 µm.
LEW Exposure Caused an Increase in the Epithelial Degradation and Induced Epithelial-Stromal Separation in the Rabbit Cornea
Our completed clinical study has shown that LEW exposure causes corneal ulceration (Tewari-Singh et al., 2016). Hence, we next examined the H&E stained corneal tissue sections for the effect of LEW on the corneal epithelial layer. LEW exposures led to an increase in corneal epithelial degradation which was prominent at day 3 post-exposure with the most profound effect at day 7 post-exposure (Figs. 1A and 1B). As compared with 2.5 min exposure, the effect of 7.5 min LEW exposure at day 7 was more damaging and resulted in a maximal (2.2-fold) decrease in epithelial thickness depicting membrane degradation (Figs. 2B and 2C). At both the exposure durations, LEW-induced epithelial degradation started to resolve by day 14 postexposure. However, it completely resolved by day 28 postexposure in 2.5 min exposed corneas but the injury was still evident in corneas exposed for 7.5 min duration (Figs. 2A–C).
Figure 2.
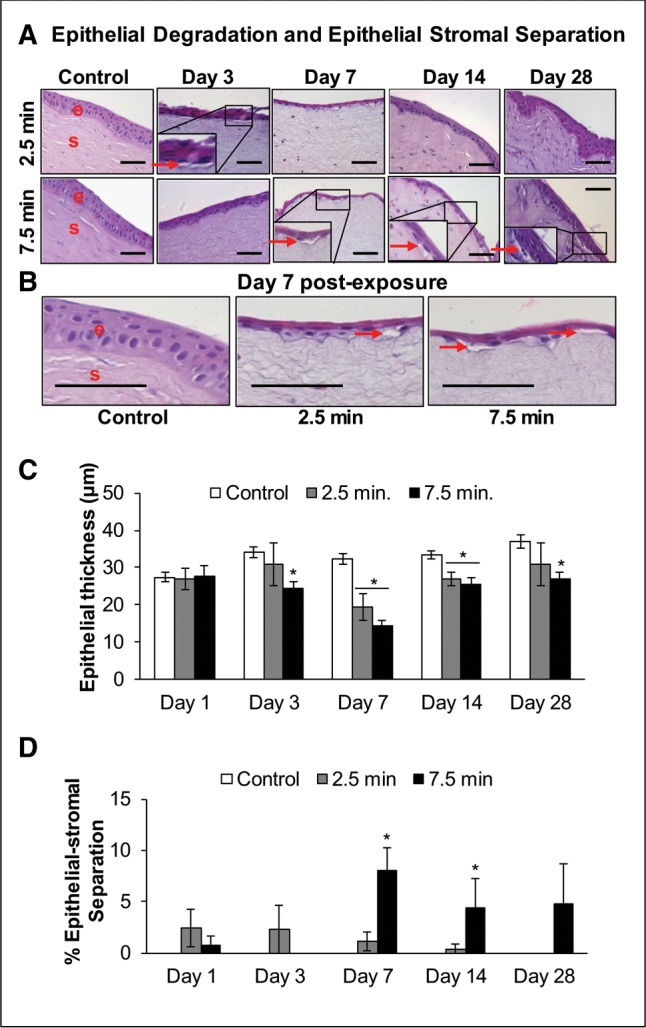
Ocular LEW exposure causes an increase in the epithelial-degradation and induces epithelial-stromal separation in the rabbit cornea. New Zealand white rabbits were exposed to either dilution air (control; left eye) or LEW (0.2 mg/L; right eye) vapor for either 2.5 or 7.5 min. The rabbits were euthanized; the corneal tissue was dissected from eye at days 1, 3, 7, 14, and 28 post-LEW exposure and fixed for histopathologic evaluation. The corneal sections were H&E stained; epithelial thickness was measured to examine degradation and epithelial-stromal separation was quantified as detailed under the “Materials and Methods” section. Representative images showing epithelial-degradation and -stromal separation (enlarged inset; A). Bar diagram showing quantification of the epithelial thickness (B) and epithelial-stromal separation (C). Data presented are mean ± SEM (n = 3–5). *P < 0.05 compared with the control group; e, epithelium; s, stroma; arrows, epithelial-stromal separation; size bar in representative images, 50 µm.
LEW exposure did not induce very large epithelial-stromal separations (vesications) but resulted in small separated areas appearing like bullae (Figs. 2A and 2B; red arrows). LEW exposure at 7.5 min exposure duration induced significant (P < .05) epithelial-stromal separations at days 7 and 14 post-exposure (Figure 2D). This injurious effect of LEW exposure was also most prominent at day 7 post-exposure (Figure 2D).
LEW Exposure Caused an Increase in the Inflammatory Cells, and Blood Capillaries and Vessels in the Stroma of the Rabbit Cornea
Next, we examined whether the erythema and inflammation observed clinically in the rabbit eyes following LEW exposure (Tewari-Singh et al., 2016) was associated histologically with an inflammatory response. The H&E stained corneal sections showed that LEW exposure at both the exposure durations induced an increase in inflammatory cells in the corneal stroma starting at day 1 post-exposure, which peaked at days 3–7 postexposure (Figure 3A; red arrows). This LEW-induced increase in the inflammatory cells was resolved by day 28 post-exposure in the corneas exposed to LEW for 2.5 min duration (Figure 3B). However, following 7.5 min LEW exposure, the increase in inflammatory cells was maximal (4.2-fold) at day 7 post-exposure and was somewhat lowered by days 14 and 28 post-exposure (Figure 3B).
Figure 3.
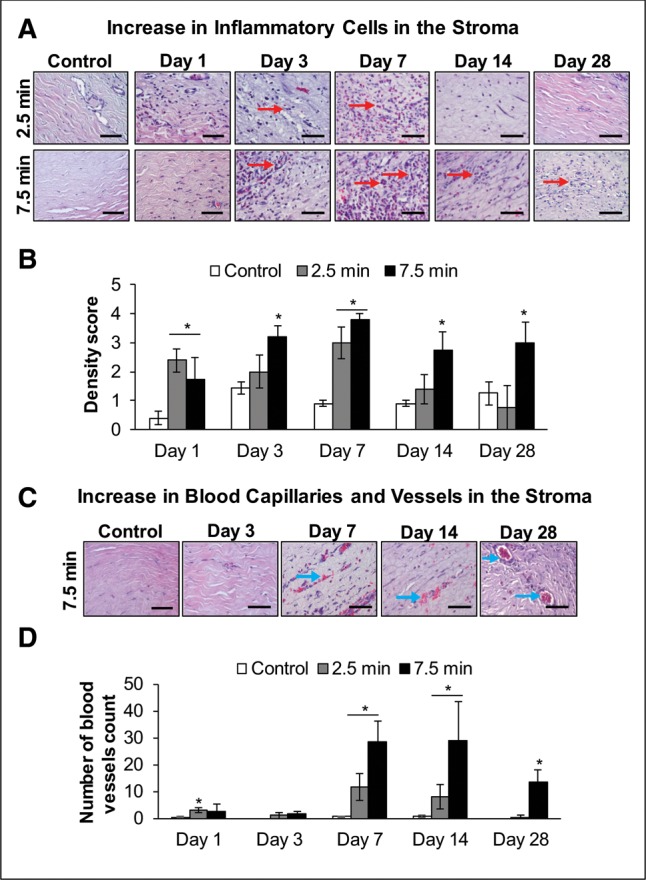
Ocular LEW exposure causes an increase in inflammatory cells and number of blood vessels in the stroma of rabbit cornea. New Zealand white rabbits were exposed to either dilution air (control; left eye) or LEW (0.2 mg/L; right eye) vapor for either 2.5 or 7.5 min. The rabbits were euthanized; the corneal tissue was dissected from eye at days 1, 3, 7, 14, and 28 post-LEW exposure and fixed for histopathologic evaluation. The corneal sections were H&E stained; inflammatory cells and blood vessels were quantified as detailed under the “Materials and Methods” section. Representative images showing an increase in the inflammatory cells in the corneal stroma following LEW exposure (A) and bar diagram showing the quantification of inflammatory cells (B). Representative images showing an increase in number of blood vessels in the corneal stroma following 7.5 min LEW exposure (C) and bar diagram showing the quantification of blood vessels (D). Data presented are mean ± SEM (n = 3–5). *P < 0.05 compared with the control group; e, epithelium; s, stroma; arrows, inflammatory cells or blood vessels; size bar in representative images, 50 µm.
LEW exposure in rabbits also resulted in the presence of blood capillaries and vessels in the stroma of the cornea with marked dose-dependent 47.3- and 36.5-fold increases at days 7 and 14 post-exposure, respectively (Figs. 3A and 3B).
LEW Exposure Caused a Decrease in the Number of Keratocytes in the Stroma of the Rabbit Cornea
The examination of the corneal stroma in the H&E stained corneal tissue sections also demonstrated a noticeable LEW-induced decrease in the number of keratocytes in the stroma of the corneas at days 1–28 post-LEW exposure (Figure 4A). At day 7 post-LEW exposure, a maximal keratocyte decrease of 2.8- and 8.0-fold, compared with controls, in corneas exposed for 2.5 and 7.5 min to LEW, respectively, was observed (Figure 4B).
Figure 4.

Ocular LEW exposure causes a decrease in the number of keratocytes (keratocyte cell death) in the stroma of rabbit cornea. New Zealand white rabbits were exposed to either dilution air (control; left eye) or LEW (0.2 mg/L; right eye) vapor for either 2.5 or 7.5 min. The rabbits were euthanized; the corneal tissue was dissected from eye at days 1, 3, 7, 14, and 28 post-LEW exposure and fixed for histopathologic evaluation. The corneal sections were H&E stained; keratocytes were quantified as detailed under the “Materials and Methods” section. Representative images showing a decrease in keratocytes in the corneal stroma following LEW exposure (A) and bar diagram showing the quantification of keratocytes (B). Data presented are mean ± SEM (n = 3–5). *P < 0.05 compared with the control group; arrows, keratocytes; size bar in representative images, 50 µm.
LEW Exposure Caused an Increase in the Expression of COX-2, IL-8, MMP-9, and VEGF in the Rabbit Cornea
Inflammatory mediators: because an inflammatory response was observed in our completed clinical evaluation and an increase in inflammatory cells in the H&E stained stromal tissue was evidenced; we next analyzed the expression of COX-2 (an inducible enzyme expressed in mononuclear phagocytes and neutrophils that is involved in pro-inflammatory prostaglandins synthesis) and changes in other inflammatory cytokines via cytokine array. Although an increase in the COX-2 expression was observed staring at day 1 post-LEW exposure, most prominent immunohistochemistry staining intensity for COX-2 compared with control was observed at days 7 and 14 post-LEW exposure at both the exposure durations (Figure 5A). Scoring of the COX-2 expression intensity exhibited a maximal increase of 2.4- and 3.0-fold in corneas exposed to LEW for 2.5 and 7.5 min, respectively, at day 7 postexposure (Figure 5B). At day 7 postexposure, 7.5 min LEW exposure also resulted in a 29.4-fold increase in cytokine IL-8 levels (Figure 5C).
MMP-9 is reported to be an important mediator in vesicating agents-induced blister formation and could play a vital role in LEW-induced epithelial-stromal separation observed following its exposure in our H&E stained corneas. Matrix metalloproteinase-9 expression is mostly observed in the epithelial basement membrane zone and anterior stroma (Gordon et al., 2010). A significant (P < 0.05) increase in the intensity of MMP-9 staining was observed at days 1–28 post-LEW exposure at both the exposure durations, mainly in the epithelial layer with further pronounced staining at the basement zone (Figs. 6A and 6B; red arrows). At longer LEW exposure duration of 7.5 min, a maximal increase of 2.9-fold was observed at day 7 post-exposure (Figure 6B). Cytokine array further confirmed this LEW-induced increase in MMP-9 levels where the longer 7.5 min LEW exposure duration showed an even higher 9.0-fold increase in MMP-9 level (Figure 6C).
Neovascularization observed following LEW exposure in rabbit eyes (Tewari-Singh et al., 2016) could be associated with angiogenic factor VEGF, which is reported to promote ocular neovascularization in animal ocular exposures including mustard vesicating agents (Goswami et al., 2016; Kadar et al., 2001; McNutt et al., 2012a; Petrali et al., 2000). At the lower exposure duration of 2.5 min, significant (P < .05) LEW-induced increases in the VEGF expression was observed from days 3 to 14 postexposure (Figs. 7A and 7B). A significant (P < .05) increase in the VEGF expression following 7.5 min LEW exposure was observed at all the study time points from days 1 to 28 postexposure, which was maximal (3.6-fold increase) at day 14 postexposure (Figure 7B).
Figure 5.
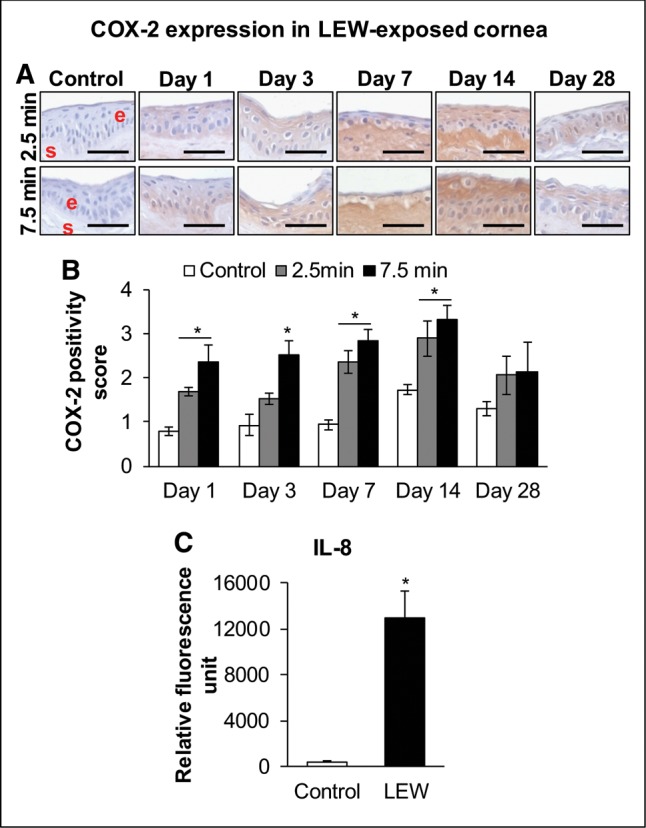
Ocular LEW exposure causes an increase in the expression of cyclooxygenase-2 (COX-2) and IL-8 in the rabbit cornea. New Zealand white rabbits were exposed to either dilution air (control; left eye) or LEW (0.2 mg/L; right eye) vapor for either 2.5 or 7.5 min. The rabbits were euthanized; the corneal tissue was dissected from eye at days 1, 3, 7, 14, and 28 post-LEW exposure and either fixed for immunohistochemistry (IHC) evaluation or frozen for cytokine array analysis. The corneal sections were IHC stained and the COX-2 expression was quantified as detailed under the “Materials and Methods” section. Representative images showing an increase in COX-2 expression following LEW exposure (A) and bar diagram showing the COX-2 positivity score (B). The corneal protein was also subjected to cytokine array analysis and relative fluorescence units for IL-8 were calculated as detailed under the “Materials and Methods” section. Bar diagram showing the IL-8 relative fluorescence units (C). Data presented are mean ± SEM (n = 3–5). *P < 0.05 compared with the control group; e, epithelium; s, stroma; size bar in representative images, 50 µm.
Figure 6.
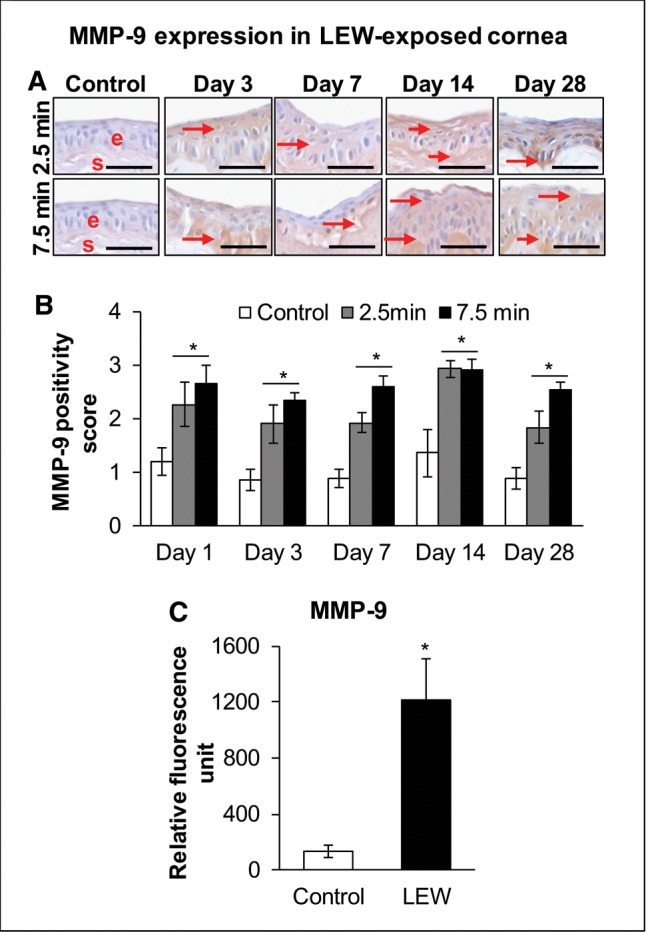
Ocular LEW exposure causes an increase in the expression of matrix metalloproteinase-9 (MMP-9) in the rabbit cornea. New Zealand white rabbits were exposed to either dilution air (control; left eye) or LEW (0.2 mg/L; right eye) vapor for either 2.5 or 7.5 min. The rabbits were euthanized; the corneal tissue was dissected from eye at days 1, 3, 7, 14, and 28 post-LEW exposure and either fixed for IHC evaluation or frozen for cytokine array analysis. The corneal sections were IHC stained and the MMP-9 expression was quantified as detailed under the “Materials and Methods” section. Representative images showing an increase in MMP-9 expression following LEW exposure (A) and bar diagram showing the MMP-9 positivity score (B). The corneal protein was also subjected to cytokine array analysis and relative fluorescence units for MMP-9 were calculated as detailed under the “Materials and Methods” section. Bar diagram showing the MMP-9 relative fluorescence units (C). Data presented are mean ± SEM (n = 3–5). *P < 0.05 compared with the control group; e, epithelium; s, stroma; size bar in representative images, 50 µm; arrows, MMP-9 staining.
Figure 7.
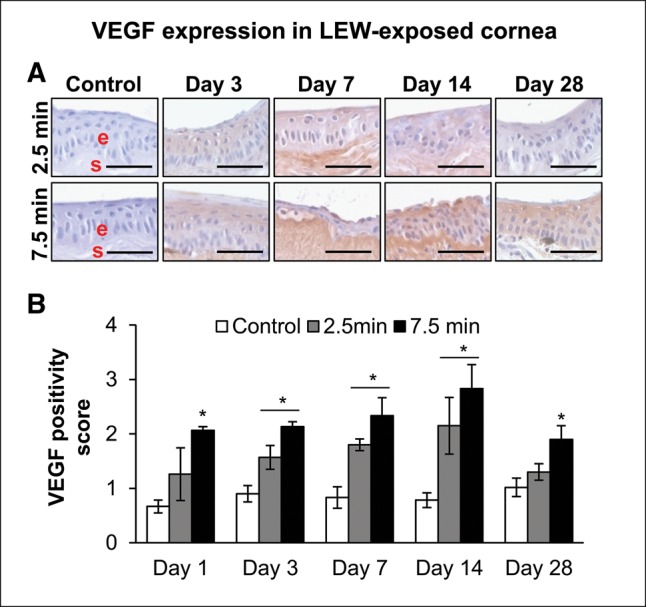
Ocular LEW exposure causes an increase in the expression of vascular endothelial growth factor (VEGF) in the rabbit cornea. New Zealand white rabbits were exposed to either dilution air (control; left eye) or LEW (0.2 mg/L; right eye) vapor for either 2.5 or 7.5 min. The rabbits were euthanized; the corneal tissue was dissected from eye at days 1, 3, 7, 14, and 28 post-LEW exposure and either fixed for IHC evaluation or frozen for cytokine array analysis. The corneal sections were IHC stained and the VEGF expression was quantified as detailed under the “Materials and Methods” section. Representative images showing an increase in VEGF expression following LEW exposure (A) and bar diagram showing the VEGF positivity score. Data presented are mean ± SEM (n = 3–5). *P < 0.05 compared with the control group; e, epithelium; s, stroma; size bar in representative images, 50 µm.
DISCUSSION
LEW, the most devastating agent amongst the organoarsenical vesicant warfare agents, is a potential threat of warfare, terrorism, and accidental exposure due to ease in manufacture, possible stockpiles, and quick onset of action (Li et al., 2016a; Noort et al., 2002). Eye is the most sensitive organ to vesicant exposure and LEW-induced ocular damage is instant which can result in massive necrosis and eventual blindness. However, LEW-induced clinical and pathologic ocular changes and biomarkers for mechanistic and efficacy studies are not well-characterized (Hughes, 1946). This impediment is mainly attributed to the development of antidote BAL, which shows some efficacy against LEW-induced tissue injury; however, its use has significant drawbacks due to its inherent toxicity, limited treatment window, and difficult administration (Boyd et al., 1989; Hughes, 1946; Mouret et al., 2013; Sahu et al., 2013). Hence, to identify effective targeted therapies that can be easily delivered to counter LEW-induced ocular injury, study of its molecular pathogenesis to establish relevant biomarkers is necessary. Our efforts in this direction provide a detailed assessment of histopathological and molecular changes related to our earlier reported clinical effects of LEW exposure in the rabbit cornea, which is the most susceptible ocular tissue to vesicant LEW exposure (Tewari-Singh et al., 2016). Additionally, these biologically relevant injury biomarkers are established in a rabbit ocular injury model relevant to human exposure (Anumolu et al., 2010; Kadar et al., 2001; Mann et al., 1947; Milhorn et al., 2010). Our follow-up study outcomes presented here suggest a role of COX-2, IL-8, MMP-9, and VEGF in LEW-induced corneal inflammation, vesication, and neovascularization. We believe that this first time detailed pathophysiological analysis following LEW ocular exposure will be valuable for screening and identifying therapies against LEW-induced corneal injuries.
The histopathological changes and the molecular events observed in LEW-induced corneal injury related to inflammation, vesication, and neovascularization mostly paralleled another of our recently completed studies with mustard vesicating agent SM in the rabbit in vivo corneal injury model (unpublished) and with SM analog nitrogen mustard (NM) in ex vivo rabbit corneal injury model (Goswami et al., 2016). These studies indicate that the pathophysiology of mustard vesicating agents-induced corneal injury is similar to LEW-induced ocular injury, though severity of damage and early occurrence of symptom following LEW exposure might differ. These outcomes also corroborate with the reports on clinical lesions from LEW exposure (Lindsay et al., 2004; Mann et al., 1946; Tewari-Singh et al., 2016) and supported by an earlier reported study where LEW, SM, and NM exposures induced common histologic events (Adler et al., 1947). Furthermore, increases in corneal edema, epithelial and stromal cell death, and infiltration of inflammatory cells, epithelial-degradation/denudation, and epithelial-stromal separation have been observed following SM exposure in rabbit corneas, which is in conjunction with our observations in this study following LEW exposure in the rabbit corneas starting at day 1 postexposure (Amir et al., 2000; Gordon et al., 2010; Milhorn et al., 2010; McNutt et al., 2012a,b). In addition, histopathological changes following LEW exposure in skin are reported to be similar to those observed with SM (Rice and Brown, 1999). Following LEW exposure, the maximal effect on different pathophysiological parameters was observed from days 3 to 14 post-exposure and most effects peaked at day 7 post-exposure. Because LEW exposure caused epithelial degradation, the observed LEW-induced increase in corneal thickness is resulting from the induced stromal thickness. Corneal thickness depends on the hydration state of the cornea. The primary factors that affect corneal hydration/corneal oedema are stromal swelling pressure, barrier function of the epithelium and endothelium, endothelial pump, tear evaporation, and intraocular pressure. A lot of mechanical, dystrophic, inflammatory, and toxic causes can result in failure of the cornea and could result in increased corneal thickness (Burcham et al., 2012). The increase in corneal thickness observed with LEW exposure could be due to the loss of barrier epithelium function and influx of inflammatory cells. In the absence of corneal epithelium, the stromal thickness could increase within hours of contact with tears and toxic substances (Burcham et al., 2012).
COX-2 is reported to play an important role in mustard vesicating agents- and LEW-mediated skin injury. COX-2 is an enzyme involved in prostaglandin biosynthesis, which in turn causes the influx of inflammatory cells at the site of the damage (Kehe et al., 2008; Kumar et al., 2015; Li et al., 2016b; Paromov et al., 2007; Shakarjian et al., 2010; Wormser et al., 2004). Our completed studies have reported the contribution of COX-2 in mediating vesicant-induced inflammatory responses in ex vivo rabbit cornea (Goswami et al., 2016; Tewari-Singh et al., 2012). This study further supports the role of COX-2 in arsenical vesicating agent LEW-induced corneal inflammation. Several cytokines including interleukin levels have been shown to be involved in LEW-induced skin inflammation and SM-induced ocular injury (Ghasemi et al., 2009; Nguon et al., 2014; Ruff et al., 2013). In this study, an increase in IL-8 levels was observed following LEW exposure indicating its role in LEW-induced corneal injury. Matrix metalloproteinases are regulators of inflammatory and immune responses and play a significant role in degradation of extracellular matrix causing vesication and cell death (Shakarjian et al., 2010). MMP-9 is reported to be a key mediator in SM- and LEW-induced skin vesication and SM-induced epithelial-stromal separation (Ghasemi et al., 2009; Gordon et al., 2010; Horwitz et al., 2014; Kehe et al., 2008; Kumar et al., 2015; Nguon et al., 2014; Ruff et al., 2013; Shakarjian et al., 2006, 2010; Tewari-Singh and Agarwal, 2016). This study also suggests that MMP-9 plays a key role in LEW-induced epithelial-stromal separation and inflammation in the cornea. Neovascularization, a major lesion resulting from vesicant exposure, is a late event and angiogenic factor VEGF is reported to promote corneal neovascularization in animal eyes exposed to SM (Gordon et al., 2010; Kadar et al., 2001; McNutt et al., 2012a; Petrali et al., 2000; Safarinejad et al., 2001). LEW exposure also resulted in visible neovascularization at day 7 post-exposure which was persistent till day 28 post-exposure (Tewari-Singh et al., 2016), and results hereby indicate that it is associated with increased VEGF levels that peaked at day 14 post-exposure. The increase in VEGF levels might be playing a part in repair mechanisms in vesicant-induced corneal injury by increasing the limbal vessel permeability and attracting monocytes in addition to inducing angiogenesis (Kadar et al., 2014; Philipp et al., 2000).
The outcomes from this study are highly significant because although the clinical ocular lesions from LEW exposure appear to be comparable to SM, their pathology and progress could be altered due to additional arsenic poisoning and inhibition of carbohydrate metabolism which are reported to be involved in LEW injury (Kehe et al., 2001; Mouret et al., 2013; Nelson et al., 2006). Following LEW exposure, unfolded protein response, inflammatory response, apoptosis, and changes in inflammatory cytokines such as Tumor Necrosis Factor-alpha (TNF-α) are also reported to play a significant role in its skin effects (Arroyo et al., 2004; Li et al., 2016b). The ability of LEW to cause toxic effects is reported to occur due to its ability to combine with thiol groups, react with biological sulfhydryl groups, release of hydrochloric acid, nonalkylating properties could have additional mechanisms of altered histopathology (Goldman and Dacre, 1989; Lindsay et al., 2004). Arsenic also binds to lipoic acid, a dithiol 8-carbon component of the pyruvate dehydrogenase complex, resulting in accumulation of pyruvate. This results in the inhibition of glycolysis. Some other enzymes affected include amylases, lipases, cholinesterase, and some ATP enzymes. In addition, reduction in cellular glutathione levels, oxidative stress, and lipid peroxidation also contributes to LEW-induced injury (Augerson, 2000). Hence, further “omics” and systems analysis approaches could be helpful in delineating the mechanisms and pathways associated with LEW-induced corneal injury to assist in developing targeted therapies.
Although there are no antidotes available for SM-induced injuries, an effective antidote, BAL, is available against LEW-induced injury since World War II (Mouret et al., 2013). BAL binds to arsenicals and leads to complete recovery if given immediately after exposure suggesting heavy-metal induced toxicity playing a key role in LEW-induced injury. However, BAL is relatively toxic and has low water solubility. Less toxic water soluble analogs, such as meso-2,3-dimercaptosuccinic acid (DMSA) and 2,3-dimercaptopropane sulfonic acid (DMPS), have been developed over the years. These alternatives have lesser toxicity and higher therapeutic index (Kosnett, 2013; Mouret et al., 2013).The antidotes (DMSA and DMPS) reverses the inhibitory effect of arsenite on pyruvate dehydrogenase. However, there are still limitations in the use of these agents due to their toxicity, narrow therapeutic window, and difficulty in administration (Boyd et al., 1989; Mouret et al., 2013).Therefore, to develop safe, mechanism-driven antidotes for LEW-induced ocular injury, we have developed a relevant in vivo ocular injury model to understand the injury mechanism and to screen and identify effective therapies.
FUNDING
This work was supported by the Countermeasures Against Chemical Threats (CounterACT). Program, Office of the Director National Institutes of Health (NIH) and the National Eye Institute (Grant Number U01EY023143).
REFERENCES
- Adler F. H., Fry W. E., Leopold I. H. (1947). Pathologic study of ocular lesions due to lewisite (B-chlorovinyldichloroarsine) changes with and without BAL (2,3-dimercaptopropanol) therapy. Arch. Ophthal. 38, 89–108. [DOI] [PubMed] [Google Scholar]
- Amir A., Turetz J., Chapman S., Fishbeine E., Meshulam J., Sahar R., Liani H., Gilat E., Frishman G., Kadar T. (2000). Beneficial effects of topical anti-inflammatory drugs against sulfur mustard-induced ocular lesions in rabbits. J. Appl. Toxicol. 20(Suppl. 1), S109–S114. [DOI] [PubMed] [Google Scholar]
- Anumolu S. S., DeSantis A. S., Menjoge A. R., Hahn R. A., Beloni J. A., Gordon M. K., Sinko P. J. (2010). Doxycycline loaded poly(ethylene glycol) hydrogels for healing vesicant-induced ocular wounds. Biomaterials 31, 964–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo C. M., Burman D. L., Kahler D. W., Nelson M. R., Corun C. M., Guzman J. J., Smith M. A., Purcell E. D., Hackley B. E. Jr, Soni S. D., et al. (2004). TNF-alpha expression patterns as potential molecular biomarker for human skin cells exposed to vesicant chemical warfare agents: Sulfur mustard (HD) and lewisite (L). Cell Biol. Toxicol. 20, 345–359. [DOI] [PubMed] [Google Scholar]
- Augerson W. (2000). A Review of the Scientific Literature as It Pertains to Gulf War Illnesses: Volume 5: Chemical and Biological Warfare Agents. Skin Damaging Agents 15–52. RAND Corporation, Santa Monica, CA. [Google Scholar]
- Beebe G. W. (1960). Lung cancer in World War I veterans: Possible relation to mustard-gas injury and 1918 influenza epidemic. J. Natl. Cancer Inst. 25, 1231–1252. [PubMed] [Google Scholar]
- Boyd V. L., Harbell J. W., O’Connor R. J., McGown E. L. (1989). 2,3-Dithioerythritol, a possible new arsenic antidote. Chem. Res. Toxicol. 2, 301–306. [DOI] [PubMed] [Google Scholar]
- Burcham P. C., Raso A., Kaminskas L. M. (2012). Chaperone heat shock protein 90 mobilization and hydralazine cytoprotection against acrolein-induced carbonyl stress. Mol. Pharmacol. 82, 876–886. [DOI] [PubMed] [Google Scholar]
- Ghasemi H., Ghazanfari T., Yaraee R., Ghassemi-Broumand M., Soroush M. R., Pourfarzam S., Masdari Z., Faghihzadeh S., Babaei M., Javadi M. A., et al. (2009). Evaluation of relationship between the serum levels of inflammatory mediators and ocular injuries induced by sulfur mustard: Sardasht-Iran cohort study. Int. Immunopharmacol. 9, 1494–1498. [DOI] [PubMed] [Google Scholar]
- Goldman M., Dacre J. C. (1989). Lewisite: Its chemistry, toxicology, and biological effects. Rev. Environ. Contam. Toxicol. 110, 75–115. [DOI] [PubMed] [Google Scholar]
- Gordon M. K., Desantis A., Deshmukh M., Lacey C. J., Hahn R. A., Beloni J., Anumolu S. S., Schlager J. J., Gallo M. A., Gerecke D. R., et al. (2010). Doxycycline hydrogels as a potential therapy for ocular vesicant injury. J. Ocul. Pharmacol. Ther. 26, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami D. G., Tewari-Singh N., Dhar D., Kumar D., Agarwal C., Ammar D. A., Kant R., Enzenauer R. W., Petrash J. M., Agarwal R. (2016). Nitrogen mustard-induced corneal injury involves DNA damage and pathways related to inflammation, epithelial-stromal separation, and neovascularization. Cornea 35, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz V., Dachir S., Cohen M., Gutman H., Cohen L., Fishbine E., Brandeis R., Turetz J., Amir A., Gore A., et al. (2014). The beneficial effects of doxycycline, an inhibitor of matrix metalloproteinases, on sulfur mustard-induced ocular pathologies depend on the injury stage. Curr. Eye Res. 39, 803–812. [DOI] [PubMed] [Google Scholar]
- Hotta N. (1997). New concepts and insights on pathogenesis and treatment of diabetic complications: Polyol pathway and its inhibition. Nagoya J. Med. Sci. 60, 89–100. [PubMed] [Google Scholar]
- Hughes W. F., Jr. (1946). Clinical uses of 2,3-dimercaptopropanol (BAL); the treatment of lewisite burns of the eye with BAL. J. Clin. Invest. 25, 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes W. F., Jr. (1947). Treatment of lewisite burns of the eye with dimercaprol (BAL). Arch. Ophthal. 37, 25–41. [DOI] [PubMed] [Google Scholar]
- Kadar T., Amir A., Cohen L., Cohen M., Sahar R., Gutman H., Horwitz V., Dachir S. (2014). Anti-VEGF therapy (bevacizumab) for sulfur mustard-induced corneal neovascularization associated with delayed limbal stem cell deficiency in rabbits. Curr. Eye Res. 39, 439–450. [DOI] [PubMed] [Google Scholar]
- Kadar T., Turetz J., Fishbine E., Sahar R., Chapman S., Amir A. (2001). Characterization of acute and delayed ocular lesions induced by sulfur mustard in rabbits. Curr. Eye Res. 22, 42–53. [DOI] [PubMed] [Google Scholar]
- Kehe K., Balszuweit F., Emmler J., Kreppel H., Jochum M., Thiermann H. (2008). Sulfur mustard research-strategies for the development of improved medical therapy. Eplasty 8, e32.. [PMC free article] [PubMed] [Google Scholar]
- Kehe K., Flohe S., Krebs G., Kreppel H., Reichl F. X., Liebl B., Szinicz L. (2001). Effects of lewisite on cell membrane integrity and energy metabolism in human keratinocytes and SCL II cells. Toxicology 163, 137–144. [DOI] [PubMed] [Google Scholar]
- King J. R., Riviere J. E., Monteiro-Riviere N. A. (1992). Characterization of lewisite toxicity in isolated perfused skin. Toxicol. Appl. Pharmacol. 116, 189–201. [DOI] [PubMed] [Google Scholar]
- Kosnett M. J. (2013). The role of chelation in the treatment of arsenic and mercury poisoning. J. Med. Toxicol. 9, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D., Tewari-Singh N., Agarwal C., Jain A. K., Inturi S., Kant R., White C. W., Agarwal R. (2015). Nitrogen mustard exposure of murine skin induces DNA damage, oxidative stress and activation of MAPK/Akt-AP1 pathway leading to induction of inflammatory and proteolytic mediators. Toxicol. Lett. 235, 161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Srivastava R. K., Athar M. (2016a). Biological and environmental hazards associated with exposure to chemical warfare agents: Arsenicals. Ann. N. Y. Acad. Sci. 1378, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Srivastava R. K., Weng Z., Croutch C. R., Agarwal A., Elmets C. A., Afaq F., Athar M. (2016b). Molecular mechanism underlying pathogenesis of lewisite-induced cutaneous blistering and inflammation: Chemical chaperones as potential novel antidotes. Am. J. Pathol. 186, 2637–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay C. D., Hambrook J. L., Brown R. F., Platt J. C., Knight R., Rice P. (2004). Examination of changes in connective tissue macromolecular components of large white pig skin following application of lewisite vapour. J. Appl. Toxicol. 24, 37–46. [DOI] [PubMed] [Google Scholar]
- Mann I., Pirie A., Pullinger B. D. (1946). A study of lewisite lesions of the eyes of rabbits. Am. J. Ophthalmol. 29, 1215–1227. [DOI] [PubMed] [Google Scholar]
- Mann I., Pirie A., Pullinger B. D. (1947). The treatment of lewisite and other arsenical vesicant lesions of the eyes of rabbits with British anti-lewisite (BAL). Am. J. Ophthalmol. 30, 421–435. [DOI] [PubMed] [Google Scholar]
- McNutt P., Hamilton T., Nelson M., Adkins A., Swartz A., Lawrence R., Milhorn D. (2012a). Pathogenesis of acute and delayed corneal lesions after ocular exposure to sulfur mustard vapor. Cornea 31, 280–290. [DOI] [PubMed] [Google Scholar]
- McNutt P., Lyman M., Swartz A., Tuznik K., Kniffin D., Whitten K., Milhorn D., Hamilton T. (2012b). Architectural and biochemical expressions of mustard gas keratopathy: Preclinical indicators and pathogenic mechanisms. PLoS One 7, e42837.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milhorn D., Hamilton T., Nelson M., McNutt P. (2010). Progression of ocular sulfur mustard injury: Development of a model system. Ann. N. Y. Acad. Sci. 1194, 72–80. [DOI] [PubMed] [Google Scholar]
- Mouret S., Wartelle J., Emorine S., Bertoni M., Nguon N., Clery-Barraud C., Dorandeu F., Boudry I. (2013). Topical efficacy of dimercapto-chelating agents against lewisite-induced skin lesions in SKH-1 hairless mice. Toxicol. Appl. Pharmacol. 272, 291–298. [DOI] [PubMed] [Google Scholar]
- Nelson P., Hancock J. R., Sawyer T. W. (2006). Therapeutic effects of hypothermia on lewisite toxicity. Toxicology 222, 8–16. [DOI] [PubMed] [Google Scholar]
- Nguon N., Clery-Barraud C., Vallet V., Elbakdouri N., Wartelle J., Mouret S., Bertoni M., Dorandeu F., Boudry I. (2014). Time course of lewisite-induced skin lesions and inflammatory response in the SKH-1 hairless mouse model. Wound Repair Regen. 22, 272–280. [DOI] [PubMed] [Google Scholar]
- Noort D., Benschop H. P., Black R. M. (2002). Biomonitoring of exposure to chemical warfare agents: A review. Toxicol. Appl. Pharmacol. 184, 116–126. [PubMed] [Google Scholar]
- Olajos E. J., Olson C. T., Salem H., Singer A. W., Hayes T. L., Menton R. G., Miller T. L., Rosso T., MacIver B. (1998). Evaluation of neutralized chemical agent identification sets (CAIS) for skin injury with an overview of the vesicant potential of agent degradation products. J. Appl. Toxicol. 18, 409–420. [DOI] [PubMed] [Google Scholar]
- Paromov V., Suntres Z., Smith M., Stone W. L. (2007). Sulfur mustard toxicity following dermal exposure: Role of oxidative stress, and antioxidant therapy. J. Burns Wounds 7, e7.. [PMC free article] [PubMed] [Google Scholar]
- Petrali J. P., Dick E. J., Brozetti J. J., Hamilton T. A., Finger A. V. (2000). Acute ocular effects of mustard gas: Ultrastructural pathology and immunohistopathology of exposed rabbit cornea. J. Appl. Toxicol. 20(Suppl. 1), S173–S175. [DOI] [PubMed] [Google Scholar]
- Philipp W., Speicher L., Humpel C. (2000). Expression of vascular endothelial growth factor and its receptors in inflamed and vascularized human corneas. Invest. Ophthalmol. Vis. Sci. 41, 2514–2522. [PubMed] [Google Scholar]
- Rice P., Brown R. F. (1999). The development of lewisite vapour induced lesions in the domestic, white pig. Int. J. Exp. Pathol. 80, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff A. L., Jarecke A. J., Hilber D. J., Rothwell C. C., Beach S. L., Dillman J. F. III. (2013). Development of a mouse model for sulfur mustard-induced ocular injury and long-term clinical analysis of injury progression. Cutan. Ocul. Toxicol. 32, 140–149. [DOI] [PubMed] [Google Scholar]
- Safarinejad M. R., Moosavi B. G. S. A., Montazeri B. (2001). Ocular injuries caused by mustard gas: Diagnosis, treatment, and medical defense. Mil. Med. 166, 67–70. [PubMed] [Google Scholar]
- Sahu C., Pakhira S., Sen K., Das A. K. (2013). A computational study of detoxification of lewisite warfare agents by British anti-lewisite: Catalytic effects of water and ammonia on reaction mechanism and kinetics. J. Phys. Chem. A 117, 3496–3506. [DOI] [PubMed] [Google Scholar]
- Shakarjian M. P., Bhatt P., Gordon M. K., Chang Y. C., Casbohm S. L., Rudge T. L., Kiser R. C., Sabourin C. L., Casillas R. P., Ohman-Strickland P., et al. (2006). Preferential expression of matrix metalloproteinase-9 in mouse skin after sulfur mustard exposure. J. Appl. Toxicol. 26, 239–246. [DOI] [PubMed] [Google Scholar]
- Shakarjian M. P., Heck D. E., Gray J. P., Sinko P. J., Gordon M. K., Casillas R. P., Heindel N. D., Gerecke D. R., Laskin D. L., Laskin J. D. (2010). Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol. Sci. 114, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari-Singh N., Agarwal R. (2016). Mustard vesicating agent-induced toxicity in the skin tissue and silibinin as a potential countermeasure. Ann. N. Y. Acad. Sci. 1374, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari-Singh N., Croutch C. R., Tuttle R., Goswami D. G., Kant R., Peters E., Culley T., Ammar D. A., Enzenauer R. W., Petrash J. M., et al. (2016). Clinical progression of ocular injury following arsenical vesicant lewisite exposure. Cutan. Ocul. Toxicol. 35, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari-Singh N., Jain A. K., Inturi S., Ammar D. A., Agarwal C., Tyagi P., Kompella U. B., Enzenauer R. W., Petrash J. M., Agarwal R. (2012). Silibinin, dexamethasone, and doxycycline as potential therapeutic agents for treating vesicant-inflicted ocular injuries. Toxicol. Appl. Pharmacol. 264, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilensky J. A., Redman K. (2003). British anti-lewisite (dimercaprol): An amazing history. Ann. Emerg. Med. 41, 378–383. [DOI] [PubMed] [Google Scholar]
- Wormser U., Langenbach R., Peddada S., Sintov A., Brodsky B., Nyska A. (2004). Reduced sulfur mustard-induced skin toxicity in cyclooxygenase-2 knockout and celecoxib-treated mice. Toxicol. Appl. Pharmacol. 200, 40–47. [DOI] [PubMed] [Google Scholar]